

Abstract
The total chemical synthesis of lacto-N-tetraose (LNT) has been completed using both convergent and linear strategies. Similarly to that of our previous HMO syntheses, the donor-acceptor protecting-leaving group combinations were found to be of paramount significance to achieving successful glycosylations and minimizing side reactions.
Graphical Abstract
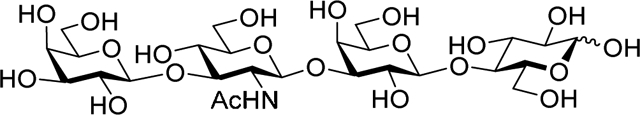
Carbohydrates are essential biomolecules that become our first food.1 Oligosaccharides present in human milk (HMO) can supply building blocks for the development of the infants’ cognition,2 act as prebiotics3 and antimicrobials.4,5 Thanks to advances in glycol-sciences, chemical structures of 162 HMO have been elucidated to date,6,7 but our understanding of how HMO function is incomplete.8,9 All HMO are composed of five monosaccharides: including glucose (Glc), galactose (Gal), N-acetylglucosamine (Glc-NAc), fucose, and sialic acid.10 Many efforts to prepare HMO enzymatically or chemically have been reported,11–13 and importance of including HMO in infant formulas has been acknowledged.14
The total amount and composition of HMO varies between women and are dependent on maternal genetics, environment, and geographic location.15 Lacto-N-tetraose (LNT) 1 represents one of the most common and abundant core structures and is classified as a type I HMO. It comprises a Galβ1→3GlcNAcβ1→3Galβ1→4Glc sequence shown in Scheme 1. More specifically, LNT is a linear tetrasaccharide wherein the reducing end lactose disaccharide (Galβ1→4Glc) is elongated with lacto-N-biose disaccharide residue (Galβ1→3GlcNAc). Chemical16–18 and enzymatic syntheses19 of LNT have been reported, and several of its derivatives have been synthesized using chemical synthesis in solution and on solid phase.20–24 Despite being one of the most abundant HMO core structures in human milk, LNT is not yet available in large quantities and at reasonable prices for research and application.
Scheme 1.

Retrosynthesis analysis of LNT 1.
Previously, we reported the total synthesis of lacto-N-neotetraose that has been completed using both linear and convergent approaches.25 Along the way, we developed the synthesis of key building blocks, accessed scalability, and refined coupling procedures to obtain different glycosidic linkages and sequences. Notably, the donor and acceptor protecting/leaving group combinations were found to be key parameters. In further synthetic studies of HMO in our lab reported herein is the synthesis of LNT 1. First, we decided to investigate a convergent (2+2) synthetic strategy, according to which we chose to converge the protected lacto-N-biose donor 2 and lactose acceptor 3.25 Based on our previous synthetic endeavors and preliminary refinement of reaction conditions,25 for the synthesis of disaccharide 2 we chose superarmed S-benzoxazolyl (SBox) galactosyl donor 429,30 and glucosamine acceptor 5. SBox galactosyl donor 4 was very instrumental in avoiding the unwanted aglycone transfer side reaction12,26 that was taking place in other previously investigated building blocks.25 As in our previous HMO synthesis, we chose benzyl ethers as semi-permanent protecting groups.
The synthesis of glucosamine thioglycoside acceptor 5 was achieved from known building block 6 as depicted in Scheme 2.27 First, precursor 6 was reacted with tert-butyldimethylsilyl chloride (TBDMSCl) in the presence of imidazole in DMF at 90 °C to obtain compound 7 in 97% yield. The latter was then converted to intermediate 8 via the reductive regioselective opening of the benzylidene acetal by reaction with 1 M BH3 in THF in the presence of catalytic TMSOTf in 82% yield. Subsequently, 6-OH in compound 8 was benzylated with BnBr in the presence of NaH in DMF to afford compound 9 in 77% yield. In order to minimize the formation of side products, temperature control is highly important in this reaction (see the experimental part for further details). Finally, the silyl group of compound 9 was removed with BF3-Et2O in CH3CN at 0 °C to afford the desired glucosamine thioglycoside acceptor 5 in 87% yield.
Scheme 2.

Convergent synthesis of protected LNT 11.
We next turned our attention to the assembly of LNB disaccharide 2. Selective activation of the SBox leaving group in glycosyl donor 4 over thioethyl anomeric moiety of acceptor 5 was achieved in the presence of silver trifluoromethanesulfonate (AgOTf) to afford β-linked disaccharide 2 in excellent yield of 99%. Direct coupling of disaccharide 2 with acceptor 3 was proven inefficient, perhaps due to a fairly unreactive donor and acceptor combination. We consistently saw incomplete reactions leading to very low yields of the tetrasaccharide product. To bypass this, we chose to employ the corresponding phosphate donor 10 that was obtained from thioglycoside 2 in 90% yield via an efficient one-step protocol developed by Seeberger and co-workers.28 The coupling of phosphate donor 10 with lactose acceptor 3 in the presence of TMSOTf was more successful, and tetrasaccharide 11 was obtained in a moderate yield of 51% with complete β-stereoselectivity.
Despite numerous attempts involving modifying the reaction condition, and replacing the glycosyl donor with O- and S-imidoyl leaving groups, we failed to improve the outcome of this reaction. We were also unable to elucidate structures of byproducts forming alongside the desired tetrasaccharide 11. Not being satisfied with the outcome of the convergent synthesis, we were curious to investigate whether the linear approach would be more successful in achieving a better outcome. Only a minimal strategic adjustment was required, and this involved conversion of building block 5 into its 4-O-Fmoc derivative 12 that was subsequently transformed into phosphate donor 13. Glycosylation between donor 13 and lactose acceptor 3 in the presence of TMSOTf afforded trisaccharide 14 in 86% yield. The Fmoc protecting group was removed with 30% Et3N in CH2Cl2 and glycosylation of the resulting trisaccharide acceptor 15 with SBox donor 4 in the presence of AgOTf afforded the desired β-linked tetrasaccharide 11 in 89% yield. Overall, the three-step linear assembly of 11 involving glycosylation of acceptor 3 with glycosyl donor 13, interim Fmoc deprotection, followed by glycosylation with donor 4 proceeded with 68% overall yield for the synthesis of tetrasaccharide 11. In contrast, the convergent approach was much less efficient, 45% over three steps, primarily due to the very low-yielding last coupling step between disaccharides 3 and 10.
With the key tetrasaccharide intermediate 11 we endeavored to carry out its deprotection steps to obtain the target LNT tetrasaccharide 1. Deprotection of the phthalimido and the ester groups was performed in the presence of NH2NH2-H2O in refluxing MeOH. Subsequent N-acetylation with acetic anhydride in MeOH furnished tetrasaccharide intermediate 16 in 92% yield. Subsequently, benzyl ethers were hydrogenated the presence of 10% Pd/C in wet ethanol to afford the target trisaccharide 1 in 81% yield.
