

Abstract
Cells of the developing central nervous system are particularly susceptible to formation of double-stranded DNA breaks (DSBs) arising from physiological and/or environmental insults. Therefore, efficient repair of DSBs is especially vital for maintaining cellular health and proper functioning in the developing brain. Here, increased expression of DSB initiating and nonhomologous end joining repair machinery in newborn neurons in the developing brains of both mouse and human are demonstrated. In parallel, the first characterization is provided of the brain phenotype in the Lig4R278H/R278H (Lig4R/R) mouse model of DNA Ligase 4 (LIG4) syndrome, in which a hypomorphic Lig4 mutation, originally identified in patients, impedes nonhomologous end joining. It is shown that Lig4R/R mice develop nonprogressive microcephaly, resulting primarily from apoptotic death of newborn neurons that is both spatially and temporally specific during peak cortical neurogenesis. This apoptosis leads to a reduction in neurons throughout the postnatal cerebral cortex, but with a more prominent impact on those of the lower cortical layers. Together, these findings begin to uncover the pathogenesis of microcephaly in LIG4 syndrome and open avenues to more focused investigations on the critical roles of DSB formation and repair in vulnerable neuronal populations of the brain.
Maintenance of genomic integrity is essential for normal cellular function, but it is of particular importance in the developing brain. On the one hand, when DNA damage goes unrepaired, the central nervous system is disproportionately affected in comparison to other organ systems.1,2 On the other hand, recent evidence suggests that double-strand break (DSB) formation is also a physiological event in both neural progenitors3,4 and postmitotic neurons,5,6 potentially promoting neuronal diversity and altering gene expression.1,7 Ultimately, an inability to repair these DSBs is detrimental, resulting in p53-mediated, apoptotic cell death.8,9 Together, these findings underscore the delicate balance that cells within the central nervous system must strike between DNA plasticity and integrity to maintain cellular heath and function.
Ligase 4 (LIG4) syndrome, a condition in which reduced LIG4 activity impedes nonhomologous end joining (NHEJ)-mediated DSB repair, serves as a devastating example of the consequences of unrepaired DNA damage.10,11 Along with immunodeficiency, microcephaly is a hallmark feature of LIG4 syndrome in patients.12,13 Phenotypic variability of the disease has been reported in patients, and it may reflect differences in the severity of the underlying gene defect.14 The nonprogressive nature of microcephaly in LIG4 syndrome suggests that the pathology arises during neural development, but the mechanisms by which microcephaly arises and the associated neurodevelopmental defects remain understudied. Systemic Lig4−/− mice exhibit profound cell death and are embryonic lethal.9,15,16 To date, two additional mouse models of LIG4 syndrome have been published. The first of these, Lig4Y288C, was identified from a mutagenesis screen and shows the immunodeficiency, radiation sensitivity, and growth retardation features associated with LIG4 syndrome.17,18 The second, Lig4R278H, contains a knock-in of the hypomorphic mutation first described in a LIG4 syndrome patient, who manifested primarily with leukemia and increased cellular radiosensitivity and developmental delay, but no overt immunodeficiency.10,19,20 The Lig4R278H mutation occurs in the highly conserved active site of the LIG4 enzyme, significantly reducing its enzymatic activity.10,20 Lig4R278H/R278H (Lig4R/R) mice demonstrate many features of LIG4 syndrome.21 Here, the brain phenotype in the Lig4R/R knock-in mouse model is characterized, and a link between the mouse and human neural development is established. Moreover, the effects of the Lig4R278H mutation were compared with those previously reported for the Lig4Y288C variant18 on brain development. This work uncovers a prominent spatiotemporal gradient of cell death affecting a select population of newborn cortical neurons, ultimately leading to microcephaly, and identifies distinctive features that characterize brain pathology in different models of the disease. These findings open new avenues for investigating the pathophysiology of LIG4 syndrome, as well as other syndromes characterized by unrepaired DNA damage in the developing central nervous system.
Materials and Methods
MRI
Detailed clinical information for this case was previously described,22 where it was noted that the individual was homozygous for LIG4 mutation leading to a histidine to arginine amino acid substitution at position 282 (H282R) in the LIG4 active site. Magnetic resonance imaging (MRI) was reviewed by S.K. and I.R.
Animals
Lig4R278H/R278H mice (referred to as Lig4R/R) and littermate controls were obtained through heterozygous breedings.21 For embryonic mice, the day of plug was designated as embryonic day (E) 0.5. For postnatal mice, the day of birth was designated as postnatal day (P) 0. Wild-type (Lig4+/+), heterozygous (Lig4R/+), and knock-in (Lig4R/R) mice were collected from each litter. All analyses were performed using male and female mice. All animal experimentation was performed under protocols (13-08-2472R; 17-10-3547R) approved by the institutional animal care and use committee of Boston Children's Hospital.
