

Abstract
Cyclopropanations of benzene rings by oxidatively generated α-oxo gold carbenes are for the first time demonstrated in a Buchner reaction, in which readily available propargyl benzyl ethers are converted in one-pot to tetrahydropyranone-fused cycloheptatrienes via sequential oxidative gold catalysis and base-promoted isomerization. Additional examples of arene cyclopropanations without fragmentation of the cyclopropane ring are also realized.
Keywords: Cyclopropanation, Gold Carbene, Oxidation, Buchner reaction, Cycloheptatriene
Graphical Abstract
Gold-catalyzed intermolecular oxidation of alkynes[1] has become a valuable synthetic strategy since the original report in 2010,[2] owing to the efficient and expedient access to α-oxo gold carbene intermediates and/or their precursors and their exceptional reactivities and synthetic versatilities. A diverse range of functional structures of significant synthetic utilities have been developed.[2–4] Comparing to the Rh/Cu carbene chemistry based on diazo carbonyl compounds,5 this chemistry employs benign and readily available alkynes substrates and hence circumvents the use of hazardous and potentially explosive α-diazo ketone precursors (Scheme 1A). These α-oxo gold carbene intermediates typically display exceptional electrophilicities, and can be trapped by tethered or external nucleophiles, including cyclopropanation of tethered alkenes.[3e, 3g, 4a, 4n, 6] However, cyclopropanation of the more challenging benzene ring,[7] owing to the loss of aromaticity, has so far not been demonstrated in the oxidative gold catalysis, despite the reports of Rh/Cu catalysis based on hazardous diazo carbonyl substrates.[5b, 8]
Scheme 1.

Background and Design.
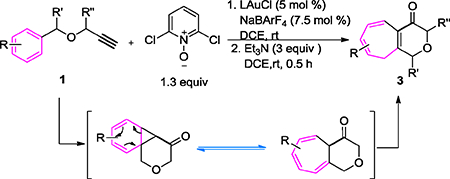
We surmised, as shown in Scheme 1B, that a readily available benzyl propargyl ether 1 could enable us to study cyclopropanations of arenes by α-oxo gold carbenes of type A. Our prior studies have revealed that the gold carbene generated from propargylic ethers[9] can readily undergo cyclization. The norcaradiene intermediate 4 would then be formed and undergo known reversible electrocyclic ring opening to afford the tetrahydropyranone-fused cycloheptatriene 3’ in the form of a Buchner reaction,[10] which could isomerize to the conjugated enone 3, or undergo alternative ring opening under acidic conditions to afford the Friedel-Crafts products 5. Notably, the related Rh-catalyzed Buchner reaction using the related diazo ketone counterpart of 1 (Scheme 1C) has only been reported once,[11] where the tetrahydropyranone-fused cycloheptatriene product was formed in a low yield.
Herein, we report the implementation of our design, which results in rapid access to the bicyclic heptatrienes.