In summary, the total synthesis of lacto-N-tetraose has been completed using both linear and convergent synthesis approaches. The linear approach was significantly more effective in this application. Along the way, we have developed new synthetic protocols for different glycosidic linkages. Notably, the donor and acceptor protecting group and the leaving group combinations were found to be of paramount significance to successful glycosylations. The protecting groups in precursors used for the synthesis of the key building block 3 were chosen to provide access to variable glycosylation sites. In this application, 3’-OH acceptor 3 was achieved via the Fmoc group removal,25 but the same precursors could also be used to achieve 6’-OH via the OPico group removal, or provide access to 3’,6’-diol for the synthesis of branched HMO. Further synthetic studies of HMO are underway in our laboratory. We expect that new methods for obtaining individual HMO will boost practical applications of these important biomolecules.
Experimental
General methods.
The reactions were performed using commercial reagents and the ACS grade solvents were purified and dried according to standard procedures. Column chromatography was performed on silica gel 60 (70–230 mesh) and Sephadex G-25 size exclusion resin, reactions were monitored by TLC on Kieselgel 60 F254. The compounds were detected by examination under UV light and by charring with 10% sulfuric acid in methanol. Solvents were removed under reduced pressure at <40 °C. CH2Cl2 was distilled from CaH2 directly prior to application. Molecular sieves (3Å), used for reactions, were crushed and activated in vacuo at 390 °C during 8 h in the first instance and then for 2–3 h at 390 °C directly prior to application. AgOTf was co-evaporated with toluene (3 × 10 mL) and dried in vacuo for 2–3 h directly prior to application. Optical rotations were measured using a Jasco polarimeter. 1H NMR spectra were recorded at 300 MHz or 600 MHz, and 13C NMR spectra were recorded at 75 MHz or 151 MHz. The 1H chemical shifts are referenced to the signal of the residual TMS (δH = 0.00 ppm) for solutions in CDCl3 or the signal of the residual D2O (δH = 4.79 ppm) for solutions in D2O. The 13C chemical shifts are referenced to the central signal of CDCl3 (δC = 77.16 ppm) for solutions in CDCl3 or the central signal of CD3COCD3 δC = 29.84 ppm) for solutions in D2O. Accurate mass spectrometry determinations were performed using Agilent 6230 ESI TOF LCMS mass spectrometer.
Preparation of monosaccharide building blocks
Ethyl 4,6-O-benzylidene-3-O-tert-butyldimethylsilyl-2-deoxy-2-phthalimido-1-thio-β-D-glucopyranoside (7).
TBDMSCl (0.37 g, 2.44 mmol) and imidazole (0.16 g, 2.44 mmol) were added to a solution of ethyl 4,6-O-benzylidene-2-deoxy-2-phthalimido-1-thio-β-D-glucopyranoside27 (6, 0.54 g, 1.22 mmol) in DMF (7.0 mL) and the resulting mixture was heated at 90 °C for 3 h. After that, the reaction mixture was cooled to rt, diluted with CH2Cl2 (~250 mL) and washed with water (40 mL), sat. aq. NaHCO3 (40 mL), and water (40 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate - hexane gradient elution) to afford the title compound as a white form in 97% yield (0.65 g, 1.18 mmol). Analytical data for 7: Rf = 0.60 (ethyl acetate/hexane, 3/7, v/v); [α]D23 −2.5 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, −0.28, −0.12 (2 s, 6H, 2 x SiCH3), 0.59 (s, 9H, SitBu), 1.19 (t, 3H, J = 7.4 Hz, CH2CH3), 2.69 (m, 2H, CH2CH3), 3.59 (t, 1H, J4,5 = 8.8 Hz, H-4), 3.71 (m, 1H, J5,6a = 10.0 Hz, J5,6b = 4.4 Hz, H-5), 3.81 (dd, 1H, J6a,6b = 10.0 Hz, H-6a), 4.32 (dd, 1H, J2,3 = 9.6 Hz, H-2), 4.39 (dd, 1H, H-6b), 4.67 (dd, 1H, J3,4 = 8.8 Hz, H-3), 5.37 (d, 1H, J1,2 = 10.7 Hz, H-1), 5.54 (s, 1H, CHPh), 7.31–7.94 (m, 9H, aromatic) ppm; 13C NMR (75 MHz, CDCl3): δ, −5.1, −4.0, 15.0, 17.8, 24.2, 25.5 (×3), 56.7, 68.8, 70.6, 70.7, 81.9, 82.8, 102.1, 123.3, 123.8, 126.5 (×2), 128.3 (×2), 129.2, 131.7, 131.9, 134.3, 134.4, 137.2, 167.7, 168.4 ppm; ESI TOF LCMS [M+Na]+ calcd for C29H37NNaO6SSi 578.2009, found 578.1997.
Ethyl 4-O-benzyl-2-deoxy-2-phthalimido-3-O-tertbutyldimethylsilyl-1-thio-β-D-glucopyranoside (8).
A 1 M solution of BH3 in THF (43 mL, 43 mmol) was added to a solution of 7 (4.80 g, 8.66 mmol) in CH2Cl2 (45 mL). The resulting solution was cooled to 0 °C, TMSOTf (0.78 mL, 4.33 mmol) was added, and the resulting mixture was stirred for 5 h while the reaction temperature was allowed to gradually increase to rt. After that, the reaction was quenched with Et3N (~2 mL) and MeOH (~5 mL), and the volatiles were removed in vacuo. The residue was diluted with CH2Cl2 (~500 mL), washed with sat. aq. NaHCO3 (50 mL) and water (2 × 50 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate - hexane gradient elution) to afford the title compound as a clear syrup in 82% yield (3.80 g, 7.06 mmol). Analytical data for 8: Rf = 0.80 (ethyl acetate/hexane, 2/3, v/v); [α]D23 +27.4 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, −0.38, 0.00 (2 s, 6H, 2 x SiCH3), 0.76 (s, 9H, SitBu), 1.20 (t, 3H, J = 7.4 Hz, CH2CH3), 2.21 (m, 1H, OH), 2.68 (m, 2H, CH2CH3), 3.52–3.67 (m, 2H, H-4, 5), 3.73 (m, 1H, H-6a), 3.94 (m, 1H, H-6b), 4.27 (dd, 1H, J2,3 = 10.6 Hz, H-2), 4.56 (dd, 1H, J3,4 = 8.0 Hz, H-3), 4.80 (dd, 2H, 2J = 11.7 Hz, CH2Ph), 5.40 (d, 1H, J1,2 = 10.6 Hz, H-1), 7.27–7.95 (m, 9H, aromatic) ppm; 13C NMR (75 MHz, CDCl3): δ, −4.6, −4.0, 15.0, 17.7, 24.3, 25.7 (×3), 56.8, 62.0, 73.3, 74.7, 79.7 (×2), 81.2, 123.3, 123.7, 127.3 (×2), 127.6, 128.4 (×2), 131.7, 132.1, 134.3 (×2), 138.1, 167.6, 168.8 ppm; ESI TOF LCMS [M+Na]+ calcd for C29H39NNaO6SSi 580.2165, found 580.2163.
