

Abstract
Background and aim
Overdose of acetaminophen (APAP) leads to liver injury, which is one of the most common causes of liver failure in the United States. We previously demonstrated that pharmacological activation of autophagy protects against APAP-induced liver injury in mice via removal of damaged mitochondria and APAP-adducts (APAP-ADs). Using an image-based high-throughput screening for autophagy modulators, we recently identified that chlorpromazine (CPZ), a dopamine inhibitor used for anti-schizophrenia, is a potent autophagy inducer in vitro. Therefore, the aim of the present study is to determine whether CPZ may protect against APAP-induced liver injury via inducing autophagy.
Methods
Wild type C57BL/6J mice were injected with APAP to induce liver injury. CPZ was administrated either at the same time with APAP (co-treatment) or 2 h later after APAP administration (post-treatment). Hemotoxyline and eosin (H&E) staining of liver histology, terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick end labeling (TUNEL) staining of necrotic cell death as well as serum levels of alanine aminotransferase (ALT) were used to monitor liver injury.
Results
We found that CPZ markedly protected against APAP-induced liver injury as demonstrated by decreased serum levels of ALT, liver necrotic areas as well as TUNEL-positive cells in mice that were either co-treated or post-treated with CPZ. Mechanistically, we observed that CPZ increased the number of autolysosomes and decreased APAP-induced c-Jun N-terminal kinase activation without affecting the metabolic activation of APAP. Pharmacological inhibition of autophagy by chloroquine partially weakened the protective effects of CPZ against APAP-induced liver injury.
Conclusions
Our results indicate that CPZ ameliorates APAP-induced liver injury partially via activating hepatic autophagy and inhibiting JNK activation.
Keywords: Chlorpromazine (CPZ), Acetaminophen (APAP), Drug-induced liver injury, Necrosis, Autolysosome, Hepatotoxicity, Glutathione (GSH), Oxidant stress
1. Introduction
Acetaminophen (APAP) is a common over-the-counter analgesic prescription. APAP overdose leads to severe hepatotoxicity and remains one of the top causes of acute liver failure in the United States.1 A large portion of APAP in liver undergoes glucuronidation and sulfation and is secreted into bile and plasma, while a small portion is metabolized by cytochrome P450 2E1 (CYP2E1) and generates a reactive metabolite, N-acetyl-p-benzoquinone imine (NAPQI).2 The highly reactive electrophile NAPQI depletes liver glutathione (GSH), and excessive NAPQI then covalently binds to intracellular proteins and nucleic acids to form APAP-adducts (APAP-ADs). The excessive NAPQI and subsequent formation of APAP-ADs are believed to be the trigger of mitochondrial dysfunction and the generation of reactive oxygen species (ROS) resulting in hepatocellular necrosis.3,4 Current treatment options for APAP-induced acute liver failure are still limited. N-acetylcysteine (NAC), the current standard of care treatment for patients with APAP overdose, restores the hepatic GSH content and therefore enhances the capacity of the liver to detoxify NAPQI during the metabolism phase and also the capacity to scavenge ROS and peroxynitrite during the progression phase of the injury.5,6 Hence, NAC is most effective when administered as early as possible after an overdose with declining efficacy when patients present at later time points.7 Thus, novel treatments for APAP-induced liver injury are still needed.
c-Jun N-terminal kinase (JNK) is a serine/threonine kinase of the mitogen-activated protein kinase (MAPK) family. JNK has three isoforms JNK1, 2, 3, and JNK1 and JNK2 are universally expressed including in the liver.8 Under various stress stimuli, JNK is phosphorylated, activated and subsequently induces or inhibits its downstream proteins controlling cell survival and cell death. Prolonged activation of JNK is involved in liver inflammation, steatosis, fibrosis and hepatocellular carcinoma.9,10 Previous studies have shown JNK phosphorylation and mitochondrial translocation of phospho-JNK (p-JNK) after APAP administration.11,12 Mice deficient of JNK1 or JNK2 are not protected against APAP-induced liver injury, possibly due to the compensatory effect of the alternative JNK protein.13 However, knockdown with antisense oligonucleotides or pharmacological inhibition of both JNK1 and JNK2 protects against APAP-induced hepatotoxicity in mice and in primary human hepatocytes.12,14,15 The mechanistic role of JNK in the development of APAP-induced cell death is the amplification of the mitochondrial oxidant stress, which is a central event in the pathophysiology.16
Macroautophagy/autophagy is a highly conserved lysosomal degradation pathway regulated by a series of autophagy-related genes. Unselective autophagy, activated by nutrient deprivation, breaks down cellular components to provide sources of energy and nutrients to survive.17 Selective autophagy, activated under nutrient-sufficient and -insufficient conditions, removes damaged or excessive organelles and protein aggregates as a protective mechanism.18–22 Autophagy is crucial in many physiological and pathological conditions, including aging, liver diseases, heart diseases, myopathies, cancers, adipocyte differentiation, and immune responses.23–29 We recently demonstrated that the activation of autophagy protects against APAP-induced hepatotoxicity by removing APAP-ADs and damaged mitochondria in vivo and in vitro.30–34
Researchers have been actively seeking autophagy inducers and inhibitors for clinical applications. Previously we developed a cell-based high-throughput image screening system and identified a few autophagy inducers.35 Among them chlorpromazine (CPZ) induces autophagy in mouse embryonic fibroblasts and two other human cancer cell lines, human colorectal tumor (HCT) 116 and A549. CPZ is a classic antipsychotic drug, and its mechanism of action is still poorly understood. In the present study, we aimed to determine whether CPZ would protect against APAP-induced liver injury in mice and its possible underlying mechanisms. We found co-treatment or post-treatment with CPZ markedly decreased APAP-induced liver injury in mice. CPZ increased the number of autolysosomes and decreased APAP-induced JNK activation without affecting APAP metabolism.
