

Abstract
The innate immune system is the first line of host defense against invading microorganisms. Polymorphonuclear leukocytes (PMNs or neutrophils) are the most abundant leukocyte in humans and essential to the innate immune response against invading pathogens. Compared to the acquired immune response, which requires time to develop and is dependent on previous interaction with specific microbes, the ability of neutrophils to kill microorganisms is immediate, non-specific, and not dependent on previous exposure to microorganisms. Historically, studies of PMN-pathogen interaction focused on the events leading to killing of microorganisms, such as recruitment/chemotaxis, transmigration, phagocytosis, and activation, whereas post-phagocytosis sequelae were infrequently considered. In addition, it was widely accepted that human neutrophils possessed limited capacity for new gene transcription and thus, relatively little biosynthetic capacity. This notion has changed dramatically within the past 20 years. Further, there is now more effort directed to understand the events occurring in PMNs after killing of microbes. Herein we give an updated review of the systems biology-level approaches that have been used to gain an enhanced view of the role of neutrophils during host-pathogen interaction and neutrophil-mediated diseases. We anticipate that these and future systems-level studies will continue to provide information important for understanding, treatment, and control of diseases caused by pathogenic microorganisms.
Keywords: Neutrophil, granulopoiesis, phagocytosis, microarray, transcriptome, inflammation, apoptosis, systems biology
1 |. INTRODUCTION
Polymorphonuclear leukocytes (PMNs or neutrophils) are the most abundant cellular component of the host immune system and primary mediators of the innate immune response to invading microorganisms. The ability of neutrophils to rapidly kill invading microbes is indispensable for maintaining host health. Defects in neutrophil microbicidal processes or an overall decrease in PMN abundance are deleterious to human health and often result in severe and recurrent infections. In this regard, the neutrophil has garnered much respect and notoriety for its principal role as executioner. Although the net result of neutrophil function is killing of invading microorganisms, the steps leading up to the final act require careful orchestration and yield many options based on complex signal transduction events. Inasmuch as neutrophils have significant potential to damage host cells and tissues, PMNs ultimately have go-no-go decisions that are important for the resolution of the inflammatory response. Therefore, an enhanced understanding of molecular signaling pathways induced in PMNs during these processes is critical for improving treatment and outcome of infectious diseases. To that end, systems biology-level approaches (e.g., transcriptomics, genomics, proteomics, and lipidomics) have yielded significant insight into essential neutrophil processes such as granulopoiesis, host defense, and resolution of the inflammatory response (Figure 1).
Figure 1.
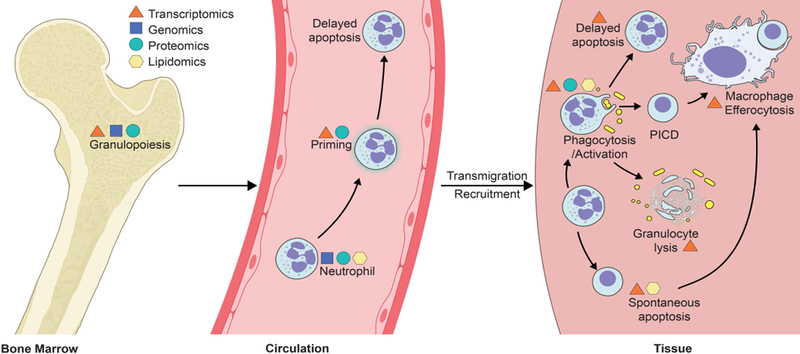
Overview of neutrophil functions/processes that have been investigated using proteomics, transcriptomics, genomics and lipidomics. PICD, phagocytosis-induced cell death. See text for details.
The goal of systems level studies is to integrate comprehensive biological data sets from diverse experimental systems to understand complex interactions at the molecular level. In more simplistic terms, systems-level studies provide the prediction of phenotype changes in biological systems from a defined stimulus. In this regard, cells of the innate immune system (leukocytes) are highly amenable to systems biology approaches. In fact, the term systems immunology has emerged more recently to describe the holistic approach aiming at understanding the complex interactions of the immune system in particular (Davis, Tato, & Furman, 2017). Although leukocytes often overlap in function, there are distinct differences among cell types such as those pertaining to turnover and production of immunoregulators. Each type of leukocyte has a unique role in the immune response and generates lineage-specific gene expression patterns. Thus, a unified approach using a single representative cell type is unrealistic. Nevertheless, systems biology-level studies have provided important insight into the complex signal transduction pathways in both neutrophils and cells of the monocyte lineage. Interestingly, comparative analysis of transcripts and DNA methylation patterns of different human immune cell types revealed the highest variability in PMNs (Ecker et al., 2017). Neutrophils have the highest number of cell type specific hypervariable regions compared with T cells and monocytes, and PMN transcripts and DNA methylation patterns have the greatest interindividual variability (Ecker et al., 2017). This might reflect the plasticity required of neutrophils as the first line of cellular defense and account in part for susceptibilities to disease and differences in disease outcomes. These attributes make neutrophils an interesting target for further systems biology approaches. Herein, we review neutrophil functions and the systems-level approaches used to better understand these processes. Such approaches are an important step toward developing a comprehensive view of host-pathogen interactions.
2 |. GENE TRANSCRIPTION IN NEUTROPHILS
Inasmuch as the primary function of neutrophils is to protect the host from invading pathogens, significant emphasis has been placed on processes relating to microbicidal activity. Neutrophils are able to initiate phagocytosis, degranulation, and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-dependent killing without new synthesis of proteins (Kasprisin & Harris, 1978). However, Kasprisin and Harris demonstrated that new gene transcription and protein synthesis are required to maintain full capacity for human neutrophil phagocytosis and associated bactericidal activity (Kasprisin & Harris, 1977, 1978). Despite these early studies, it has often been assumed that mature neutrophils possess limited transcriptional potential. In fact, those isolated from venous blood constitutively express several thousand transcripts (Holland et al., 2007). The PMN nucleus undergoes progressive chromatin condensation during the post-mitotic phase of neutrophil differentiation. In addition, mature cells contain significant quantities of heterochromatin (Bainton, Ullyot, & Farquhar, 1971), a property that suggests transcriptional repression. Conversely, early studies of RNA metabolism in mature PMNs demonstrated RNA biosynthesis increases following phagocytosis (Cline, 1966a, 1966b). It is now clear that mature neutrophils express transcription factors that in turn regulate the expression of numerous genes involved in a diversity of important cellular processes. Elucidation of mechanisms driving the transcriptional regulation of molecular pathways involved in PMN microbicidal activity, inflammation, and apoptosis are currently areas of investigation.
Mature neutrophils are exposed to numerous stimuli in the context of the inflammatory milieu. In general, exposure of PMNs to pro-inflammatory cytokines such as interferon (IFN)-γ and granulocyte/macrophage-colony stimulating factor (GM-CSF) induces activation of STAT transcription factor members, whereas tumor necrosis factor (TNF)-α and interleukin (IL)-1β induce those of the NFκB family. NFκB activation is also promoted by lipopolysaccharide (LPS), platelet-activating factor, N-formyl peptides (e.g., fMLF), leukotriene B4 (LTB4), reactive oxygen species (ROS), phagocytosis, and apoptosis (McDonald, 2004). κB-binding motifs are present in the promoter regions of several genes encoding inflammatory mediators including IL-1α/β, TNFα, IL-8, IL-12, CCL4, CCL20, and CXCL1. In addition, NFκB-mediated inhibition of neutrophil apoptosis in hypoxic environments is further regulated by the hypoxia-inducible factor (HIF) transcriptional complex (Walmsley et al., 2005). Inasmuch as apoptosis is a complex process, additional transcription factors are likely involved in regulating this critical neutrophil process. For example, the FOXO forkhead subfamily member FOXO3A has been shown to directly suppress neutrophil FASL transcription, thereby limiting the FAS-mediated death receptor pathway of apoptosis (Jonsson, Allen, & Peng, 2005). Although significant progress has been made, our understanding of the transcriptional regulation of signal transduction networks that govern neutrophil function is incomplete.
2.1 |. Transcriptome analyses of granulocyte development
Cells of the immune system originate from common hematopoietic progenitor cells in bone marrow. The greatest percentage of hematopoiesis is committed to production of neutrophils and granulocyte precursors comprise ~60% of the nucleated cells in bone marrow (Bainton et al., 1971). Human PMN turnover is 50–340 × 107cells/kg/day (~0.4–3 × 1011 cells per day in a 75 kg person) (Athens et al., 1961; Cline, 1975). The plasticity of the acute inflammatory response depends, in part, on the ability of the host to regulate neutrophil production based on demand. Thus, regulation and execution of granulopoiesis are highly structured and complex processes. Granulocyte differentiation and development are influenced by the concerted activities of myeloid colony stimulating factors such as granulocyte colony-stimulating factor (G-CSF) and GM-CSF (Dale, Liles, Llewellyn, & Price, 1998). PMN maturation occurs in distinct niches within the bone marrow microenvironment prior to migration towards venous sinuses. As myeloid precursors become mature neutrophils, they sequentially acquire features necessary for microbicidal activity, including receptors for phagocytosis and signaling, granule components, and NADPH oxidase proteins. PMN maturation is also accompanied by morphological changes (nuclear segmentation) and increased motility and chemotactic responsiveness. This post-mitotic maturation time is approximately 6–8 days under basal conditions (Dancey, Deubelbeiss, Harker, & Finch, 1976). Mature PMNs are released into the bloodstream where they circulate with a typical half-life of 6–8 hours (Athens et al., 1961; Cline, 1975; Cronkite & Fliedner, 1964; Fliedner, Cronkite, & Robertson, 1964). Morphological analysis of circulating neutrophils indicates there is a low basal level of immature cells, e.g., band cells are ~1–3 % of peripheral leukocytes (Cronkite & Fliedner, 1964). However, induction of neutrophilia during acute inflammation accelerates recruitment from bone marrow. Depletion of the stored basal reserves results in a decrease in post-mitotic maturation time to 3–4 days (Fliedner, Cronkite, Killmann, & Bond, 1964) and elicits additional release of functionally competent immature forms (Orr et al., 2007). Granulopoiesis and PMN egress from bone marrow is enhanced by several immunomodulatory factors including GM-CSF, G-CSF, IL-8, and glucocorticoids (Aglietta et al., 1989; Dale et al., 1998; Laterveer et al., 1996).
Transcriptional processes that occur during granulopoiesis have been investigated intensely. The transcriptional regulation of neutrophil development is mediated partly by the coordinated activities of several transcription factors and repressors. For example, CCAAT/enhancer binding protein alpha (C/EBPα), PU.1, RAR, CBF, and c-MYB are involved in early granulopoiesis, whereas C/EBPε, PU.1, SP1, CDP, HOXA10, signal transducer and activator of transcription (STAT)1, STAT3, STAT5 and GFI-1 are involved in terminal neutrophil differentiation (Friedman, 2002; McDonald, 2004). Early systematic studies of myeloid differentiation focused on transcriptome and proteome analyses using in vitro-differentiated murine cell lines (Iida, Kohro, Kodama, Nagata, & Fukunaga, 2005; Lian et al., 2002; Lian et al., 2001). Importantly, these studies provided the fundamental observation that a substantial proportion of neutrophil protein change is a consequence of mRNA transcription and served as the premise for additional PMN microarray studies. Subsequent transcriptome studies of neutrophil development evaluated changes in gene expression in differentiating HL60 cells (Mollinedo, Lopez-Perez, & Gajate, 2008), or investigated gene expression among immature neutrophils and neutrophil precursor cells isolated from bone marrow (Martinelli et al., 2004; Theilgaard-Monch et al., 2005). It is noteworthy that differentiation from common myeloid progenitor cells yields lineage-specific gene expression patterns (Kluger et al., 2004). Here, we provide a few examples of systems-level studies on granulopoiesis and also refer the reader to a review on the topic (Theilgaard-Monch, Porse, & Borregaard, 2006).
In general, transcriptome studies of granulopoiesis demonstrate that PMN inflammatory molecules and receptors for numerous inflammatory mediators are up-regulated throughout development, suggesting increased functional capacity upon maturation. In addition to the noted upregulation of receptors and proinflammatory molecules during granulocyte maturation, Theilgaard-Monch et al. identified previously unknown granule protein candidates, including protease inhibitors, proteases, signaling molecules, and acute-phase proteins (Theilgaard-Monch et al., 2005). These authors also found that there was temporal regulation of transcripts encoding granule proteins, consistent with the “targeting by timing” hypothesis put forth for proteins of granule subsets (Le Cabec, Cowland, Calafat, & Borregaard, 1996). By contrast, gene expression patterns in mature PMNs indicate there is a decreased capacity for synthesis of granule proteins (Martinelli et al., 2004). Functional studies with these newly identified putative granule proteins may provide additional insight into neutrophil microbicidal activity in mature cells and the process of granule exocytosis. More recent studies have evaluated the influence of selected cytokines on the granulocyte transcriptome during granulopoiesis. For example, Pederson et al. investigated changes in gene expression among neutrophil precursor cells following treatment with G-CSF in vivo (Pedersen et al., 2016). These studies were highly innovative in that they modeled emergency granulopoiesis in humans in vivo. Ellison et al. discovered that prolonged culture of differentiating myeloid precursor cells (PLB-985) with IFN-γ induces differential expression of genes encoding molecules that are involved in key basic neutrophil functions (Ellison, Gearheart, Porter, & Ambruso, 2017). Collectively, these studies provide additional insight into the regulation of granulopoiesis by inflammatory cytokines.
Recent evidence also suggests epigenetic processes and chromosomal organization play a role in the transcriptional regulation of neutrophil development (Kosak et al., 2007). In this regard, post translational modifications such as DNA methylation have become an increasing subject of investigation. Zilbauer et al. created genome-wide methylome maps of human leukocytes and found that hypomethylation of DNA in PMNs correlated with gene transcription levels (Zilbauer et al., 2013). The level of PMN hypomethylation was much greater than that in other leukocytes, thus providing support to the idea that neutrophils have a high level of genetic and/or phenotypic plasticity. Recent epigenetic studies have also revealed cell maturation-dependent changes in the methylation patterns of transcription factors that drive granulopoiesis (Ronnerblad et al., 2014). Ronnerblad et al. found concordance between altered methylation of key transcription factors, such as PU.1, GATA2, GFI1 and ETS1, and changes in gene expression during maturation of granulocytes. In general, methylation correlated inversely with gene expression. These studies provide a comprehensive view of epigenetic changes and changes in gene expression during human granulopoiesis (Ronnerblad et al., 2014).
In addition to DNA methylation, microRNA-mediated RNA interference (Johnnidis et al., 2008) and signal-dependent translation of constitutive mRNA (Lindemann et al., 2004; Yost et al., 2004) are emerging as critical regulatory elements in neutrophil development and function. Gene transcription is subject to further regulation and fine tuning by microRNAs, and these molecules are known to play a role in neutrophil development and differentiation (Fazi et al., 2005; Gantier, 2013; Johnnidis et al., 2008; Qin et al., 2017; Wong et al., 2014). Larsen et al. used a microarray-based approach to identify miRNAs that were differentially regulated during granulopoiesis, and most of these miRNAs were differentially expressed during differentiation of human myeloblasts/promyelocytes to myelocytes/metamyelocytes (Larsen et al., 2013). For example, miRs-130a, 155 and 146a, which may regulate transcripts encoding molecules involved in TGF-β signaling, were more highly expressed in promyelocytes/myelocytes compared with cells at later stages of granulocyte differentiation and maturation (Larsen et al., 2013). miR-223 is one of the better-characterized neutrophil miRNAs—it negatively regulates granulopoiesis and neutrophil function (Johnnidis et al., 2008). We refer the reader to Gurol et al. (Gurol, Zhou, & Deng, 2016) for a recent review of the role of miRNAs in neutrophil development and function.
3 |. NEUTROPHILS IN THE INFLAMMATORY RESPONSE
3.1 |. Recruitment and priming
Following release from bone marrow, neutrophils circulate in the vasculature prior to extravasation, i.e., movement of cells out of circulation to peripheral tissue. Rapid recruitment of neutrophils to tissues is fundamentally important to the innate immune system. This multi-step process involves mobilization of PMNs from bone marrow reserves, accelerated hematopoiesis and recruitment from marginated pools in response to host- and pathogen-derived chemotactic stimuli. PMN migration from the vasculature to the extravascular milieu is dependent on receptor-mediated contact with endothelia of post capillary venules and signals received through soluble mediators. Marginating granulocytes slowly roll along the endothelial surface through interactions of a family of C-type lectin glycoproteins known as selectins (Lawrence & Springer, 1991). L-selectin is constitutively expressed on the surface of neutrophils and mediates a low-affinity adhesive interaction. Stimulation of endothelial cells leads to the transient expression of both E- and P-selectin. In the presence of inflammatory mediators, adherence rapidly switches to a high-affinity interaction that is mediated by activation of PMN β2-integrins and endothelial cell intracellular adhesion molecule (ICAM)-1 and ICAM-2. Once firmly bound, several neutrophil surface molecules, including CD31 (Muller, Weigl, Deng, & Phillips, 1993), CD54 (Diamond et al., 1990), CD44 (Khan et al., 2004), and CD47 (Cooper, Lindberg, Gamble, Brown, & Vadas, 1995), facilitate transmigration through the endothelium into tissues.
The ability of neutrophils to localize to sites of infection is a key component of the acute inflammatory response. Neutrophil migration through tissue is influenced by chemoattractants produced by the host during inflammation and pathogen-derived factors. There are numerous host-derived factors that enhance the recruitment of neutrophils. IL-8 is one of the most potent neutrophil chemoattractants (Walz, Peveri, Aschauer, & Baggiolini, 1987; Yoshimura et al., 1987). This chemokine is produced in response to pro-inflammatory stimuli by a diversity of cell types including mononuclear phagocytes, neutrophils, mast cells, epithelial cells, keratinocytes, fibroblasts, and endothelial cells. Neutrophils are also recruited efficiently by leukotrienes and prostaglandins generated exogenously at sites of infection and by the complement component C5a (Ehrengruber, Geiser, & Deranleau, 1994). Many products of bacteria, such as fMLF, peptidoglycan, and phenol-soluble modulins (PSMs), are chemoattractants and contribute directly to PMN recruitment (Schmeling et al., 1979; Showell et al., 1976). In addition to facilitating neutrophil migration, chemoattractants can also prime PMNs for enhanced function. Neutrophil recruitment to sites of infection can also be influenced by microRNA interference. For example, miR-223 can dampen the influx of PMNs to the lungs of tuberculosis patients via direct interaction with chemoattractants CXCL2, CCL3, and IL‐6 (Dorhoi et al., 2013).
Given the high level of cytotoxic molecules produced by- or contained within PMNs, it is perhaps not surprising that these cells are intimately associated with the pathogenesis of tissue injury and trauma associated with inflammatory diseases (Boxer, Axtell, & Suchard, 1990; Edwards & Hallett, 1997). In order to moderate neutrophil-mediated tissue destruction, PMN activation most appropriately occurs in regions proximal to the infected tissue. However, the ability of neutrophils to respond rapidly at the infection site is essential to host defense. Thus, it is reasonable to believe that the ability of the neutrophil to reside in a state intermediate to circulating quiescence and complete activation is optimal for plasticity. To that end, neutrophil priming is a reversible process that can enhance cell functions and limit the potential for indiscriminate host tissue damage (Swain, Rohn, & Quinn, 2002). The original description of PMN priming reported the ability of a primary agonist, typically at sub-stimulatory concentration, to enhance superoxide production triggered by a second stimulus (Guthrie, McPhail, Henson, & Johnston, 1984). Neutrophils can be primed by numerous host factors and processes including cytokines, chemokines, growth-factors, chemotactic factors, leukotrienes, ROS, adherence, and cellular contact (Swain et al., 2002). Although priming classically implies augmentation of superoxide-generating potential, PMN exposure to priming agents can also promote adherence, chemotaxis, cytokine secretion, phagocytosis, degranulation, and bactericidal activity (reviewed in (Kobayashi, Voyich, Burlak, & DeLeo, 2005)). Indeed, priming typically includes mobilization of secretory vesicles and some specific granules, and secretion of cytokines, but fails to induce complete degranulation or elicit production of superoxide (DeLeo et al., 1998). Enhanced ROS production in primed PMNs is facilitated by partial assembly of the NADPH-oxidase complex. However, neutrophil exposure to different priming agents may result in heterogeneous changes in the structural organization of the oxidase (described below). Many priming agents produced by microorganisms are Toll-like receptor (TLR) agonists and may enable enhanced surveillance and clearance of pathogens from infection sites. The specific molecular mechanisms responsible for priming are unclear, but recent RNA Seq-based studies revealed that different priming agents activate divergent transcription pathways in PMNs. For example, TNF-α mediates priming via the NFκB pathway, whereas GM-CSF signals via JAK/STAT (Wright, Thomas, Moots, & Edwards, 2013).