At the outset, we employed the parent propargyl benzyl ether, i.e., 1a, for reaction discovery and condition optimization, some results of which are shown in Table 1. Initially, the sterically highly hindered Me4tBuXPhos was employed as the ligand as it was previously proved to be highly effective in promoting cyclization of α-oxo gold carbenes to tethered benzene rings and C-C triple bonds.[9] With NaBARF (7.5 mol %) as the chloride scavenger and 2-bromopyridine N-oxide (2a, 1.3 equiv) as the oxidant, the reaction was carried out in 1,2-dichloroethane (DCE) at room temperature for 2.5 h, and the resulting mixture was then treated with triethylamine (3 equiv) in order to isomerize 3’ into 3 (see Scheme 1B) and hence abolish the equilibrium between 4 and 3’. To our delight, the desired bicyclic heptatriene 3a was indeed formed in 7 % NMR yield (entry 1). Screening of various other pyridine/quinoline N-oxides (entries 2–6)[12] revealed that sterically hindered oxidants derived from electron-deficient pyridine ring, i.e., 2,6-dichloropyridine N-oxide (2c, entry 3), 2,6-dibromopyridine N-oxide (2d, entry 4) and the Hantzsch ester N-oxide 2g (entry 6)[3c] are among the most effective, with the halogenated ones being the best and equally effective and affording the desired product 3a in around 72% NMR yield. A range of other gold catalysts derived from typical ligands such as Ph3P, IPr, JohnPhos (entry 7) and tBuXPhos (entry 8) were ineffective, affording trace amount (<5%) of 3a. Sterically demanding BrettPhos (entry 9) and AdBrettPhos (entry 10) faired better, but are still substantially inferior to even more hindered Me4tBuXPhos, attesting the importance of ligand bulkiness in this chemistry. AntPhosAuCl/NaBARF4 was also tested but led to no reaction (entry 11). Deviation from the best conditions in entry 3 by lowering the reaction temperature to 0 ˚C led to a lower yield (entry 12), and by changing the chloride scanvenger from NaBArF4 to AgNTf2 resulted in predominant formation of the Friedel-Crafts product 5a (entry 13). In this latter case, the slightly exccess of acidic AgNTf2 is likely enough to promote the alternatively cyclopropane ring opening during the gold catalysis (Scheme 1B, from 4 to 5).
Table 1.
Initial discovery of gold-catalyzed oxidation of benzyl aryl ether 1a and screening of reaction conditions. [a]
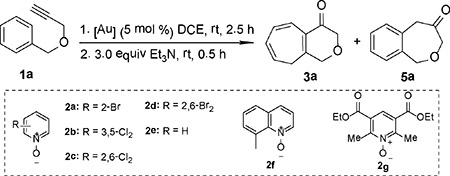 | |||||
|---|---|---|---|---|---|
| Entry | N-oxide | Gold catalyst (5 mol %) | Additives (7.5mol %) | Yield[b] |
|
| 3a | 5a | ||||
| 1 | 2a | Me4tBuXPhosAuCl | NaBArF4 | 7% | <2% |
| 2 | 2b | Me4tBuXPhosAuCl | NaBArF4 | 15% | <2% |
| 3 | 2c | Me4tBuXPhosAuCl | NaBArF4 | 72% [c] | <2% |
| 4 | 2d | Me4tBuXPhosAuCl | NaBArF4 | 71% | <2% |
| 5 | 2f | Me4tBuXPhosAuCl | NaBArF4 | 5% | <2% |
| 6 | 2g | Me4tBuXPhosAuCl | NaBArF4 | 52% | <2% |
| 7 | 2c | JohnPhosAuCl | NaBArF4 | 3% | <2% |
| 8 | 2c | tBuXPhosAuCl | NaBArF4 | 4% | <2% |
| 9 | 2c | BrettPhosAuCl | NaBArF4 | 32% | <2% |
| 10 | 2c | AdBrettPhosAuCl | NaBArF4 | 25% | <2% |
| 11 | 2c | AntPhosAuCl | NaBArF4 | nr [d] | |
| 12[e] | 2c | Me4tBuXPhosAuCl | NaBArF4 | 65% | <2% |
| 13[f] | 2c | Me4tBuXPhosAuCl | AgNTf2 | <2% | 49% |
[1a] = 0.05M, and 1.3 equiv of the oxidant.
NMR yield using diethyl phthalate as the internal reference.
Isolated yield is 69%. DCE = 1,2-dichloroethane.
95% of SM was recovered.
The reaction was carried out at 0°C for 6 h.
49% of 4a was isolated after 2.5 h.