Immunostaining
Paraffin sections (5 μm) of brains were dehydrated, and antigen retrieval was performed using Antigen Unmasking Solution (Vector Laboratories, Burlingame, CA). Sections were blocked and permeabilized (0.04% Tween-20 in PBS, 5% serum), followed by primary antibody incubation overnight at 4°C. Cryosections (embryonic samples at 5 μm or 14 μm, and postnatal samples at 14 μm) were washed in 1× PBS followed by antigen retrieval using boiling HistoVT One (Nicalai Tesque, Kyoto, Japan) or 100 mmol/L sodium citrate with 0.05% Tween-20. Samples were blocked and permeabilized (0.3% Triton X-100 in PBS, 5% serum), then incubated with primary antibodies overnight at 4°C (antibodies diluted in 0.3% Triton X-100 in PBS, 5% serum). After washing, slides were incubated with fluorescent secondary antibodies (Alexa Fluor series; dilution 1:500; Invitrogen, Carlsbad, CA), counterstained with Hoechst 33258 (H3570; dilution 1:10,000; Fisher Scientific, Hampton, NH), and then mounted with Fluoromount-G (SouthernBiotech, Birmingham, AL). The following primary antibodies were used: anti-53BP1 (4937; dilution: 1:100; Cell Signaling Technology, Danvers, MA), anti–5-bromo-2′-deoxyuridine (BrdU) (MCA2060; dilution: 1:200; Bio-Rad, Hercules, CA), anti-cleaved caspase 3 (9661; dilution: 1:100; Cell Signaling Technology), anti-Ctip2 (ab18465; dilution: 1:200; Abcam, Cambridge, MA), anti-Cux1 (sc-13024; dilution: 1:200; Santa Cruz Biotechnology, Dallas, TX), anti-Ki67 (550609; dilution: 1:50; BD Biosciences, San Jose, CA), anti-Topoisomerase II β (TOP2B) (ab109524; dilution: 1:100; Abcam), and anti-Tuj1 (801202; dilution: 1:200; BioLegend, San Diego, CA). All image analyses were performed using FIJI (ImageJ software version 2.0.0-rc-54; NIH, Bethesda, MD) and statistical analyses were performed using Prism software version 7.04 (GraphPad Software, La Jolla, CA).
E14.5 BrdU Pulse
Timed-pregnant females at E13.5 were injected intraperitoneally with 50 mg/kg BrdU (Millipore Sigma, Burlington, MA) and sacrificed 24 hours later. Embryos were fixed in 4% paraformaldehyde in PBS for 2 hours at room temperature followed by sucrose gradient and embedding in OCT as described.23 Brains were sectioned at 5-μm thickness. Following immunostaining, FIJI (ImageJ version 2.0.0-rc-54) was used to perform cell counts and quantification from four consecutive sections from both hemispheres, and statistical analyses were performed using Prism software version 7.04 (GraphPad Software) comparing averaged counts from each brain.
Analysis of Human Single-Cell RNA-Sequencing Data
Data set was provided by Alex Pollen.24 Because these data were zero-inflated (many cells had zero counts per million reads for the genes analyzed), to perform statistical analyses, a two-part Wilcoxon test was used to test differences in gene expression25 as described.26 Statistical analyses were performed using the R statistical software RStudio version 1.0.143 (R Foundation for Statistical Computing, Vienna, Austria).
Results
Patients with LIG4 syndrome are reported to have small head circumference, though MRI images have never been reported. A brain MRI of a 3-month–old LIG4 syndrome patient, with a head circumference of 33 cm (normal range for an unaffected age-matched female is 36.8 to 41.7 cm) was obtained. The MRI confirmed microcephaly and revealed no gross malformations, atrophy, or tissue loss (Figure 1A). The sulci appeared relatively normal with no enlargements, and the gyral pattern was not unaltered.
Figure 1.

Newborn neurons are uniquely equipped with nonhomologous end joining (NHEJ) machinery during development. A: Transverse T2-weighted (left) and coronal fluid-attenuated inversion recovery (FLAIR) (right) brain MRIs of patient with homozygous LIG4 mutation leading to H282R substitution reveal microcephaly with no gross malformations. B: Schematic of NHEJ versus homologous recombination (HR) repair pathway choice, demonstrating interaction of TOP2B, γH2AX, 53BP1, BRCA1, and LIG4. C: Single-cell sequencing data of human radial glia (RG), intermediate progenitor cells (IPCs), and neurons at gestational weeks 16 to 1824 demonstrate that TOP2B expression is increased among neurons, whereas 53BP1 and BRCA1 expression is increased among progenitor cells. D: Expression of TOP2B (green) at E14.5 is enhanced in newborn neurons in the cortical plate of the embryonic mouse cortex. The boxed area corresponds to the higher-magnification image on the right. E: Expression of 53BP1 (red) is enhanced in the TUJ1+ (green) region that represents the cortical plate of the embryonic mouse cortex at E14.5. Asterisks indicate nonspecific labeling of vasculature. The boxed area corresponds to the higher-magnification image on the right. F:Brca1 RNA expression is enhanced among cortical progenitors in the ventricular and subventricular zones of the developing mouse cortex at E14.5. Image acquired from GenePaint (http://gp3.mpg.de, last accessed August 9, 2019) (set ID: MH2991). Data are expressed as means ± SEM (C). **P < 0.01, ****P < 0.0001 (two-part Wilcoxon with Bonferroni correction). Scale bars: 200 μm (D and E, left panels); 50 μm (D and E, right panels). CP, cortical plate; GW, gestational weeks; scSeq, single-cell sequencing; VZ, ventricular zone.