2. Materials and methods
2.1. Reagents
For animal experiments, stock solutions of CPZ hydrochloride, chloroquine (CQ), and buthionine sulphoximine (BSO) were prepared in double distilled water (ddH2O), and diluted with saline before use. APAP (Sigma) was dissolved in saline and warmed up to ensure its complete dissolution prior to injection. All reagents were either from Sigma or Thermal Fisher Scientific.
2.2. Antibodies
The rabbit anti-APAP-AD antibody was a kind gift from Dr. Lance Pohl (National Heart, Lung and Blood Institute).31 The other antibodies were: CYP2E1 (Abcam, #ab28146),p62 (Abnova, #H00008878-M01), phospho-eukaryotic initiation factor 4E-binding protein 1 (p-4EBP1) (Cell Signaling, #9451), total 4EBP1 (Cell Signaling, #9452), p-JNK (Cell Signaling, #4668), total JNK (BD Pharmingen, #554285), β-actin (Sigma, #a5541), and glyceralde-hyde phosphate dehydrogenase (GAPDH) (Cell Signaling, #2118). The microtubule-associated protein 1 light chain 3 (LC3) antibody was developed as described previously.36 The secondary antibodies were horseradish peroxidase (HRP)-conjugated goat-anti-rabbit (Jackson ImmunoResearch, #111–035-045), and HRP-conjugated goat-anti-mouse (Jackson ImmunoResearch, #115-035-062).
2.3. Animals
Two-month old (C57BL/6J) male mice were caged with free access to chow food and water in 12/12 light cycle. Mice were injected with 500 mg/kg of APAP intraperitoneally (i.p.) in the morning. In co-treatment model, 6 mg/kg of CPZ was injected i.p. at the same time with APAP, and the mice were euthanized at 0.5, 2, 6, or 24 h after treatment. In post-treatment model, 6 mg/kg of CPZ was injected i.p. 2 h after APAP treatment, and the mice were euthanized 6 h after APAP. In post-treatment + CQ model, CQ (60 mg/kg) was injected i.p. at the same time with APAP. BSO (2 mmol/kg) was injected i.p. 1 h after APAP treatment. Matched volume of saline was injected as control. All animal protocols were approved by the Institutional Animal Care and Use Committee of the University of Kansas Medical Center.
2.4. Immunoblot
For total liver lysates, livers were dissected and snap-frozen in liquid nitrogen after the mice were euthanized, and liver tissues were homogenized in radioimmunoprecipitation assay buffer. For nuclear/cytosolic fractionation, frozen liver tissues were processed using a NE/PER kit (Thermo Fisher) following the manufacturer’s instructions. All the protein lysates were supplied with a protease inhibitor cocktail (BioTool), mixed with sodium dodecyl sulfate (SDS) loading buffer containing dithiothreitol and boiled at 95 °C for 10 min. Thirty microgram of protein was loaded for SDS-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membrane. The membranes were blocked in 5% milk in Tris-buffered saline with 0.1% Tween 20 buffer, and then incubated with primary antibody and secondary antibody prepared in 5% milk in Tris-buffered saline with 0.1% Tween 20. The signals were detected with SuperSignal™ West Pico Chemiluminescent Substrate (Thermal) and/or Immobilon Western HRP Substrate (Millipore). Densitometry was analyzed with Image J software (National Institutes of Health (NIH), USA).
2.5. Histology analysis
Fresh liver tissues were kept in 10% formaldehyde overnight and transferred to 70% ethanol for at least 24 h. Then the tissues were dehydrated, embedded in paraffin, and cut into 5 μm slices. For general morphology, slides were stained with hemotoxyline and eosin (H&E). For terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling (TUNEL) assay, slides were stained with the In Situ Cell Death Detection Kit, AP (Roche Diagnostics) following the instruction manual. The percentage of necrotic area was measured with Image J software.
2.6. Electron microscopy (EM) analysis
For EM studies, fresh liver tissues were fixed with EM fixation buffer (2.5% glutaraldehyde, 1% OsO4, 100 mM sodium cacodylate buffer, pH 7.4). The tissues were further dehydrated, cut into thin sections and stained with uranyl acetate and lead citrate. All the images were obtained using a JEM 1016CX electron microscope with a digital camera. Autophagic vesicles were counted in at least 15 cells in each group. The area of cytoplasm was measured with Image J software.