With the optimal conditions (Table 1, entry 3) in hand, the reaction scope was examined. As shown in Table 2, entries 1 and 3, benzylic ethers possessing an electron-donating 4-Me or 4-MeO group at the benzene ring reacted as expected, despite exhibiting moderate yields. A poor regioselectivity along with a low yield was detected in the case of a meta-methyl group (entry 2). On the other hand, electron-withdrawing arene substituents are detrimental to the gold catalysis and, for example, little desired product was formed with an ortho-Br and para-CN. This can be attributed to the decreased nucleophilicity of the benzene ring. Interestingly, aliphatic substitutions at the substrate benzyl position including methyl (entry 4), phenethyl (entry 5) and the sterically demanding isopropyl (entry 6) and cyclohexyl group (entry 7) are readily accommodated and moreover, the corresponding gold catalyses afforded the bicyclic cycloheptatrienes (i.e., 3e-h) in good yields. An allyl and a benzyl group are also readily tolerated at the benzyl position (entries 8 and 9), and their π bonds do not noticeably interfere with the desired cyclopropanation, likely due to the formation of larger rings and hence slower cyclization kinetics. Moreover, double substitutions at the benzyl position with aliphatic, aromatic and/or allyl group were uneventful (entries 10–12). On the other hand, a methyl substitution at the propargylic position, as in entry 13, led to 3n in only 40% yield.
Table 2.
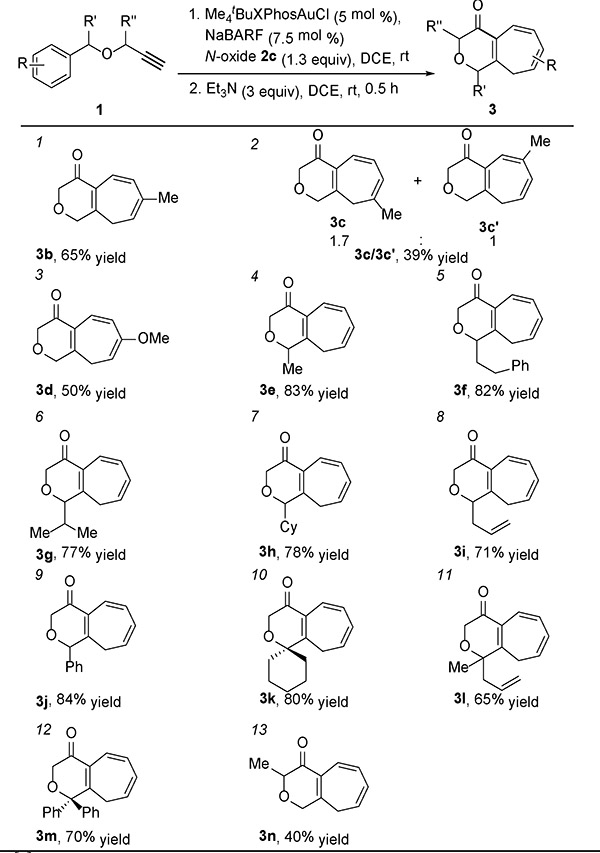 |
The reactions were run in a vial without exclusion of air and moisture, and the substrate concentration was 0.05 M.
Yields of isolated products are reported.
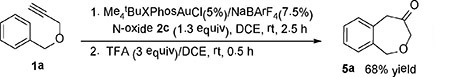
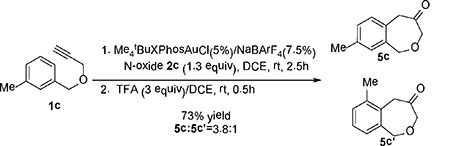
As the norcaradiene intermediate 4 (Scheme 1B) can undergo alternative cyclopropane ring opening under acidic conditions, we treated the resulting mixture of 1a upon the oxidative gold catalysis with trifluoroacetic acid (3 equiv) instead of Et3N. As expected, the 1,5-dihydrobenzo[c]oxepin-4(3H)-one 5a[13] was formed in 68% isolated yield. On the other hand, treatment of 3a with trifluoroacetic acid for much longer 10 h expectedly resulted in no formation of 5a. Likewise, substrate 1c with a meta-methyl substitution on the benzene ring also led to the oxepinones 5c/5c’ in 73% combined yield [Eqn (2)]. However, the inferred cyclopropanation regioselectivity, i.e., para:ortho = 3.8:1, is higher than that observed upon base treatment (1.7:1, Table 2, entry 2). Considering the difference in yields, it is likely that the base treatment decomposed some of the para-norcaradiene isomer.