3.2 |. Systems biology-level approaches
3.2.1 |. Chemotaxis and transmigration
Active recruitment of neutrophils to the inflammatory milieu is a sequential process, resulting in the convergence of complex signals that ultimately enhance PMN function. Microarray analysis of PMNs exposed to the potent chemoattractant fMLF revealed a number of up-regulated transcripts encoding pro-inflammatory molecules, such as CCL2, CCL3, CCL4, VEGF, CXCL1, CXCL2, IL1B, IL8, and TNF (Zhang et al., 2004). In addition, several genes encoding factors involved in cytoskeletal regulation, adhesion, and motility were up-regulated following fMLF stimulation including ICAM1, PLAU, PLAUR, TUBB, TUBB2, TUBB5, ACTG1, LAMB3, CD47, and TPM4. Consistent with these findings, analysis of the neutrophil proteome following stimulation through the related formyl peptide receptor-like 1 (FPRL-1) identified several proteins involved in the remodeling of the cytoskeleton (Boldt, Rist, Weiss, Weith, & Lenter, 2006). Differentially expressed or modified protein candidates such as the actin- and tubulin-interacting proteins, L-plastin, cofilin, moesin, and stathmin, were identified by 2-D difference gel electrophoresis (DIGE) analysis (Boldt et al., 2006). Thus, these in vitro studies demonstrate that selective stimulation of neutrophil formyl-peptide receptors induces a limited number of changes that likely enhance both chemotaxis and pro-inflammatory capacity. Two recent studies investigated neutrophil transcriptional changes following migration to aseptic skin lesions (Theilgaard-Monch, Knudsen, Follin, & Borregaard, 2004) and LPS-induced alveolar transmigration (Coldren et al., 2006). Although neutrophil migration to sites of infection is a complex multi-faceted process, the neutrophil transcriptomes delineated in these in vivo studies were remarkably similar to those observed in vitro using various PMN priming agents. In general, there were increases in transcripts encoding molecules of neutrophil pathways that regulate pro-inflammatory capacity, adherence, and migration. On the other hand, there was down-regulation of transcripts involved in pathways that promote apoptosis (Theilgaard-Monch et al., 2004). These findings confirm and extend previous reports that indicate the pro-survival and pro-inflammatory effects of priming agents on neutrophil function. More recent studies investigated the influence of miRNAs on neutrophil chemotaxis and inflammatory response and identified miR-223 as a negative regulator of these processes. miR-223 knockout mice have hyperactive neutrophils with an increased inflammatory response (Johnnidis et al., 2008), whereas overexpression of miR-223 decreases inflammasome activity and IL‐1β production (Bauernfeind et al., 2012).
3.2.2 |. Priming with G-CSF and GM-CSF
GM-CSF and G-CSF are multi-functional colony stimulating factors that not only participate in myelopoiesis, but emerging evidence suggests these important molecules enhance the cellular response at sites of inflammation. Similar to other neutrophil priming agents, both G-CSF and GM-CSF have been shown to delay neutrophil spontaneous apoptosis and this property is consistent with an overall role in promoting the inflammatory response. Microarray studies of the effects of G-CSF on mature human neutrophils demonstrated increased expression of genes encoding plasminogen activator urokinase (PLAU), IL-1α, granulocyte chemotactic protein-2 (GCP-2), TLR2, and epithelial cell-derived neutrophil attractant-78 (ENA-78) (Suzuki et al., 2002). ENA-78 was subsequently confirmed to enhance chemotaxis of mature neutrophils, thereby providing evidence that G-CSF stimulates mobilization of PMNs to inflammatory sites.
The effects of GM-CSF on neutrophil gene expression have been evaluated previously by microarray analyses (Kobayashi, Voyich, Whitney, & DeLeo, 2005; Martinelli et al., 2004). Martinelli et al. (Martinelli et al., 2004) evaluated differences in relative PMN gene expression following 7 h of in vitro stimulation with GM-CSF, whereas Kobayashi et al. (Kobayashi, Voyich, Whitney, et al., 2005) performed an extended time course (24 h) of GM-CSF treatment to delineate effects on cell fate. Martinelli et al. noted a comparatively high level of IFN-regulated genes in mature PMNs (versus immature cells) that decreased significantly following GM-CSF treatment. Further investigation revealed that type I- and type II IFN signal transduction is more efficient in mature PMNs, and they proposed that IFN-signaling potentiates bactericidal activity through formation of neutrophil extracellular traps. Since relatively little is known about the role of IFN-signaling in neutrophil function, additional studies are needed to elucidate this interesting hypothesis. By comparison, Kobayashi et al. found a dramatic increase in transcripts encoding proteins that facilitate host defense or are central to the inflammatory response including CD14, CD32, CD44, CD54, CD66A, CD69, CD74, CD89, CD119, CCR1 (encoding chemokine (C-C motif) receptor 1), CYBB (encoding gp91phox), IL1B, IL3RA, IL1R2, and TLR2 (Kobayashi, Voyich, Whitney, et al., 2005). Although GM-CSF was known to enhance proinflammatory capacity, the number of host defense molecules found to be up-regulated by this cytokine is remarkable. Two other findings from this study were unexpected and made possible only by the availability of genome-wide approaches. First, transcripts encoding dozens of ribosomes and translation factors were increased 18–24 h after treatment with GM-CSF. This observation is consistent with the idea that human neutrophils have significant biosynthetic capacity. Second, there was an increase in mRNAs encoding major histocompatibility (MHC) class II proteins, which suggests that neutrophils have potential as antigen presenting cells (Kobayashi, Voyich, Whitney, et al., 2005). This notion is underscored by the recent finding that human neutrophils pulsed with cytomegalovirus or influenza hemagglutinin can present these antigens to CD4+ T cells, albeit the ability of neutrophils to present antigen is less than that of traditional Ag presenting cells (e.g., dendritic cells and monocytes) (Vono et al., 2017). Similarly, neutrophils primed by cytokines (a combination of GM-CSF, IFNγ, and IL-3) have the ability to present antigen during allergic inflammation (Polak et al., 2019). Inasmuch as GM-CSF extends the PMNs lifespan significantly, the ability of human PMNs to function as antigen presenting cells could be important in this context. We refer the interested reader to a recent article by Lin and Loré for a more comprehensive review of granulocytes as antigen presenting cells (Lin & Lore, 2017).
In the absence of GM-CSF stimulation, the expression of dozens of genes encoding PMN proinflammatory molecules diminishes over 24 h in culture and this phenomenon correlates well with induction of spontaneous neutrophil apoptosis. Neutrophil apoptosis is accompanied by decreased functional capacity (e.g., phagocytic capacity) (Whyte, Meagher, MacDermot, & Haslett, 1993), a finding consistent with down-regulation of receptors and diminished expression of transcripts encoding CD11b, CD16, CD18, CD32, CD35, and CD64 (Kobayashi, Voyich, Whitney, et al., 2005). In addition, down-regulation of genes encoding antiapoptotic proteins, such as myeloid cell leukemia sequence 1 (MCL1), caspase 8 and FAS-associated via death domain-like apoptosis regulator (CFLAR), B cell chronic lymphocytic leukemia/lymphoma 2 (BCL2/adenovirus E1B 19 kDa-interacting protein 2 (BNIP2) and serum/glucocorticoid-regulated kinase (SGK) coincided with spontaneous apoptosis. In stark contrast, GM-CSF treatment delayed neutrophil apoptosis and prevented down-regulation of transcripts encoding pro-inflammatory factors (Kobayashi, Voyich, Whitney, et al., 2005). These findings, coupled with those by Pedersen et al. (Pedersen et al., 2016), provide a comprehensive view of the processes that regulate neutrophil survival and apoptosis in the context of GM-CSF.
3.2.3 |. Priming with LPS
Studies on the immunomodulatory effects of bacterial endotoxin are of historical importance for description of neutrophil function in general. In 1960, Cohn and Morse reported that LPS treatment enhanced neutrophil bactericidal activity (Cohn & Morse, 1960). In the early 1980’s, Dahinden et al. demonstrated the ability of LPS to alter PMN adhesion, respiratory burst, degranulation, and motility (Dahinden, Galanos, & Fehr, 1983). These studies were followed by evidence that LPS is a potent neutrophil priming agent (Guthrie et al., 1984). The discovery that a TLR4/MD2/CD14 complex recognizes LPS facilitated further investigation into the pathways governing effects on neutrophil function (Sabroe, Jones, Usher, Whyte, & Dower, 2002). Since then, several investigators have used transcriptome and proteome analyses to elucidate further the neutrophil LPS-mediated signal transduction pathways (Fessler, Malcolm, Duncan, & Worthen, 2002; Malcolm, Arndt, Manos, Jones, & Worthen, 2003; O’Neill et al., 2004; Silva et al., 2007; Tamassia et al., 2007; Tsukahara et al., 2003; Zhang et al., 2004). Although there were variations in both microarray platforms used and LPS dose levels, there was a general concordance of gene expression patterns that support increased pro-inflammatory capacity of LPS-stimulated neutrophils. For example, Rel/NFκB family members, NFKB1, NFKB2, and RELA, were up-regulated in response to LPS treatment. In addition, transcripts encoding IL-1β, IL-6, MIP-1β, MIP-3α, MCP-1, GRO3, IL-10RA, TNF-α, and HM74 were up-regulated. Studies with LPS-stimulated PMNs performed by Worthen and colleagues revealed a surprising increase in expression of IFN-stimulated genes (ISG), such as ISG15, MX1, IFI56, IFIT4, IFI54, IFI58, and IFP35 (Fessler et al., 2002). A follow-up study by the same laboratory investigated the mechanisms for induction of ISGs in LPS-stimulated neutrophils (Malcolm et al., 2003). In contrast to monocytes, neutrophil expression levels of IFNA and IFNB remained unchanged and phosphorylation of STAT proteins was not detected. These authors concluded that neutrophil ISG expression following LPS-stimulation proceeds in an IFN-independent fashion. By contrast, Tamassia et al. did not detect LPS-mediated induction of ISGs, although expression IFNB was similarly absent in PMNs and present in monocytes (Tamassia et al., 2007). The reason for the discrepancy between studies is unclear. However, the divergence in TLR4 signaling pathways between neutrophils and monocytes is intriguing and warrants further investigation.
3.3 |. Neutrophil phagocytosis and activation
Neutrophils bind and ingest invading microorganisms at sites of infection through a process known as phagocytosis (Figure 2). Bacteria contain a diversity of evolutionarily-conserved structures that facilitate direct recognition by neutrophils. Molecules such as LPS, lipoprotein, lipoteichoic acid (LTA), and flagellin comprise pathogen-associated molecular patterns (PAMPs) that interact with receptors on the surface of neutrophil membranes. Neutrophils express TLRs 1, 2, 4–10, and additional receptors such as the peptidoglycan recognition protein (Hayashi, Means, & Luster, 2003). In general, ligation of the neutrophil pattern recognition receptors activates signal transduction pathways that ultimately prolong cell survival (Sabroe et al., 2003), facilitate adhesion (Sabroe et al., 2003) and phagocytosis (Hayashi et al., 2003), enhance release of cytokines, chemokines and ROS (Hayashi et al., 2003; Kurt-Jones et al., 2002; Sabroe et al., 2003), and promote degranulation (Bellocchio et al., 2004; Lotz et al., 2004), thereby contributing to innate host defense. PMN phagocytosis is promoted efficiently by opsonization of microbes with host proteins such as antibody and complement. Specific antibody recognizes epitopes on microbial surfaces and promotes the deposition of complement components through the classic pathway of complement activation. Antibodies bound to the surface of microbes are recognized by neutrophil receptors specific for the Fc-region of antibody, including CD64 (FcγRI, IgG receptor), CD32 (FcγRIIa, low-affinity IgG receptor) (DeLeo & Nauseef, 2014; Mantovani, 1975), CD16 (FcγRIIIb, low-affinity IgG receptor) (Fleit, Wright, & Unkeless, 1982; Mantovani, 1975), CD89 (FcαR, IgA receptor) (Albrechtsen, Yeaman, & Kerr, 1988), and CD23 (FcεRI, IgE receptor) (Gounni et al., 2001). Microbes opsonized with complement are efficiently recognized by PMN surface receptors, such as ClqR (Eggleton et al., 1995), CD35 (CR1) (Rabellino, Ross, & Polley, 1978; Ross, Jarowski, Rabellino, & Winchester, 1978), CD11b/CD18 (CR3) (Dana, Todd, Pitt, Springer, & Arnaout, 1984; Hickstein et al., 1987) and CD11c/CD18 (CR4) (Myones, Dalzell, Hogg, & Ross, 1988). Ligation of these membrane-bound opsonin receptors initiates changes in the cytoskeleton that execute the physical process of phagocytosis. Polymerization of PMN actin microfilaments proximal to receptor ligation facilitates plasma membrane flow around the microbial surface until completion of the engulfment process. Ingested microbes are thus sequestered within membrane-bound vacuoles known as phagosomes (Figure 2).
Figure 2.
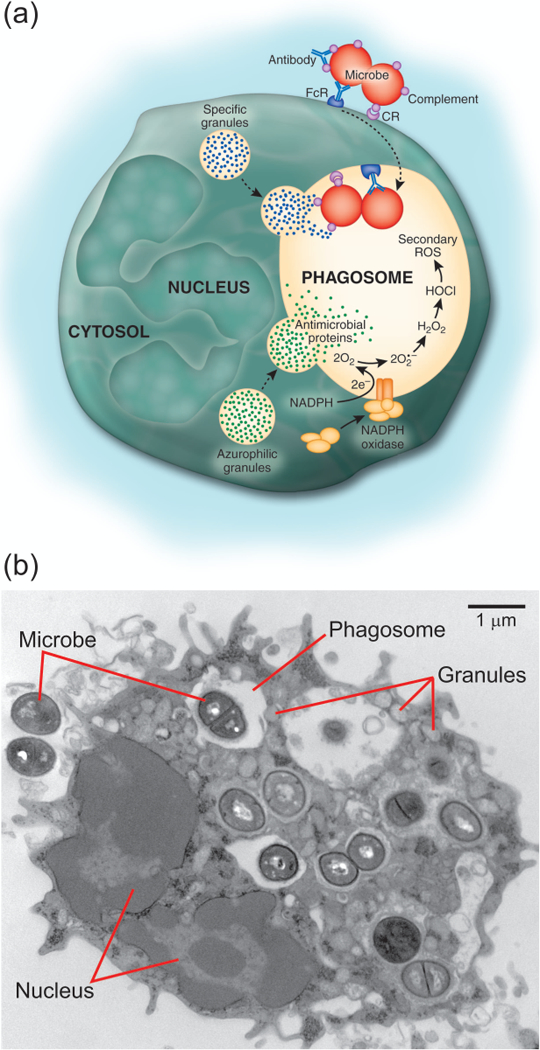
Neutrophil phagocytosis and microbicidal process. Panel A illustrates binding and phagocytosis of a microbe opsonized with antibody or serum complement. Phagocytosis triggers production of superoxide (O2•–) from which other secondarily derived ROS are formed, including hydrogen peroxide (H2O2) and hypochlorous acid (HOCl). Panel B is a transmission electron micrograph of a human neutrophil that has phagocytosed numerous Staphylococcus aureus (Microbe).
PMN phagocytosis initiates sequential execution of the battery of neutrophil microbicidal mechanisms. Neutrophil antimicrobial activity is generated from two primary sources: 1) production of superoxide radicals and other secondarily-derived reactive oxygen species (ROS), and 2) granules containing antimicrobial peptides, proteins, and degradative enzymes (Figure 2). Neutrophil activation triggers an abrupt increase in oxygen consumption, a process classically termed the respiratory burst. The generation of ROS is mediated by a multi-component membrane-bound complex, NADPH-dependent oxidase (Thomas, 2017). In unactivated neutrophils, components of the NADPH oxidase are compartmentalized to either the cytosol (p40phox, p47phox, p67phox, and the GTPase Rac2) or membranes (flavocytochrome b558, comprised of gp91phox and p22phox). The majority of flavocytochrome b558 resides in the membranes of specific granules, gelatinase granules, and secretory vesicles, whereas the remainder is found in the plasma membrane. PMN priming stimuli alter structure and subcellular distribution of NADPH oxidase proteins. In general, the majority of neutrophil priming agents, including LPS (DeLeo et al., 1998), IL-8 (Guichard et al., 2005), GM-CSF (Dang et al., 1999), and TNF-α (Brown et al., 2004), induce a modest increase in phosphorylation of p47phox, although only LPS and IL-8 cause translocation (limited) of p47phox from cytosol to the plasma membrane (DeLeo et al., 1998; Guichard et al., 2005). PMN priming with LPS also results in partial redistribution of flavocytochrome b558 from the specific granules to the plasma membrane (DeLeo et al., 1998). The assembly of NADPH oxidase is accomplished by translocation of the cytosolic components to the plasma- or phagosome membrane and association with flavocytochrome b558. The oxidase mediates electron transfer from cytosolic NADPH to intraphagosomal molecular oxygen, thereby forming superoxide anion. Superoxide anion is short-lived and dismutates rapidly to hydrogen peroxide and forms other secondary reactive products, such as hypochlorous acid, hydroxyl radical, and singlet oxygen, which are effective microbicidal compounds (Klebanoff, 1968, 1974; Rosen & Klebanoff, 1977, 1979).
Concomitant with assembly of the NADPH-oxidase, neutrophil phagocytosis triggers degranulation, which involves fusion of cytoplasmic granules with the plasma- and/or phagosome membrane (reviewed in ref. (Cowland & Borregaard, 2016)). Peroxidase negative granules, i.e., secretory vesicles, gelatinase granules, and specific granules, serve as a reservoir of functionally important membrane proteins such as CR3, formyl peptide receptor, flavocytochrome b558, and β2-integrins. Fusion of primary/azurophilic granules (peroxidase-positive granules) with phagosomes enriches the vacuole lumen with antimicrobial agents including α-defensins, cathepsins, proteinase-3, elastase, azurocidin, lysozyme, and bactericidal-permeability-increasing protein. The cumulative antimicrobial activity of neutrophils is thus comprised of ROS and a broad range of antimicrobial peptides and enzymes. Inasmuch as many of these agents do not discriminate between host and microbe, and neutrophils also contain enzymes that degrade host tissues (e.g., elastase, gelatinase, and collagenase), a mechanism must exist to limit damage to host cells and tissues during and after the inflammatory response.
3.3.1 |. Transcriptome analyses of neutrophil phagocytosis
Although ingestion of microbes is mediated by interactions with neutrophil Fc- and complement receptors, the microbial surface contains a diversity of structures that are both phylogenetically conserved and species-specific. Pathogens are also capable of secreting putative virulence factors that interact with human cells, including those of the innate immune system. Thus, PMN response to bacterial pathogens involves complex signals induced by ligation of multiple receptors in addition to those participating in Fc- and complement-mediated phagocytosis.