2.7. GSH/glutathione disulfide (GSSG) measurement
GSH and GSSG levels in liver tissue were measured as previously described.37 For GSH measurement, frozen liver tissues were homogenized in 3% sulfosalicylic acid, centrifuged, and diluted in 0.01 N hydrochloric acid for GSH measurement with the modified Tietze assay. Another aliquot was added to potassium phosphate buffer containing N-ethylmaleimide to trap GSH for GSSG measurement. The residual N-ethylmaleimide was removed by a Sep-Pak column and GSSG was measured by a modified Tietze assay.37
2.8. Serum alanine aminotransferase (ALT) measurement
Blood samples were collected from auxiliary artery after the mice were euthanized. Samples were allowed to sit for 30 min, then centrifuged at 3000 rpm at 4 °C for 10 min, and the supernatant serum was collected. ALT activities were measured using the ALT (SGPT) Reagent Set (POINTE Scientific) following the instruction manual at λ = 340 nm. Millipore water was used as blank control.
2.9. Statistical analysis
Data were presented as the mean ± standard error of the mean (SEM). Experimental data were subjected to Student’s t-test when 2 groups are compared or one-way analysis of variance (ANOVA) where appropriate. P < 0.05 was considered statistically significant. All statistical analyses were performed using IBM SPSS software (IBM, USA).
3. Results
3.1. CPZ co-treatment and post-treatment protect against APAP-induced liver injury
To determine whether CPZ would protect against APAP-induced liver injury, fed C57BL/6J mice were treated with CPZ i.p. at the same time when APAP was administered (Fig. 1A). APAP led to significantly elevated serum ALT levels at 6 and 24 h (Fig. 1B). Liver H&E staining and TUNEL staining at 6 h showed severe centrilobular necrosis, hemorrhage and increased TUNEL-positive cells in pericentral vein areas, which was almost abolished in the presence of CPZ (Figs. 1C–E). Consistent with the histological changes and TUNEL staining, the serum levels of ALT were also markedly lower in the CPZ-treated animals for up to 24 h (Fig. 1B). To test whether CPZ post-treatment would also have a protective effect against APAP-induced liver injury, mice were treated with CPZ i.p. 2 h after APAP administration (Fig. 2A), at which time most of the APAP was already metabolized. CPZ post-treatment significantly inhibited APAP-induced serum ALT elevation (Fig. 2B), liver necrosis and TUNEL-positive cells (Figs. 2C–E). These results indicate that both co-treatment and post-treatment with CPZ protect against APAP-induced liver injury.
Fig. 1. CPZ co-treatment protects against APAP-induced liver injury.

(A) Schematic illustration of APAP treatment and CPZ co-treatment in vivo. Mice were injected i.p. with APAP at 500 mg/kg and CPZ at 6 mg/kg. Serum and livers were collected at 0.5, 2, 6 and 24 h respectively. (B) Serum ALT at different time points (N = 3–7). (C) Representative images (× 20) of liver H&E staining at 6 h after treatment. Dashed line encloses necrotic area. (D) Representative images (× 20) of liver TUNEL staining at 6 h after treatment. (E) Percentage of necrotic area based on H&E staining (N = 4). Data are presented as the mean ± SEM. Student’s t-test, *P < 0.05 (APAP vs. APAP + CPZ). Abbreviations: CPZ, chlorpromazine; APAP, acetaminophen; ALT, alanine aminotransferase; H&E, hematoxylin and eosin; TUNEL, terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick end labeling; i.p., intraperitoneally; SEM, standard error of the mean.
Fig. 2. CPZ post-treatment protects against APAP-induced liver injury.

(A) Schematic illustration of APAP treatment and CPZ post-treatment in vivo. Mice were treated i.p. with APAP (500 mg/kg) and after 2 h with CPZ (6 mg/kg) or saline. Serum and livers were collected at 6 h after APAP treatment. (B) Serum ALT at 6 h. (C) Representative images ( × 20) of liver H&E staining at 6 h after treatment. Dashed line encloses necrotic area. (D) Representative images ( × 20) of liver TUNEL staining at 6 h after treatment. (E) Percentage of necrotic area based on H&E staining (N = 4). Data are presented as the mean±SEM. Student’s t-test, *P<0.05. Abbreviations: CPZ, chlorpromazine; APAP, acetaminophen; ALT, alanine aminotransferase; H&E, hematoxylin and eosin; TUNEL, terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick end labeling; i.p., intraperitoneally; SEM, standard error of the mean.
3.2. CPZ does not affect APAP metabolism in mouse livers
APAP is mainly metabolized by liver CYP2E1, and its metabolite NAPQI binds to various intracellular proteins and forms APAP-ADs. After CPZ co-treatment, neither the levels of liver CYP2E1 nor APAP-ADs were altered at 6 h compared with APAP treatment alone (Figs. 3A and B), suggesting that CPZ did not inhibit CYP2E1-mediated APAP metabolism. Consistent with the CYP2E1 and APAP-ADs data, hepatic GSH levels were depleted to the same levels regardless of the presence or absence of CPZ after mice were treated with APAP for 2 h. The levels of hepatic GSH started to recover at 6 h and completely recovered to the basal levels after APAP treatment for 24 h in the presence or absence of CPZ (Fig. 3C). The GSSG levels and the GSSG/GSH ratios were significantly higher at 24 h in APAP group, which was markedly decreased in APAP and CPZ co-treatment group. These data suggest that CPZ does not affect APAP metabolism but inhibits APAP-induced oxidative stress at the late phase in mouse livers. CPZ increases the number of autolysosomes and autophagic flux in APAP-treated mouse livers.