Ethyl 4,6-di-O-benzyl-2-deoxy-2-phthalimido-3-O-tertbutyldimethylsilyl-1-thio-β-D-glucopyranoside (9).
NaH (0.51 g, 0.021 mmol) was added portionwise to a cooled (−20 °C) solution of 8 (3.80 g, 7.06 mmol) in DMF (30 mL) and the resulting mixture was stirred under argon at −20 °C until gas evolution has ceased. After that, BnBr (1.06 mL, 9.18 mmol) was added and the resulting mixture was stirred for 6 h at −15 °C. The reaction mixture was cooled to −40 °C and glacial acetic acid (~2 mL) was added dropwise. The resulting mixture was allowed to attain rt, then diluted with EtOAc (~500 mL) and washed with water (50 mL), sat. aq. NaHCO3 (50 mL) and water (2 × 50 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate - hexane gradient elution) to afford the title compound as a colorless syrup in 77% yield (3.65 g, 5.42 mmol). Analytical data for 9: Rf = 0.80 (ethyl acetate/hexane,3/7, v/v); [α]D23 +39.3 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, −0.36, 0.00 (2 s, 6H, 2 x SiCH3), 0.79 (s, 9H, SitBu), 1.26 (t, 3H, J = 7.4 Hz, CH2CH3), 2.73 (m, 2H, CH2CH3), 3.63–3.71 (m, 2H, H-4, 5), 3.77–3.86 (br d, 2H, H-6a, 6b), 4.35 (dd, 1H, J2,3 = 10.0 Hz, H-2), 4.58 (dd, 1H, J3,4 = 7.6 Hz, H-3), 4.64 (d, 2H, 2J = 9.6 Hz, CH2Ph), 4.79 (dd, 2H, 2J = 12.0 Hz, CH2Ph), 5.38 (d, 1H, J1,2 = 10.5 Hz, H-1), 7.21–7.99 (m, 14H, aromatic) ppm; 13C NMR (75 MHz, CDCl3): δ, −4.6, −4.1, 15.1, 17.7, 23.9, 25.8 (×3), 56.8, 69.0, 73.4, 73.5, 74.6, 79.5, 80.0, 80.8, 123.2, 123.7, 127.1 (×2), 127.4, 127.6, 127.8 (×2), 128.4 (×4), 131.8, 132.2, 134.3 (×2), 138.3, 138.4, 167.7, 168.8 ppm; ESI TOF LCMS [M+Na]+ calcd for C36H45NNaO6SSi 670.2635, found 670.2633.
Ethyl 4,6-di-O-benzyl-2-deoxy-2-phthalimido-1-thio-β-D-glucopyranoside (5).
BF3-Et2O (1.05 mL, 8.29 mmol) was added to a solution of 9 (5.07 g, 7.54 mmol) in dry CH3CN (90 mL) and the resulting mixture was stirred under argon for 20 min at 0 °C. After that, the reaction was quenched with sat. aq. NaHCO3 (5 mL), and the volatiles were removed in vacuo. The residue was diluted with CH2Cl2 (~500 mL) and washed with brine (2 × 50 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate - hexane gradient elution) to afford the title compound as a white amorphous solid in 87% yield (3.49 g, 6.54 mmol). Analytical data for 5: Rf = 0.30 (ethyl acetate/hexane, 3/7, v/v); [α]D22 +7.3 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 1.20 (t, 3H, J = 7.4 Hz, CH2CH3). 2.36 (d, 1H, J = 4.5 Hz, OH), 2.67 (m, 2H, CH2CH3), 3.58–3.69 (m, 2H, H-4, 5), 3.75–3.85 (m, 2H, H-6a, 6b), 4.24 (dd, 1H, J2,3 = 10.4 Hz, H-2), 4.48 (m, 1H, H-3), 4.63 (dd, 2H, 2J = 12.1 Hz, CH2Ph), 4.70 (m, 2H, 2J = 11.5 Hz, CH2Ph), 5.29 (d, 1H, J1,2 = 10.4 Hz, H-1), 7.15–7.90 (m, 14H, aromatic) ppm; 13C NMR (75 MHz, CDCl3): δ, 15.1, 24.1, 55.7, 69.0, 72.8, 73.6, 74.8, 79.3 (×2), 81.2, 123.4, 123.8, 127.8, 127.9 (×2), 128.0 (×2), 128.1, 128.5 (×2), 128.7 (×2), 131.7, 131.8, 134.2 (×2), 138.2 (×2), 168.1, 168.3 ppm; ESI TOF LCMS [M+Na]+ calcd for C30H31NNaO6S 556.1770, found 556.1767.
Ethyl 4,6-di-O-benzyl-2-deoxy-3-O-fluorenylmethoxycarbonyl-2-phthalimido-1-thio-β-D-glucopyranoside (12).
FmocCl (3.88 g, 15.04 mmol) was added to a solution of 5 (2.08 g, 3.90 mmol) in CH2Cl2 (50 mL) and pyridine (1.72 mL) and the resulting mixture was stirred under argon for 2 h at rt. After that, the reaction mixture was diluted with CH2Cl2 (~500 mL) and washed with 1 M aq. HCl (50 mL) and water (2 × 50 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate - hexane gradient elution) to afford the title compound as a white amorphous solid in 92% yield (2.70 g, 3.58 mmol). Analytical data for 12: Rf = 0.40 (ethyl acetate/hexane,2/3, v/v); [α]D23 +55.2 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 1.22 (t, 3H, J = 7.4 Hz, CH2CH3), 2.70 (m, 2H, CH2CH3), 3.73–3.86 (m, 4H, H-5, 6a, 6b, OCOCH2CH), 3.91–4.16 (m, 3H, H-4, OCOCH2CH), 4.47 (dd, 1H, J2,3 = 10.5 Hz, H-2), 4.62 (dd, 2H, 2J = 12.1 Hz, CH2Ph), 4.64 (dd, 2H, 2J = 11.2 Hz, CH2Ph), 5.46 (d, 1H, J1,2 = 10.5 Hz, H-1), 5.75 (dd, 1H, J3,4 = 8.9 Hz, H-3), 7.07–7.88 (m, 26H, aromatic) ppm; 13C NMR (75 MHz, CDCl3): δ, 15.1, 24.2, 46.5, 54.1, 68.8, 70.3, 73.6, 75.0, 76.6, 78.5, 79.2, 81.0, 120.0 (×2), 123.7, 123.8, 125.1, 125.3, 127.3 (×2), 127.8 (×4), 127.9 (×4), 128.4 (×2), 128.5 (×2), 131.3, 131.8, 134.1, 134.4, 137.8, 138.2, 141.1, 141.2, 143.0, 143.3, 154.8, 167.5, 168.0 ppm; ESI TOF LCMS [M+Na]+ calcd for C45H41NNaO8S 778.2451, found 778.2451.