Multiple studies have used microarray analysis to gain insight into molecular determinants that mediate events accompanying PMN phagocytosis. Early transcriptome studies demonstrated that the process of PMN phagocytosis per se induces numerous transcriptional changes that are consistent with increased production of pro-inflammatory mediators not unlike that observed following priming (Kobayashi, Voyich, Buhl, Stahl, & DeLeo, 2002; Subrahmanyam et al., 2001). For example, PMNs up-regulate transcripts early that encode important inflammatory mediators, such as CCL3, CCL4, CCL20, oncostatin M, CXCL2, CXCL3, VEGF, IL-6, and TNF-α, following Fc- and complement receptor-mediated phagocytosis. However, it is also evident that binding and ingestion of microorganisms results in a complex series of neutrophil signal transduction events including TLR activation and the production of additional inflammatory mediators that are potentially pathogen-specific. Genome-wide PMN transcription analyses have been performed following phagocytosis of several microorganisms including Escherichia coli K12 (Subrahmanyam et al., 2001; Zhang et al., 2004), attenuated Yersinia pestis KIM5 and KIM6 (Subrahmanyam et al., 2001), Staphylococcus aureus (Borjesson et al., 2005; Kobayashi et al., 2010; Kobayashi, Braughton, et al., 2003), Streptococcus pyogenes (Kobayashi, Braughton, et al., 2003), Burkholderia cepacia (Kobayashi, Braughton, et al., 2003), Listeria monocytogenes (Kobayashi, Braughton, et al., 2003), Borrelia hermsii (Kobayashi, Braughton, et al., 2003), Mycobacterium bovis (Suttmann, Lehan, Bohle, & Brandau, 2003), Candida albicans (Fradin et al., 2007; Mullick et al., 2004), and Anaplasma phagocytophilum (Borjesson et al., 2005; Lee & Goodman, 2006; Lee, Kioi, Han, Puri, & Goodman, 2008; Sukumaran, Carlyon, Cai, Berliner, & Fikrig, 2005). Inasmuch as differences in pathogen-induced PMN transcript levels occur in signal transduction mediators and prominent transcription factors, it is not surprising that the PMN response is not entirely conserved. Thus, identification of pathogen-specific PMN transcriptional profiles is a sound approach toward elucidation of mechanisms of bacterial and fungal pathogenesis. Although detailed dissection of pathogen-specific PMN profiles is beyond the scope of this review, specific examples are provided below.
3.3.2 |. Proteome analysis of neutrophil phagosomes
Compared with transcriptome-based studies of PMNs, there are fewer neutrophil proteome studies. Burlak et al. used high-resolution subcellular proteomics to generate a comprehensive view of proteins associated with human neutrophil phagosomes (Burlak, Whitney, Mead, Hackstadt, & DeLeo, 2006). Many of the proteins found to be associated with neutrophil phagosomes are also known to be associated with phagosomes of macrophages (Garin et al., 2001). These include proteins comprising the actin-based cytoskeleton or those involved in cell motility, such as actin, α-actinin, subunits of the actin-related protein 2/3 complex, myosin, tropomyosin, and vimentin. Somewhat unexpectedly, proteins typically found in the endoplasmic reticulum, including calnexin, ERp29, GRP58/ERp57, GRP78/Ig-heavy chain binding protein (BIP) and protein disulfide isomerase, were associated with neutrophil phagosomes. Previous studies by Garin et al. and Gagnon et al. were the first to demonstrate that molecular chaperones are associated with phagosomes of macrophages and proposed that the endoplasmic reticulum contributes to phagocytosis (Gagnon et al., 2002; Garin et al., 2001). The role of endoplasmic reticulum in macrophage phagocytosis was recently a topic of intense debate (Gagnon, Bergeron, & Desjardins, 2005; Touret, Paroutis, & Grinstein, 2005; Touret, Paroutis, Terebiznik, et al., 2005), and the precise function of these phagosomal chaperones remains to be determined. However, recent work by several laboratories indicates that antigens processed within phagosomes undergo cross presentation by MHC class I molecules (Ackerman, Kyritsis, Tampe, & Cresswell, 2003; Gagnon et al., 2002; Guermonprez et al., 2003; Houde et al., 2003). Based on these studies, it was proposed that protein processing and quality control machinery, which includes molecular chaperones and proteasome subunits, function within phagosomes to promote antigen presentation (Houde et al., 2003). Burlak et al. identified 11 molecular chaperones and 5 proteasome subunits associated with human neutrophil phagosomes, and thus the function of these molecules on phagosomes is perhaps conserved between macrophages and neutrophils. Another unexpected finding from the proteomics analysis by Burlak et al. was that proteins typically found in the mitochondria, including F1 ATPase, prohibitin, and peroxiredoxin 3, were also associated with phagosomes (Burlak et al., 2006). Although the function of these proteins at neutrophil phagosomes is unknown, the results of these systems biology-level studies suggest that human PMNs have functions beyond killing of microorganisms, including potential for antigen processing.
3.3.3 |. Proteins of neutrophil granules
To better understand mechanisms of granule exocytosis, Lominadze et al. performed a comprehensive analysis of proteins comprising human neutrophil granule subsets (Lominadze et al., 2005). These studies identified 286 proteins in gelatinase, specific and azurophil granules, and provided the first comprehensive view of proteins associated with these important neutrophil organelles. Subsequent work by Jethwaney et al. reported the proteomes of human neutrophil plasma membranes and secretory vesicles (Jethwaney et al., 2007). Each study identified proteins not previously known to be associated with the secretory vesicles (e.g., 5-lipoxygenase-activating protein and dysferlin) or neutrophil granules (e.g., calreticulin) (Jethwaney et al., 2007; Lominadze et al., 2005). More recently, Rorvig et al. performed proteome profiling of neutrophil granule subsets, secretory vesicles and the plasma membrane and compared protein distribution in each location with PMN transcriptome data (Rorvig, Ostergaard, Heegaard, & Borregaard, 2013). This interesting comparison enabled the authors to identify novel proteins not formerly associated with neutrophils, and notably, revealed an overall 64% correspondence between proteome and transcriptome data. Taken together, these studies are an important step toward developing a comprehensive repository of neutrophil proteins that can be used for systems biology-level analyses.
4 |. NEUTROPHIL APOPTOSIS
Apoptosis is an important mechanism for regulating homeostasis in cells of the immune system. Normal turnover of aging neutrophils occurs in the absence of activation through a process known as spontaneous apoptosis (Savill, 1997; Savill et al., 1989). The intrinsic ability of aged neutrophils to undergo apoptosis is essential for maintaining appropriate cell numbers in circulation. In addition, neutrophil apoptosis plays a central role in resolution of the acute inflammatory response. The indiscriminate release of highly toxic microbicidal components as a consequence of PMN lysis can lead to host tissue damage, which is exacerbated by prolonged inflammation. The association between prolonged neutrophil survival and clinically apparent inflammatory disorders is well documented (Boxer et al., 1990; Edwards & Hallett, 1997). Apoptotic PMNs are recognized and cleared by macrophages in a process known as efferocytosis, thereby preventing excessive host tissue damage by limiting inflammation at sites of infection (Savill et al., 1989). Notwithstanding, the inflammatory milieu is a complex mixture of microbe- and host-derived factors and PMN apoptosis is not an immutable process. Determination of neutrophil fate in such a confounded environment requires simultaneous deciphering of both pro- and anti-apoptotic signals. Furthermore, stimuli that are potent inducers of apoptosis in other cell types can have the opposite effect on neutrophils. For example, exposure of neutrophils to corticosteroids (Meagher, Cousin, Seckl, & Haslett, 1996), hypoxic environments (Hannah et al., 1995), TNF-α (Murray et al., 1997), and conditions that elevate intracellular Ca2+ (Whyte, Hardwick, Meagher, Savill, & Haslett, 1993) and ATP levels (Bours, Swennen, Di Virgilio, Cronstein, & Dagnelie, 2006) inhibit rather than promote neutrophil apoptosis. Nevertheless, it is increasingly clear that the extension of neutrophil survival promotes enhanced clearance of invading microbial pathogens, whereas induction of PMN apoptosis facilitates cell turnover and resolution of inflammation.
4.1 |. Constitutive (spontaneous) neutrophil apoptosis
PMN senescence induces characteristic morphological and physiological changes including cell shrinkage, nuclear condensation, vacuolated cytoplasm, DNA fragmentation, and mitochondrial depolarization (Savill et al., 1989) (Figure 3). In addition, neutrophil apoptosis is associated with an overall diminished functional capacity including impaired chemotaxis, phagocytosis, respiratory burst, and degranulation (Kobayashi, Voyich, Whitney, et al., 2005; Whyte, Meagher, et al., 1993). The overall decrease in neutrophil function is in part a result of down-regulation or shedding of neutrophil surface receptors (Hart, Ross, Ross, Haslett, & Dransfield, 2000; Homburg et al., 1995; Kobayashi, Voyich, Braughton, & DeLeo, 2003). Apoptotic neutrophils also expose phosphatidylserine on the cell surface, which triggers specific recognition and removal by macrophages (Fadok et al., 1992; Henson, 2017). Similar to other cells of the immune system, neutrophils are susceptible to both intrinsic and extrinsic pathways of apoptosis. The intrinsic ‘stress-induced’ pathway of apoptosis is accompanied by reduction in mitochondrial membrane potential, release of cytochrome c and activation of caspase 9. The extrinsic pathways of apoptosis are triggered by engagement of death receptors of the TNF family, which leads to the activation of caspase 8. The majority of studies demonstrate that spontaneous PMN apoptosis occurs primarily through intrinsic pathways and there is little evidence that death-receptors participate in this process. Although the mechanisms for initiation of PMN constitutive apoptosis are unknown, it is well accepted that neutrophil apoptosis is a highly complex process and reports of PMN-specific variations in conventional pathways are common.
Figure 3.
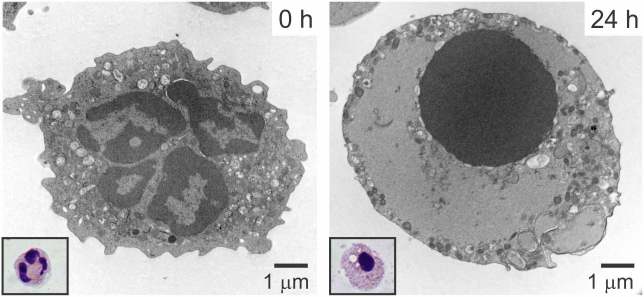
Neutrophil apoptosis. Human neutrophils were isolated from venous blood and then processed for transmission electron microscopy or stained with Wright-Giemsa (inset) after purification (0 h) or 24 h in culture.
Mitochondria are a central component to intrinsic apoptosis pathways. However, neutrophil mitochondria are somewhat of an enigma. Neutrophil processes such as chemotaxis consume large quantities of ATP, which are almost exclusively generated through glycolysis (Lane & Lamkin, 1984). Formation of respiratory chain supercomplexes, which are important for oxidative phosphorylation, is lacking in human neutrophils (van Raam et al., 2008). Early ultrastructural studies of neutrophil mitochondria reveal these organelles to be few in number and small in size (Bainton et al., 1971). However, more recent neutrophil imaging studies using fluorescent probes to measure transmembrane potential indicate PMNs contain a complex tubular network of mitochondria (Fossati et al., 2003; Maianski, Mul, van Buul, Roos, & Kuijpers, 2002). Mitochondrial outer membrane permeabilization is an early event in the intrinsic apoptosis pathway and is facilitated by the activation of the pro-apoptotic BCL2-associated × protein (BAX) (Maianski et al., 2002; Pryde, Walker, Rossi, Hannah, & Haslett, 2000). During apoptosis, mitochondria release additional pro-apoptotic factors into the cytoplasm, such as cytochrome c, Smac/DIABLO (Maianski et al., 2004), Omi/HtrA2 (Maianski et al., 2004), EndoG (El Kebir, Jozsef, Khreiss, & Filep, 2006), and apoptosis-inducing factor (AIF) (El Kebir et al., 2006). Cytosolic cytochrome c, although significantly reduced in neutrophils (Maianski et al., 2004; Murphy, O’Neill, Adrain, Watson, & Martin, 2003), interacts with apoptotic protease-activating factor 1 (APAF-1) in the presence of dATP to trigger formation of a complex called the apoptosome (Hill, Vaidyanathan, Ramos, Ginsberg, & Werner, 2002). The apoptosome recruits, binds, and activates the apoptosis initiator caspase-9. Caspase activation is additionally controlled by the X-linked inhibitor of apoptosis (XIAP) (Maianski et al., 2004), which is rapidly recruited to the apoptosome complex. Displacement of XIAP by Smac/DIABLO potentiates caspase activation, including the apoptosis executioner caspase-3.
Activation of the apoptosis initiator caspase-8 is involved in neutrophil spontaneous apoptosis. Although activation of caspase-8 is commonly associated with apoptosis induced by ligation of death receptors, caspase-8 is peculiarly activated at times early (prior to caspase-3) in neutrophil spontaneous apoptosis (Brown & Savill, 1999; Khwaja & Tatton, 1999; Scheel-Toellner, Wang, Craddock, et al., 2004). Ligation of death receptors, such as TNF-α receptor (Murray et al., 1997), FAS (Liles, Kiener, Ledbetter, Aruffo, & Klebanoff, 1996), and TRAIL receptor (Renshaw et al., 2003), initiates the formation of a death-inducing signaling complex (DISC) and subsequent activation of caspase-8. However, caspase-8 activation during neutrophil spontaneous apoptosis occurs independently of death receptor ligation, which suggests the presence of an alternative activation pathway. Two distinct mechanisms have been proposed to account for the activation of caspase-8 during neutrophil spontaneous apoptosis. The first mechanism involves acid sphingomyelinase-induced clustering of death receptors in lipid rafts, spontaneous DISC assembly and subsequent activation of caspase-8 (Scheel-Toellner, Wang, Assi, et al., 2004). The second mechanism provides for the direct activation of caspase-8 by the neutrophil aspartic acid protease cathepsin D that is released from azurophilic granules (Conus et al., 2008). Provocatively, ROS are important for both death receptor clustering and permeabilization of azurophilic granules during neutrophil spontaneous apoptosis. However, the mechanism for ROS production in non-activated neutrophils is unclear. ROS are clearly important for activation of caspase-8 during phagocytosis-induced neutrophil programmed cell death (Zhang, Hirahashi, Cullere, & Mayadas, 2003). Caspase-8 is also involved in cleavage of the BCL-2 family protein BID that participates in mitochondrial membrane permeabilization by a mechanism similar to BAX (Baumann et al., 2003). The activity of caspases can also be impaired through direct modifications such as phosphorylation of caspase-8 and caspase-3 by p38-mitogen-activated protein kinase (MAPK) (Alvarado-Kristensson et al., 2004). There are numerous additional pro- and anti-apoptotic factors that have been implicated in spontaneous apoptosis of neutrophils (DeLeo, 2004). The existence of multiple pathways governing neutrophil cell fate highlights the importance of apoptosis as a regulatory rheostat between homeostasis and host defense. However, determination of the hierarchy for execution of distinct pathways in neutrophil apoptosis remains to be elucidated. Given the complexity of apoptosis pathways, studies of neutrophil apoptosis are ideally suited to interrogation by genome-wide approaches, as comprehensive understanding of these mechanisms using traditional single molecule studies would be time-consuming.
4.2 |. Extracellular factors
The ability of neutrophils to receive and respond to environmental signals is prerequisite for participation in host defense. Microbial infections are accompanied by important host responses, including release of pro-inflammatory cytokines, which alter neutrophil apoptosis. In general, the majority of inflammatory mediators are associated with delaying neutrophil spontaneous apoptosis and include factors, such as IL-1β (Colotta, Re, Polentarutti, Sozzani, & Mantovani, 1992), IL-6 (Biffl, Moore, Moore, & Barnett, 1995; Colotta et al., 1992), IL-8 (Kettritz et al., 1998), GROα (Dunican, Leuenroth, Ayala, & Simms, 2000), platelet-activating factor (Murray et al., 1997), IFN-γ (Colotta et al., 1992; Sakamoto et al., 2005), G-CSF (Colotta et al., 1992), C5a (Lee, Whyte, & Haslett, 1993) and GM-CSF (Colotta et al., 1992). Notably, most of the host-derived cytokines that delay apoptosis are also neutrophil priming agents. However, reports on the effects of the potent PMN priming agent TNF-α on neutrophil survival are more variable. TNF-α has both pro- and anti-apoptotic effects on neutrophils. The divergent effects of TNF-α on neutrophil survival are related to concentration (van den Berg, Weyer, Weening, Roos, & Kuijpers, 2001), duration of stimulus (Murray et al., 1997), and functional state of the cell (Avdi et al., 2001). High concentrations of TNF-α induce spontaneous neutrophil apoptosis associated with increased levels of ROS, whereas prolonged survival correlates with activation of NFκB (Ward, Chilvers, et al., 1999). The in vivo relevance of this phenomenon is unclear. In addition to effects of pro-inflammatory cytokines on PMN fate, prolonged neutrophil survival is enhanced by neutrophil adhesion to ligands of β2-integrin (Whitlock, Gardai, Fadok, Bratton, & Henson, 2000). Ligation of CD11b with soluble fibrinogen delays neutrophil apoptosis and triggers NFκB activation through an ERK1/2-dependent pathway (Rubel et al., 2003). Antibody cross-linking of β2-integrin induces the activation of survival pathways through Akt and MAPK-ERK (Whitlock et al., 2000). However, subsequent stimulation with TNF-α leads to decreased Akt activity through SH2-domain-containing inositol phosphatase (SHIP) activation and results in rapid progression of neutrophil apoptosis (Gardai et al., 2002).
Bacterial-derived products such as LPS, LTA, and secreted toxins are known to delay neutrophil apoptosis (DeLeo, 2004). Inasmuch as several TLR ligands are potent PMN priming agents, it follows that TLR-mediated signal transduction pathways influence neutrophil fate. However, the majority of evidence suggests that only TLR4 and TLR2 directly modulate PMN survival. Stimulation of neutrophils with either purified LPS (TLR4) (Sabroe et al., 2003) or LTA (TLR2) (Lotz et al., 2004) delays spontaneous apoptosis. Inhibition of NFκB activation is associated with enhanced TLR4-mediated neutrophil survival (Sabroe et al., 2003). Studies on TLR-inhibition of neutrophil apoptosis in whole blood indicate that phosphatidylinositol 3-kinase (PI3K) activation may also be involved in enhanced survival (Francois et al., 2005). The addition of monocytes to TLR4-stimulated PMNs also prolongs neutrophil survival (Sabroe et al., 2003), suggesting a possible synergy with cytokine-signaling pathways. Mononuclear cell-dependent enhancement of neutrophil survival has also been demonstrated following exposure to staphylococcal enterotoxins (Moulding, Walter, Hart, & Edwards, 1999). Several other bacterial toxins have been shown to directly promote neutrophil survival, such as Escherichia coli verotoxin (Liu et al., 1999), Shiga toxin (Brigotti et al., 2008), and Staphylococcus epidermidis PSMs (Liles, Thomsen, O’Mahony, & Klebanoff, 2001). Together, these observations suggest that enhanced neutrophil survival is a desirable consequence during early stages of inflammation and promotes the clearance of bacterial pathogens.