Fig. 3. CPZ does not affect APAP metabolism.

Mice were treated i.p. with APAP (500 mg/kg) and CPZ (6mg/kg) simultaneously, and livers were collected at 0.5, 2, 6 and 24h respectively. (A) Representative immunoblot of CYP2E1 and APAP-ADs in liver collected at 6 h after treatment. β-actin was used as loading control. (B) Densitometry of (A). (C) Total GSH and GSSG were measured, and GSSG/GSH ratios were calculated (N = 3—4). Data are presented as the mean ± SEM. Student’s t-test, *P< 0.05 (APAP vs. APAP + CPZ). Abbreviations: CPZ, chlorpromazine; APAP, acetaminophen; i.p., intraperitoneally; APAP-ADs, acetaminophen adducts; GSH, glutathione; GSSG, glutathione disulfide; CYP2E1, cytochrome P450 2E1; SEM, standard error of the mean.
3.3. CPZ increases the number of autolysosomes and autophagic flux in APAP-treated mouse livers
We previous reported that APAP increased autophagosome formation in mouse livers.30,38 Consistent with these findings, we found that APAP treatment increased LC3-II levels (Figs. 4A and B) and the number of autophagic vacuoles in mouse livers (Figs. 5A and B). Interestingly, the p62 protein expression was also increased after APAP treatment, possibly due to autophagy-independent transcriptional upregulation (data not shown). CPZ alone decreased LC3-I expression, increased LC3-II expression and significantly increased late autophagic vacuoles. Co-treatment of CPZ with APAP decreased the levels of LC3-II and p62 compared with APAP treatment alone (Figs. 4A and B), suggesting a possible increased autophagic degradation of both LC3-II and p62 in the cotreatment group. Indeed, EM studies also revealed that co-treatment of CPZ with APAP increased the degradative autolysosome (AVd) numbers compared with APAP treatment alone (Figs. 5A and B). Consistent with our previous findings in cultured non-hepatocytes, CPZ alone increased the levels of phosphorylated 4EBP1, a substrate protein of mammalian target of rapamycin (mTOR), suggesting CPZ-increased autophagy is mTOR-independent.35 APAP treatment decreased levels of 4EBP1 phosphorylation, which was not significantly affected by the cotreatment with CPZ (Figs. 4A and B). To further determine the role of autophagy in the protection of APAP-induced liver injury by CPZ, we treated the mice with the autophagy inhibitor CQ with APAP followed by CPZ treatment (Fig. 6A). As can be seen in Fig. 6B, post-treatment of CPZ markedly decreased the serum levels of ALT induced by APAP. Co-treated in mice with CQ significantly increased serum ALT levels compared with APAP + CPZ group, suggesting that the protective effects of CPZ against APAP-induced liver injury is partially via autophagy induction. Indeed, the hepatic levels of LC3-II were higher in the APAP + CPZ + CQ group than either in APAP alone or in APAP + CPZ group, suggesting that CPZ enhances hepatic autophagic flux in APAP-treated mice (Figs. 6C and D). Together, these results suggest that APAP and CPZ co-treatment increases numbers of AVd and autophagic flux and protects against APAP-induced liver injury in mice.
Fig. 4. CPZ co-treatment decreases hepatic levels of p62 and LC3-II.

Mice were treated i.p. with APAP (500 mg/kg) and CPZ (6 mg/kg) simultaneously, and livers were collected at 6 h. (A) Representative immunoblot of liver p62, LC3, p-4EBP1 and total 4EBP1. β-actin was used as loading control. (B) Densitometry of (A) (N = 3—4). Abbreviations: CPZ, chlorpromazine; APAP, acetaminophen; i.p., intraperitoneally; LC3, microtubule-associated protein 1 light chain 3; 4EBP1, eukaryotic initiation factor 4E-binding protein 1.
Fig. 5. CPZ increases the number of autolysosomes in APAP-treated mouse livers.

Mice were treated i.p. with APAP (500 mg/kg) and CPZ (6 mg/kg) simultaneously, and liver sections were fixed for EM analysis. (A) Representative EM images. Results are presented as the mean ± SEM. AVi (white arrow), initiative autophagic vesicle; AVii (red arrow), intermediate autophagic vesicle; AVd (black arrow), degradative autophagic vesicle, also referred to autolysosome. Scale bar: 500 nm. (B) Quantification of autophagic vesicles in EM images. At least 15 cells were counted in each group. One-way ANOVA, *P < 0.05 (vs. saline control), #P < 0.05 (vs. APAP). Abbreviations: CPZ, chlorpromazine; APAP, acetaminophen; i.p., intraperitoneally; EM, electron microscopy; AVd, degradative autolysosome; M, mitochondria; N, nucleus; SEM, standard error of the mean; ANOVA, analysis of variance.
Fig. 6. CQ co-treatment partially ameliorates the protective effects of CPZ against APAP-induced liver injury.