 |
(1) |
 |
(2) |
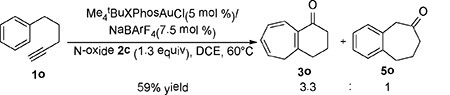
We attempted to extend the chemistry to the all-carbon counterpart of 1a, i.e., 1o. As shown in Eqn (3), the desired Buchner product 3o and the Friedel-Crafts benzocycloheptanone 5o were formed in a combined 59% isolated yields with a moderate regioselectivity.[14] Notably, the reaction required heating (60 ˚C) and no acid or base treatment was needed.
 |
(3) |
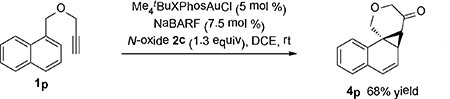
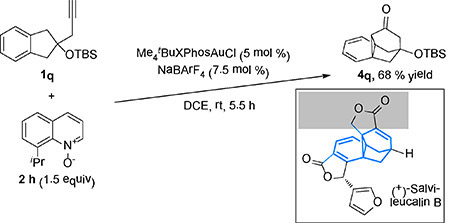
We are interesting in systems where the electrocyclic ring opening of the norcaradiene 4 is retarded. As such, the cyclopropanation of a benzene ring by in situ-generated α-oxo gold carbenes can be confirmed beyond doubt. We examined the propargyl 1-naphthylmethyl ether 1p. It is known in metal carbene chemistry[15] that vicinal-benzene-fused norcaradienes (i.e., the benzene ring next to the cyclopropane ring) do not readily undergo electrocyclic ring-opening due to the necessity of loss of aromaticity in the process. Indeed, our gold catalysis cleanly affords the cyclopropane product 4p [Eqn (4)]. With the substrate 1q easily prepared from 2-indanone, the benzene cyclopropanation product 4q was readily formed in a decent yield [Eqn (5)], the stability of which can be readily attributed to the avoidance of bridgehead double bonds in the ring open form. Interestingly, 4q possesses the core structure of salvileucalin B, [16] thus revealing the synthetic potential of the oxidative gold catalysis.
These experimental results point to [17] a reaction mechanism consistent with that shown in Scheme 1B, where α-oxo gold carbenes readily cyclopropanate benzene rings.
 |
(4) |
 |
(5) |
In summary, we have realized cyclopropanations of benzene rings by oxidatively generated α-oxo gold carbene intermediates. From readily available propargyl benzyl ethers, this chemistry enables an expedient synthesis of various tetrahydropyranone-fused cycloheptatrienes upon basic treatment, while avoiding the use of hazardous diazo carbonyl substrates. Alternative acidic workup provides rapid access to 3, 5-dihydrobenzo[c]oxepin-4(1H)-ones. We have also demonstrated that some norcaradiene intermediates, e.g. 4p and 4q, can be isolated, thereby confirming the arene cyclopropanation in these oxidative gold catalysis.
Experimental Section
General procedure for one-pot gold-catalyzed oxidative propargyl benzyl ethers to tetrahydropyranone-fused cycloheptatrienes 3:
2, 6-dichloropyridine N-oxide 2c (42.4 mg, 0.26 mmol), and Me4t-BuXPhosAuCl (7.1 mg, 0.010 mmol, 5 mol %) and NaBArF4 (13 mg, 0.015 mmol, 7.5 mol %) were added sequentially to a solution of the propargyl benzyl ethers 1 (0.20 mmol) in DCE (4 mL. 0.05M) at room temperature. The resulting reaction mixture was stirred at RT, and the progress of the reaction was monitored by TLC. The reaction typically took 1–3 h. Upon completion, the reaction mixture was added 3 equiv. Et3N and stirred for 0.5 h at room temperature. The reaction mixture was concentrated under vacuum. The residue was purified by chromatography on silica gel (eluent: hexanes/ethyl acetate) to afford the desired products 3.