4.3 |. Influence of bacteria on PMN survival
The ability of inflammatory mediators and bacterial-derived products to support neutrophil survival preceding phagocytosis is important for maintenance of optimal microbicidal potential. In contrast, phagocytosis-induced cell death (PICD) is a mechanism to clear tissues of effete neutrophils containing killed microbes, thereby facilitating the resolution of infection. As with spontaneous neutrophil apoptosis, timely removal of neutrophils—although in this case after phagocytosis—would prevent release of cytotoxic molecules into surrounding host tissues, a phenomenon caused by necrotic lysis. Activation of human neutrophils by phagocytosis can significantly accelerate the rate of apoptosis (Coxon et al., 1996; Gamberale, Giordano, Trevani, Andonegui, & Geffner, 1998; Kobayashi et al., 2002; Watson, Redmond, Wang, Condron, & Bouchier-Hayes, 1996; Zhang et al., 2003). Consistent with this idea, neutrophil ingestion of Escherichia coli, Streptococcus pneumoniae, Streptococcus pyogenes, Candida albicans, Staphylococcus aureus, Mycobaterium tuberculosis, Burkholderia cepacia, Borrelia hermsii, and Listeria monocytogenes significantly accelerates the rate of PMN apoptosis (DeLeo, 2004; Kobayashi, Braughton, et al., 2003; Watson et al., 1996). Importantly, phagocytosis significantly increases the rate of PMN apoptosis irrespective of any delay in cell fate imparted by cytokines or bacteria-derived factors (Engelich, White, & Hartshorn, 2001; Hart et al., 2000; Watson et al., 1996). Thus, PMN apoptosis plays a central role not only in regulation of cell turnover, but also in termination of the inflammatory process.
Although, PICD can be desirable for the resolution of infection and prevention of prolonged inflammation, this process is not immutable. The relatively short life-span of neutrophils is not amenable to the long-term survival strategies employed by most intracellular pathogens; however, several bacterial pathogens are capable of exploiting apoptosis/PICD as a potential mechanism of pathogenesis. To that end, these microorganisms either accelerate or delay neutrophil apoptosis. A limited number of bacterial pathogens have been shown conclusively to delay human neutrophil apoptosis. For example, Anaplasma phagocytophilum, the causative agent of human granulocytic anaplasmosis, was the first bacterial pathogen reported to delay PMN apoptosis (Yoshiie, Kim, Mott, & Rikihisa, 2000). This delay in apoptosis extends the neutrophil lifespan such that the pathogen is able to replicate within an endosomal compartment (Yoshiie et al., 2000). Conversely, bacterial pathogens such as Staphylococcus aureus and Streptococcus pyogenes cause direct PMN lysis and/or accelerate bacteria-induced apoptosis to the point of secondary necrosis (Kobayashi, Braughton, et al., 2003; Voyich et al., 2005). The deleterious effects on neutrophil cell fate may be mediated in part by bacterial toxins. For example, some staphylococci produce leukotoxins that promote apoptosis at sublytic levels (Genestier et al., 2005) or profoundly affect neutrophil integrity (Ventura et al., 2010; Wang et al., 2007). In addition, Pseudomonas aeruginosa pyocyanin (Usher et al., 2002) and Aspergillus fumigatus gliotoxin (Ward, Dransfield, Chilvers, Haslett, & Rossi, 1999) have been shown to promote neutrophil apoptosis. It is likely that accelerated PMN lysis contributes to the overall levels of tissue necrosis associated with these infections.
4.4 |. Neutrophil apoptosis differentiation program
The importance of neutrophil spontaneous apoptosis in maintenance of cellular homeostasis is well recognized and considerable effort has been expended towards determining the molecular mechanisms governing this essential process. In addition, we now know that neutrophil activation has a direct impact on apoptosis and regulation of PICD differs from that of spontaneous neutrophil apoptosis. Reports of additional factors and conditions that modulate neutrophil fate have increased steadily over the past decade, but we are still far from a complete understanding of the specific pathways that regulate neutrophil apoptosis. That said, systems biology-level approaches have been instrumental in re-shaping our view of the role of neutrophils in the resolution of the inflammatory response. Notably, genome-wide studies have been a crucial guide in our investigation of the molecular determinants that regulate neutrophil PICD and have provided new insight into mechanisms by which bacteria exploit this important process.
Within a few hour hours after neutrophil phagocytosis, the number of differentially expressed apoptosis-related genes increases dramatically. For example, genes encoding apoptosis mediators, such as BAX, BCL2A1, CFLAR, and nuclear orphan receptors TR3 (NR4A1), NURR1 (NR4A2), and NOR1 (NR4A3), are up-regulated prior to initiation of PICD (Kobayashi et al., 2002). In addition, phagocytosis of bacterial pathogens up-regulates PMN transcripts encoding several key mediators of TNF- and TLR2-signaling, including TNF, IRAK1, and TNFAIP8 (Kobayashi, Braughton, et al., 2003). Several lines of evidence indicate that proinflammatory capacity of human PMNs is regulated at the level of gene expression during PICD. For instance, the initial stages of PMN apoptosis are accompanied by changes in the expression of genes encoding important regulators of detoxification and redox pathways, such as those governing glutathione-, thioredoxin-, and heme metabolism (Kobayashi, Voyich, Somerville, et al., 2003). Thus, transcriptional regulation of PMN detoxification pathways provides a secondary mechanism to limit inflammatory potential should neutrophils undergo premature lysis. Importantly, progression of neutrophil apoptosis correlates with down-regulation of genes encoding proinflammatory mediators, phosphoinositide metabolism, and calcium signal transduction (Kobayashi, Voyich, Braughton, et al., 2003; Kobayashi, Voyich, Somerville, et al., 2003). Although it is appreciated that neutrophil apoptosis is characterized by an overall decrease in functional capacity, the sheer number of transcripts identified by genome-wide analyses as down-regulated during PICD was somewhat unexpected (Kobayashi, Braughton, et al., 2003; Kobayashi, Voyich, Braughton, et al., 2003; Kobayashi et al., 2002). For example, dozens of transcripts encoding key proinflammatory mediators and signal transduction molecules are down-regulated during the initiation of phagocytosis-induced apoptosis (Kobayashi, Braughton, et al., 2003; Kobayashi, Voyich, Braughton, et al., 2003; Kobayashi et al., 2002; Kobayashi, Voyich, Somerville, et al., 2003). In addition, the vast majority of genes encoding proinflammatory molecules that were up-regulated at times early following phagocytosis decreased significantly following induction of PICD. Based on these findings and the integration of functional studies using human neutrophils, we proposed that bacteria induce an apoptosis differentiation program in human neutrophils that facilitates PMN turnover and resolution of the inflammatory response (Kobayashi & Deleo, 2004). Thus, accelerated neutrophil apoptosis after phagocytosis and the accompanying down-regulation of inflammatory capacity is an outcome that contributes to the resolution of infection. Consistent with this hypothesis, PMN phagocytosis of such different bacteria as Borrelia hermsii, Burkholderia cepacia, Listeria monocytogenes, Staphylococcus aureus, and Streptococcus pyogenes elicit similar patterns of gene expression that collectively form a common transcriptome (apoptosis differentiation program) (Kobayashi, Braughton, et al., 2003). The neutrophil apoptosis differentiation program includes molecules involved in multiple biological processes, culminating with down-regulation of proinflammatory capacity and concomitant induction of apoptosis/PICD.
Inasmuch as neutrophil apoptosis is a critical process for resolution of inflammation, it is not surprising that some bacterial pathogens alter the normal PMN apoptosis differentiation program as a mechanism of pathogenesis. For instance, the ability of Streptococcus pyogenes to cause rapid PICD and ultimately PMN lysis is reflected by and/or results from the changes in neutrophil gene expression (Kobayashi, Braughton, et al., 2003). The ability of Streptococcus pyogenes to exploit PMN fate pathways is likely an important component of streptococcal pathogenesis and is a potential contributor to fulminant tissue destruction observed in invasive disease (Musser & DeLeo, 2005). It is important to note that ability of Streptococcus pyogenes to induce comparatively more rapid PICD and neutrophil lysis was discovered with a transcriptome approach, as those results directed subsequent in vitro studies. A similar approach has been used to better understand Staphylococcus aureus-induced PMN lysis after phagocytosis (Kobayashi et al., 2010).
By contrast, the obligate intracellular pathogen Anaplasma phagocytophilum inhibits neutrophil apoptosis to survive and thereby cause disease. To investigate the molecular basis of Anaplasma phagocytophilum survival within neutrophils, Borjesson et al. used oligonucleotide microarrays to measure global changes in human PMN gene expression following infection with Anaplasma phagocytophilum (Borjesson et al., 2005). Functional analysis of infected PMNs confirmed previous reports that Anaplasma phagocytophilum fails to trigger neutrophil production of ROS (Carlyon, Abdel-Latif, Pypaert, Lacy, & Fikrig, 2004; Mott & Rikihisa, 2000) and the pathogen delays PMN spontaneous apoptosis (Yoshiie et al., 2000). In addition, PMN uptake of Anaplasma phagocytophilum occurs at a slow rate compared to that observed with other bacteria (Borjesson et al., 2005). Consistent with the functional studies, infection of human PMNs with Anaplasma phagocytophilum delayed up-regulation of transcripts involved in the acute inflammatory response, such as TNF, IL1B, CXCL1, CXCL2, CXCL3, CCL3, CCL4, and CD54 (Borjesson et al., 2005). Similar gene expression profiles from Anaplasma phagocytophilum-infected neutrophils were reported in subsequent studies (Lee & Goodman, 2006; Lee et al., 2008; Sukumaran et al., 2005). A key finding of these studies related to the levels of PMN transcripts encoding NADPH oxidase proteins. Previous studies using promyelocytic HL60 cells, an immortalized cell line sometimes used as a model for human neutrophils, had suggested that the ability of Anaplasma phagocytophilum to inhibit ROS production was due to down-regulation of transcripts encoding NADPH oxidase proteins (Banerjee, Anguita, Roos, & Fikrig, 2000; Carlyon, Chan, Galan, Roos, & Fikrig, 2002). In contrast, a microarray-based approach using human neutrophils indicated that these transcripts remained unchanged or increased in expression following ingestion of Anaplasma phagocytophilum (Borjesson et al., 2005). This finding resolved controversy in part about the mechanism underlying inhibition of ROS in Anaplasma phagocytophilum-infected neutrophils. In addition, Anaplasma phagocytophilum-infected PMNs increased expression levels of several anti-apoptosis genes including BIRC2, BIRC3, CFLAR, TNFAIP8, and TNIP2, and decreased expression of numerous apoptosis-inducing factors. Moreover, interaction of live or dead Anaplasma phagocytophilum with PMNs results in the inability to induce neutrophil apoptosis via ligation of the FAS death receptor (Borjesson et al., 2005), and indicate that pre-existing surface molecules facilitate survival during Anaplasma phagocytophilum infection, presumably to promote bacterial replication and persistence.
Francisella tularensis and Francisella novicida each inhibit PMN apoptosis and thereby prolong the PMN lifespan (Kinkead, Fayram, & Allen, 2017; Schwartz et al., 2012). Schwartz et al. used oligonucleotide microarrays and a qPCR approach to determine that infection of human PMNs with F. tularensis causes differential expression of over 3000 PMN genes (Schwartz et al., 2013). A subset of these genes, such as those encoding caspases, BAX, BCL2A1 and X-linked inhibitor of apoptosis, are involved in intrinsic and extrinsic apoptosis pathways (Schwartz et al., 2013). Inasmuch as Francisella tularensis can escape from phagosomes and survive within the neutrophil cytoplasm (McCaffrey & Allen, 2006), the ability of these bacterial pathogens to delay PMN turnover is likely important for the success of this organism as a human pathogen.
The ability of pathogens to alter neutrophil fate by either blocking apoptosis to facilitate intracellular pathogen survival or promoting rapid lysis to eliminate neutrophils represent plausible mechanisms of virulence. The genome-wide analyses with human neutrophils and bacteria, coupled with the results of many targeted studies using neutrophils and animal models, led to the hypothesis that there are two possible outcomes of PMN-bacteria interactions (Figure 4). On one hand, phagocytosis activates neutrophils and ingested microorganisms are killed, thus leading to PICD and removal of effete PMNs by macrophages. This process leads to the resolution of infection and is healthy for the host. Alternatively, pathogens are ingested but not killed, and neutrophils either lyse or have delayed turnover that promotes pathogen replication (Figure 4). Either of these latter outcomes is counterproductive for the resolution of infection and potentially results in disease. Although systems biology-level approaches have been critical to enhancing our understanding of the molecular mechanisms of neutrophil PICD and bacterial pathogenesis, the complex association of neutrophils, macrophages, and secreted pathogen and host factors at infection sites complicate this rather simplistic model. Furthermore, it has been suggested recently that bacterial pathogens alter the process of efferocytosis by influencing secretion of TNF-α from macrophages (Persson, Blomgran-Julinder, Rahman, Zheng, & Stendahl, 2008), which may in turn impact the resolution of inflammation (McPhillips et al., 2007; Michlewska, Dransfield, Megson, & Rossi, 2009). The development of complex in vitro and in vivo model systems, combined with genome-wide approaches, will be the next important step towards a complete understanding of the role of neutrophil apoptosis in bacterial pathogenesis.
Figure 4.
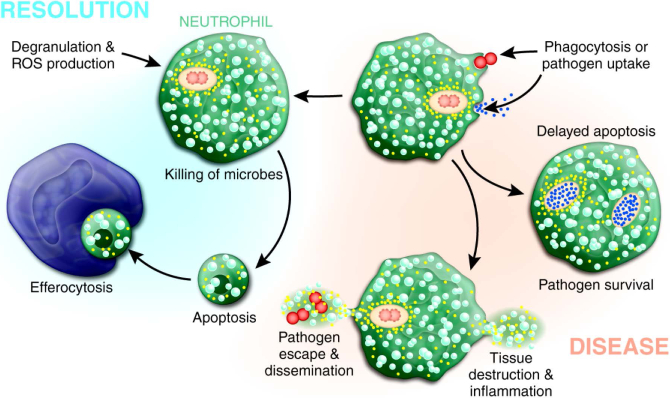
A schematic that illustrates possible outcomes of microbe-neutrophil interaction. See text for details.
5 |. DISEASES
Neutropenia and inherent deficiencies in neutrophil function are important predisposing risk factors for development of life-threatening bacterial and fungal infections. The majority of congenital neutrophil disorders are relatively rare, thereby confounding characterization of disease. Genome-wide expression analyses have provided new insight into the pathophysiology of disease or served as an important first step toward identifying the molecular genetic basis of disease. As a recent example, Grabowski et al. developed a novel proteomics approach to identify and characterize monogenic neutrophil diseases (Grabowski et al., 2019). The method has the potential to provide information important for the identification and diagnosis of immune disorders. Here we review select diseases involving neutrophils that have been studied using a systems biology approach.
5.1 |. Chronic granulomatous disease
Leukocytes from patients with X-linked chronic granulomatous disease (XCGD) are defective in their ability to produce ROS due to defined heterogeneous mutations that preclude assembly of the NADPH-oxidase (Curnutte, Scott, & Mayo, 1989; Hamers, de Boer, Meerhof, Weening, & Roos, 1984; Nunoi, Rotrosen, Gallin, & Malech, 1988; Royer-Pokora et al., 1986; Volpp, Nauseef, & Clark, 1988). Although the genetic and cellular basis of XCGD has long been known (reviewed in refs. (Lekstrom-Himes & Gallin, 2000; Roos, 2016)), the molecular basis of associated pathophysiologies, such as formation of granulomas, remains undetermined. We previously used gene expression profiling to obtain a comprehensive global view of the impact of PMN-derived oxidants on the resolution of inflammation (Kobayashi et al., 2004). Analysis of PMNs from XCGD patients revealed that these cells had increased levels of transcripts encoding proinflammatory molecules and decreased expression of genes encoding anti-inflammation mediators, consistent with a previous report showing excessive IL-8 production by CGD neutrophils (Lekstrom-Himes, Kuhns, Alvord, & Gallin, 2005). In addition, neutrophil transcripts involved in PICD are differentially expressed between activated PMNs from healthy control subjects and XCGD patients and coincide with a delay in apoptosis. Taken together, these findings provide support to the hypothesis that reduced or absent PICD in XCGD neutrophils results in delayed resolution of inflammation, which could contribute to the formation of granulomas in XCGD patients.
5.2 |. Specific granule deficiency
Neutrophil specific granule deficiency (SGD) is a rare congenital disorder characterized by susceptibility to recurrent pyogenic infections. Neutrophils from SGD patients have impaired bactericidal activity due to deficiencies in specific granule contents, in addition to selective absence of azurophilic- and tertiary granule constituents. The molecular basis of SGD involves germ-line mutation of the transcription factor, C/EBPε(Gombart et al., 2001; Lekstrom-Himes, Dorman, Kopar, Holland, & Gallin, 1999). Preceding characterization of the human defect, studies with mice deficient in C/EBPε revealed that the transcription factor has a defined role in hematopoiesis, and mature neutrophils are functionally impaired (Yamanaka et al., 1997). In subsequent studies, gene expression profiling was used to provide additional insight into the pathophysiology of SGD in a mouse model of disease (Gombart et al., 2005). This study demonstrated myeloid cell transcriptional changes in genes corresponding to a broad range of cellular processes. Dysregulation of gene expression was appropriately observed in key inflammatory response regulators, such as CCL2, CCL4, CCL7, IL6, IL1B, CCR1, IL8RB, and CSF3R. In addition, there was dysregulation of genes encoding cytoskeletal structural and regulatory proteins, potentially explaining neutrophil defects in chemotaxis, phagocytosis and superoxide production. Further investigation into the role of C/EBPε in regulating the transcription of genes identified in those studies will likely provide additional insight into the pathophysiology of SGD.
5.3 |. Hyper-IgE Syndrome (HIES)/Job’s Syndrome
HIES or Job’s syndrome is a rare immunological disorder characterized by dermatitis, recurrent staphylococcal skin abscesses, cyst-forming pneumonias, elevated serum IgE levels, and multiple developmental abnormalities. Although HIES was originally described by Davis et al. in 1966 (Davis, Schaller, & Wedgwood, 1966), the molecular genetic basis of the disease was identified only recently (Holland et al., 2007; Minegishi et al., 2007). As a first step toward identifying the cause of HIES and to gain insight into the molecular processes that contribute to the pathophysiology of the disease, Holland et al. compared global gene expression in leukocytes from HIES patients and healthy control individuals (Holland et al., 2007). Transcriptome analysis of unstimulated and stimulated PMNs from HIES patients revealed that there were elevated levels of transcripts encoding proinflammatory molecules in patient cells, including at least thirty transcripts encoding proteins related to IFN signal transduction. Collectively, the PMN transcriptome data suggested that signal transduction involving IFNs and STATs was altered in HIES patients. Consistent with those results, HIES leukocytes had increased production of IFN-γ and TNF-α following stimulation. However, leukocytes from these patients also displayed impaired stimulus-dependent signaling through the IL-6 receptor (Holland et al., 2007). DNA sequence analysis revealed distinct mutations in the DNA-binding region and SH2 domain of STAT3 in HIES patients (Holland et al., 2007). These studies clearly demonstrate that autosomal dominant and sporadic forms of HIES are caused by mutations in the gene encoding STAT3, a key transcription activator following cytokine- or growth factor stimulation, thereby explaining the diverse nature of the disease. Given the involvement of STAT3-SOCS3 in recruitment of myeloid cells and execution of anti-inflammatory responses in mature immune cells (O’Shea & Murray, 2008), the finding is of significant importance for increased understanding of the regulation of inflammation in general. Moreover, the studies strong provide support to the idea that microarray-based studies can be used as a first-tier approach for the identification of the genetic basis of hereditary immune disorders.
5.4 |. Acute respiratory distress syndrome (ARDS)
ARDS is a condition characterized by exasperated inflammation of the lungs and can be the result of a variety of underlying conditions such as pancreatitis, sepsis, trauma, aspiration or severe burns. During ARDS, the alveolar-capillary barrier is disrupted, resulting in lung edema and reduced gas exchange. ARDS is characterized by increased accumulation of PMNs in the microvasculature and interstitial space of the lung. A study comparing the transcriptional profile of blood PMNs from ARDS patients and healthy control donors treated with recombinant human GM-CSF reported more than 1300 differentially expressed PMN genes, including those involved in chemotaxis and apoptosis (Juss et al., 2015; Juss et al., 2016). These authors showed further that blood and alveolar PMNs from ARDS patients were primed for enhanced production of reactive oxygen species, and constitutive apoptosis was delayed (Juss et al., 2016). The authors used a microarray approach as a step to identify novel therapeutic interventions.