(A) Mice were treated i.p. with APAP (500 mg/kg) and CQ (60 mg/kg) and 2 h later these mice were further treated with CPZ (6 mg/kg, i.p.) for another 4 h. (B) Serum ALT levels were analyzed. (C) Representative immunoblot of LC3. β-Actin was used as loading control. (D) Densitometry of (C) (N = 3). Results are presented as the mean ± SEM. Student’s t-test, *P< 0.05 (vs. saline control), #P< 0.05 (vs. APAP). Abbreviations: CPZ, chlorpromazine; APAP, acetaminophen; i.p., intraperitoneally; CQ chloroquine; ALT, alanine aminotransferase; LC3, microtubule-associated protein 1 light chain 3; SEM, standard error of the mean.
3.4. Depletion of GSH by BSO abolishes the protective effects of CPZ against APAP-induced liver injury
To determine whether the levels of hepatic GSH would affect the protective effects of CPZ against APAP-induced liver injury, we co-treated mice with BSO 1 h post APAP administration but 1 h prior to CPZ injection (Fig. 7A). We found BSO alone significantly lowered the hepatic GSH levels compared with control group (Fig. 7B). BSO together with APAP and CPZ also further decreased hepatic GSH levels compared with APAP + CPZ or APAP alone group. Interestingly, the decreased ALT levels in APAP + CPZ group were almost completely reversed by BSO (Fig. 7C). These results indicate that the protective effects of CPZ against APAP-induced liver injury can be abolished by further depletion of hepatic GSH levels.
Fig. 7. BSO co-treatment abolishes the protective effects of CPZ against APAP-induced liver injury.

(A) Mice were treated i.p. with APAP (500 mg/kg), and 1 h and 2 h later these mice were further treated with BSO (2 mmol/kg) and CPZ (6 mg/kg) respectively, and livers were collected 6 h after treatment. (B) Total hepatic GSH and (C) serum ALT levels were measured. Results are presented as the mean ± SEM (N = 3—4). Student’s t-test, *P< 0.05 (vs. saline control), #P < 0.05 (vs. APAP). Abbreviations: BSO, buthionine sulphoximine; CPZ, chlorpromazine; APAP, acetaminophen; i.p., intraperitoneally; GSH, glutathione; ALT, alanine aminotransferase; SEM, standard error of the mean.
3.5. CPZ co-treatment inhibits JNK activation
It has been suggested that increased JNK phosphorylation can amplify mitochondrial production of ROS and peroxynitrite and increase mitochondria membrane permeability transition resulting in APAP-induced cell death.39 We found that APAP treatment increased levels of phosphorylated total JNK to almost 15-fold of control mice, which was markedly decreased by co-treatment with CPZ in mouse livers (Figs. 8A and B). These results suggest that CPZ co-treatment in part reduces APAP-induced JNK activation in mouse livers.
Fig. 8. CPZ co-treatment partially decreases JNK phosphorylation.
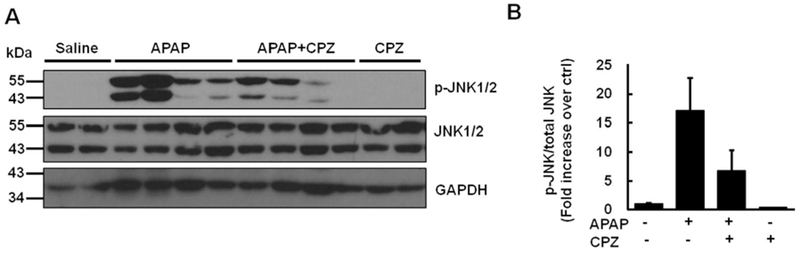
Mice were treated i.p. with APAP (500 mg/kg) and CPZ (6 mg/kg) simultaneously, and livers were collected at 6 h. (A) Representative immunoblot of total JNK and p-JNK. GAPDH was used as loading control. (B) Densitometry of (A) (N = 3—4). Results are presented as the mean ± SEM. Abbreviations: CPZ, chlorpromazine; JNK, c-Jun N-terminal kinase; p-JNK, phospho-C-Jun N-terminal kinase; APAP, acetaminophen; i.p., intraperitoneally; GAPDH, glyceraldehyde phosphate dehydrogenase; SEM, standard error of the mean.
4. Discussion
CPZ is a dopamine receptor antagonist and a calcium channel blocker.40–43 Previously, a few papers studied the protective effects of CPZ against APAP-induced liver injury in mice. Saville et al.44 treated fed and fasted mice with CPZ (6 mg/kg) 1 h prior to APAP treatment and observed complete protection up to 24 h. They found that CPZ pre-treatment inhibited APAP-induced elevation in phosphorylase α activity, suggesting a possible inhibition of cytosolic calcium level. A study with similar conditions found that CPZ pre-treatment (6 mg/kg CPZ, 1 h pre-treatment) inhibited APAP-induced decrease in mitochondrial calcium sequestration, suggesting a restoration of mitochondrial calcium homeostasis.45 Another study confirmed that CPZ pre-treatment (25 mg/kg CPZ, 1 h or 2 h pre-treatment) decreased nuclear calcium level and nuclear DNA fragmentation.46 Later the same group showed CPZ post-treatment (25 mg/kg CPZ, 1 h post-treatment) also led to protection up to 24 h, and it inhibited APAP-induced lipid peroxidation and DNA fragmentation.47 Our study administered CPZ at a low concentration (6 mg/kg) and added a later time point (2 h post-treatment) when a substantial fraction of the APAP dose was already metabolized, suggesting a greater potential for translation into clinical application, considering most APAP overdose patients will only receive treatment many hours after APAP consumption. Though there is abundant evidence showing that CPZ intervention is associated with decreased cytosolic calcium level, whether APAP-induced calcium efflux is a major cause of cell death or a secondary effect of the injury is still debatable.