To gain more insight into LIG4 activity requirements during maturation of the developing cortex, spatial distribution in expression of machinery involved in DSB formation and NHEJ DNA repair in developing human brain cells was first examined (Figure 1B). Analysis of a single-cell RNA sequencing data set from the developing human brain at 16 to 18 weeks gestation24 demonstrated varying expression levels of DSB repair genes in progenitors and newborn neurons. For example, TOP2B, a protein responsible for regulating transcription through DSB induction in gene promoter regions,27, 28, 29 was expressed most highly in newborn neurons (Figure 1C). TP53-binding protein 1 (53BP1) plays a vital role in DSB repair pathway choice by binding to DNA damage sites and recruiting additional DNA repair proteins that block resection of the DNA ends, which ultimately promotes the NHEJ pathway while preventing homologous recombination (HR).30 Surprisingly, 53BP1 expression was slightly increased in radial glia compared with neurons, but expressed throughout the developing cortex (Figure 1C). However, BRCA1, which antagonizes 53BP1 at DSB sites to inhibit NHEJ and promote HR,30 also showed increased expression in radial glial progenitors in the developing human brain but was virtually absent from neurons (Figure 1C). No difference in expression levels of the core NHEJ machinery proteins (Ku70, Ku80, Artemis, DNA-PKcs) was observed between radial glia, intermediate progenitor cells, and neurons (data not shown). Together, these data support the model that although NHEJ-pathway genes are expressed throughout the cortex, newborn neurons simultaneously have reduced expression of genes important for HR, suggesting that these cells likely heavily rely on NHEJ for DNA repair.
During peak neurogenesis in the developing mouse cerebral cortex (E14.5), expression pattern and localization of the NHEJ-specific machinery recapitulated what was observed in the human cerebral cortex. Top2b was robustly expressed among neurons populating the developing cortical plate compared with progenitors along the ventricular zone (Figure 1D). 53bp1 expression was similarly concentrated among Tuj1-positive neurons (Figure 1E), whereas Brca1 expression was limited to progenitors in the ventricular and subventricular zones (Figure 1F). This increased expression of Brca1 in progenitor cells may be due to its role in promoting HR (Figure 1B), its role in regulation of cell cycle progression, particularly through S and G2 phases,31 or a combination of both. Taken together, these data suggest that newborn cortical neurons may incur Top2b-mediated endogenous DSBs, and regardless of timing of DSB formation, any DSBs present in postmitotic neurons are preferentially repaired via NHEJ, a biological process that is conserved across species and for which enzymatically active LIG4 is vital.
Homozygosity for the hypomorphic LIG4 R278H mutation in humans has been reported in a patient with leukemia, increased cellular radiosensitivity, and global developmental delay, but no obvious immunodeficiency.10,19,20 In a knock-in mouse for the homozygous Lig4 R278H mutation (Lig4R/R), the immune deficiency, radiosensitivity, and developmental delay have been carefully characterized,21,32 but the brain phenotype, an important component of this devastating syndrome, has remained uncharacterized. No differences in overall brain size were observed between Lig4R/R and wild-type mice at E14.5; however, brain size was markedly reduced by P8 (Figure 2A). Brain weight and body weight were also reduced by P8 in knock-in mice compared to wild-type and heterozygous littermates (Figure 2, B and C). Although P8 is a postnatal age in mice, it corresponds to approximately 36 to 40 gestational weeks in human brain development,33 suggesting that the microcephaly observed at P8 Lig4R/R mice pathologically recapitulates primary microcephaly in patients. Additionally, the decreased brain weight of knock-in mice at P8 was nonprogressive, as the brain weights of all three genotypes followed the same growth trajectory postnatally until P28 (Figure 2B). At P28, Lig4R/R brains were smaller than wild-type and heterozygous littermates (Figure 2A). P28 cerebral cortical length and hemispheric surface area were also reduced in Lig4R/R mice compared with littermate controls (Figure 2, D–F). However, both P8 and P28 heterozygous brains were not microcephalic compared with wild-type (Figure 2, B, D, and E), suggesting that a single allele provides sufficient Lig4 expression for relatively normal brain development. Overall, these observations parallel findings from LIG4 syndrome patients, of which 26 of 28 patients presented with primary, nonprogressive microcephaly,13 suggesting that the mechanisms underlying the phenotype are activated primarily during fetal development.
Figure 2.

Lig4R/R mice recapitulate microcephaly and growth delay observed in patients with LIG4 syndrome. A: E14.5, P8, and P28 brains from wild-type (Lig4+/+) and knock-in (Lig4R/R) mice. Dashed lines indicate the anterior and posterior ends of the cortex of the wild-type (Lig4+/+) mice at each age. At P8 and P28, the cortices of knock-in (Lig4R/R) mice show a marked decrease in size compared to the wild-type (Lig4+/+) littermate controls. B:Lig4R/R mice show decreased brain weight postnatally compared to wild-type (Lig4+/+) and heterozygous (Lig4R/+) littermates. C:Lig4R/R mice have reduced body weight at P28. D: Schematics demonstrate cortical length and area measurements taken from P28 gross brain samples. E: Cerebral cortical length in P28 Lig4R/R knock-in mice is reduced compared with wild-type (Lig4+/+) and heterozygous (Lig4R/+) littermates. F: Cerebral cortical hemispheric surface area in P28 Lig4R/R knock-in mice is reduced compared with heterozygous (Lig4R/+) and wild-type (Lig4+/+) littermates. Data are expressed as means ± SEM (B, C, E, and F). **P < 0.01, ***P < 0.001, and ****P < 0.0001 (two-way analysis of variance); †P < 0.05, ††P < 0.01 (one-way analysis of variance). Scale bars = 1 mm (A, E14.5 brains); 2 mm (A, P8 and P28 brains).