Supplementary Material
Acknowledgements
This work is supported financially by NIH (R01GM123342). K. Ji thanks the National Natural Science Foundation of China (NSF-21502150) Northwest A&F University Youth Training Programme (Z109021601) for support. K. Ji also thanks Professor Wenjun Tang for providing the ligand AntPhos.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/adsc.201######.
Contributor Information
Kegong Ji, Email: jikegong@nwsuaf.edu.cn.
Liming Zhang, Email: zhang@chem.ucsb.edu.
References
- [1].Zhang L, Acc. Chem. Res, 2014, 47, 877–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Ye L, He W and Zhang L, J. Am. Chem. Soc, 2010, 132, 8550–8551; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ye L, Cui L, Zhang G and Zhang L, J. Am. Chem. Soc, 2010, 132, 3258–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].For additional work by us, see:He W, Li C and Zhang L, J. Am. Chem. Soc, 2011, 133, 8482–8485;Luo Y, Ji K, Li Y and Zhang L, J. Am. Chem. Soc, 2012, 134, 17412–17415;Wang Y, Ji K, Lan S and Zhang L, Angew. Chem., Int. Ed, 2012, 51, 1915–1918;Ji K, Zhao Y and Zhang L, Angew. Chem., Int. Ed, 2013, 52, 6508–6512;Ji K and Zhang L, Org. Chem. Front, 2014, 1, 34–38;Chen H and Zhang L, Angew. Chem., Int. Ed, 2015, 54, 11775–11779;Ji K, Zheng Z, Wang Z and Zhang L, Angew. Chem., Int. Ed, 2015, 54, 1245–1249.
- [4].For selected work by other research groups, see:Vasu D, Hung H-H, Bhunia S, Gawade SA, Das A and Liu R-S, Angew. Chem., Int. Ed, 2011, 50, 6911–6914;Davies PW, Cremonesi A and Martin N, Chem. Commun, 2011, 47, 379–381;Qian D and Zhang J, Chem. Commun, 2012, 48, 7082–7084;Xu M, Ren T-T and Li C-Y, Org. Lett, 2012, 14, 4902–4905;Bhunia S, Ghorpade S, Huple DB and Liu R-S, Angew. Chem., Int. Ed, 2012, 51, 2939–2942;Ghorpade S, Su M-D and Liu R-S, Angew. Chem., Int. Ed, 2013, 52, 4229–4234;Ling H-B, Chen Z-S, Yang F, Xu B, Gao J-M, Ji K, J. Org. Chem. 2017, 82, 7070−7076;Henrion G, Chavas TEJ, Le Goff X and Gagosz F, Angew. Chem., Int. Ed, 2013, 52, 6277–6282;Fu J, Shang H, Wang Z, Chang L, Shao W, Yang Z and Tang Y, Angew. Chem., Int. Ed, 2013, 52, 4198–4202;Sun N, Chen M and Liu Y, J. Org. Chem, 2014, 79, 4055–4067;Chen M, Chen Y, Sun N, Zhao J, Liu Y and Li Y, Angew. Chem., Int. Ed, 2015, 1200–1204;Shu C, Liu R, Liu S, Li J-Q, Yu Y-F, He Q, Lu X and Ye L-W, Chem. - Asian J, 2015, 10, 91–95;Ferrer S and Echavarren AM, Angew. Chem., Int. Ed, 2016, 55, 11178–11182.Yao X, Wang T, Zhang X, Wang P, Zhang B, Wei J, Zhang Z, Adv. Synth. Catal 2016, 358, 1534–1539.