6 |. SINGLE CELL GENOMICS
Single cell genomics has emerged as a high-resolution approach to dissect heterogeneity within host cell populations (Stubbington, Rozenblatt-Rosen, Regev, & Teichmann, 2017). The method is based on single-cell RNA sequencing (scRNA-seq) and has provided the means to identify, characterize, and classify cell populations and associated biomarkers in multiple scientific disciplines (Chen, Ye, & Guo, 2019; Chu et al., 2016; Muller & Diaz, 2017; Ofengeim, Giagtzoglou, Huh, Zou, & Yuan, 2017; Stubbington et al., 2017). Macrophages, dendritic cells, and T cells are the immune cell subsets that have been most widely studied by scRNA-seq (Avraham et al., 2015; Gaublomme et al., 2015; Saliba et al., 2016; Shalek et al., 2013; Zheng et al., 2017). For example, Avraham et al. employed scRNA-seq in combination with fluorescent markers to study the response of individual macrophages to ingested Salmonella typhimurium (Avraham et al., 2015). These authors demonstrated that the macrophage type I IFN response is varied depending on bacterial PhoP activity. Subsequent studies by Saliba et al. used scRNA-seq to show that Salmonella typhimurium-containing macrophages display either pro- or anti-inflammatory states depending on growth rate of the phagocytosed bacteria (Saliba et al., 2016). Collectively, these studies represent a major step forward in our efforts to generate a comprehensive view of host-pathogen interactions.
Although scRNA-seq has been used to investigate multiple leukocyte subsets, at present there are few such studies with neutrophils (Kippner, Kim, Gibson, & Kemp, 2014; Zhu et al., 2018; Zilionis et al., 2019). Inasmuch as neutrophils are a first line of defense against invading bacteria and fungi, it is conceivable (or even likely) that the response to these pathogens is varied from cell to cell, and might account for differential disease outcomes. Previous studies using gradient density separation and/or flow cytometry identified PMN subsets in peripheral blood, but the extent of the heterogeneity has remained incompletely defined. As a step toward addressing this deficiency in knowledge, Kippner et al. used single cell sorting and nanoscale qRT-PCR to identify two distinct neutrophil subtypes isolated from peripheral blood of healthy volunteers using (Kippner et al., 2014). Subsequent studies by Zhu et al. combined cytometry time of flight (CyTOF) and scRNA-seq to identify an early unipotent neutrophil progenitor in mouse and human bone marrow that is different from the common granulocyte and monocyte progenitor GMP (Zhu et al., 2018). Most recently, Zilionis et al. used scRNA-seq to determine the transcriptome of tumor-infiltrating myeloid cells in non-small cell lung cancer patients and in a mouse cancer model (Zilionis et al., 2019). The authors discovered that although there are clear differences between mouse and human neutrophil subsets, some of the tumor-associated phenotypes of neutrophils are conserved across species (Zilionis et al., 2019). One of the caveats associated with single cell technology for study of neutrophils is the limited level of transcript typically present in mature peripheral blood neutrophils. Although these and other technical challenges must be overcome, scRNA-seq remains a promising technique for future studies of neutrophil biology.
7 |. CONCLUSIONS
The implementation of systems biology technologies has provided a means to globally interrogate the molecular events governing host-pathogen interactions. Because of these genome-wide approaches, we now know that neutrophils have significant biosynthetic potential and therefore, their role in the innate immune system is being reconsidered more broadly. Systems biology-level approaches have enabled molecular dissection of key neutrophil processes or functions, including post-phagocytosis sequelae and many new discoveries have been made (Table 1). The idea that neutrophils actively participate in processes beyond phagocytosis and killing of microbes is a relatively recent concept and there is now considerable effort directed to understand the role of PMNs in the resolution of inflammation.
TABLE 1.
Selected attributes, processes, and functions of neutrophils discovered using genome-wide approaches.
| Attribute, function or process | Discovery or conclusion | Ref. |
|---|---|---|
| Granulopoiesis | Positive correlation between protein and mRNA levels during neutrophil development | (Iida et al., 2005; Lian et al., 2002; Lian et al., 2001) |
| Temporal regulation of transcripts encoding granule proteins during neutrophilic differentiation of human primary bone marrow progenitors | (Theilgaard-Monch et al., 2005) | |
| Lineage specificity in gene expression among hematopoietic cells | (Kluger et al., 2004) | |
| Mature neutrophils have increased capacity to respond to interferon-gamma | (Martinelli et al., 2004) | |
| Coordinate gene regulation during cellular differentiation is influenced by chromosomal organization | (Kosak et al., 2007) | |
| DNA methylation patterns differ during stages of granulopoesis and mature PMNs | (Ronnerblad et al., 2014) | |
| miRNAs influence key transcription factors during granulopoesis | (Larsen et al., 2013; Yuan et al., 2018) | |
| In mature neutrophils, G-CSF treatment as a model of emergency granulopoesis leads to reduced transcripts of four major granule proteins involved in bactericidal activity | (Pedersen et al., 2016) | |
| Identification of a unipotent neutrophil progenitor | (Zhu et al., 2018) | |
| Heterogeneity | Identification of neutrophil subsets in populations of tumor-infiltrating myeloid cells; identification of neutrophil subsets in peripheral blood of healthy subjects | (Kippner et al., 2014; Zilionis et al., 2019) |
| Recruitment | ||
| Chemotaxis | Regulation of chemotaxis by miRNA | (Bauernfeind et al., 2012; Dorhoi et al., 2013; Johnnidis et al., 2008) |
| Priming | Comprehensive view of the similarities and differences between priming agents and phagocytic stimuli | (Wright et al., 2013; Zhang et al., 2004) |
| Lipopolysaccharide elicits changes in expression of interferon-stimulated genes (ISGs) | (Fessler et al., 2002; Malcolm et al., 2003) | |
| Transmigration | Regulates cell fate and promotes wound healing | (Theilgaard-Monch et al., 2004) |
| Spontaneous apoptosis | Comprehensive view of molecules involved in the loss of neutrophil function during apoptosis. This study reveals the remarkable number of host defense-related molecules that are down-regulated during constitutive (spontaneous) neutrophil apoptosis. | (Kobayashi, Voyich, Whitney, et al., 2005) |
| PMNs undergoing apoptosis exhibit a pro resolving lipid mediator profile | (Dalli & Serhan, 2012) | |
| Phagocytosis | ||
| Phagocytosis-induced cell death (PICD) | Phagocytosis induces a neutrophil apoptosis differentiation program, a final stage of transcriptionally regulated PMN maturation. Neutrophil programmed cell death is regulated at the level of gene expression, and thus PMN gene regulation facilitates resolution of inflammation. | (Kobayashi et al., 2002) |
| Comprehensive view of the down-regulation of pro-inflammatory capacity during PICD. The study highlights the sheer magnitude of change in proinflammatory molecules during apoptosis. | (Kobayashi, Voyich, Braughton, et al., 2003) | |
| Bacterial pathogens modulate the neutrophil apoptosis differentiation program. This work forms the basis of a paradigm for the resolution of infection. The discovery is that there are two possible outcomes of neutrophil-bacteria interactions; one leads to resolution of infection and the other to disease (see Figure 4). | (Kobayashi, Braughton, et al., 2003) | |
| PICD is delayed in neutrophils from chronic granulomatous disease patients and ROS are critical for PICD. | (Kobayashi et al., 2004) | |
| Neutrophil phagosomes contain proteins typically associated with the endoplasmic reticulum. This finding suggests neutrophil phagosomes have potential for antigen processing | (Burlak et al., 2006) | |
| Delayed apoptosis | A. phagocytophilum fails to induce the apoptosis differentiation program in human neutrophils. | (Borjesson et al., 2005; Lee & Goodman, 2006; Lee et al., 2008; Sukumaran et al., 2005) |
| Leishmania major and A. phagocytophilum enhance release of pro-inflammatory LTB4, and concomitantly decrease production of pro-resolving LXA4 | (Plagge & Laskay, 2017) | |
| GM-CSF enhances neutrophil pro-inflammatory capacity by up-regulating dozens of molecules involved in host defense. Neutrophils have potential for antigen presentation. | (Kobayashi, Voyich, Whitney, et al., 2005; Lin & Lore, 2017; Polak et al., 2019; Vono et al., 2017) |
Current genome-wide approaches with neutrophils primarily involve analysis of transcriptome or proteome. However, more recent systems-level studies include analysis of lipid mediators, such as lipoxins, resolvins, and protectins, which have an essential role in the resolution of inflammation (Serhan, Lu, Hong, & Yang, 2007). Indeed, metabolome studies by Serhan and colleagues revealed that human PMNs convert resolvin E1 to novel metabolic products that have potent anti-inflammatory activities (Hong et al., 2008). This line of investigation is especially intriguing for system-level analyses, since it is possible to predict synthesis of these novel compounds using lipidomics and then test function (Serhan, Hong, & Lu, 2006; Serhan et al., 2007). Lipid mediator (LM) metabololipidomics profiling of neutrophils, zymosan-stimulated neutrophils and neutrophils undergoing apoptosis revealed distinct LM patterns (Dalli & Serhan, 2012). For example, PMNs undergoing apoptosis have a pro-resolving LM profile, including production of prostaglandin E2, lipoxin B4 and RvE2, whereas activated PMNs have a proinflammatory LM profile predominated by leukotriene B4 (Dalli & Serhan, 2012). More recently, metabololipidomics was coupled with a liquid chromatography tandem-mass cytometry approach to show that lipid mediators RvE1, RvD1, RvD5, LXB4, and MaR1 directly target neutrophils (and monocytes) and that stimulation of these leukocytes with LMs increases phagocytosis (Norris, Libreros, Chiang, & Serhan, 2017). Infection of human neutrophils with Leishmania major or Anaplasma phagocytophilum, two pathogens with known ability to survive in PMNs and prolong their survival, promoted release of pro-inflammatory LTB4 but decreased production of pro-resolving LXA4 (Plagge & Laskay, 2017). These findings indicate that pathogens have the ability to alter neutrophil lipid mediator signaling and thereby influence the outcome of infection.
There is little doubt that whole-genome sequencing will continue to play a prominent role in studies directed to elucidate the molecular genetic basis of immune disorders, including those that involve neutrophils and host defense. Systems-level approaches have been implemented for discovery of tumor-specific somatic mutations; for example, Ley et al. used whole-genome sequencing to identify eight new mutations associated with acute myeloid leukemia (Ley et al., 2008). For a true systems biology analysis of the role of neutrophils in innate immunity, it will be important to develop tools that integrate data sets from multiple laboratories into a user-friendly platform from which to ask important questions. There is a significant challenge in the integration of enormous data sets generated across a diversity of platforms. Comparison of systems-level studies in neutrophils is further confounded by apparent functional discrepancies between primary human neutrophils, in vitro cell lines (e.g. HL-60), and experimental animal model systems. Despite these caveats, significant progress has been made toward a comprehensive understanding of neutrophil functions and the role of these cells in innate immunity.
FUNDING INFORMATION
The authors are supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
Contributor Information
Viktoria Rungelrath, Laboratory of Bacteriology, Rocky Mountain Laboratories, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Scott D. Kobayashi, Laboratory of Bacteriology, Rocky Mountain Laboratories, National Institute of Allergy and Infectious Diseases, National Institutes of Health
Frank R. DeLeo, Laboratory of Bacteriology, Rocky Mountain Laboratories, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
REFERENCES
- Ackerman AL, Kyritsis C, Tampe R, & Cresswell P (2003). Early phagosomes in dendritic cells form a cellular compartment sufficient for cross presentation of exogenous antigens. Proc Natl Acad Sci U S A, 100(22), 12889–12894. doi: 10.1073/pnas.1735556100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aglietta M, Piacibello W, Sanavio F, Stacchini A, Apra F, Schena M, … Gavosto F (1989). Kinetics of human hemopoietic cells after in vivo administration of granulocyte-macrophage colony-stimulating factor. J Clin Invest, 83(2), 551–557. doi: 10.1172/JCI113917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrechtsen M, Yeaman GR, & Kerr MA (1988). Characterization of the IgA receptor from human polymorphonuclear leucocytes. Immunology, 64(2), 201–205. [PMC free article] [PubMed] [Google Scholar]
- Alvarado-Kristensson M, Melander F, Leandersson K, Ronnstrand L, Wernstedt C, & Andersson T (2004). p38-MAPK signals survival by phosphorylation of caspase-8 and caspase-3 in human neutrophils. J Exp Med, 199(4), 449–458. doi: 10.1084/jem.20031771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athens JW, Haab OP, Raab SO, Mauer AM, Ashenbrucker H, Cartwright GE, & Wintrobe MM (1961). Leukokinetic studies. IV. The total blood, circulating and marginal granulocyte pools and the granulocyte turnover rate in normal subjects. J Clin Invest, 40, 989–995. doi: 10.1172/JCI104338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avdi NJ, Nick JA, Whitlock BB, Billstrom MA, Henson PM, Johnson GL, & Worthen GS (2001). Tumor necrosis factor-alpha activation of the c-Jun N-terminal kinase pathway in human neutrophils. Integrin involvement in a pathway leading from cytoplasmic tyrosine kinases apoptosis. J Biol Chem, 276(3), 2189–2199. doi: 10.1074/jbc.M007527200 [DOI] [PubMed] [Google Scholar]
- Avraham R, Haseley N, Brown D, Penaranda C, Jijon HB, Trombetta JJ, … Hung DT (2015). Pathogen cell-to-cell variability drives heterogeneity in host immune responses. Cell, 162(6), 1309–1321. doi: 10.1016/j.cell.2015.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainton DF, Ullyot JL, & Farquhar MG (1971). The development of neutrophilic polymorphonuclear leukocytes in human bone marrow. J Exp Med, 134(4), 907–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R, Anguita J, Roos D, & Fikrig E (2000). Cutting edge: infection by the agent of human granulocytic ehrlichiosis prevents the respiratory burst by down-regulating gp91phox. J Immunol, 164(8), 3946–3949. [DOI] [PubMed] [Google Scholar]
- Bauernfeind F, Rieger A, Schildberg FA, Knolle PA, Schmid-Burgk JL, & Hornung V (2012). NLRP3 inflammasome activity is negatively controlled by miR-223. J Immunol, 189(8), 4175–4181. doi: 10.4049/jimmunol.1201516 [DOI] [PubMed] [Google Scholar]
- Baumann R, Casaulta C, Simon D, Conus S, Yousefi S, & Simon HU (2003). Macrophage migration inhibitory factor delays apoptosis in neutrophils by inhibiting the mitochondria-dependent death pathway. FASEB J, 17(15), 2221–2230. doi: 10.1096/fj.03-0110com [DOI] [PubMed] [Google Scholar]
- Bellocchio S, Moretti S, Perruccio K, Fallarino F, Bozza S, Montagnoli C, … Romani L (2004). TLRs govern neutrophil activity in aspergillosis. J Immunol, 173(12), 7406–7415. [DOI] [PubMed] [Google Scholar]
- Biffl WL, Moore EE, Moore FA, & Barnett CC Jr. (1995). Interleukin-6 suppression of neutrophil apoptosis is neutrophil concentration dependent. J Leukoc Biol, 58(5), 582–584. [DOI] [PubMed] [Google Scholar]
- Boldt K, Rist W, Weiss SM, Weith A, & Lenter MC (2006). FPRL-1 induces modifications of migration-associated proteins in human neutrophils. Proteomics, 6(17), 4790–4799. doi: 10.1002/pmic.200600121 [DOI] [PubMed] [Google Scholar]
- Borjesson DL, Kobayashi SD, Whitney AR, Voyich JM, Argue CM, & DeLeo FR (2005). Insights into pathogen immune evasion mechanisms: Anaplasma phagocytophilum fails to induce an apoptosis differentiation program in human neutrophils. J Immunol, 174(10), 6364–6372. [DOI] [PubMed] [Google Scholar]
- Bours MJ, Swennen EL, Di Virgilio F, Cronstein BN, & Dagnelie PC (2006). Adenosine 5’-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther, 112(2), 358–404. doi: 10.1016/j.pharmthera.2005.04.013 [DOI] [PubMed] [Google Scholar]
- Boxer LA, Axtell R, & Suchard S (1990). The role of the neutrophil in inflammatory diseases of the lung. Blood Cells, 16(1), 25–40; discussion 41–22. [PubMed] [Google Scholar]
- Brigotti M, Carnicelli D, Ravanelli E, Barbieri S, Ricci F, Bontadini A, … Tazzari PL (2008). Interactions between Shiga toxins and human polymorphonuclear leukocytes. J Leukoc Biol, 84(4), 1019–1027. doi: 10.1189/jlb.0308157 [DOI] [PubMed] [Google Scholar]
- Brown GE, Stewart MQ, Bissonnette SA, Elia AE, Wilker E, & Yaffe MB (2004). Distinct ligand-dependent roles for p38 MAPK in priming and activation of the neutrophil NADPH oxidase. J Biol Chem, 279(26), 27059–27068. doi: 10.1074/jbc.M314258200 [DOI] [PubMed] [Google Scholar]
- Brown SB, & Savill J (1999). Phagocytosis triggers macrophage release of Fas ligand and induces apoptosis of bystander leukocytes. J Immunol, 162(1), 480–485. [PubMed] [Google Scholar]
- Burlak C, Whitney AR, Mead DJ, Hackstadt T, & DeLeo FR (2006). Maturation of human neutrophil phagosomes includes incorporation of molecular chaperones and endoplasmic reticulum quality control machinery. Mol Cell Proteomics, 5(4), 620–634. doi: 10.1074/mcp.M500336-MCP200 [DOI] [PubMed] [Google Scholar]
- Carlyon JA, Abdel-Latif D, Pypaert M, Lacy P, & Fikrig E (2004). Anaplasma phagocytophilum utilizes multiple host evasion mechanisms to thwart NADPH oxidase-mediated killing during neutrophil infection. Infect Immun, 72(8), 4772–4783. doi: 10.1128/IAI.72.8.4772-4783.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlyon JA, Chan WT, Galan J, Roos D, & Fikrig E (2002). Repression of rac2 mRNA expression by Anaplasma phagocytophila is essential to the inhibition of superoxide production and bacterial proliferation. J Immunol, 169(12), 7009–7018. [DOI] [PubMed] [Google Scholar]