Di-O-butyl 4,6-di-O-benzyl-2-deoxy-3-O-fluorenylmethoxycarbonyl-2-phthalimido-β-D-glucopyranosyl phosphate (13).
A mixture containing thioglycoside 12 (0.50 g, 0.66 mmol), dibutyl hydrogen phosphate (0.39 mL, 1.99 mmol), and molecular sieves (3 Å, 1.0 g) in CH2Cl2 (10 mL) was stirred under argon for 1h. The mixture was cooled to 0 °C, NIS (0.29 g, 1.32 mmol) and TfOH (10 μL, 0.13 mmol) were added, and the resulting mixture was stirred under argon for 20 min at 0 °C. After that, the solids were filtered off and washed successively with CH2Cl2. The combined filtrate (~100 mL) was washed with 10% aq. Na2S2O3 (15 mL), sat. aq. NaHCO3 (15 mL), and water (2 × 15 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (acetone - toluene gradient elution) to afford the title compound as a white form in 94% yield (0.56 g, 0.62 mmol). Analytical data for 13: Rf = 0.45 (ethyl acetate/hexane,1/4, v/v); [α]D23 +43.0 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 0.71, 0.85 (2 t, 6H, 2 x O(CH2)3CH3), 1.07 (m, 2H, O(CH2)2CH2CH3), 1.22–1.37 (m, 4H, OCH2CH2CH2CH3), 1.48–1.60 (m, 2H, OCH2CH2CH2CH3), 3.67–4.17 (m, 11H, 2 x OCH2CH2CH2CH3, H-4, 5, 6a, 6b, OCOCH2CH, OCOCH2CH), 4.49 (dd, 1H, J2,3 = 10.6 Hz, H-2), 4.60 (dd, 2H, 2J = 11.9 Hz, CH2Ph), 4.64 (dd, 2H, 2J =11.2 Hz, CH2Ph), 5.80 (dd, 1H, J3,4 = 8.9 Hz, H-3), 6.02 (d, 1H, J1,2 = 8.3, H-1), 6.97–7.86 (m, 22H, aromatic) ppm; 13C NMR (75 MHz, CDCl3): δ, 13.5, 13.6, 18.4, 18.6, 31.9 (d, J = 7.0 Hz), 32.0 (d, J = 7.2 Hz), 46.4, 55.3 (d, J = 8.9 Hz). 67.9 (d, J 6.1 Hz), 68.1 (d, J = 6.0 Hz), 70.3, 73.6, 74.9, 75.2, 76.0, 76.8, 77.3, 93.9 (d, J = 4.6 Hz), 120.0 (×2), 123.6, 125.0, 125.2, 125.4, 127.2 (×2), 127.7 (×3), 127.8 (×3), 127.9 (×3), 128.3, 128.4 (×3), 128.5 (×3), 129.1, 134.2, 137.6, 137.9, 141.1, 141.2, 143.0, 143.2, 154.6 ppm; ESI TOF LCMS [M+Na]+ calcd for C51H54NNaO12P 926.3281, found 926.3285.
Synthesis of oligosaccharides
Ethyl O-(2-O-benzoyl-3,4,6-tri-O-benzyl-β-D-galactopyranosyl)-(1→3)-4,6-di-O-benzyl-2-deoxy-2-phthalimido-1-thio-β-D-glucopyranoside (2).
A mixture of benzoxazolyl 2-O-benzoyl-3,4,6-tri-O-benzyl-1-thio-β-D-galactopyranoside29,30 (4, 0.20 g, 0.29 mmol), acceptor 5 (0.12 g, 0.22 mmol), and freshly activated molecular sieves (3Å, 600 mg) in CH2Cl2 (7 mL) was stirred under argon for 2 h. The reaction mixture was cooled to −30 °C, and freshly conditioned AgOTf (0.15 g, 0.58 mmol) was added. The resulting mixture was stirred for 15 min while the temperature was allowed to increase gradually. The reaction mixture was then diluted with CH2Cl2, the solids were filtered off, and rinsed successively with CH2Cl2. The combined filtrate (~50 mL) was washed with sat. aq. NaHCO3 (10 mL) and water (2 × 10 mL) The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (acetone - toluene gradient elution) to afford the title compound as a white foam in 99% yield (0.23 g, 0.22 mmol). Analytical data for 2: Rf = 0.55 (acetone/toluene, 1/9 v/v); [α]D23 +34.1 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 1.09 (t, 3H, J = 7.4 Hz, CH2CH3). 2.56 (m, 2H, CH2CH3), 3.30–3.51 (m, 4H, H-3ʹ, 5ʹ, 6aʹ, 6bʹ), 3.57–3.72 (m, 2H, H-4, 5), 3.77 (br d, 2H, J = 2.1 Hz, H-6a, 6b), 3.94 (br d, 1H, J3ʹ,4ʹ = 2.5 Hz, H-4ʹ), 4.26 (dd, 2H, 2J = 11.6 Hz, CH2Ph), 4.29 (d, 1H, J = 12.0 Hz, CHPh), 4.30 (dd, 1H, J2,3 = 10.3, H-2), 4.44 (d, 1H, J1ʹ,2ʹ = 7.9 Hz, H-1ʹ), 4.454.64 (m, 5H, 5 × CHPh), 4.83 (dd, 1H, J3,4 = 8.0 Hz, H-3), 4.91 (d, 1H, J = 11.3 Hz, CHPh), 5.06 (d, 1H, J1,2 = 10.4 Hz, H-1), 5.09 (d, 1H, J = 10.5 Hz, CHPh), 5.52 (dd, 1H, J2ʹ,3ʹ = 9.9 Hz, H-2ʹ), 6.92–7.78 (m, 34H, aromatic) ppm; 13C NMR (75 MHz, CDCl3): δ, 14.9, 23.7, 54.8, 67.8, 69.2, 71.7, 72.5, 72.8, 73.3, 73.4, 73.5, 74.8, 75.0, 77.4, 77.8, 79.5, 80.3, 81.0, 100.7, 127.2, 127.5 (×3), 127.6 (×2), 127.8 (×4), 128.0 (×3), 128.1 (×6), 128.2 (×3), 128.3 (×3), 128.4 (×3), 128.5 (×3), 130.0 (×2), 130.3, 131.5, 132.8, 134.0, 137.5, 138.0, 138.3, 138.7 (×2), 165.4 ppm; ESI TOF LCMS [M+Na]+ calcd for C64H63NNaO12S 1092.3969, found 1092.3981.
Di-O-butyl O-(2-O-benzoyl-3,4,6-tri-O-benzyl-β-D-galactopyranosyl)-(1→3)-4,6-di-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl phosphate (10).