Here we reported that several novel mechanisms may account for the protective effects of CPZ against APAP-induced liver injury. Firstly, we previously identified CPZ as a potent autophagy inducer via a high-throughput imaging screening in cultured cells.35 CPZ may protect against APAP-induced liver injury via enhanced auto-phagy by targeting APAP-induced damaged mitochondria. Indeed, co-treatment of CPZ with APAP increased the degradation of LC3-II and p62 protein, supporting a possible increased autophagic flux in mouse livers. The increased numbers of AVd by EM studies may help to explain the decreased rather than increased LC3-II levels in the co-treatment of CPZ and APAP group. Treatment with the lysosomal inhibitor CQ in the mouse livers together with CPZ and APAP further confirmed that CPZ increases autophagic flux in this model. However, based on the partially increased serum ALT levels by CQ and previous findings, increased autophagy may have an impact on the pathophysiology but cannot account for the entire mechanism of protection by CPZ.30,31,48
Secondly, APAP overdose triggers MAPK cascade and induces JNK activation in mouse hepatocytes.49 Sustained JNK activation loop well correlates to APAP-induced acute injury.12 Recently it is found that phosphorylated JNK binds to Src homology 3 domain (SH3) homology associated Bruton tyrosine kinase (BTK) binding protein (Sab) on the mitochondria outer membrane.50 The interaction between JNK and Sab dephosphorylates and inactivates intramitochondrial Src in a Src homology region 2 domain-containing phosphatase-1 (SHP-1)-dependent, docking protein 4 (DOK4)-dependent manner, and leads to electron transport inhibition and further ROS release.50 Here we confirmed that APAP overdose at 6 h triggers JNK phosphorylation in liver. There were no reports about whether CPZ affects JNK activation yet. In our hands at 6 h, CPZ alone did not affect JNK phosphorylation, while CPZ cotreatment appeared to partially attenuate APAP-induced JNK phosphorylation. Interestingly, after CPZ co-treatment, the expression of phosphorylated JNK varied among the mice that we had assessed but all of these mice showed no signs of liver injury based on the serum levels of ALT and liver histology of necrosis. These results suggest that CPZ may work mainly downstream of JNK activation to block APAP-induced liver injury. It would be interesting to test whether mitochondrial Sab phosphorylation and intramitochondrial Src phosphorylation are affected by CPZ in the future.
Thirdly, it is well documented that increased oxidative stress plays a critical role in APAP-induced liver injury.34,51 CPZ has no effects on APAP-induced depleted GSH levels, so the protective effect of CPZ against APAP-induced liver injury is likely to be GSH-independent. Considering the decreased GSSG/GSH ratio at late time point by CPZ and the abolishment of the protective effects of CPZ against APAP-induced liver injury by BSO, CPZ may also regulate the oxidative stress in the liver. In future it will be interesting to test the detailed mechanisms of how CPZ affects hepatic oxidative stress.
Caution needs to be taken when the mechanisms mentioned above are weighed. On one hand, we performed a complete GSH depletion by BSO in the presence of CPZ and most of the mice had severe liver injury. These data suggest that once hepatic GSH levels are depleted to a certain level, the protective effects of CPZ will be lost. On the other hand, pharmacological inhibition of autophagy by CQ only partially reduced the protective effects of CPZ, and this was likely due to the incomplete inhibition of autophagy by CQ in the mouse livers. Depletion of cellular GSH by BSO has been shown to enhance starvation-induced autophagy in vitro.52 Therefore, we would conclude that BSO-induced GSH depletion resulted in more severe injury than CQ-induced autophagy inhibition did. GSH replenishment (by some other reagents like NAC) together with CPZ thus may offer better protection against APAP-induced liver injury. It should also be noted that CPZ may activate other pathways that contribute to its protection against APAP-induced liver injury such as blocking calcium channel in addition to autophagy induction as discussed above. Since we did not observe GSH replenishment of CPZ, we think that CPZ-induced autophagy would be less likely associated with the GSH levels.
5. Conclusions
We here reported the protective effects of both co-treatment and post-treatment of CPZ against APAP-induced liver injury in mice. CPZ-induced protection against APAP-induced liver injury is associated with increased autolysosome numbers and autophagic flux as well as reduced JNK activation.
Acknowledgements
This research was funded by the USA NIH R01 AA 020518, R01 DK 102142, U01 AA 024733, P20 GM 103549 (COBRE), and P30 GM 118247 (COBRE). The author Yuan Li is a recipient of Biomedical Research Training Program funded by University of Kansas Medical Center. We thank Margitta Lebofsky for technical assistance for the measurement of hepatic GSH.