- Chen H, Ye F, & Guo G (2019). Revolutionizing immunology with single-cell RNA sequencing. Cell Mol Immunol, 16(3), 242–249. doi: 10.1038/s41423-019-0214-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu LF, Leng N, Zhang J, Hou Z, Mamott D, Vereide DT, … Thomson JA (2016). Single-cell RNA-seq reveals novel regulators of human embryonic stem cell differentiation to definitive endoderm. Genome Biol, 17(1), 173. doi: 10.1186/s13059-016-1033-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline MJ (1966a). Phagocytosis and synthesis of ribonucleic acid in human granulocytes. Nature, 212(5069), 1431–1433. [DOI] [PubMed] [Google Scholar]
- Cline MJ (1966b). Ribonucleic acid biosynthesis in human leukocytes: the fate of rapidly labeled RNA in normal and abnormal leukocytes. Blood, 28(5), 650–664. [PubMed] [Google Scholar]
- Cline MJ (1975). Production and distribution of neutrophils In Cline MJ (Ed.), The White Cell (pp. 22–38). Cambridge, MA: Harvard University Press. [Google Scholar]
- Cohn ZA, & Morse SI (1960). Functional and metabolic properties of polymorphonuclear leucocytes. II. The influence of a lipopolysaccharide endotoxin. J Exp Med, 111, 689–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coldren CD, Nick JA, Poch KR, Woolum MD, Fouty BW, O’Brien JM, … Geraci MW (2006). Functional and genomic changes induced by alveolar transmigration in human neutrophils. Am J Physiol Lung Cell Mol Physiol, 291(6), L1267–1276. doi: 10.1152/ajplung.00097.2006 [DOI] [PubMed] [Google Scholar]
- Colotta F, Re F, Polentarutti N, Sozzani S, & Mantovani A (1992). Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood, 80(8), 2012–2020. [PubMed] [Google Scholar]
- Conus S, Perozzo R, Reinheckel T, Peters C, Scapozza L, Yousefi S, & Simon HU (2008). Caspase-8 is activated by cathepsin D initiating neutrophil apoptosis during the resolution of inflammation. J Exp Med, 205(3), 685–698. doi: 10.1084/jem.20072152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper D, Lindberg FP, Gamble JR, Brown EJ, & Vadas MA (1995). Transendothelial migration of neutrophils involves integrin-associated protein (CD47). Proc Natl Acad Sci U S A, 92(9), 3978–3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowland JB, & Borregaard N (2016). Granulopoiesis and granules of human neutrophils. Immunol Rev, 273(1), 11–28. doi: 10.1111/imr.12440 [DOI] [PubMed] [Google Scholar]
- Coxon A, Rieu P, Barkalow FJ, Askari S, Sharpe AH, von Andrian UH, … Mayadas TN (1996). A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity, 5(6), 653–666. [DOI] [PubMed] [Google Scholar]
- Cronkite EP, & Fliedner TM (1964). Granulocytopoiesis. N Engl J Med, 270, 1403–1408 CONCL. doi: 10.1056/NEJM196406252702608 [DOI] [PubMed] [Google Scholar]
- Curnutte JT, Scott PJ, & Mayo LA (1989). Cytosolic components of the respiratory burst oxidase: resolution of four components, two of which are missing in complementing types of chronic granulomatous disease. Proc Natl Acad Sci U S A, 86(3), 825–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahinden C, Galanos C, & Fehr J (1983). Granulocyte activation by endotoxin. I. Correlation between adherence and other granulocyte functions, and role of endotoxin structure on biologic activity. J Immunol, 130(2), 857–862. [PubMed] [Google Scholar]
- Dale DC, Liles WC, Llewellyn C, & Price TH (1998). Effects of granulocyte-macrophage colony-stimulating factor (GM-CSF) on neutrophil kinetics and function in normal human volunteers. Am J Hematol, 57(1), 7–15. [DOI] [PubMed] [Google Scholar]
- Dalli J, & Serhan CN (2012). Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood, 120(15), e60–72. doi: 10.1182/blood-2012-04-423525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dana N, Todd RF 3rd, Pitt J, Springer TA, & Arnaout MA (1984). Deficiency of a surface membrane glycoprotein (Mo1) in man. J Clin Invest, 73(1), 153–159. doi: 10.1172/JCI111186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancey JT, Deubelbeiss KA, Harker LA, & Finch CA (1976). Neutrophil kinetics in man. J Clin Invest, 58(3), 705–715. doi: 10.1172/JCI108517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang PM, Dewas C, Gaudry M, Fay M, Pedruzzi E, Gougerot-Pocidalo MA, & El Benna J (1999). Priming of human neutrophil respiratory burst by granulocyte/macrophage colony-stimulating factor (GM-CSF) involves partial phosphorylation of p47(phox). J Biol Chem, 274(29), 20704–20708. [DOI] [PubMed] [Google Scholar]
- Davis MM, Tato CM, & Furman D (2017). Systems immunology: just getting started. Nat Immunol, 18(7), 725–732. doi: 10.1038/ni.3768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis SD, Schaller J, & Wedgwood RJ (1966). Job’s Syndrome. Recurrent, “cold”, staphylococcal abscesses. Lancet, 1(7445), 1013–1015. [DOI] [PubMed] [Google Scholar]
- DeLeo FR (2004). Modulation of phagocyte apoptosis by bacterial pathogens. Apoptosis, 9(4), 399–413. doi: 10.1023/B:APPT.0000031448.64969.fa [DOI] [PubMed] [Google Scholar]
- DeLeo FR, & Nauseef WM (2014). Granulocytic Phagocytes In Bennett JE, Dolin R, & Blaser MJ (Eds.), Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases (8th ed., Vol. 1, pp. 78–92). Philadelphia, PA USA: Elsevier Saunders. [Google Scholar]
- DeLeo FR, Renee J, McCormick S, Nakamura M, Apicella M, Weiss JP, & Nauseef WM (1998). Neutrophils exposed to bacterial lipopolysaccharide upregulate NADPH oxidase assembly. J Clin Invest, 101(2), 455–463. doi: 10.1172/JCI949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Staunton DE, de Fougerolles AR, Stacker SA, Garcia-Aguilar J, Hibbs ML, & Springer TA (1990). ICAM-1 (CD54): a counter-receptor for Mac-1 (CD11b/CD18). J Cell Biol, 111(6 Pt 2), 3129–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorhoi A, Iannaccone M, Farinacci M, Fae KC, Schreiber J, Moura-Alves P, … Kaufmann SH (2013). MicroRNA-223 controls susceptibility to tuberculosis by regulating lung neutrophil recruitment. J Clin Invest, 123(11), 4836–4848. doi: 10.1172/JCI67604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunican AL, Leuenroth SJ, Ayala A, & Simms HH (2000). CXC chemokine suppression of polymorphonuclear leukocytes apoptosis and preservation of function is oxidative stress independent. Shock, 13(3), 244–250. [DOI] [PubMed] [Google Scholar]
- Ecker S, Chen L, Pancaldi V, Bagger FO, Fernandez JM, Carrillo de Santa Pau E, … Paul DS (2017). Genome-wide analysis of differential transcriptional and epigenetic variability across human immune cell types. Genome Biol, 18(1), 18. doi: 10.1186/s13059-017-1156-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SW, & Hallett MB (1997). Seeing the wood for the trees: the forgotten role of neutrophils in rheumatoid arthritis. Immunol Today, 18(7), 320–324. [DOI] [PubMed] [Google Scholar]
- Eggleton P, Ghebrehiwet B, Sastry KN, Coburn JP, Zaner KS, Reid KB, & Tauber AI (1995). Identification of a gC1q-binding protein (gC1q-R) on the surface of human neutrophils. Subcellular localization and binding properties in comparison with the cC1q-R. J Clin Invest, 95(4), 1569–1578. doi: 10.1172/JCI117830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrengruber MU, Geiser T, & Deranleau DA (1994). Activation of human neutrophils by C3a and C5A. Comparison of the effects on shape changes, chemotaxis, secretion, and respiratory burst. FEBS Lett, 346(2–3), 181–184. [DOI] [PubMed] [Google Scholar]
- El Kebir D, Jozsef L, Khreiss T, & Filep JG (2006). Inhibition of K+ efflux prevents mitochondrial dysfunction, and suppresses caspase-3-, apoptosis-inducing factor-, and endonuclease G-mediated constitutive apoptosis in human neutrophils. Cell Signal, 18(12), 2302–2313. doi: 10.1016/j.cellsig.2006.05.013 [DOI] [PubMed] [Google Scholar]
- Ellison MA, Gearheart CM, Porter CC, & Ambruso DR (2017). IFN-gamma alters the expression of diverse immunity related genes in a cell culture model designed to represent maturing neutrophils. PLoS One, 12(10), e0185956. doi: 10.1371/journal.pone.0185956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelich G, White M, & Hartshorn KL (2001). Neutrophil survival is markedly reduced by incubation with influenza virus and Streptococcus pneumoniae: role of respiratory burst. J Leukoc Biol, 69(1), 50–56. [PubMed] [Google Scholar]
- Fadok VA, Savill JS, Haslett C, Bratton DL, Doherty DE, Campbell PA, & Henson PM (1992). Different populations of macrophages use either the vitronectin receptor or the phosphatidylserine receptor to recognize and remove apoptotic cells. J Immunol, 149(12), 4029–4035. [PubMed] [Google Scholar]
- Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, Nervi C, & Bozzoni I (2005). A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell, 123(5), 819–831. doi: 10.1016/j.cell.2005.09.023 [DOI] [PubMed] [Google Scholar]
- Fessler MB, Malcolm KC, Duncan MW, & Worthen GS (2002). A genomic and proteomic analysis of activation of the human neutrophil by lipopolysaccharide and its mediation by p38 mitogen-activated protein kinase. J Biol Chem, 277(35), 31291–31302. doi: 10.1074/jbc.M200755200 [DOI] [PubMed] [Google Scholar]
- Fleit HB, Wright SD, & Unkeless JC (1982). Human neutrophil Fc gamma receptor distribution and structure. Proc Natl Acad Sci U S A, 79(10), 3275–3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliedner TM, Cronkite EP, Killmann SA, & Bond VP (1964). Granulocytopoiesis. Ii. Emergence and Pattern of Labeling of Neutrophilic Granulocytes in Humans. Blood, 24, 683–700. [PubMed] [Google Scholar]
- Fliedner TM, Cronkite EP, & Robertson JS (1964). Granulocytopoiesis. I. Senescence and Random Loss of Neutrophilic Granulocytes in Human Beings. Blood, 24, 402–414. [PubMed] [Google Scholar]
- Fossati G, Moulding DA, Spiller DG, Moots RJ, White MR, & Edwards SW (2003). The mitochondrial network of human neutrophils: role in chemotaxis, phagocytosis, respiratory burst activation, and commitment to apoptosis. J Immunol, 170(4), 1964–1972. [DOI] [PubMed] [Google Scholar]
- Fradin C, Mavor AL, Weindl G, Schaller M, Hanke K, Kaufmann SH, … Hube B (2007). The early transcriptional response of human granulocytes to infection with Candida albicans is not essential for killing but reflects cellular communications. Infect Immun, 75(3), 1493–1501. doi: 10.1128/IAI.01651-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francois S, El Benna J, Dang PM, Pedruzzi E, Gougerot-Pocidalo MA, & Elbim C (2005). Inhibition of neutrophil apoptosis by TLR agonists in whole blood: involvement of the phosphoinositide 3-kinase/Akt and NF-kappaB signaling pathways, leading to increased levels of Mcl-1, A1, and phosphorylated Bad. J Immunol, 174(6), 3633–3642. [DOI] [PubMed] [Google Scholar]
- Friedman AD (2002). Transcriptional regulation of granulocyte and monocyte development. Oncogene, 21(21), 3377–3390. doi: 10.1038/sj.onc.1205324 [DOI] [PubMed] [Google Scholar]
- Gagnon E, Bergeron JJ, & Desjardins M (2005). ER-mediated phagocytosis: myth or reality? J Leukoc Biol, 77(6), 843–845. doi: 10.1189/jlb.0305129 [DOI] [PubMed] [Google Scholar]
- Gagnon E, Duclos S, Rondeau C, Chevet E, Cameron PH, Steele-Mortimer O, … Desjardins M (2002). Endoplasmic reticulum-mediated phagocytosis is a mechanism of entry into macrophages. Cell, 110(1), 119–131. [DOI] [PubMed] [Google Scholar]
- Gamberale R, Giordano M, Trevani AS, Andonegui G, & Geffner JR (1998). Modulation of human neutrophil apoptosis by immune complexes. J Immunol, 161(7), 3666–3674. [PubMed] [Google Scholar]
- Gantier MP (2013). The not-so-neutral role of microRNAs in neutrophil biology. J Leukoc Biol, 94(4), 575–583. doi: 10.1189/jlb.1012539 [DOI] [PubMed] [Google Scholar]
- Gardai S, Whitlock BB, Helgason C, Ambruso D, Fadok V, Bratton D, & Henson PM (2002). Activation of SHIP by NADPH oxidase-stimulated Lyn leads to enhanced apoptosis in neutrophils. J Biol Chem, 277(7), 5236–5246. doi: 10.1074/jbc.M110005200 [DOI] [PubMed] [Google Scholar]
- Garin J, Diez R, Kieffer S, Dermine JF, Duclos S, Gagnon E, … Desjardins M (2001). The phagosome proteome: insight into phagosome functions. J Cell Biol, 152(1), 165–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaublomme JT, Yosef N, Lee Y, Gertner RS, Yang LV, Wu C, … Regev A (2015). Single-Cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell, 163(6), 1400–1412. doi: 10.1016/j.cell.2015.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genestier AL, Michallet MC, Prevost G, Bellot G, Chalabreysse L, Peyrol S, … Genestier L (2005). Staphylococcus aureus Panton-Valentine leukocidin directly targets mitochondria and induces Bax-independent apoptosis of human neutrophils. J Clin Invest, 115(11), 3117–3127. doi: 10.1172/JCI22684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gombart AF, Krug U, O’Kelly J, An E, Vegesna V, & Koeffler HP (2005). Aberrant expression of neutrophil and macrophage-related genes in a murine model for human neutrophil-specific granule deficiency. J Leukoc Biol, 78(5), 1153–1165. doi: 10.1189/jlb.0504286 [DOI] [PubMed] [Google Scholar]
- Gombart AF, Shiohara M, Kwok SH, Agematsu K, Komiyama A, & Koeffler HP (2001). Neutrophil-specific granule deficiency: homozygous recessive inheritance of a frameshift mutation in the gene encoding transcription factor CCAAT/enhancer binding protein--epsilon. Blood, 97(9), 2561–2567. [DOI] [PubMed] [Google Scholar]
- Gounni AS, Lamkhioued B, Koussih L, Ra C, Renzi PM, & Hamid Q (2001). Human neutrophils express the high-affinity receptor for immunoglobulin E (Fc epsilon RI): role in asthma. FASEB J, 15(6), 940–949. [DOI] [PubMed] [Google Scholar]
- Grabowski P, Hesse S, Hollizeck S, Rohlfs M, Behrends U, Sherkat R, … Rappsilber J (2019). Proteome analysis of human neutrophil granulocytes from patients with monogenic disease using data-independent acquisition. Mol Cell Proteomics. doi: 10.1074/mcp.RA118.001141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guermonprez P, Saveanu L, Kleijmeer M, Davoust J, Van Endert P, & Amigorena S (2003). ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature, 425(6956), 397–402. doi: 10.1038/nature01911 [DOI] [PubMed] [Google Scholar]
- Guichard C, Pedruzzi E, Dewas C, Fay M, Pouzet C, Bens M, … Elbim C (2005). Interleukin-8-induced priming of neutrophil oxidative burst requires sequential recruitment of NADPH oxidase components into lipid rafts. J Biol Chem, 280(44), 37021–37032. doi: 10.1074/jbc.M506594200 [DOI] [PubMed] [Google Scholar]
- Gurol T, Zhou W, & Deng Q (2016). MicroRNAs in neutrophils: potential next generation therapeutics for inflammatory ailments. Immunol Rev, 273(1), 29–47. doi: 10.1111/imr.12450 [DOI] [PubMed] [Google Scholar]
- Guthrie LA, McPhail LC, Henson PM, & Johnston RB Jr. (1984). Priming of neutrophils for enhanced release of oxygen metabolites by bacterial lipopolysaccharide. Evidence for increased activity of the superoxide-producing enzyme. J Exp Med, 160(6), 1656–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamers MN, de Boer M, Meerhof LJ, Weening RS, & Roos D (1984). Complementation in monocyte hybrids revealing genetic heterogeneity in chronic granulomatous disease. Nature, 307(5951), 553–555. [DOI] [PubMed] [Google Scholar]
- Hannah S, Mecklenburgh K, Rahman I, Bellingan GJ, Greening A, Haslett C, & Chilvers ER (1995). Hypoxia prolongs neutrophil survival in vitro. FEBS Lett, 372(2–3), 233–237. [DOI] [PubMed] [Google Scholar]
- Hart SP, Ross JA, Ross K, Haslett C, & Dransfield I (2000). Molecular characterization of the surface of apoptotic neutrophils: implications for functional downregulation and recognition by phagocytes. Cell Death Differ, 7(5), 493–503. doi: 10.1038/sj.cdd.4400680 [DOI] [PubMed] [Google Scholar]
- Hayashi F, Means TK, & Luster AD (2003). Toll-like receptors stimulate human neutrophil function. Blood, 102(7), 2660–2669. doi: 10.1182/blood-2003-04-1078 [DOI] [PubMed] [Google Scholar]
- Henson PM (2017). Cell Removal: Efferocytosis. Annu Rev Cell Dev Biol, 33, 127–144. doi: 10.1146/annurev-cellbio-111315-125315 [DOI] [PubMed] [Google Scholar]
- Hickstein DD, Ozols J, Williams SA, Baenziger JU, Locksley RM, & Roth GJ (1987). Isolation and characterization of the receptor on human neutrophils that mediates cellular adherence. J Biol Chem, 262(12), 5576–5580. [PubMed] [Google Scholar]
- Hill JM, Vaidyanathan H, Ramos JW, Ginsberg MH, & Werner MH (2002). Recognition of ERK MAP kinase by PEA-15 reveals a common docking site within the death domain and death effector domain. EMBO J, 21(23), 6494–6504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, … Grimbacher B (2007). STAT3 mutations in the hyper-IgE syndrome. N Engl J Med, 357(16), 1608–1619. doi: 10.1056/NEJMoa073687 [DOI] [PubMed] [Google Scholar]
- Homburg CH, de Haas M, von dem Borne AE, Verhoeven AJ, Reutelingsperger CP, & Roos D (1995). Human neutrophils lose their surface Fc gamma RIII and acquire Annexin V binding sites during apoptosis in vitro. Blood, 85(2), 532–540. [PubMed] [Google Scholar]
- Hong S, Porter TF, Lu Y, Oh SF, Pillai PS, & Serhan CN (2008). Resolvin E1 metabolome in local inactivation during inflammation-resolution. J Immunol, 180(5), 3512–3519. [DOI] [PubMed] [Google Scholar]
- Houde M, Bertholet S, Gagnon E, Brunet S, Goyette G, Laplante A, … Desjardins M (2003). Phagosomes are competent organelles for antigen cross-presentation. Nature, 425(6956), 402–406. doi: 10.1038/nature01912 [DOI] [PubMed] [Google Scholar]
- Iida S, Kohro T, Kodama T, Nagata S, & Fukunaga R (2005). Identification of CCR2, flotillin, and gp49B genes as new G-CSF targets during neutrophilic differentiation. J Leukoc Biol, 78(2), 481–490. doi: 10.1189/jlb.0904515 [DOI] [PubMed] [Google Scholar]
- Jethwaney D, Islam MR, Leidal KG, de Bernabe DB, Campbell KP, Nauseef WM, & Gibson BW (2007). Proteomic analysis of plasma membrane and secretory vesicles from human neutrophils. Proteome Sci, 5, 12. doi: 10.1186/1477-5956-5-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnnidis JB, Harris MH, Wheeler RT, Stehling-Sun S, Lam MH, Kirak O, … Camargo FD (2008). Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature, 451(7182), 1125–1129. doi: 10.1038/nature06607 [DOI] [PubMed] [Google Scholar]
- Jonsson H, Allen P, & Peng SL (2005). Inflammatory arthritis requires Foxo3a to prevent Fas ligand-induced neutrophil apoptosis. Nat Med, 11(6), 666–671. doi: 10.1038/nm1248 [DOI] [PubMed] [Google Scholar]