The microcephaly observed postnatally in mice as well as in patients may be ascribed to decreased proliferation, increased rate of differentiation, and/or increased cell death. Peak neurogenesis in the mouse cerebral cortex occurs at E14.5, when global Lig4−/− mice have been reported to undergo extensive cell death.9,15 Although gross comparison of E14.5 embryos and brains from knock-in mice revealed no differences in brain weight or body weight at E14.5 (Figure 2, B and C), evidence of activated pathological mechanisms at this early stage were still investigated. Analyses of cleaved caspase 3 (CC3) expression in E12.5, E14.5, and E16.5 cerebral cortices revealed a peak of cell death at E14.5 in Lig4R/R mice compared with littermate controls (Figure 3, A and B). No differences in cell death between Lig4R/R mice and controls were observed at E12.5, consistent with normal brain size early on in forebrain development. By contrast, some cell death persisted at E16.5 (Figure 3B). Many of the dying cells were positioned in the developing cortical plate, and some retained Tuj1 expression (Figure 3, C and D). Most remarkably, the cell death occurred in a sweeping medial-to-lateral gradient across the cerebral cortex, with more cell death occurring laterally (Figure 3, E and F). Together, these findings suggest both temporal and spatial differences in vulnerability to cell death with impaired Lig4 activity.
Figure 3.

Lig4R/R mutation leads to increased cell death in newborn neurons during cerebral cortical development, contributing to microcephaly phenotype. A and B: Cleaved caspase 3 (CC3, red) staining reveals increased cell death in Lig4R/R knock-in mice at E14.5. C and D: In the E14.5 dorsolateral cortex, increase in CC3+ (red) cells primarily occurs in Tuj1+ (green) neurons in the cortical plate. E and F: CC3 (red) staining reveals a medial-to-lateral gradient of cell death with the majority of CC3+ (red) cells in the lateral cortex. Cortex was divided into six evenly spaced bins for quantification in F. Data are expressed as means ± SEM. *P < 0.05, **P < 0.01 (unpaired, two-tailed t-test); †P < 0.05 (one-way analysis of variance). Scale bars: 50 μm (A); 100 μm (C and E). L, lateral; M, medial.
It was tested whether alterations in proliferation and cell cycle exit may also contribute to the smaller brain phenotype by pairing a 24-hour BrdU pulse with Ki-67 immunostaining (Figure 4A). Although no difference was observed in total BrdU+ cells (Figure 4B), quantification of the ratio of BrdU+/Ki-67+ to total BrdU+ cells suggested a modest increase in proliferative fraction in the E14.5 Lig4R/R dorsolateral cortex compared with wild-type littermate controls (Figure 4C). However, the cell death observed in newborn neurons in Lig4R/R mice (Figure 3), which would constitute the BrdU+Ki-67− population, may confound this type of analysis. Together, these results were interpreted to indicate that the primary cause of microcephaly observed in Lig4R/R mice is apoptotic cell death of newborn neurons.
Figure 4.

Lig4R/R mice maintain normal progenitor proliferation rate. A: BrdU (red) and Ki-67 (green) colabeling in E14.5 dorsolateral cortex following 24-hour BrdU pulse. B: No change is observed in total number of BrdU+ cells in the Lig4R/R knock-in cortex compared with wild-type (Lig4+/+). BrdU+ cells were counted in a 200-μm–wide region of the dorsolateral cortex. C: Quantification of proliferative fraction in the dorsolateral cortex reveals increased cell cycle re-entry of Lig4R/R neural progenitors. Proliferative fraction: BrdU+Ki-67+ cells/total Ki-67+ cells in 200-μm–wide region. Data are expressed as means ± SEM. *P < 0.05 (unpaired two-tailed t-test). Scale bar = 100 μm.
It was next tested whether the enhanced cell death observed in Lig4R/R mice during embryonic development altered later cortical lamination. Staining was performed for Cux1, which marks the upper cortical layers II to IV,34,35 and Ctip2, which labels subcortical projection neurons residing primarily in the deep cortical layers,36,37 at P8, when cortical lamination is complete. Cux1+ and Ctip2+ cells in the dorsolateral region of the anterior cortex were quantified by partitioning the cortex into six equally sized regions (bins), because cortical thickness may vary across individual sections and samples (Figure 5A). The percentages of Cux1+ cells in each bin were slightly, but not significantly, reduced in Lig4R/R mice compared with wild-type littermate controls, whereas percentages of Ctip2+ cells were decreased in Lig4R/R mice to a greater degree. A large reduction in total number of nuclei across all bins was also observed (n = 5 from 2 litters) (Figure 5, B–D), Together, these data indicate a reduction in the numbers of neurons populating all layers of the cerebral cortex, with neurons of the lower layers being more severely affected. No evidence of disrupted migration was observed. Together, our findings indicate that Lig4 activity is critical for maintaining cellular health, particularly in a select subset of newborn neurons, destined to populate the entire radial extent of the cerebral cortex.
Figure 5.
