

Abstract
The Human Proteome Project (HPP) annually reports on progress made throughout the field in credibly identifying and characterizing the complete human protein parts list and making proteomics an integral part of multiomics studies in medicine and the life sciences. NeXtProt release 2019−01−11 contains 17 694 proteins with strong protein-level evidence (PE1), compliant with HPP Guidelines for Interpretation of MS Data v2.1; these represent 89% of all 19 823 neXtProt predicted coding genes (all PE1,2,3,4 proteins), up from 17 470 one year earlier. Conversely, the number of neXtProt PE2,3,4 proteins, termed the “missing proteins” (MPs), has been reduced from 2949 to 2129 since 2016 through efforts throughout the community, including the chromosome-centric HPP. PeptideAtlas is the source of uniformly reanalyzed raw mass spectrometry data for neXtProt; PeptideAtlas added 495 canonical proteins between 2018 and 2019, especially from studies designed to detect hard-to-identify proteins. Meanwhile, the Human Protein Atlas has released version 18.1 with immunohistochemical evidence of expression of 17 000 proteins and survival plots as part of the Pathology Atlas. Many investigators apply multiplexed SRM-targeted proteomics for quantitation of organ-specific popular proteins in studies of various human diseases. The 19 teams of the Biology and Disease-driven B/D-HPP published a total of 160 publications in 2018, bringing proteomics to a broad array of biomedical research.
Keywords: neXtProt protein existence metrics, missing proteins (MPs), unannotated protein existence 1 (uPE1), neXt-MP50 and CP50 challenges, Human Proteome Project (HPP) Guidelines 3.0, Chromosome-centric HPP (C-HPP), Biology and Disease-HPP (B/D-HPP), PeptideAtlas, Human Protein Atlas
Graphical Abstract
PROGRESS ON THE HUMAN PROTEOME PARTS LIST: NEXTPROT AND PEPTIDEATLAS
Since its launch in September 2010, the HUPO Human Proteome Project (HPP) has provided a productive framework for international communication, collaboration, quality assurance, guideline generation, data sharing and reanalysis, and acceleration of progress in building and utilizing proteomic knowledge globally. The HPP has 50 collaborating research teams organized by chromosome (1−22, X, Y) and mitochondria (C-HPP), biological processes and disease categories (B/D-HPP), and resource pillars for antibody-based protein localization, mass spectrometry, knowledgebases, and pathology.
Remarkable progress has been documented on the chromosome-centric human proteome parts list, as officially curated by neXtProt.1–7 Table 1 shows the increase from 13 975 PE1 proteins in 2012 to 17 694 in release 2019−01. This PE1 total now represents 89% of the PE1,2,3,4 proteins predicted to be coded in the latest version of the human genome (GRCh38). Conversely, the number of yet-to-be detected proteins, termed missing proteins (MPs), are those whose evidence is limited to transcript expression (PE2), homology from other species (PE3), or gene models (PE4). MPs have declined from 5511 in 2012 to 2129 in 2019. Splice variants, sequence variants, and some post-translational modifications (PTMs) are tabulated for each protein entry.8 We acknowledge that neXtProt continues to carry a category PE5, which represents dubious or uncertain genes, including many pseudogenes. However, the HPP in 2013 removed PE5 from the denominator of all predicted proteins to be found3 and in 2016 excluded these from the MP-50 Challenge.7
Table 1.
neXtProt Protein Evidence Levels from 2012 to 2019: Progress in Identifying PE1 Proteinsa and PE2,3,4 Missing Proteinsb
| PE Level | 2012–02 | 2013–09 | 2014–10 | 2016–01 | 2017–01 | 2018–01 | 2019–01 |
|---|---|---|---|---|---|---|---|
| 1: Evidence at protein level | 13 975 | 15 646 | 16 491 | 16 518 | 17 008 | 17 470 | 17 694 |
| 2: Evidence at transcript level | 5205 | 3570 | 2647 | 2290 | 1939 | 1660 | 1548 |
| 3: Inferred from homology | 218 | 187 | 214 | 565 | 563 | 452 | 510 |
| 4: Predicted | 88 | 87 | 87 | 94 | 77 | 74 | 71 |
| 5: Uncertain or dubious | 622 | 638 | 616 | 588 | 512 | 57 | 516 |
| Human PeptideAtlas canonical proteins | 12 509 | 13 377 | 14 928 | 14 569 | 15 173 | 15 798 | 16 293 |
PE1/PE1+2+3+4 = 17,694/19,823 = 89.3%
PE 2+3+4 = 2129 “missing proteins” as of 2019–01.
As shown at the bottom of Table 1, there has been a corresponding increase in the number of proteins in the human build of PeptideAtlas. PeptideAtlas provides an essential HPP function by uniformly processing raw MS data/metadata registered in ProteomeXchange through its Trans-Proteomic Pipeline (TPP)9,10 and applying the communally agreed HPP Guidelines 2.1 for Interpretation of Mass Spectrometry Data.11 The number of proteins in PeptideAtlas that are assessed as canonical has increased from 12 509 to 16 293, despite the implementation of more stringent guidelines, which accounts for the observed dip between 2014 and 2016. We present the main sources of the increment of 495 canonical proteins from 2018 to 2019 in Figure 3, below.
Figure 3.