A mixture of compound 2 (0.236 g, 0.234 mmol), dibutyl hydrogen phosphate (0.14 mL, 0.802 mmol), and freshly activated molecular sieves (3 Å, 0.5 g) in CH2Cl2 (5.0 mL) was stirred under argon for 1 h at rt. The mixture was cooled to 0 °C, NIS (0.104 g, 0.468 mmol) and TfOH (4.15 μL, 0.047 mmol) were added, and the resulting mixture was stirred for 20 min at 0 °C. After that, the solids were filtered off and rinsed successively with CH2Cl2. The combined filtrate (~100 mL) was washed with 10% aq. Na2S2O3 (15 mL) and sat. aq. NaHCO3 (15 mL), and water (2 × 15 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (acetone - toluene gradient elution) to afford the title compound as an oily syrup in 94% yield (0.267 g, 0.219 mmol). Analytical data for 10: Rf = 0.35 (acetone/toluene,1/9, v/v); [α]D23 +26.2 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 0.67 (t, 3H, J = 7.2 Hz, O(CH2)3CH3), 0.81 (t, 3H, J = 7.3 Hz, O(CH2)3CH3), 0.90–1.06 (m, 2H, O(CH2)2CH2CH3), 1.11–1.31 (m, 4H, O(CH2)2CH2CH3, OCH2CH2CH2CH3), 1.38–1.53 (m, 2H, OCH2CH2CH2CH3), 3.31–3.96 (m, 13H, H-3ʹ, 4, 4ʹ, 5, 5ʹ, 6a, 6b, 6aʹ, 6bʹ, 2 x OCOCH2(CH)2CH3), 4.20 (d, 1H, 2J = 11.7 Hz, CHPh), 4.24–4.32 (m, 3H, H-2, 2 x CHPh), 4.43–4.54 (m, 5H, H-1, 4 x CHPh), 4.59 (d, 1H, 2J = 12.0 Hz, CHPh), 4.91 (m, 2H, H-3, CHPh), 5.09 (d, 1H, 2J = 10.5 Hz, CHPh), 5.51 (dd, 1H, J2ʹ,3ʹ = 8.8 Hz, H-2ʹ), 5.69 (dd, 1H, J1,2 = 7.4 Hz, H-1) 6.94–7.74 (m, 36H, aromatic) ppm; 13C NMR (75 MHz, CDCl3): δ, 13.5, 13.6, 18.3, 18.6, 31.8 (d, J = 7.1 Hz). 32.0 (d, J = 7.2 Hz), 56.1, 56.2, 67.6, 67.8 (d, J = 4.5 Hz), 68.0 (d, J = 6.4 Hz), 68.6, 71.7, 72.5, 72.8, 73.3, 73.5 (×2), 74.8, 74.9, 75.6, 76.2, 80.2, 94.1 (d, J = 4.6 Hz), 100.8, 123.5, 127.3, 127.5 (×2), 127.6 (×3), 127.7, 127.8 (×3), 128.0 (×7), 128.1 (×4), 128.2 (×4), 128.3 (×2), 128.4 (×2), 128.5 (×2), 130.0, 130.2, 131.5, 132.7, 134.0, 137.5, 138.0, 138.1, 138.6, 138.7, 165.3 ppm; ESI TOF LCMS [M+Na]+ calcd for C70H76NNaO16P 1240.4799, found 1240.4829.
Benzyl O-(4,6-di-O-benzyl-2-deoxy-3-O-fluorenylmethoxycarbonyl-2-phthalimido-β-D-glucopyranosyl)-(1→3)-O-(2-O-benzoyl-4-O-benzyl-6-O-picoloyl-β-D-galactopyranosyl)-(1→4)-2,3,6-tri-O-benzyl-β-D-glucopyranoside (14).
A mixture of donor 13 (0.14 g, 0.15 mmol), acceptor 325 (0.12 g, 0.12 mmol) (reference from LNnT paper), and freshly activated molecular sieves (3Å, 450 mg) in CH2Cl2 (7.0 mL) was stirred under argon for 2 h. The mixture was cooled to −30 °C, TMSOTf (56 μL, 0.31 mmol) was added, and the resulting mixture was stirred for 15 min while the temperature was allowed to increase gradually. The reaction mixture was then diluted with CH2Cl2, the solids were filtered off and rinsed successively with CH2Cl2. The combined filtrate (~50 mL) was washed with sat. aq. NaHCO3 (10 mL) and water (2 × 10 mL) The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (acetone - toluene gradient elution) to afford the title compound as a white foam in 86% yield (0.17 g, 0.10 mmol). Analytical data for 14: Rf = 0.45 (acetone/toluene, 1/4 v/v); [α]D23 +11.3 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3): δ, 2.91 (ddd, 1H, J = 1.8, 3.0, 9.9 Hz, H-5), 3.27 (dd, 1H, J = 9.5 Hz, H-6a), 3.33 (dd, 1H, J2,3 = 8.4 Hz, H-2), 3.38–3.42 (m, 2H, H-3, 6b), 3.69–3.75 (m, 3H, H-3ʹ, 5ʹ, OCOCH2CH), 3.79–3.85 (m, 4H, H-4, 5ʹʹ, 6aʹʹ, 6bʹʹ), 3.87–3.93 (m, 2H, H-4ʹʹ, OCOCH2CH), 4.02 (dd, 1H, J = 10.5, 7.2 Hz, OCOCH2CH), 4.11 (br d, 1H, J = 2.4 Hz, H-4ʹ), 4.18–4.24 (m, 2H, H-6aʹ, CHPh), 4.27 (d, 1H, J1,2 = 7.7 Hz, H-1), 4.34–4.40 (m, 2H, H-2ʹʹ, 6bʹ), 4.42–4.49 (m, 3H, J1ʹ,2ʹ = 8.1 Hz, H-1ʹ, 2 x CHPh), 4.5–4.69 (m, 7H, 7 x CHPh), 4.80 (d, 1H, 2J = 12.0 Hz, CHPh), 4.81 (d, 1H, 2J = 12.0 Hz, CHPh), 4.92 (d, 1H, 2J = 10.4 Hz, CHPh), 5.12 (d, 1H, 2J = 11.5 Hz, CHPh), 5.36 (dd, 1H, J2ʹ,3ʹ = 10.1 Hz, H-2ʹ), 5.44 (d, 1H, J1ʹʹ,2ʹʹ = 8.3 Hz, H-1ʹʹ), 5.66 (dd, 1H, J = 8.9, 10.7 Hz, H-3ʹʹ), 6.81–8.73 (m, 56H, aromatic) ppm; 13C NMR (151 MHz, cdcl3): δ, 46.4, 55.2, 63.8, 67.6, 68.9, 70.3, 71.0, 71.9, 72.0, 73.5 (×2), 73.6 (×2), 74.4, 74.7, 75.0 (×2), 75.2, 75.6, 76.2, 76.6, 80.6, 81.7, 82.6, 99.4, 100.4, 102.6, 120.0, 125.0, 125.3, 125.5, 126.9, 127.2 (×2), 127.3, 127.6 (×2), 127.7 (×3), 127.8 (×3), 127.9 (×5), 128.0, 128.1 (×9), 128.3 (×6), 128.4 (×3), 128.5 (×3), 128.6 (×9), 128.7 (×3), 129.4, 129.8, 132.9, 137.0, 137.6 (×2), 137.9, 138.3, 138.7 (×2), 139.0, 141.1, 141.2, 142.9, 143.3, 147.8, 150.0, 154.6, 164.5, 164.5 ppm; ESI TOF LCMS [M+Na]+ calcd for C103H94N2NaO21 1718.6280, found 1718.6222.