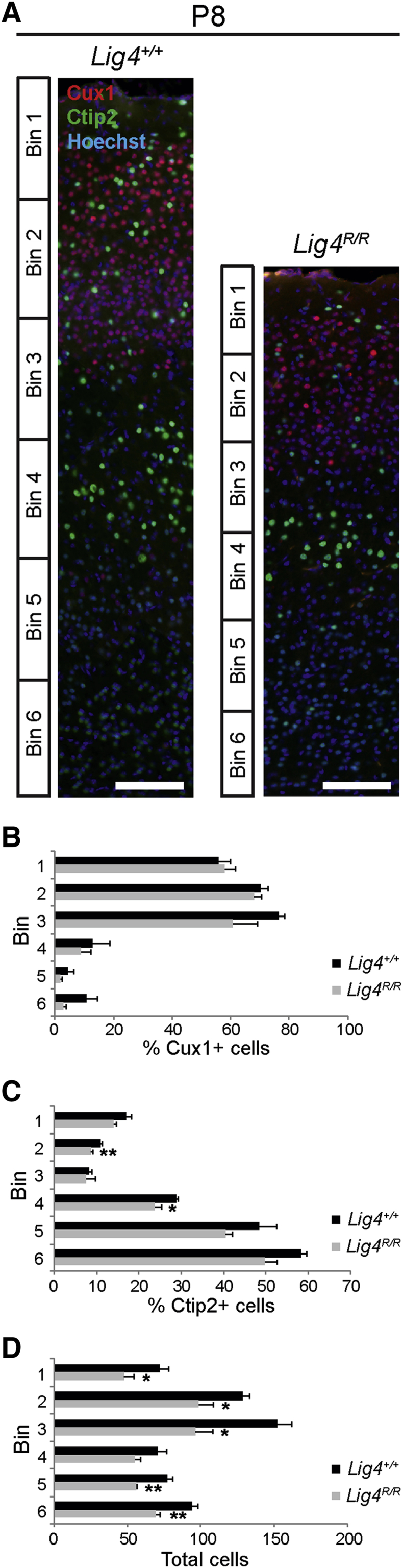
P8 Lig4R/R mice show reduction in cortical thickness postnatally with decreased cellularity but largely intact laminar architecture. A: Staining with Cux1 (upper layers, red) and Ctip2 (lower layers, green) at P8 reveal intact lamination of the anterior dorsolateral cortex but overall decrease in total number of cells across the entire thickness of the cortex in Lig4R/R mice. B: No significant reduction in percentage of Cux1+ cells is found in the anterior dorsolateral cortex of Lig4R/R mice at P8. C: Mild reduction in percentage of Ctip2+ cells was found in bins 2 and 4 of the anterior dorsolateral cortex of Lig4R/R mice at P8. D: Total cell number decreases throughout the dorsolateral cortex of Lig4R/R mice at P8. Cortex was divided into six evenly spaced bins for quantification in B–D. Data are expressed as means ± SEM. *P < 0.05, **P < 0.01 versus wild type control (unpaired two-tailed t-test). Scale bar = 100 μm.
Discussion
Our work demonstrates that the Lig4R/R knock-in mouse model recapitulates the primary, nonprogressive microcephaly observed in human LIG4 syndrome. The microcephaly can be attributed largely to a spatiotemporal gradient of apoptotic neuronal cell death during critical stages of cerebral cortical development. The selective vulnerability of neurons to NHEJ DNA repair deficiency is matched by expression levels of DNA repair machinery in the developing mouse and human brain, in which postmitotic neurons have increased expression of NHEJ machinery compared with proliferating radial glia and intermediate progenitor cells.
Apoptosis likely serves a protective mechanism that aids proper brain development by preventing neurons with persistent DNA damage from integrating into brain circuitry.1 In the developing neocortex, a marked increase in apoptosis was observed at E14.5, a time when layer IV pyramidal neurons are being born and deeper layer V and VI neurons are migrating to their final destinations in the neocortex.38 Compared with these findings, Lig4Y288C mice display a smaller brain, with thinner neural epithelium and increased apoptosis in the ventricular, subventricular zones and intermediate zones already at day E14.5 to E15.5,18 indicating an earlier and more severe impact of the p.Y288C Lig4 variant. On the other hand, Lig4R/R mice displayed increased 53BP1 expression in cortical neurons, and a similar gradient (from lower to higher expression in the ventricular, subventricular, intermediate zones and in the cortical plate) had been previously reported in Lig4Y288C mice,18 suggesting that in both strains, repair of DNA DSBs by NHEJ occurs predominantly in nonreplicating cells.
Recent work has shown that neurons of different layers differ in their activity patterns in response to stimulation, with neurons of deeper layers activating gene expression quickly but briefly.39 DSB generation may represent one mechanism by which neurons can induce expression of early response genes.5,6 Thus, it is likely that DSB formation may play a role in facilitating the ability of these deep-layer neurons to rapidly induce gene expression, which may selectively predispose this neuronal population to apoptotic cell death in the absence of DNA repair machinery. This hypothesis is consistent with our observation that, though Lig4R/R mice suffer loss of neurons throughout the cortex, the Ctip2+ deep-layer neurons were disproportionately affected (Figure 5C). Production of reactive oxygen species provides another interesting hypothesis for the particular susceptibility of Lig4-deficient deep-layer neurons to apoptosis. Reactive oxygen species are known to both induce DSBs40 and play physiological roles in neuronal differentiation, maturation, and plasticity.41 It remains to be determined whether neurons born at different times and destined for different cortical layers have varying metabolic demands that may result in the differential production of reactive oxygen species, ultimately leading to apoptosis in the absence of sufficient NHEJ.
Though an increase was observed in cell death in neurons destined for lower layers, many neurons in these layers nevertheless survive. Paired with the observed spatiotemporal gradient of cell death, these data raise intriguing questions regarding cell vulnerability. Both Lig4 splice variants contain the full coding region of the gene, including the portion manipulated in the Lig4R/R mouse, suggesting that differential expression of Lig4 isoforms is not likely the cause of the observed gradient of apoptosis across neurons of the developing cortex. However, many questions remain regarding both differential susceptibility to DNA damage under homeostatic conditions as well as the ability of surviving neurons to employ alternative, noncanonical DNA repair mechanisms that do not require Lig4, such as microhomology-mediated end joining.42 Future studies should focus on these selectively vulnerable neuronal populations to begin to unravel the molecular pathogenesis of Lig4 syndrome. High-throughput, genome-wide, translocation sequencing, a technique widely used in cancerous cells43,44 and recently used in cultured neural progenitor cells,3,4 may prove useful for identifying genomic locations of endogenous DSBs in vivo in the developing central nervous system.