Footnotes
Edited by Yuxia Jiang, Peiling Zhu and Genshu Wang.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Mowry JB, Spyker DA, Brooks DE, Zimmerman A, Schauben JL. 2015 Annual report of the American association of poison control centers’ National Poison Data System (NPDS): 33rd annual report. Clin Toxicol. 2016;54:924–1109. [DOI] [PubMed] [Google Scholar]
- 2.Dahlin DC, Miwa GT, Lu AY, Nelson SD. N-acetyl-p-benzoquinone imine: a cytochrome P-450-mediated oxidation product of acetaminophen. Proc Natl Acad Sci U S A. 1984;81:1327–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McGill MR, Sharpe MR, Williams CD, Taha M, Curry SC, Jaeschke H. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J Clin Invest. 2012;122:1574–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hinson JA, Roberts DW, James LP. Mechanisms of acetaminophen-induced liver necrosis. Handb Exp Pharmacol. 2010:369–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Corcoran GB, Racz WJ, Smith CV, Mitchell JR. Effects of N-acetylcysteine on acetaminophen covalent binding and hepatic necrosis in mice. J Pharmacol Exp Ther. 1985;232:864–872. [PubMed] [Google Scholar]
- 6.Saito C, Zwingmann C, Jaeschke H. Novel mechanisms of protection against acetaminophen hepatotoxicity in mice by glutathione and N-acetylcysteine. Hepatology. 2010;51:246–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heard KJ. Acetylcysteine for acetaminophen poisoning. N Engl J Med. 2008;359: 285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gupta S, Barrett T, Whitmarsh AJ, et al. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996;15:2760–2770. [PMC free article] [PubMed] [Google Scholar]
- 9.Seki E, Brenner DA, Karin M. A liver full of JNK: Signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology. 2012;143:307–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Du K, Xie Y, McGill MR, Jaeschke H. Pathophysiological significance of c-jun N-terminal kinase in acetaminophen hepatotoxicity. Expert Opin Drug Metab Toxicol. 2015;11:1769–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gandhi A, Guo T, Ghose R. Role of c-Jun N-terminal kinase (JNK) in regulating tumor necrosis factor-alpha (TNF-alpha) mediated increase of acetaminophen (APAP) and chlorpromazine (CPZ) toxicity in murine hepatocytes. J Toxicol Sci. 2010;35:163–173. [DOI] [PubMed] [Google Scholar]
- 12. Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, Kaplowitz N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J Biol Chem. 2008;283: 13565–13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gunawan BK, Liu ZX, Han D, Hanawa N, Gaarde WA, Kaplowitz N. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology. 2006;131:165–178. [DOI] [PubMed] [Google Scholar]
- 14.Latchoumycandane C, Goh CW, Ong MM, Boelsterli UA. Mitochondrial protection by the JNK inhibitor leflunomide rescues mice from acetaminophen-induced liver injury. Hepatology. 2007;45:412–421. [DOI] [PubMed] [Google Scholar]
- 15.Xie Y, McGill MR, Dorko K, et al. Mechanisms of acetaminophen-induced cell death in primary human hepatocytes. Toxicol Appl Pharmacol. 2014;279: 266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Du K, Ramachandran A, Jaeschke H. Oxidative stress during acetaminophen hepatotoxicity: sources, pathophysiological role and therapeutic potential. Redox Biol. 2016;10:148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizushima N Autophagy: process and function. Genes Dev. 2007;21: 2861–2873. [DOI] [PubMed] [Google Scholar]
- 18.Weidberg H, Shvets E, Elazar Z. Lipophagy: selective catabolism designed for lipids. Dev Cell. 2009;16:628–630. [DOI] [PubMed] [Google Scholar]
- 19.Bernales S, Schuck S, Walter P. ER-phagy: selective autophagy of the endoplasmic reticulum. Autophagy. 2007;3:285–287. [DOI] [PubMed] [Google Scholar]
- 20. Lemasters JJ Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005;8:3–5. [DOI] [PubMed] [Google Scholar]
- 21.Dunn WA Jr, Cregg JM, Kiel JA, et al. Pexophagy: the selective autophagy of peroxisomes. Autophagy. 2005;1:75–83. [DOI] [PubMed] [Google Scholar]
- 22.Williams JA, Ding WX. Mechanisms, pathophysiological roles and methods for analyzing mitophagy-recent insights. Biol Chem. 2018;399:147–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ueno T, Komatsu M. Autophagy in the liver: functions in health and disease. Nat Rev Gastroenterol Hepatol. 2017;14:170–184. [DOI] [PubMed] [Google Scholar]
- 24.Maejima Y, Chen Y, Isobe M, Gustafsson ÅB, Kitsis RN, Sadoshima J. Recent progress in research on molecular mechanisms of autophagy in the heart. Am J Physiol Heart Circ Physiol. 2015;308:H259–H268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sandri M, Coletto L, Grumati P, Bonaldo P. Misregulation of autophagy and protein degradation systems in myopathies and muscular dystrophies. J Cell Sci. 2013;126:5325–5333. [DOI] [PubMed] [Google Scholar]