- Juss J, Herre J, Begg M, Bradley G, Lennon M, Amour A, … Chilvers ER (2015). Genome-wide transcription profiling in neutrophils in acute respiratory distress syndrome. Lancet, 385 Suppl 1, S55. doi: 10.1016/S0140-6736(15)60370-1 [DOI] [PubMed] [Google Scholar]
- Juss JK, House D, Amour A, Begg M, Herre J, Storisteanu DM, … Chilvers ER (2016). Acute Respiratory Distress Syndrome Neutrophils Have a Distinct Phenotype and Are Resistant to Phosphoinositide 3-Kinase Inhibition. Am J Respir Crit Care Med, 194(8), 961–973. doi: 10.1164/rccm.201509-1818OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasprisin DO, & Harris MB (1977). The role of RNA metabolism in polymorphonuclear leukocyte phagocytosis. J Lab Clin Med, 90(1), 118–124. [PubMed] [Google Scholar]
- Kasprisin DO, & Harris MB (1978). The role of protein synthesis in polymorphonuclear leukocyte phagocytosis II. Exp Hematol, 6(7), 585–589. [PubMed] [Google Scholar]
- Kettritz R, Gaido ML, Haller H, Luft FC, Jennette CJ, & Falk RJ (1998). Interleukin-8 delays spontaneous and tumor necrosis factor-alpha-mediated apoptosis of human neutrophils. Kidney Int, 53(1), 84–91. doi: 10.1046/j.1523-1755.1998.00741.x [DOI] [PubMed] [Google Scholar]
- Khan AI, Kerfoot SM, Heit B, Liu L, Andonegui G, Ruffell B, … Kubes P (2004). Role of CD44 and hyaluronan in neutrophil recruitment. J Immunol, 173(12), 7594–7601. [DOI] [PubMed] [Google Scholar]
- Khwaja A, & Tatton L (1999). Caspase-mediated proteolysis and activation of protein kinase Cdelta plays a central role in neutrophil apoptosis. Blood, 94(1), 291–301. [PubMed] [Google Scholar]
- Kinkead LC, Fayram DC, & Allen LH (2017). Francisella novicida inhibits spontaneous apoptosis and extends human neutrophil lifespan. J Leukoc Biol, 102(3), 815–828. doi: 10.1189/jlb.4MA0117-014R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kippner LE, Kim J, Gibson G, & Kemp ML (2014). Single cell transcriptional analysis reveals novel innate immune cell types. PeerJ, 2, e452. doi: 10.7717/peerj.452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebanoff SJ (1968). Myeloperoxidase-halide-hydrogen peroxide antibacterial system. J Bacteriol, 95(6), 2131–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebanoff SJ (1974). Role of the superoxide anion in the myeloperoxidase-mediated antimicrobial system. J Biol Chem, 249(12), 3724–3728. [PubMed] [Google Scholar]
- Kluger Y, Tuck DP, Chang JT, Nakayama Y, Poddar R, Kohya N, … Weissman SM (2004). Lineage specificity of gene expression patterns. Proc Natl Acad Sci U S A, 101(17), 6508–6513. doi: 10.1073/pnas.0401136101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi SD, Braughton KR, Palazzolo-Ballance AM, Kennedy AD, Sampaio E, Kristosturyan E, … DeLeo FR (2010). Rapid neutrophil destruction following phagocytosis of Staphylococcus aureus. J Innate Immun, 2(6), 560–575. doi: 10.1159/000317134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi SD, Braughton KR, Whitney AR, Voyich JM, Schwan TG, Musser JM, & DeLeo FR (2003). Bacterial pathogens modulate an apoptosis differentiation program in human neutrophils. Proc Natl Acad Sci U S A, 100(19), 10948–10953. doi: 10.1073/pnas.1833375100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi SD, & Deleo FR (2004). An apoptosis differentiation programme in human polymorphonuclear leucocytes. Biochem Soc Trans, 32(Pt3), 474–476. doi: 10.1042/BST0320474 [DOI] [PubMed] [Google Scholar]
- Kobayashi SD, Voyich JM, Braughton KR, & DeLeo FR (2003). Down-regulation of proinflammatory capacity during apoptosis in human polymorphonuclear leukocytes. J Immunol, 170(6), 3357–3368. [DOI] [PubMed] [Google Scholar]
- Kobayashi SD, Voyich JM, Braughton KR, Whitney AR, Nauseef WM, Malech HL, & DeLeo FR (2004). Gene expression profiling provides insight into the pathophysiology of chronic granulomatous disease. J Immunol, 172(1), 636–643. [DOI] [PubMed] [Google Scholar]
- Kobayashi SD, Voyich JM, Buhl CL, Stahl RM, & DeLeo FR (2002). Global changes in gene expression by human polymorphonuclear leukocytes during receptor-mediated phagocytosis: cell fate is regulated at the level of gene expression. Proc Natl Acad Sci U S A, 99(10), 6901–6906. doi: 10.1073/pnas.092148299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi SD, Voyich JM, Burlak C, & DeLeo FR (2005). Neutrophils in the innate immune response. Arch Immunol Ther Exp (Warsz), 53(6), 505–517. [PubMed] [Google Scholar]
- Kobayashi SD, Voyich JM, Somerville GA, Braughton KR, Malech HL, Musser JM, & DeLeo FR (2003). An apoptosis-differentiation program in human polymorphonuclear leukocytes facilitates resolution of inflammation. J Leukoc Biol, 73(2), 315–322. [DOI] [PubMed] [Google Scholar]
- Kobayashi SD, Voyich JM, Whitney AR, & DeLeo FR (2005). Spontaneous neutrophil apoptosis and regulation of cell survival by granulocyte macrophage-colony stimulating factor. J Leukoc Biol, 78(6), 1408–1418. doi: 10.1189/jlb.0605289 [DOI] [PubMed] [Google Scholar]
- Kosak ST, Scalzo D, Alworth SV, Li F, Palmer S, Enver T, … Groudine M (2007). Coordinate gene regulation during hematopoiesis is related to genomic organization. PLoS Biol, 5(11), e309. doi: 10.1371/journal.pbio.0050309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurt-Jones EA, Mandell L, Whitney C, Padgett A, Gosselin K, Newburger PE, & Finberg RW (2002). Role of toll-like receptor 2 (TLR2) in neutrophil activation: GM-CSF enhances TLR2 expression and TLR2-mediated interleukin 8 responses in neutrophils. Blood, 100(5), 1860–1868. [PubMed] [Google Scholar]
- Lane TA, & Lamkin GE (1984). A reassessment of the energy requirements for neutrophil migration: adenosine triphosphate depletion enhances chemotaxis. Blood, 64(5), 986–993. [PubMed] [Google Scholar]
- Larsen MT, Hother C, Hager M, Pedersen CC, Theilgaard-Monch K, Borregaard N, & Cowland JB (2013). MicroRNA profiling in human neutrophils during bone marrow granulopoiesis and in vivo exudation. PLoS One, 8(3), e58454. doi: 10.1371/journal.pone.0058454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laterveer L, Lindley IJ, Heemskerk DP, Camps JA, Pauwels EK, Willemze R, & Fibbe WE (1996). Rapid mobilization of hematopoietic progenitor cells in rhesus monkeys by a single intravenous injection of interleukin-8. Blood, 87(2), 781–788. [PubMed] [Google Scholar]
- Lawrence MB, & Springer TA (1991). Leukocytes roll on a selectin at physiologic flow rates: distinction from and prerequisite for adhesion through integrins. Cell, 65(5), 859–873. [DOI] [PubMed] [Google Scholar]
- Le Cabec V, Cowland JB, Calafat J, & Borregaard N (1996). Targeting of proteins to granule subsets is determined by timing and not by sorting: The specific granule protein NGAL is localized to azurophil granules when expressed in HL-60 cells. Proc Natl Acad Sci U S A, 93(13), 6454–6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A, Whyte MK, & Haslett C (1993). Inhibition of apoptosis and prolongation of neutrophil functional longevity by inflammatory mediators. J Leukoc Biol, 54(4), 283–288. [PubMed] [Google Scholar]
- Lee HC, & Goodman JL (2006). Anaplasma phagocytophilum causes global induction of antiapoptosis in human neutrophils. Genomics, 88(4), 496–503. doi: 10.1016/j.ygeno.2006.06.002 [DOI] [PubMed] [Google Scholar]
- Lee HC, Kioi M, Han J, Puri RK, & Goodman JL (2008). Anaplasma phagocytophilum-induced gene expression in both human neutrophils and HL-60 cells. Genomics, 92(3), 144–151. doi: 10.1016/j.ygeno.2008.05.005 [DOI] [PubMed] [Google Scholar]
- Lekstrom-Himes JA, Dorman SE, Kopar P, Holland SM, & Gallin JI (1999). Neutrophil-specific granule deficiency results from a novel mutation with loss of function of the transcription factor CCAAT/enhancer binding protein epsilon. J Exp Med, 189(11), 1847–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lekstrom-Himes JA, & Gallin JI (2000). Immunodeficiency diseases caused by defects in phagocytes. N Engl J Med, 343(23), 1703–1714. doi: 10.1056/NEJM200012073432307 [DOI] [PubMed] [Google Scholar]
- Lekstrom-Himes JA, Kuhns DB, Alvord WG, & Gallin JI (2005). Inhibition of human neutrophil IL-8 production by hydrogen peroxide and dysregulation in chronic granulomatous disease. J Immunol, 174(1), 411–417. [DOI] [PubMed] [Google Scholar]
- Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, … Wilson RK (2008). DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature, 456(7218), 66–72. doi: 10.1038/nature07485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian Z, Kluger Y, Greenbaum DS, Tuck D, Gerstein M, Berliner N, … Newburger PE (2002). Genomic and proteomic analysis of the myeloid differentiation program: global analysis of gene expression during induced differentiation in the MPRO cell line. Blood, 100(9), 3209–3220. doi: 10.1182/blood-2002-03-0850 [DOI] [PubMed] [Google Scholar]
- Lian Z, Wang L, Yamaga S, Bonds W, Beazer-Barclay Y, Kluger Y, … Weissman SM (2001). Genomic and proteomic analysis of the myeloid differentiation program. Blood, 98(3), 513–524. [DOI] [PubMed] [Google Scholar]
- Liles WC, Kiener PA, Ledbetter JA, Aruffo A, & Klebanoff SJ (1996). Differential expression of Fas (CD95) and Fas ligand on normal human phagocytes: implications for the regulation of apoptosis in neutrophils. J Exp Med, 184(2), 429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liles WC, Thomsen AR, O’Mahony DS, & Klebanoff SJ (2001). Stimulation of human neutrophils and monocytes by staphylococcal phenol-soluble modulin. J Leukoc Biol, 70(1), 96–102. [PubMed] [Google Scholar]
- Lin A, & Lore K (2017). Granulocytes: new members of the antigen-presenting cell family. Front Immunol, 8, 1781. doi: 10.3389/fimmu.2017.01781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindemann SW, Yost CC, Denis MM, McIntyre TM, Weyrich AS, & Zimmerman GA (2004). Neutrophils alter the inflammatory milieu by signal-dependent translation of constitutive messenger RNAs. Proc Natl Acad Sci U S A, 101(18), 7076–7081. doi: 10.1073/pnas.0401901101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Akahoshi T, Sasahana T, Kitasato H, Namai R, Sasaki T, … Kondo H (1999). Inhibition of neutrophil apoptosis by verotoxin 2 derived from Escherichia coli O157:H7. Infect Immun, 67(11), 6203–6205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lominadze G, Powell DW, Luerman GC, Link AJ, Ward RA, & McLeish KR (2005). Proteomic analysis of human neutrophil granules. Mol Cell Proteomics, 4(10), 1503–1521. doi: 10.1074/mcp.M500143-MCP200 [DOI] [PubMed] [Google Scholar]
- Lotz S, Aga E, Wilde I, van Zandbergen G, Hartung T, Solbach W, & Laskay T (2004). Highly purified lipoteichoic acid activates neutrophil granulocytes and delays their spontaneous apoptosis via CD14 and TLR2. J Leukoc Biol, 75(3), 467–477. doi: 10.1189/jlb.0803360 [DOI] [PubMed] [Google Scholar]
- Maianski NA, Geissler J, Srinivasula SM, Alnemri ES, Roos D, & Kuijpers TW (2004). Functional characterization of mitochondria in neutrophils: a role restricted to apoptosis. Cell Death Differ, 11(2), 143–153. doi: 10.1038/sj.cdd.4401320 [DOI] [PubMed] [Google Scholar]
- Maianski NA, Mul FP, van Buul JD, Roos D, & Kuijpers TW (2002). Granulocyte colony-stimulating factor inhibits the mitochondria-dependent activation of caspase-3 in neutrophils. Blood, 99(2), 672–679. [DOI] [PubMed] [Google Scholar]
- Malcolm KC, Arndt PG, Manos EJ, Jones DA, & Worthen GS (2003). Microarray analysis of lipopolysaccharide-treated human neutrophils. Am J Physiol Lung Cell Mol Physiol, 284(4), L663–670. doi: 10.1152/ajplung.00094.2002 [DOI] [PubMed] [Google Scholar]
- Mantovani B (1975). Different roles of IgG and complement receptors in phagocytosis by polymorphonuclear leukocytes. J Immunol, 115(1), 15–17. [PubMed] [Google Scholar]
- Martinelli S, Urosevic M, Daryadel A, Oberholzer PA, Baumann C, Fey MF, … Yousefi S (2004). Induction of genes mediating interferon-dependent extracellular trap formation during neutrophil differentiation. J Biol Chem, 279(42), 44123–44132. doi: 10.1074/jbc.M405883200 [DOI] [PubMed] [Google Scholar]
- McCaffrey RL, & Allen LA (2006). Francisella tularensis LVS evades killing by human neutrophils via inhibition of the respiratory burst and phagosome escape. J Leukoc Biol, 80(6), 1224–1230. doi: 10.1189/jlb.0406287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald PP (2004). Transcriptional regulation in neutrophils: teaching old cells new tricks. Adv Immunol, 82, 1–48. doi: 10.1016/S0065-2776(04)82001-7 [DOI] [PubMed] [Google Scholar]
- McPhillips K, Janssen WJ, Ghosh M, Byrne A, Gardai S, Remigio L, … Henson P (2007). TNF-alpha inhibits macrophage clearance of apoptotic cells via cytosolic phospholipase A2 and oxidant-dependent mechanisms. J Immunol, 178(12), 8117–8126. [DOI] [PubMed] [Google Scholar]
- Meagher LC, Cousin JM, Seckl JR, & Haslett C (1996). Opposing effects of glucocorticoids on the rate of apoptosis in neutrophilic and eosinophilic granulocytes. J Immunol, 156(11), 4422–4428. [PubMed] [Google Scholar]
- Michlewska S, Dransfield I, Megson IL, & Rossi AG (2009). Macrophage phagocytosis of apoptotic neutrophils is critically regulated by the opposing actions of pro-inflammatory and anti-inflammatory agents: key role for TNF-alpha. FASEB J, 23(3), 844–854. doi: 10.1096/fj.08-121228 [DOI] [PubMed] [Google Scholar]
- Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, … Karasuyama H (2007). Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature, 448(7157), 1058–1062. doi: 10.1038/nature06096 [DOI] [PubMed] [Google Scholar]
- Mollinedo F, Lopez-Perez R, & Gajate C (2008). Differential gene expression patterns coupled to commitment and acquisition of phenotypic hallmarks during neutrophil differentiation of human leukaemia HL-60 cells. Gene, 419(1–2), 16–26. doi: 10.1016/j.gene.2008.04.015 [DOI] [PubMed] [Google Scholar]
- Mott J, & Rikihisa Y (2000). Human granulocytic ehrlichiosis agent inhibits superoxide anion generation by human neutrophils. Infect Immun, 68(12), 6697–6703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulding DA, Walter C, Hart CA, & Edwards SW (1999). Effects of staphylococcal enterotoxins on human neutrophil functions and apoptosis. Infect Immun, 67(5), 2312–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S, & Diaz A (2017). Single-Cell mRNA Sequencing in Cancer Research: Integrating the Genomic Fingerprint. Front Genet, 8, 73. doi: 10.3389/fgene.2017.00073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller WA, Weigl SA, Deng X, & Phillips DM (1993). PECAM-1 is required for transendothelial migration of leukocytes. J Exp Med, 178(2), 449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullick A, Elias M, Harakidas P, Marcil A, Whiteway M, Ge B, … Thomas DY (2004). Gene expression in HL60 granulocytoids and human polymorphonuclear leukocytes exposed to Candida albicans. Infect Immun, 72(1), 414–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy BM, O’Neill AJ, Adrain C, Watson RW, & Martin SJ (2003). The apoptosome pathway to caspase activation in primary human neutrophils exhibits dramatically reduced requirements for cytochrome C. J Exp Med, 197(5), 625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray J, Barbara JA, Dunkley SA, Lopez AF, Van Ostade X, Condliffe AM, … Chilvers ER (1997). Regulation of neutrophil apoptosis by tumor necrosis factor-alpha: requirement for TNFR55 and TNFR75 for induction of apoptosis in vitro. Blood, 90(7), 2772–2783. [PubMed] [Google Scholar]
- Musser JM, & DeLeo FR (2005). Toward a genome-wide systems biology analysis of host-pathogen interactions in group A Streptococcus. Am J Pathol, 167(6), 1461–1472. doi: 10.1016/S0002-9440(10)61232-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myones BL, Dalzell JG, Hogg N, & Ross GD (1988). Neutrophil and monocyte cell surface p150,95 has iC3b-receptor (CR4) activity resembling CR3. J Clin Invest, 82(2), 640–651. doi: 10.1172/JCI113643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris PC, Libreros S, Chiang N, & Serhan CN (2017). A cluster of immunoresolvents links coagulation to innate host defense in human blood. Sci Signal, 10(490). doi: 10.1126/scisignal.aan1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunoi H, Rotrosen D, Gallin JI, & Malech HL (1988). Two forms of autosomal chronic granulomatous disease lack distinct neutrophil cytosol factors. Science, 242(4883), 1298–1301. [DOI] [PubMed] [Google Scholar]
- O’Neill AJ, Doyle BT, Molloy E, Watson C, Phelan D, Greenan MC, … Watson RW (2004). Gene expression profile of inflammatory neutrophils: alterations in the inhibitors of apoptosis proteins during spontaneous and delayed apoptosis. Shock, 21(6), 512–518. [DOI] [PubMed] [Google Scholar]
- O’Shea JJ, & Murray PJ (2008). Cytokine signaling modules in inflammatory responses. Immunity, 28(4), 477–487. doi: 10.1016/j.immuni.2008.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofengeim D, Giagtzoglou N, Huh D, Zou C, & Yuan J (2017). Single-Cell RNA Sequencing: Unraveling the Brain One Cell at a Time. Trends Mol Med, 23(6), 563–576. doi: 10.1016/j.molmed.2017.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr Y, Wilson DP, Taylor JM, Bannon PG, Geczy C, Davenport MP, & Kritharides L (2007). A kinetic model of bone marrow neutrophil production that characterizes late phenotypic maturation. Am J Physiol Regul Integr Comp Physiol, 292(4), R1707–1716. doi: 10.1152/ajpregu.00627.2006 [DOI] [PubMed] [Google Scholar]
- Pedersen CC, Borup R, Fischer-Nielsen A, Mora-Jensen H, Fossum A, Cowland JB, & Borregaard N (2016). Changes in Gene Expression during G-CSF-Induced Emergency Granulopoiesis in Humans. J Immunol, 197(5), 1989–1999. doi: 10.4049/jimmunol.1502690 [DOI] [PubMed] [Google Scholar]
- Persson YA, Blomgran-Julinder R, Rahman S, Zheng L, & Stendahl O (2008). Mycobacterium tuberculosis-induced apoptotic neutrophils trigger a pro-inflammatory response in macrophages through release of heat shock protein 72, acting in synergy with the bacteria. Microbes Infect, 10(3), 233–240. doi: 10.1016/j.micinf.2007.11.007 [DOI] [PubMed] [Google Scholar]