Beyond genetic contributions to microcephaly, environmental triggers, including viral infections (eg, Zika and Dengue viruses), have suggested up-regulation of DNA repair pathway machinery.45,46 The Lig4R/R mouse may, therefore, serve as an ideal model for investigating and therapeutically combating microcephaly pathogenesis in a variety of contexts.
Acknowledgments
We thank members of the Lehtinen and Notarangelo laboratories; Alec Walker, Roberto Chiarle, and Taek-Chin Cheong for helpful discussions; Alex Pollen for sharing the human scSeq data set; and Rebeca Borges for sharing R code and statistical expertise.
Footnotes
Supported by the New York Stem Cell Foundation (M.K.L.); BCHIDDRC1U54HD090255; and the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, NIH (L.D.N.). M.K.L. is a New York Stem Cell Foundation–Robertson Investigator.
M.P.L. and M.L.S. contributed equally to this work.
Disclosures: None declared.
References
- 1.Alt F.W., Schwer B. DNA double-strand breaks as drivers of neural genomic change, function, and disease. DNA Repair (Amst) 2018;71:158–163. doi: 10.1016/j.dnarep.2018.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McKinnon P.J. Genome integrity and disease prevention in the nervous system. Genes Dev. 2017;31:1180–1194. doi: 10.1101/gad.301325.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwer B., Wei P.C., Chang A.N., Kao J., Du Z., Meyers R.M., Alt F.W. Transcription-associated processes cause DNA double-strand breaks and translocations in neural stem/progenitor cells. Proc Natl Acad Sci U S A. 2016;113:2258–2263. doi: 10.1073/pnas.1525564113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wei P.C., Chang A.N., Kao J., Du Z., Meyers R.M., Alt F.W., Schwer B. Long neural genes harbor recurrent DNA break clusters in neural stem/progenitor cells. Cell. 2016;164:644–655. doi: 10.1016/j.cell.2015.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Madabhushi R., Gao F., Pfenning A.R., Pan L., Yamakawa S., Seo J., Rueda R., Phan T.X., Yamakawa H., Pao P.C., Stott R.T., Gjoneska E., Nott A., Cho S., Kellis M., Tsai L.H. Activity-induced DNA breaks govern the expression of neuronal early-response genes. Cell. 2015;161:1592–1605. doi: 10.1016/j.cell.2015.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suberbielle E., Sanchez P.E., Kravitz A.V., Wang X., Ho K., Eilertson K., Devidze N., Kreitzer A.C., Mucke L. Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-beta. Nat Neurosci. 2013;16:613–621. doi: 10.1038/nn.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meisenberg C., Pinder S.I., Hopkins S.R., Wooller S.K., Benstead-Hume G., Pearl F.M.G., Jeggo P.A., Downs J.A. Repression of transcription at DNA breaks requires cohesin throughout interphase and prevents genome instability. Mol Cell. 2019;73:212–223.e7. doi: 10.1016/j.molcel.2018.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao Y., Sun Y., Frank K.M., Dikkes P., Fujiwara Y., Seidl K.J., Sekiguchi J.M., Rathbun G.A., Swat W., Wang J., Bronson R.T., Malynn B.A., Bryans M., Zhu C., Chaudhuri J., Davidson L., Ferrini R., Stamato T., Orkin S.H., Greenberg M.E., Alt F.W. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell. 1998;95:891–902. doi: 10.1016/s0092-8674(00)81714-6. [DOI] [PubMed] [Google Scholar]
- 9.Frank K.M., Sharpless N.E., Gao Y., Sekiguchi J.M., Ferguson D.O., Zhu C., Manis J.P., Horner J., DePinho R.A., Alt F.W. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol Cell. 2000;5:993–1002. doi: 10.1016/s1097-2765(00)80264-6. [DOI] [PubMed] [Google Scholar]
- 10.O'Driscoll M., Cerosaletti K.M., Girard P.M., Dai Y., Stumm M., Kysela B., Hirsch B., Gennery A., Palmer S.E., Seidel J., Gatti R.A., Varon R., Oettinger M.A., Neitzel H., Jeggo P.A., Concannon P. DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol Cell. 2001;8:1175–1185. doi: 10.1016/s1097-2765(01)00408-7. [DOI] [PubMed] [Google Scholar]
- 11.Girard P.M., Kysela B., Harer C.J., Doherty A.J., Jeggo P.A. Analysis of DNA ligase IV mutations found in LIG4 syndrome patients: the impact of two linked polymorphisms. Hum Mol Genet. 2004;13:2369–2376. doi: 10.1093/hmg/ddh274. [DOI] [PubMed] [Google Scholar]
- 12.Chistiakov D.A., Voronova N.V., Chistiakov A.P. Ligase IV syndrome. Eur J Med Genet. 2009;52:373–378. doi: 10.1016/j.ejmg.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 13.Altmann T., Gennery A.R. DNA ligase IV syndrome; a review. Orphanet J Rare Dis. 2016;11:137. doi: 10.1186/s13023-016-0520-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Felgentreff K., Baxi S.N., Lee Y.N., Dobbs K., Henderson L.A., Csomos K., Tsitsikov E.N., Armanios M., Walter J.E., Notarangelo L.D. Ligase-4 deficiency causes distinctive immune abnormalities in asymptomatic individuals. J Clin Immunol. 2016;36:341–353. doi: 10.1007/s10875-016-0266-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barnes D.E., Stamp G., Rosewell I., Denzel A., Lindahl T. Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Curr Biol. 1998;8:1395–1398. doi: 10.1016/s0960-9822(98)00021-9. [DOI] [PubMed] [Google Scholar]