Benzyl O-(4,6-di-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl)-(1→3)-O-(2-O-benzoyl-4-O-benzyl-6-O-picoloyl-β-D-galactopyranosyl)-(1→4)-2,3,6-tri-O-benzyl-β-D-glucopyranoside (15).
Compound 14 (135 mg, 0.0796 mmol) was dissolved in a mixture of Et3N in CH2Cl2 (5.0 mL, 3/7, v/v) and the resulting solution was stirred for 2 h at rt. After that, the reaction mixture was concentrated in vacuo, and the residue was purified by column chromatography on silica gel (acetone - toluene gradient elution) to afford the title compound as a white foam in 89% yield (104.8 mg, 0.0711 mmol). Analytical data for 15: Rf = 0.55 (acetone/toluene, 1/4 v/v); [α]D22 −11.6 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3): δ, 2.45 (d, 1H, J = 4.6 Hz, OH), 2.94 (ddd, 1H, J = 8.9 Hz, H-5), 3.29 (dd, 1H, J = 10.3 Hz, H-6a), 3.34 (dd, 1H, J2,3 = 9.0 Hz, H-2), 3.39–3.44 (m, 2H, H-3, 6b), 3.61 (dd, 1H, J3”,4” = 9.1 Hz, J4”,5” = 9.1 Hz, H-4ʹʹ), 3.67–3.76 (m, 3H, H-3ʹ, 5ʹʹ, 6aʹʹ), 3.78–3.88 (m, 3H, H-4, 5ʹ, 6bʹʹ), 4.11 (br s, 1H, H-4ʹ), 4.13–4.26 (m, 3H, H-2ʹʹ, 6aʹ, CHPh), 4.29 (d, 1H, J1.2 = 7.7 Hz, H-1), 4.36 (dd, 1H, J = 6.1, 11.0 Hz, H-6bʹ), 4.44–4.52 (m, 4H, J1ʹ,2ʹ = 8.0 Hz, H-1ʹ, 3ʹʹ, 2 x CHPh), 4.55–4.70 (m, 6H, 6 x CHPh), 4.74 (d, 1H, 2J = 11.4 Hz, CHPh), 4.82 (d, 2H, 2J = 12.0 Hz, CHPh × 2), 4.94 (d, 1H, 2J = 10.4 Hz, CHPh), 5.14 (d, 1H, 2J = 11.5 Hz, CHPh), 5.22 (d, 1H, J1ʹʹ,2ʹʹ = 8.3 Hz, H-1ʹʹ), 5.34–5.39 (dd, 1H, J2ʹ,3ʹ = 9.9 Hz, H-2ʹ), 7.00–8.75 (m, 48H, aromatic) ppm; 13C NMR (151 MHz, CDCl3): δ, 56.8, 63.9, 67.5, 69.1, 71.0 (×2), 72.0 (×2), 73.4, 73.6 (×2), 74.3, 74.9 (×3), 75.1, 75.6, 76.1, 79.2, 80.3, 81.6, 82.6, 99.7, 100.3, 102.5, 125.4, 126.9, 127.2, 127.5, 127.7 (×3), 127.8 (×3), 127.9, 128.0 (×8), 128.1 (×4), 128.2 (×3), 128.3 (×6), 128.4 (×3), 128.5 (×3), 128.6 (×6), 128.7 (×3), 129.4, 129.6, 132.9, 133.6, 137.0, 137.5, 137.9, 138.1, 138.2, 138.6, 138.8, 139.0, 147.7, 149.9, 164.4 (×2) ppm; ESI TOF LCMS [M+H]+ calcd for C88H85N2O19 1473.5747, found 1473.5757.
Benzyl O-(2-O-benzoyl-3,4,6-tri-O-benzyl-β-D-galactopyranosyl)-(1→3)-O-(4,6-di-O-benzyl-2-deoxy-2-phthlimido-β-D-glucopyranosyl)-(1→3)-O-(2-O-benzoyl-4-O- benzyl-6-O-picoloyl-β-D-galactopyranosyl)-(1→4)-2,3,6-tri-O-benzyl-β-D-glucopyranoside (11).
Convergent method.