- 26.Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Canc. 2017;17:528–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, Ding WX. Adipose tissue autophagy and homeostasis in alcohol-induced liver injury. Liver Res. 2017;1:54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kroemer G Autophagy: a druggable process that is deregulated in aging and human disease. J Clin Invest. 2015;125:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology. 2012;55:222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ni HM, McGill MR, Chao X, et al. Removal of acetaminophen protein adducts by autophagy protects against acetaminophen-induced liver injury in mice. J Hepatol. 2016;65:354–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ding WX, Guo F, Ni HM, et al. Parkin and mitofusins reciprocally regulate mitophagy and mitochondrial spheroid formation. J Biol Chem. 2012;287: 42379–42388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams JA, Ni HM, Haynes A, et al. Chronic deletion and acute knockdown of Parkin have differential responses to acetaminophen-induced mitophagy and liver injury in mice. J Biol Chem. 2015;290:10934–10946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chao X, Wang H, Jaeschke H, Ding WX. Role and mechanisms of autophagy in acetaminophen-induced liver injury. Liver Int. 2018;38:1363–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Y, McGreal S, Zhao J, et al. A cell-based quantitative high-throughput image screening identified novel autophagy modulators. Pharmacol Res. 2016;110:35–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ding WX, Ni HM, Gao W, et al. Oncogenic transformation confers a selective susceptibility to the combined suppression of the proteasome and autophagy. Mol Canc Ther. 2009;8:2036–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jaeschke H, Mitchell JR. Use of isolated perfused organs in hypoxia and ischemia/reperfusion oxidant stress. Methods Enzymol. 1990;186, 752–729. [DOI] [PubMed] [Google Scholar]
- 38.Ni HM, Williams JA, Jaeschke H, Ding WX. Zonated induction of autophagy and mitochondrial spheroids limits acetaminophen-induced necrosis in the liver. Redox Biol. 2013;1:427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramachandran A, Jaeschke H. Acetaminophen toxicity: novel insights into mechanisms and future perspectives. Gene Expr. 2018;18:19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horn AS, Snyder SH. Chlorpromazine and dopamine: conformational similarities that correlate with the antischizophrenic activity of phenothiazine drugs. Proc Natl Acad Sci U S A. 1971;68:2325–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kwant WO, Seeman P. The displacement of membrane calcium by a local anesthetic (chlorpromazine). Biochim Biophys Acta. 1969;193:338–349. [DOI] [PubMed] [Google Scholar]
- 42.Marshak DR, Lukas TJ, Watterson DM. Drug-protein interactions: binding of chlorpromazine to calmodulin, calmodulin fragments, and related calcium binding proteins. Biochemistry. 1985;24:144–150. [DOI] [PubMed] [Google Scholar]
- 43.Ogata N, Yoshii M, Narahashi T. Differential block of sodium and calcium channels by chlorpromazine in mouse neuroblastoma cells. J Physiol. 1990;420: 165–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saville JG, Davidson CP, D’Adrea GH, Born CK, Hamrick ME. Inhibition of acetaminophen hepatotoxicity by chlorpromazine in fed and fasted mice. Biochem Pharmacol. 1988;37:2467–2471. [DOI] [PubMed] [Google Scholar]
- 45.Harris SR, Hamrick ME. Antagonism of acetaminophen hepatotoxicity by phospholipase A2 inhibitors. Res Commun Chem Pathol Pharmacol. 1993;79:23–44. [PubMed] [Google Scholar]
- 46.Ray SD, Kamendulis LM, Gurule MW, Yorkin RD, Corcoran GB. Ca2+ antagonists inhibit DNA fragmentation and toxic cell death induced by acetaminophen. FASEB J. 1993;7:453–463. [DOI] [PubMed] [Google Scholar]
- 47.Ray SD, Balasubramanian G, Bagchi D, Reddy CS. Ca(2+)-calmodulin antagonist chlorpromazine and poly (ADP-ribose) polymerase modulators 4-amino-benzamide and nicotinamide influence hepatic expression of BCL-XL and P53 and protect against acetaminophen-induced programmed and unprogrammed cell death in mice. Free Radic Biol Med. 2001;31:277–291. [DOI] [PubMed] [Google Scholar]
- 48.Ni HM, Williams JA, Jaeschke H, Ding WX. Zonated induction of autophagy and mitochondrial spheroids limits acetaminophen-induced necrosis in the liver. Redox Biol. 2013;1:427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stamper BD, Bammler TK, Beyer RP, Farin FM, Nelson SD. Differential regulation of mitogen-activated protein kinase pathways by acetaminophen and its nonhepatotoxic regioisomer 3‘-hydroxyacetanilide in TAMH Cells. Toxicol Sci. 2010;116:164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Win S, Than TA, Min RW, Aghajan M, Kaplowitz N. c-Jun N-terminal kinase mediates mouse liver injury through a novel Sab (SH3BP5)-dependent pathway leading to inactivation of intramitochondrial Src. Hepatology. 2016;63:1987–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ramachandran A, Jaeschke H. Oxidative stress and acute hepatic injury. Curr Opin Toxicol. 2018;7:17–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Desideri E, Filomeni G, Ciriolo MR. Glutathione participates in the modulation of starvation-induced autophagy in carcinoma cells. Autophagy. 2012;8:1769–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]