- 16.Frank K.M., Sekiguchi J.M., Seidl K.J., Swat W., Rathbun G.A., Cheng H.L., Davidson L., Kangaloo L., Alt F.W. Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature. 1998;396:173–177. doi: 10.1038/24172. [DOI] [PubMed] [Google Scholar]
- 17.Nijnik A., Woodbine L., Marchetti C., Dawson S., Lambe T., Liu C., Rodrigues N.P., Crockford T.L., Cabuy E., Vindigni A., Enver T., Bell J.I., Slijepcevic P., Goodnow C.C., Jeggo P.A., Cornall R.J. DNA repair is limiting for haematopoietic stem cells during ageing. Nature. 2007;447:686–690. doi: 10.1038/nature05875. [DOI] [PubMed] [Google Scholar]
- 18.Gatz S.A., Ju L., Gruber R., Hoffmann E., Carr A.M., Wang Z.Q., Liu C., Jeggo P.A. Requirement for DNA ligase IV during embryonic neuronal development. J Neurosci. 2011;31:10088–10100. doi: 10.1523/JNEUROSCI.1324-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plowman P.N., Bridges B.A., Arlett C.F., Hinney A., Kingston J.E. An instance of clinical radiation morbidity and cellular radiosensitivity, not associated with ataxia-telangiectasia. Br J Radiol. 1990;63:624–628. doi: 10.1259/0007-1285-63-752-624. [DOI] [PubMed] [Google Scholar]
- 20.Riballo E., Critchlow S.E., Teo S.H., Doherty A.J., Priestley A., Broughton B., Kysela B., Beamish H., Plowman N., Arlett C.F., Lehmann A.R., Jackson S.P., Jeggo P.A. Identification of a defect in DNA ligase IV in a radiosensitive leukaemia patient. Curr Biol. 1999;9:699–702. doi: 10.1016/s0960-9822(99)80311-x. [DOI] [PubMed] [Google Scholar]
- 21.Rucci F., Notarangelo L.D., Fazeli A., Patrizi L., Hickernell T., Paganini T., Coakley K.M., Detre C., Keszei M., Walter J.E., Feldman L., Cheng H.L., Poliani P.L., Wang J.H., Balter B.B., Recher M., Andersson E.M., Zha S., Giliani S., Terhorst C., Alt F.W., Yan C.T. Homozygous DNA ligase IV R278H mutation in mice leads to leaky SCID and represents a model for human LIG4 syndrome. Proc Natl Acad Sci U S A. 2010;107:3024–3029. doi: 10.1073/pnas.0914865107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.IJspeert H., Warris A., van der Flier M., Reisli I., Keles S., Chishimba S., van Dongen J.J., van Gent D.C., van der Burg M. Clinical spectrum of LIG4 deficiency is broadened with severe dysmaturity, primordial dwarfism, and neurological abnormalities. Hum Mutat. 2013;34:1611–1614. doi: 10.1002/humu.22436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chau K.F., Shannon M.L., Fame R.M., Fonseca E., Mullan H., Johnson M.B., Sendamarai A.K., Springel M.W., Laurent B., Lehtinen M.K. Downregulation of ribosome biogenesis during early forebrain development. Elife. 2018;7:e36998. doi: 10.7554/eLife.36998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pollen A.A., Nowakowski T.J., Chen J., Retallack H., Sandoval-Espinosa C., Nicholas C.R., Shuga J., Liu S.J., Oldham M.C., Diaz A., Lim D.A., Leyrat A.A., West J.A., Kriegstein A.R. Molecular identity of human outer radial glia during cortical development. Cell. 2015;163:55–67. doi: 10.1016/j.cell.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taylor S., Pollard K. Hypothesis tests for point-mass mixture data with application to 'omics data with many zero values. Stat Appl Genet Mol Biol. 2009;8 doi: 10.2202/1544-6115.1425. Article 8. [DOI] [PubMed] [Google Scholar]
- 26.Smith R.S., Kenny C.J., Ganesh V., Jang A., Borges-Monroy R., Partlow J.N., Hill R.S., Shin T., Chen A.Y., Doan R.N., Anttonen A.K., Ignatius J., Medne L., Bönnemann C.G., Hecht J.L., Salonen O., Barkovich A.J., Poduri A., Wilke M., de Wit M.C.Y., Mancini G.M.S., Sztriha L., Im K., Amrom D., Andermann E., Paetau R., Lehesjoki A.E., Walsh C.A., Lehtinen M.K. Sodium channel SCN3A (NaV1.3) regulation of human cerebral cortical folding and oral motor development. Neuron. 2018;99:905–913.e7. doi: 10.1016/j.neuron.2018.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bunch H., Lawney B.P., Lin Y.F., Asaithamby A., Murshid A., Wang Y.E., Chen B.P., Calderwood S.K. Transcriptional elongation requires DNA break-induced signalling. Nat Commun. 2015;6:10191. doi: 10.1038/ncomms10191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fong Y.W., Cattoglio C., Tjian R. The intertwined roles of transcription and repair proteins. Mol Cell. 2013;52:291–302. doi: 10.1016/j.molcel.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ju B.G., Lunyak V.V., Perissi V., Garcia-Bassets I., Rose D.W., Glass C.K., Rosenfeld M.G. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science. 2006;312:1798–1802. doi: 10.1126/science.1127196. [DOI] [PubMed] [Google Scholar]