- [5].a) Taber DF, in Carbon-Carbon σ-Bond Formation, ed. Pattenden G, Pergamon Press, Oxford, England; New York, 1991, vol. 3, pp. 1045–1062; [Google Scholar]; b) Doyle MP, McKervey MA and Ye T, Modern catalytic methods for organic synthesis with diazo compounds: from cyclopropanes to ylides, Wiley, New York, 1998; [Google Scholar]; c) Davies HML and Beckwith REJ, Chem. Rev, 2003, 103, 2861–2903. [DOI] [PubMed] [Google Scholar]
- [6].a) Qian D and Zhang J, Chem. Commun, 2011, 47, 11152–11154; [DOI] [PubMed] [Google Scholar]; b) Qian D, Hu H, Liu F, Tang B, Ye W, Wang Y and Zhang J, Angew. Chem., Int. Ed, 2014, 53, 13751–13755. [DOI] [PubMed] [Google Scholar]
- [7].For cyclopropanation of furans, see:Hashmi ASK, Frost TM and Bats JW, J. Am. Chem. Soc, 2000, 122, 11553–11554.Hashmi ASK, Rudolph M, Siehl H-U, Tanaka M, Bats JW, Frey W, Chem. Eur. J 2008, 14, 3703–3708. “Hashmi ASK, Angew. Chem. Int. Ed 2010, 49, 5232–5241.
- [8].Lebel H, Marcoux J-F, Molinaro C and Charette AB, Chem. Rev, 2003, 103, 977–1050. [DOI] [PubMed] [Google Scholar]
- [9].a) Ji K, Liu X, Du B, Yang F and Gao J, Chem. Commun, 2015, 51, 10318–10321; [DOI] [PubMed] [Google Scholar]; b) Zheng Z and Zhang L, Org. Chem. Front, 2015, 2, 1556–1560. [Google Scholar]
- [10].For gold-catalyzed retro-Buchner reactions, see:Solorio-Alvarado CSR, Wang Y and Echavarren AM, J. Am. Chem. Soc, 2011, 133, 11952–11955;Lauterbach T, Higuchi T, Hussong MW, Rudolph M, Rominger F, Mashima K and Hashmi ASK, 2015, 357, 775–781;Wang Y, McGonigal PR, Herlé B, Besora M and Echavarren AM, J. Am. Chem. Soc, 2014, 136, 801–809;Herlé B, Holstein PM and Echavarren AM, ACS Catal., 2017, 7, 3668–3675.
- [11].a) Doyle MP, Protopopova MN, Peterson CS, Vitale JP, McKervey MA and García CF, J. Am. Chem. Soc, 1996, 118, 7865–7866; [Google Scholar]; b) Liu Y, Deng Y, Zavalij PY, Liu R, Doyle MP, Chem. Commun, 2015, 51, 565–568; [DOI] [PubMed] [Google Scholar]; c) Xu X, Wang X, Zavalij PY, Doyle MP, Org. Lett. 2015, 17, 790−793. [DOI] [PubMed] [Google Scholar]
- [12].Wang Y and Zhang L, Synthesis, 2015, 47, 289–305. [Google Scholar]
- [13].Xu Z, Chen H, Wang Z, Ying A and Zhang L, J. Am. Chem. Soc, 2016, 138, 5515–5518. [DOI] [PubMed] [Google Scholar]
- [14].Kennedy M, McKervey MA, Maguire AR, Tuladhar SM and Twohig MF, J. Chem. Soc. Perk. T1, 1990, 1047–1054. [Google Scholar]
- [15].a) Manitto P, Monti D and Speranza G, J. Org. Chem, 1995, 60, 484–485; [Google Scholar]; b) Pérez PJ, Díaz-Requejo MM and Rivilla I, Beilstein J. Org. Chem, 2011, 7, 653–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Aoyagi Y, Yamazaki A, Nakatsugawa C, Fukaya H, Takeya K, Kawauchi S and Izumi H, Org. Lett, 2008, 10, 4429–4432; [DOI] [PubMed] [Google Scholar]; b) Levin S, Nani RR and Reisman SE, J. Am. Chem. Soc, 2011, 133, 774–776. [DOI] [PubMed] [Google Scholar]
- [17].a) Dorel R, Echavarren AM, Chem. Rev 2015, 115, 9028–9072. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hashmi ASK, Angew. Chem. Int. Ed 2010, 49, 5232–5241. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.