A mixture of donor 10 (80.0 mg, 0.065 mmol), acceptor 3 (49.8 mg, 0.050 mmol),25 and freshly activated molecular sieves (3Å, 400 mg) in CH2Cl2 (7.0 mL) was stirred under argon for 2 h. The mixture was cooled to −60 °C, TMSOTf (24 μL, 0.131 mmol) was added, and the resulting mixture was stirred for 30 min while the temperature was allowed to increase gradually. The reaction mixture was then diluted with CH2Cl2, the solids were filtered off and rinsed successively with CH2Cl2. The combined filtrate (~50 mL) was washed with sat. aq. NaHCO3 (10 mL) and water (2 × 10 mL) The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (acetone - toluene gradient elution) to afford the title compound as an off-white amorphous solid in 51% yield (66.6 mg, 0.033 mmol). Linear method. A mixture of benzoxazolyl 2-O-benzoyl-3,4,6-tri-O-benzyl-1-thio-β-D-galactopyranoside29,30 (4, 13.3 mg, 0.0194 mmol), acceptor 15 (22 mg, 0.0149 mmol), and freshly activated molecular sieves (3Å, 100 mg) in CH2Cl2 (2.0 mL) was stirred under argon for 2 h. The reaction mixture was cooled to −30 °C, freshly conditioned AgOTf (10.0 mg, 0.0387 mmol) was added, and the resulting mixture was stirred for 15 min while the temperature was allowed to increase gradually. The reaction mixture was then diluted with CH2Cl2, the solids were filtered off and was rinsed successively with CH2Cl2. The combined filtrate (~30 mL) was washed with sat. aq. NaHCO3 (7 mL) and water (2 × 7 mL) The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (acetone -toluene gradient elution) to afford the title compound as a white foam in 89% yield (26.7 mg, 0.0132 mmol). Analytical data for 11: Rf = 0.45 (acetone/toluene, 1/4, v/v); [α]D22 +2.5 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3): δ, 2.85 (ddd, 1H, J = 9.6 Hz, H-5), 3.19 (dd, 1H, J = 10.2 Hz, H-6a), 3.25–3.37 (m, 6H, H-2, 3, 3ʹʹʹ, 6aʹʹʹ, 6b, 6bʹʹʹ), 3.43 (m, 1H, H-5ʹʹʹ), 3.58–3.66 (m, 3H, H-3ʹ, 4ʹʹ, 5ʹ), 3.70 (m, 1H, H-5ʹʹ), 3.84–3.75 (m, 3H, H-4, 6aʹʹ, 6bʹʹ), 3.88 (br d, 1 H, J = 1.9 Hz, H-4ʹʹʹ), 3.98 (s, 1H, H-4ʹ), 4.14–4.30 (m, 9H, H-1, 1ʹʹʹ, 2ʹʹ, 6aʹ, 6bʹ, 4 x CHPh), 4.33 (d, 1H, J1ʹ,2ʹ = 8.0 Hz, H-1ʹ), 4.41–4.60 (m, 9H, 9 x CHPh), 4.66 (d, 1H, 2J = 11.0 Hz, CHPh), 4.78–4.82 (m, 3H, H-3ʹʹ, 2 x CHPh), 4.89 (d, 2H, 2J = 11.0 Hz, 2 x CHPh), 4.95 (d, 1H, 2J = 11.7 Hz, CHPh), 5.00 (d, 1H, J1ʹʹ,2ʹʹ = 8.3 Hz, H-1ʹʹ), 5.06 (d, 1H, 2J = 10.4 Hz, CHPh), 5.17 (dd, 1H, J2ʹ,3ʹ = 9.8 Hz, H-2ʹ), 5.45 (dd, 1H, J 2ʹʹ,3ʹʹ = 8.9 Hz, H-2ʹʹʹ), 6.75–8.69 (m, 68H, aromatic) ppm: 13C NMR (151 MHz, CDCl3): δ, 29.8, 31.1, 56.0, 63.8, 67.4, 67.8, 69.5, 71.0, 71.8, 72.0 (× 2), 72.6, 73.2, 73.5 (× 2), 73.6, 74.4, 74.8, 74.9, 75.0, 75.1, 75.2, 75.6, 75.8 (× 2), 76.1, 79.4, 80.2, 81.6, 82.6, 99.3, 100.4, 100.5, 102.6, 122.7, 123.7, 125.4, 126.8, 127.3, 127.4 (×2), 127.5 (×4), 127.6 (×2), 127.7, 127.8 (×7), 127.9, 128.0 (×3), 128.1 (×7), 128.2 (×8), 128.3 (×4), 128.4 (×7), 128.5 (×6), 128.6 (×2), 128.8 (×2), 129.6, 129.8, 130.0, 130.2, 130.9, 131.3, 132.7, 133.0, 133.4, 134.6, 136.9, 137.5, 137.6, 138.0, 138.1, 138.2, 138.5, 138.7, 138.8, 139.0, 147.8, 150.0, 164.3, 164.4, 165.5, 166.3, 168.4 ppm; ESI TOF LCMS [M+H]+calcd for C122H117N2O25 2010.7979, found 2010.7963.
Deprotection of tetrasaccharide 11
Benzyl O-(3,4,6-tri-O-benzyl-β-D-galactopyranosyl)-(1→3)-O-(3,6-di-O-benzyl-2-acetamido-2-deoxy-β-D-glucopyranosyl)-(1→3)-O-(4-O-benzyl-β-D-galactopyranosyl)-(1→4)-2,3,6-tri-O-benzyl-β-D-glucopyranoside (16).
Compound 11 (59.0 mg, 0.029 mmol) was dissolved in MeOH (3.0 mL), NH2NH2-H2O (130 μL, 2.64 mmol) was added, and the resulting mixture was heated at 90 °C for 24 h. After that, the volatiles were removed under reduced pressure, and the residue was dried in vacuo for 3 h. The crude residue was dissolved in a mixture of Ac2O and MeOH (2.0 mL, 1/1, v/v) and the resulting mixture was stirred for 12 h at rt. The volatiles were removed under reduced pressure, the residue was diluted with CH2Cl2 (50 mL), and washed with sat. aq. NaHCO3 (10 mL) and 1 M HCl (10 mL). The organic phase was separated, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (acetone - toluene gradient elution) to afford the title compound as an off-white amorphous solid in 92% yield (42.9 mg, 0.026 mmol). Analytical data for 16: Rf = 0.50 (acetone/toluene, 3/7 v/v); [α]D23 +70.8 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3): δ, 1.81 (s, 3H, CH3CO), 2.95–4.03 (m, 24H, H-2, 2ʹ, 2ʹʹ, 2ʹʹʹ, 3, 3ʹ, 3ʹʹ, 3ʹʹʹ, 4, 4ʹ, 4ʹʹ, 4ʹʹʹ, 5, 5ʹ, 5ʹʹ, 5ʹʹʹ, 6a, 6aʹ, 6aʹʹ, 6aʹʹʹ, 6b, 6bʹ, 6bʹʹ,6bʹʹʹ), 4.23 (d, 1H, 2J = 11.8 Hz, CHPh), 4.30–4.58 (m, 10H, H-1, 1ʹ, 1ʹʹʹ, 7 x CHPh), 4.60–4.96 (m, 11H, 11 x CHPh), 5.01–5.04 (m, 2H, H-1ʹʹ, CHPh), 6.48 (d, 1H, J = 6.3 Hz, NHCOCH3), 7.12–7.37 (m, 50H, aromatic) ppm; 13C NMR (151 MHz, CDCl3): δ, 23.7, 58.4, 61.9, 68.2, 68.8, 69.4, 71.3, 71.6, 71.9, 72.1, 72.8, 73.5, 73.7 (×3), 73.8, 74.3, 74.7, 74.8, 74.9, 75.1 (×2), 75.2, 75.4, 76.6, 77.1, 81.9, 82.1, 83.0, 83.1, 83.7, 101.8, 102.9, 103.1, 104.5, 127.4, 127.5, 127.6 (×3), 127.7 (×2), 127.8 (×3), 127.9 (×4), 128.0 (×7), 128.2 (×8), 128.3 (×6), 128.4 (×4), 128.5 (×6), 128.7 (×3), 128.8 (×2), 137.6, 138.0 (×2), 138.1, 138.2, 138.4, 138.6, 138.7, 138.9, 139.0, 172.3 ppm; ESI TOF LCMS [M+Na]+calcd for C96H105NNaO21 1631.7110, found 1631.7121.
O-(β-D-Galactopyranosyl)-(1→3)-O-(2-acetamido-2-deoxy-β-D-glucopyranosyl)-(1→3)-O-(β-D-galactopyranosyl)-(1→4)-D-glucopyranose (1, LNT).