- 30.Panier S., Boulton S.J. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol. 2014;15:7–18. doi: 10.1038/nrm3719. [DOI] [PubMed] [Google Scholar]
- 31.Deng C.X. BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. 2006;34:1416–1426. doi: 10.1093/nar/gkl010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park J., Welner R.S., Chan M.Y., Troppito L., Staber P.B., Tenen D.G., Yan C.T. The DNA ligase IV syndrome R278H mutation impairs B lymphopoiesis via error-prone nonhomologous end-joining. J Immunol. 2016;196:244–255. doi: 10.4049/jimmunol.1403099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Semple B.D., Blomgren K., Gimlin K., Ferriero D.M., Noble-Haeusslein L.J. Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol. 2013;106–107:1–16. doi: 10.1016/j.pneurobio.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McEvilly R.J., de Diaz M.O., Schonemann M.D., Hooshmand F., Rosenfeld M.G. Transcriptional regulation of cortical neuron migration by POU domain factors. Science. 2002;295:1528–1532. doi: 10.1126/science.1067132. [DOI] [PubMed] [Google Scholar]
- 35.Nieto M., Monuki E.S., Tang H., Imitola J., Haubst N., Khoury S.J., Cunningham J., Gotz M., Walsh C.A. Expression of Cux-1 and Cux-2 in the subventricular zone and upper layers II-IV of the cerebral cortex. J Comp Neurol. 2004;479:168–180. doi: 10.1002/cne.20322. [DOI] [PubMed] [Google Scholar]
- 36.Hevner R.F., Shi L., Justice N., Hsueh Y., Sheng M., Smiga S., Bulfone A., Goffinet A.M., Campagnoni A.T., Rubenstein J.L. Tbr1 regulates differentiation of the preplate and layer 6. Neuron. 2001;29:353–366. doi: 10.1016/s0896-6273(01)00211-2. [DOI] [PubMed] [Google Scholar]
- 37.Leid M., Ishmael J.E., Avram D., Shepherd D., Fraulob V., Dolle P. CTIP1 and CTIP2 are differentially expressed during mouse embryogenesis. Gene Expr Patterns. 2004;4:733–739. doi: 10.1016/j.modgep.2004.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Molyneaux B.J., Arlotta P., Menezes J.R., Macklis J.D. Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci. 2007;8:427–437. doi: 10.1038/nrn2151. [DOI] [PubMed] [Google Scholar]
- 39.Tyssowski K.M., DeStefino N.R., Cho J.H., Dunn C.J., Poston R.G., Carty C.E., Jones R.D., Chang S.M., Romeo P., Wurzelmann M.K., Ward J.M., Andermann M.L., Saha R.N., Dudek S.M., Gray J.M. Different neuronal activity patterns induce different gene expression programs. Neuron. 2018;98:530–546.e11. doi: 10.1016/j.neuron.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Woodbine L., Brunton H., Goodarzi A.A., Shibata A., Jeggo P.A. Endogenously induced DNA double strand breaks arise in heterochromatic DNA regions and require ataxia telangiectasia mutated and Artemis for their repair. Nucleic Acids Res. 2011;39:6986–6997. doi: 10.1093/nar/gkr331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Borquez D.A., Urrutia P.J., Wilson C., van Zundert B., Nunez M.T., Gonzalez-Billault C. Dissecting the role of redox signaling in neuronal development. J Neurochem. 2016;137:506–517. doi: 10.1111/jnc.13581. [DOI] [PubMed] [Google Scholar]
- 42.Truong L.N., Li Y., Shi L.Z., Hwang P.Y., He J., Wang H., Razavian N., Berns M.W., Wu X. Microhomology-mediated end joining and homologous recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc Natl Acad Sci U S A. 2013;110:7720–7725. doi: 10.1073/pnas.1213431110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chiarle R., Zhang Y., Frock R.L., Lewis S.M., Molinie B., Ho Y.J., Myers D.R., Choi V.W., Compagno M., Malkin D.J., Neuberg D., Monti S., Giallourakis C.C., Gostissa M., Alt F.W. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell. 2011;147:107–119. doi: 10.1016/j.cell.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Compagno M., Wang Q., Pighi C., Cheong T.C., Meng F.L., Poggio T., Yeap L.S., Karaca E., Blasco R.B., Langellotto F., Ambrogio C., Voena C., Wiestner A., Kasar S.N., Brown J.R., Sun J., Wu C.J., Gostissa M., Alt F.W., Chiarle R. Phosphatidylinositol 3-kinase delta blockade increases genomic instability in B cells. Nature. 2017;542:489–493. doi: 10.1038/nature21406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ghouzzi V.E., Bianchi F.T., Molineris I., Mounce B.C., Berto G.E., Rak M., Lebon S., Aubry L., Tocco C., Gai M., Chiotto A.M., Sgrò F., Pallavicini G., Simon-Loriere E., Passemard S., Vignuzzi M., Gressens P., Di Cunto F. ZIKA virus elicits P53 activation and genotoxic stress in human neural progenitors similar to mutations involved in severe forms of genetic microcephaly. Cell Death Dis. 2016;7:e2440. doi: 10.1038/cddis.2016.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Merfeld E., Ben-Avi L., Kennon M., Cerveny K.L. Potential mechanisms of Zika-linked microcephaly. Wiley Interdiscip Rev Dev Biol. 2017;6:e273. doi: 10.1002/wdev.273. [DOI] [PMC free article] [PubMed] [Google Scholar]