- Plagge M, & Laskay T (2017). Early Production of the Neutrophil-Derived Lipid Mediators LTB4 and LXA4 Is Modulated by Intracellular Infection with Leishmania major. Biomed Res Int, 2017, 2014583. doi: 10.1155/2017/2014583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak D, Hafner C, Briza P, Kitzmuller C, Elbe-Burger A, Samadi N, … Bohle B (2019). A novel role for neutrophils in IgE-mediated allergy: Evidence for antigen presentation in late-phase reactions. J Allergy Clin Immunol, 143(3), 1143–1152 e1144. doi: 10.1016/j.jaci.2018.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryde JG, Walker A, Rossi AG, Hannah S, & Haslett C (2000). Temperature-dependent arrest of neutrophil apoptosis. Failure of Bax insertion into mitochondria at 15 degrees C prevents the release of cytochrome c. J Biol Chem, 275(43), 33574–33584. doi: 10.1074/jbc.M001008200 [DOI] [PubMed] [Google Scholar]
- Qin Y, Wu L, Ouyang Y, Zhou P, Zhou H, Wang Y, … Shen N (2017). MiR-125a Is a critical modulator for neutrophil development. PLoS Genet, 13(10), e1007027. doi: 10.1371/journal.pgen.1007027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabellino EM, Ross GD, & Polley MJ (1978). Membrane receptors of mouse leukocytes. I. Two types of complement receptors for different regions of C3. J Immunol, 120(3), 879–885. [PubMed] [Google Scholar]
- Renshaw SA, Parmar JS, Singleton V, Rowe SJ, Dockrell DH, Dower SK, … Whyte MK (2003). Acceleration of human neutrophil apoptosis by TRAIL. J Immunol, 170(2), 1027–1033. [DOI] [PubMed] [Google Scholar]
- Ronnerblad M, Andersson R, Olofsson T, Douagi I, Karimi M, Lehmann S, … consortium F (2014). Analysis of the DNA methylome and transcriptome in granulopoiesis reveals timed changes and dynamic enhancer methylation. Blood, 123(17), e79–89. doi: 10.1182/blood-2013-02-482893 [DOI] [PubMed] [Google Scholar]
- Roos D (2016). Chronic granulomatous disease. Br Med Bull, 118(1), 50–63. doi: 10.1093/bmb/ldw009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorvig S, Ostergaard O, Heegaard NH, & Borregaard N (2013). Proteome profiling of human neutrophil granule subsets, secretory vesicles, and cell membrane: correlation with transcriptome profiling of neutrophil precursors. J Leukoc Biol, 94(4), 711–721. doi: 10.1189/jlb.1212619 [DOI] [PubMed] [Google Scholar]
- Rosen H, & Klebanoff SJ (1977). Formation of singlet oxygen by the myeloperoxidase-mediated antimicrobial system. J Biol Chem, 252(14), 4803–4810. [PubMed] [Google Scholar]
- Rosen H, & Klebanoff SJ (1979). Bactericidal activity of a superoxide anion-generating system. A model for the polymorphonuclear leukocyte. J Exp Med, 149(1), 27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross GD, Jarowski CI, Rabellino EM, & Winchester RJ (1978). The sequential appearance of Ia-like antigens and two different complement receptors during the maturation of human neutrophils. J Exp Med, 147(3), 730–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer-Pokora B, Kunkel LM, Monaco AP, Goff SC, Newburger PE, Baehner RL, … Orkin SH (1986). Cloning the gene for an inherited human disorder--chronic granulomatous disease--on the basis of its chromosomal location. Nature, 322(6074), 32–38. doi: 10.1038/322032a0 [DOI] [PubMed] [Google Scholar]
- Rubel C, Gomez S, Fernandez GC, Isturiz MA, Caamano J, & Palermo MS (2003). Fibrinogen-CD11b/CD18 interaction activates the NF-kappa B pathway and delays apoptosis in human neutrophils. Eur J Immunol, 33(5), 1429–1438. doi: 10.1002/eji.200323512 [DOI] [PubMed] [Google Scholar]
- Sabroe I, Jones EC, Usher LR, Whyte MK, & Dower SK (2002). Toll-like receptor (TLR)2 and TLR4 in human peripheral blood granulocytes: a critical role for monocytes in leukocyte lipopolysaccharide responses. J Immunol, 168(9), 4701–4710. [DOI] [PubMed] [Google Scholar]
- Sabroe I, Prince LR, Jones EC, Horsburgh MJ, Foster SJ, Vogel SN, … Whyte MK (2003). Selective roles for Toll-like receptor (TLR)2 and TLR4 in the regulation of neutrophil activation and life span. J Immunol, 170(10), 5268–5275. [DOI] [PubMed] [Google Scholar]
- Sakamoto E, Hato F, Kato T, Sakamoto C, Akahori M, Hino M, & Kitagawa S (2005). Type I and type II interferons delay human neutrophil apoptosis via activation of STAT3 and up-regulation of cellular inhibitor of apoptosis 2. J Leukoc Biol, 78(1), 301–309. doi: 10.1189/jlb.1104690 [DOI] [PubMed] [Google Scholar]
- Saliba AE, Li L, Westermann AJ, Appenzeller S, Stapels DA, Schulte LN, … Vogel J (2016). Single-cell RNA-seq ties macrophage polarization to growth rate of intracellular Salmonella. Nat Microbiol, 2, 16206. doi: 10.1038/nmicrobiol.2016.206 [DOI] [PubMed] [Google Scholar]
- Savill J (1997). Apoptosis in resolution of inflammation. J Leukoc Biol, 61(4), 375–380. [DOI] [PubMed] [Google Scholar]
- Savill JS, Wyllie AH, Henson JE, Walport MJ, Henson PM, & Haslett C (1989). Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J Clin Invest, 83(3), 865–875. doi: 10.1172/JCI113970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel-Toellner D, Wang K, Assi LK, Webb PR, Craddock RM, Salmon M, & Lord JM (2004). Clustering of death receptors in lipid rafts initiates neutrophil spontaneous apoptosis. Biochem Soc Trans, 32(Pt 5), 679–681. doi: 10.1042/BST0320679 [DOI] [PubMed] [Google Scholar]
- Scheel-Toellner D, Wang K, Craddock R, Webb PR, McGettrick HM, Assi LK, … Lord JM (2004). Reactive oxygen species limit neutrophil life span by activating death receptor signaling. Blood, 104(8), 2557–2564. doi: 10.1182/blood-2004-01-0191 [DOI] [PubMed] [Google Scholar]
- Schmeling DJ, Peterson PK, Hammerschmidt DE, Kim Y, Verhoef J, Wilkinson BJ, & Quie PG (1979). Chemotaxigenesis by cell surface components of Staphylococcus aureus. Infect Immun, 26(1), 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JT, Bandyopadhyay S, Kobayashi SD, McCracken J, Whitney AR, Deleo FR, & Allen LA (2013). Francisella tularensis alters human neutrophil gene expression: insights into the molecular basis of delayed neutrophil apoptosis. J Innate Immun, 5(2), 124–136. doi: 10.1159/000342430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JT, Barker JH, Kaufman J, Fayram DC, McCracken JM, & Allen LA (2012). Francisella tularensis inhibits the intrinsic and extrinsic pathways to delay constitutive apoptosis and prolong human neutrophil lifespan. J Immunol, 188(7), 3351–3363. doi: 10.4049/jimmunol.1102863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Hong S, & Lu Y (2006). Lipid mediator informatics-lipidomics: novel pathways in mapping resolution. AAPS J, 8(2), E284–297. doi: 10.1208/aapsj080233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Lu Y, Hong S, & Yang R (2007). Mediator lipidomics: search algorithms for eicosanoids, resolvins, and protectins. Methods Enzymol, 432, 275–317. doi: 10.1016/S0076-6879(07)32012-0 [DOI] [PubMed] [Google Scholar]
- Shalek AK, Satija R, Adiconis X, Gertner RS, Gaublomme JT, Raychowdhury R, … Regev A (2013). Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature, 498(7453), 236–240. doi: 10.1038/nature12172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Showell HJ, Freer RJ, Zigmond SH, Schiffmann E, Aswanikumar S, Corcoran B, & Becker EL (1976). The structure-activity relations of synthetic peptides as chemotactic factors and inducers of lysosomal secretion for neutrophils. J Exp Med, 143(5), 1154–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva E, Arcaroli J, He Q, Svetkauskaite D, Coldren C, Nick JA, … Abraham E (2007). HMGB1 and LPS induce distinct patterns of gene expression and activation in neutrophils from patients with sepsis-induced acute lung injury. Intensive Care Med, 33(10), 1829–1839. doi: 10.1007/s00134-007-0748-2 [DOI] [PubMed] [Google Scholar]
- Stubbington MJT, Rozenblatt-Rosen O, Regev A, & Teichmann SA (2017). Single-cell transcriptomics to explore the immune system in health and disease. Science, 358(6359), 58–63. doi: 10.1126/science.aan6828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subrahmanyam YV, Yamaga S, Prashar Y, Lee HH, Hoe NP, Kluger Y, … Weissman SM (2001). RNA expression patterns change dramatically in human neutrophils exposed to bacteria. Blood, 97(8), 2457–2468. [DOI] [PubMed] [Google Scholar]
- Sukumaran B, Carlyon JA, Cai JL, Berliner N, & Fikrig E (2005). Early transcriptional response of human neutrophils to Anaplasma phagocytophilum infection. Infect Immun, 73(12), 8089–8099. doi: 10.1128/IAI.73.12.8089-8099.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttmann H, Lehan N, Bohle A, & Brandau S (2003). Stimulation of neutrophil granulocytes with Mycobacterium bovis bacillus Calmette-Guerin induces changes in phenotype and gene expression and inhibits spontaneous apoptosis. Infect Immun, 71(8), 4647–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Kobayashi M, Chiba K, Horiuchi I, Wang J, Kondoh T, … Asaka M (2002). Autocrine production of epithelial cell-derived neutrophil attractant-78 induced by granulocyte colony-stimulating factor in neutrophils. Blood, 99(5), 1863–1865. [PubMed] [Google Scholar]
- Swain SD, Rohn TT, & Quinn MT (2002). Neutrophil priming in host defense: role of oxidants as priming agents. Antioxid Redox Signal, 4(1), 69–83. doi: 10.1089/152308602753625870 [DOI] [PubMed] [Google Scholar]
- Tamassia N, Le Moigne V, Calzetti F, Donini M, Gasperini S, Ear T, … Cassatella MA (2007). The MyD88-independent pathway is not mobilized in human neutrophils stimulated via TLR4. J Immunol, 178(11), 7344–7356. [DOI] [PubMed] [Google Scholar]
- Theilgaard-Monch K, Jacobsen LC, Borup R, Rasmussen T, Bjerregaard MD, Nielsen FC, … Borregaard N (2005). The transcriptional program of terminal granulocytic differentiation. Blood, 105(4), 1785–1796. doi: 10.1182/blood-2004-08-3346 [DOI] [PubMed] [Google Scholar]
- Theilgaard-Monch K, Knudsen S, Follin P, & Borregaard N (2004). The transcriptional activation program of human neutrophils in skin lesions supports their important role in wound healing. J Immunol, 172(12), 7684–7693. [DOI] [PubMed] [Google Scholar]
- Theilgaard-Monch K, Porse BT, & Borregaard N (2006). Systems biology of neutrophil differentiation and immune response. Curr Opin Immunol, 18(1), 54–60. doi: 10.1016/j.coi.2005.11.010 [DOI] [PubMed] [Google Scholar]
- Thomas DC (2017). The phagocyte respiratory burst: Historical perspectives and recent advances. Immunol Lett, 192, 88–96. doi: 10.1016/j.imlet.2017.08.016 [DOI] [PubMed] [Google Scholar]
- Touret N, Paroutis P, & Grinstein S (2005). The nature of the phagosomal membrane: endoplasmic reticulum versus plasmalemma. J Leukoc Biol, 77(6), 878–885. doi: 10.1189/jlb.1104630 [DOI] [PubMed] [Google Scholar]
- Touret N, Paroutis P, Terebiznik M, Harrison RE, Trombetta S, Pypaert M, … Grinstein S (2005). Quantitative and dynamic assessment of the contribution of the ER to phagosome formation. Cell, 123(1), 157–170. doi: 10.1016/j.cell.2005.08.018 [DOI] [PubMed] [Google Scholar]
- Tsukahara Y, Lian Z, Zhang X, Whitney C, Kluger Y, Tuck D, … Newburger PE (2003). Gene expression in human neutrophils during activation and priming by bacterial lipopolysaccharide. J Cell Biochem, 89(4), 848–861. doi: 10.1002/jcb.10526 [DOI] [PubMed] [Google Scholar]
- Usher LR, Lawson RA, Geary I, Taylor CJ, Bingle CD, Taylor GW, & Whyte MK (2002). Induction of neutrophil apoptosis by the Pseudomonas aeruginosa exotoxin pyocyanin: a potential mechanism of persistent infection. J Immunol, 168(4), 1861–1868. [DOI] [PubMed] [Google Scholar]
- van den Berg JM, Weyer S, Weening JJ, Roos D, & Kuijpers TW (2001). Divergent effects of tumor necrosis factor alpha on apoptosis of human neutrophils. J Leukoc Biol, 69(3), 467–473. [PubMed] [Google Scholar]
- van Raam BJ, Sluiter W, de Wit E, Roos D, Verhoeven AJ, & Kuijpers TW (2008). Mitochondrial membrane potential in human neutrophils is maintained by complex III activity in the absence of supercomplex organisation. PLoS One, 3(4), e2013. doi: 10.1371/journal.pone.0002013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura CL, Malachowa N, Hammer CH, Nardone GA, Robinson MA, Kobayashi SD, & DeLeo FR (2010). Identification of a novel Staphylococcus aureus two-component leukotoxin using cell surface proteomics. PLoS One, 5(7), e11634. doi: 10.1371/journal.pone.0011634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpp BD, Nauseef WM, & Clark RA (1988). Two cytosolic neutrophil oxidase components absent in autosomal chronic granulomatous disease. Science, 242(4883), 1295–1297. [DOI] [PubMed] [Google Scholar]
- Vono M, Lin A, Norrby-Teglund A, Koup RA, Liang F, & Lore K (2017). Neutrophils acquire the capacity for antigen presentation to memory CD4(+) T cells in vitro and ex vivo. Blood, 129(14), 1991–2001. doi: 10.1182/blood-2016-10-744441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voyich JM, Braughton KR, Sturdevant DE, Whitney AR, Said-Salim B, Porcella SF, … DeLeo FR (2005). Insights into mechanisms used by Staphylococcus aureus to avoid destruction by human neutrophils. J Immunol, 175(6), 3907–3919. [DOI] [PubMed] [Google Scholar]
- Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, … Chilvers ER (2005). Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med, 201(1), 105–115. doi: 10.1084/jem.20040624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz A, Peveri P, Aschauer H, & Baggiolini M (1987). Purification and amino acid sequencing of NAF, a novel neutrophil-activating factor produced by monocytes. Biochem Biophys Res Commun, 149(2), 755–761. [DOI] [PubMed] [Google Scholar]
- Wang R, Braughton KR, Kretschmer D, Bach TH, Queck SY, Li M, … Otto M (2007). Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med, 13(12), 1510–1514. doi: 10.1038/nm1656 [DOI] [PubMed] [Google Scholar]
- Ward C, Chilvers ER, Lawson MF, Pryde JG, Fujihara S, Farrow SN, … Rossi AG (1999). NF-kappaB activation is a critical regulator of human granulocyte apoptosis in vitro. J Biol Chem, 274(7), 4309–4318. [DOI] [PubMed] [Google Scholar]
- Ward C, Dransfield I, Chilvers ER, Haslett C, & Rossi AG (1999). Pharmacological manipulation of granulocyte apoptosis: potential therapeutic targets. Trends Pharmacol Sci, 20(12), 503–509. [DOI] [PubMed] [Google Scholar]
- Watson RW, Redmond HP, Wang JH, Condron C, & Bouchier-Hayes D (1996). Neutrophils undergo apoptosis following ingestion of Escherichia coli. J Immunol, 156(10), 3986–3992. [PubMed] [Google Scholar]
- Whitlock BB, Gardai S, Fadok V, Bratton D, & Henson PM (2000). Differential roles for alpha(M)beta(2) integrin clustering or activation in the control of apoptosis via regulation of akt and ERK survival mechanisms. J Cell Biol, 151(6), 1305–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte MK, Hardwick SJ, Meagher LC, Savill JS, & Haslett C (1993). Transient elevations of cytosolic free calcium retard subsequent apoptosis in neutrophils in vitro. J Clin Invest, 92(1), 446–455. doi: 10.1172/JCI116587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte MK, Meagher LC, MacDermot J, & Haslett C (1993). Impairment of function in aging neutrophils is associated with apoptosis. J Immunol, 150(11), 5124–5134. [PubMed] [Google Scholar]
- Wong JJ, Ritchie W, Gao D, Lau KA, Gonzalez M, Choudhary A, … Holst J (2014). Identification of nuclear-enriched miRNAs during mouse granulopoiesis. J Hematol Oncol, 7, 42. doi: 10.1186/1756-8722-7-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright HL, Thomas HB, Moots RJ, & Edwards SW (2013). RNA-seq reveals activation of both common and cytokine-specific pathways following neutrophil priming. PLoS One, 8(3), e58598. doi: 10.1371/journal.pone.0058598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka R, Barlow C, Lekstrom-Himes J, Castilla LH, Liu PP, Eckhaus M, … Xanthopoulos KG (1997). Impaired granulopoiesis, myelodysplasia, and early lethality in CCAAT/enhancer binding protein epsilon-deficient mice. Proc Natl Acad Sci U S A, 94(24), 13187–13192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiie K, Kim HY, Mott J, & Rikihisa Y (2000). Intracellular infection by the human granulocytic ehrlichiosis agent inhibits human neutrophil apoptosis. Infect Immun, 68(3), 1125–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura T, Matsushima K, Tanaka S, Robinson EA, Appella E, Oppenheim JJ, & Leonard EJ (1987). Purification of a human monocyte-derived neutrophil chemotactic factor that has peptide sequence similarity to other host defense cytokines. Proc Natl Acad Sci U S A, 84(24), 9233–9237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost CC, Denis MM, Lindemann S, Rubner FJ, Marathe GK, Buerke M, … Zimmerman GA (2004). Activated polymorphonuclear leukocytes rapidly synthesize retinoic acid receptor-alpha: a mechanism for translational control of transcriptional events. J Exp Med, 200(5), 671–680. doi: 10.1084/jem.20040224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Berg N, Lee JW, Le TT, Neudecker V, Jing N, & Eltzschig H (2018). MicroRNA miR-223 as regulator of innate immunity. J Leukoc Biol, 104(3), 515–524. doi: 10.1002/JLB.3MR0218-079R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Hirahashi J, Cullere X, & Mayadas TN (2003). Elucidation of molecular events leading to neutrophil apoptosis following phagocytosis: cross-talk between caspase 8, reactive oxygen species, and MAPK/ERK activation. J Biol Chem, 278(31), 28443–28454. doi: 10.1074/jbc.M210727200 [DOI] [PubMed] [Google Scholar]
- Zhang X, Kluger Y, Nakayama Y, Poddar R, Whitney C, DeTora A, … Newburger PE (2004). Gene expression in mature neutrophils: early responses to inflammatory stimuli. J Leukoc Biol, 75(2), 358–372. doi: 10.1189/jlb.0903412 [DOI] [PubMed] [Google Scholar]
- Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, … Zhang Z (2017). Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell, 169(7), 1342–1356 e1316. doi: 10.1016/j.cell.2017.05.035 [DOI] [PubMed] [Google Scholar]
- Zhu YP, Padgett L, Dinh HQ, Marcovecchio P, Blatchley A, Wu R, … Hedrick CC (2018). Identification of an early unipotent neutrophil progenitor with pro-tumoral activity in mouse and human bone marrow. Cell Rep, 24(9), 2329–2341 e2328. doi: 10.1016/j.celrep.2018.07.097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilbauer M, Rayner TF, Clark C, Coffey AJ, Joyce CJ, Palta P, … Smith KG (2013). Genome-wide methylation analyses of primary human leukocyte subsets identifies functionally important cell-type-specific hypomethylated regions. Blood, 122(25), e52–60. doi: 10.1182/blood-2013-05-503201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, … Klein AM (2019). Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity. doi: 10.1016/j.immuni.2019.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]