10% Pd on carbon (125 mg) was added to a solution of tetrasaccharide 16 (40 mg, 0.025 mml) in 80% aq. EtOH (5.0 mL), and the resulting mixture was stirred under hydrogen atmosphere for 24 h at rt. After that, the solids were filtered off and rinsed successively with methanol and water. The combined filtrate (~40 mL) was concentrated in vacuo. The residue was purified by size exclusion column chromatography on Sephadex G-25 using water as the eluent to afford the title compound as a white amorphous solid in 81% yield (14.3 mg, 0.020 mmol). Analytical data for 1: Rf = 0.30 (chloroform/methanol/water, 2/1/0.4, v/v/v); 1H NMR (600 MHz, D2O): δ, 2.01 (s, 3H, CH3CO), 3.24–3.51 (m, 2H), 3.49–3.96 (m, 33H), 4.14 (d, 1H, J = 3.3 Hz), 4.43 (d, 2H, J = 7.8 Hz), 4.65 (d, 1H, J = 8.0 Hz), 4.72 (dd, 1H, J = 2.5, 8.4 Hz), 5.21 (d, 1H, J = 3.8 Hz) ppm; 13C NMR (151 MHz, D2O): δ, 22.6, 55.0, 60.3, 60.4, 60.8, 61.3, 61.4, 68.7, 68.8, 68.9, 70.4, 70.5, 71.0, 71.5, 71.8, 72.8, 74.1, 74.7, 75.1, 75.2, 75.5, 75.6, 78.6, 78.7, 82.3, 82.4, 92.2, 96.0, 96.1, 102.9, 103.2, 103.3, 103.8, 175.3 ppm; ESI TOF LCMS [M+Na]+calcd for C26H45NNaO21 730.2382, found 730.2361.
Supplementary Material
Scheme 3.

The linear synthesis of tetrasaccharide 11 and its deprotection to obtain LNT 1.
The total synthesis of lacto-N-tetraose has been completed using both linear and convergent synthesis approaches;
The linear approach was proven to be more efficient in this application;
New synthetic protocols for different glycosidic linkages have been developed and refined;
New methods for obtaining individual HMO help to improve understanding their roles and boost practical applications
ACKNOWLEDGMENT
This work was supported by the National Institute of General Medical Sciences (U01GM120673).
Footnotes
Supporting Information
NMR spectra for all new compounds. This material is available free of charge via the Internet at
The authors declare no competing financial interests.
REFERENCES
- (1).Eriksen KG; Christensen SH; Lind MV; Michaelsen KF Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 200. [DOI] [PubMed] [Google Scholar]
- (2).Wu S; Grimm R; German JB; Lebrilla CB J. Proteome Res 2011, 10, 856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Thomson P; Medina DA; Garrido D Food Microbiol 2018, 75, 37. [DOI] [PubMed] [Google Scholar]
- (4).Plaza-Diaz J; Fontana L; Gil A Nutrients 2018, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Craft KM; Townsend SD ACS Infect. Dis 2018, 4, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Urashima T; Hirabayashi J; Sato S; Kobata A Trends Glycosci. Glycotechnol 2018, 30, SE51. [Google Scholar]
- (7).Remoroza CA; Mak TD; De Leoz MLA; Mirokhin YA; Stein SE Anal. Chem 2018, 90, 8977. [DOI] [PubMed] [Google Scholar]
- (8).Xu G; Davis JC; Goonatilleke E; Smilowitz JT; German JB; Lebrilla CB J. Nutr 2017, 147, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Bode L; Jantscher-Krenn E Adv. Nutr 2012, 3, 383S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wu X; Jackson RT; Khan SA; Ahuja J; Pehrsson PR Curr. Dev. Nutr 2018, 2, nzy025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Chen X Adv. Carbohydr. Chem. Biochem 2015, 72, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Xiao Z; Guo Y; Liu Y; Li L; Zhang Q; Wen L; Wang X; Kondengaden SM; Wu Z; Zhou J; Cao X; Li X; Ma C; Wang PG J. Org. Chem 2016, 81, 5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Prudden AR; Liu L; Capicciotti CJ; Wolfert MA; Wang S; Gao Z; Meng L; Moremen KW; Boons GJ Proc. Nat. Acad. Sci 2017, 114, 6954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Vandenplas Y; Berger B; Carnielli VP; Ksiazyk J; Lagstrom H; Sanchez Luna M; Migacheva N; Mosselmans JM; Picaud JC; Possner M; Singhal A; Wabitsch M Nutrients 2018, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ramani S; Stewart CJ; Laucirica DR; Ajami NJ; Robertson B; Autran CA; Shinge D; Rani S; Anandan S; Hu L; Ferreon JC; Kuruvilla KA; Petrosino JF; Venkataram Prasad BV; Bode L; Kang G; Estes MK Nature Commun 2018, 9, 5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Takamura T; Chiba T; Ishihara H; Tejima S Chem. Pharm. Bull 1980, 28, 1804. [Google Scholar]
- (17).Aly MRE; Ibrahim ESI; El Ashry ESH; Schmidt RR Carbohydr. Res 1999, 316, 121. [DOI] [PubMed] [Google Scholar]
- (18).Craft KM; Townsend SD Carbohydr. Res 2017, 440–441, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Baumgaertner F; Conrad J; Sprenger GA; Albermann C Chem Bio Chem 2014, 15, 1896. [DOI] [PubMed] [Google Scholar]
- (20).Arboe Jennum C; Hauch Fenger T; Bruun LM; Madsen R Eur. J. Org. Chem 2014, 2014, 3232. [Google Scholar]
- (21).Hahm HS; Hurevich M; Seeberger PH Nat. Commun 2016, 7, 12482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Hsu Y; Lu XA; Zulueta MM; Tsai CM; Lin KI; Hung SC; Wong CH J. Am. Chem. Soc 2012, 134, 4549. [DOI] [PubMed] [Google Scholar]
- (23).Sherman AA; Yudina ON; Shashkov AS; Menshov VM; Nifant’ev NE Carbohydr. Res 2001, 330, 445. [DOI] [PubMed] [Google Scholar]
- (24).Schmidt D; Thiem J Beilstein J. Org. Chem 2010, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Bandara MD; Stine KJ; Demchenko AV Carbohydr. Res 2019, in press; DOI: 10.1016/j.carres.2019.107743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Li Z; Gildersleeve JJ Am. Chem. Soc 2006, 128, 11612. [DOI] [PubMed] [Google Scholar]
- (27).Nagorny P; Fasching B; Li X; Chen G; Aussedat B; Danishefsky SJ J. Am. Chem. Soc 2009, 131, 5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Palmacci ER; Plante OJ; Seeberger PH Eur. J. Org. Chem 2002, 595. [Google Scholar]
- (29).Mydock LK; Demchenko AV Org. Lett 2008, 10, 2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Mydock LK; Demchenko AV Org. Lett 2008, 10, 2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.