Top 10 newly analyzed deposited ProteomeXchange data sets contributing to the increased number of canonical proteins found in PeptideAtlas between 2018 and 2019 builds. Each data set is labeled with the number of new canonical proteins, ProteomeXchange identifier (PXD), reference citation,22,24,25,37–43 and methods highlighted.
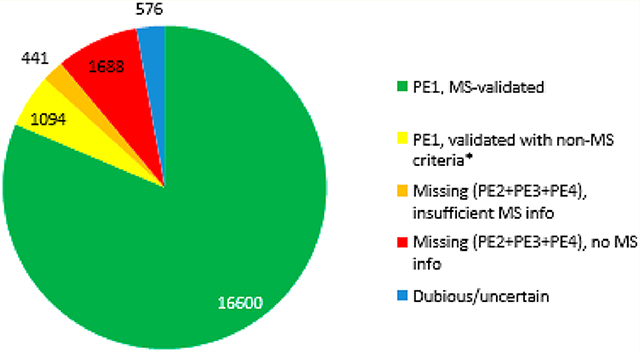
In Figure 1 we account for the types of experimental evidence upon which neXtProt has classified the human proteome. Of all 17 694 PE1 proteins, 16 600 are now based on validated mass spectrometry results (Figure 1, green). Of these, 98% represent canonical proteins in the human PeptideAtlas build. In addition, 1094 PE1 proteins (yellow) were identified based upon protein−protein interactions (388), PTMs or proteolytic processing (158), disease mutations (123), Edman sequencing (90), 3D structure (50), Ab-based data (46), or other biochemical studies (239). The 2129 PE2,3,4 MPs are comprised of 1688 with no MS data (red) plus 441 with insufficient or unconfirmed MS data (orange) that were not compliant with HPP Guidelines 2.1; the full list can be retrieved by query NXQ_00204.
Figure 1.

Pie charts showing distribution of human proteins based on the type of protein evidence data. Shown here are the numbers of PE1 proteins (green plus yellow), PE2,3,4 MPs, and PE5 entries as of neXtProt releases 2018−01 (left) and 2019−01 (right).
The year-to-year changes in neXtProt HPP metrics are quite complex to track, due to such factors as revisions of total gene entries in UniProt/SwissProt, reassessment of evidence level in neXtProt upon review of additional data, and policy changes about potential inclusion of claimed products of small open reading frames (smORFs), long noncoding RNAs, or immunoglobulin genes. The flowchart illustrating all PE level transitions from neXtProt release 2018−01 to release 2019−01 is presented in Figure 2 (right), along with the previously published transition from 2017 to 2018 for comparison (left).7
Figure 2.

These flowcharts depict the changes in neXtProt PE1−5 categories from release 2017−01 to 2018−01 (left) and from 2018−01 to 2019−01 (right).
During 2017, 431 MPs were promoted to PE1 and 44 new SwissProt proteins were added as PE1s, while 3 PE1s were demoted to MPs and 10 PE1s were deleted. During 2018, 213 MPs and 5 PE5 entries were promoted to PE1 along with 78 new proteins, while 65 PE1s were demoted and 7 PE1s deleted. This results in a net gain of 224 PE1 proteins and a net reduction of 57 PE2,3,4 MPs in the HPP neXtProt baseline for 2019. Note especially that the number of MPs was inflated by 40 new entries in 2017 and 116 in 2018. Uncertainties in the reference human genome produce a significant undertow which impacts the global efforts to reduce the number of the MPs.
Table 2 presents a detailed chromosome-by-chromosome accounting of the status of the MPs search, elaborated from neXtProt 2019−01 [https://www.nextprot.org/about/proteinexistence and ftp://ftp.nextprot.org/pub/current_release/Chr_reports/]. Column 9 shows the number of PE1,2,3,4 MPs in neXtProt 2016–01 and 2019–01 by chromosome. Also tabulated are the numbers of functionally unannotated PE1 proteins (uPE1) of the human proteome.12 Together, the PE2,3,4 MPs (NXQ_00204) and the uPE1 proteins (NXQ_00022) constitute a large part of what has been termed the “Dark Proteome” (DP).13
Table 2.
Chromosome-by-Chromosome Status of Predicted Proteins in neXtProt 2019–01a
| Chr | PE1 | PE2 | PE3 | PE4 | PE5 | PE1–4 (total) | PE1/PE1–4 | PE2,3,4 MPs 2016/2019 | uPE1 Proteins |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1796 | 150 | 66 | 8 | 49 | 2020 | 1796/2020=88.9% | 309/224 | 138 |
| 2 | 1178 | 71 | 18 | 1 | 17 | 1268 | 1178/1268=92.9% | 134/90 | 90 |
| 3 | 969 | 76 | 19 | 3 | 15 | 1067 | 969/1067= 90.8% | 141/98 | 59 |
| 4 | 678 | 46 | 21 | 2 | 21 | 747 | 678/747=90.8% | 95/69 | 50 |
| 5 | 796 | 55 | 9 | 3 | 13 | 863 | 796/863=92.2% | 122/67 | 52 |
| 6 | 983 | 79 | 12 | 6 | 30 | 1080 | 983/1080=91.0% | 136/97 | 54 |
| 7 | 808 | 89 | 41 | 5 | 42 | 943 | 808/943=85.7% | 137/135 | 50 |
| 8 | 602 | 45 | 14 | 2 | 39 | 663 | 602/663=90.8% | 95/61 | 36 |
| 9 | 671 | 71 | 21 | 12 | 35 | 775 | 671/775=86.6% | 129/104 | 58 |
| 10 | 665 | 60 | 7 | 1 | 17 | 733 | 665/733=90.7% | 115/68 | 57 |
| 11 | 1014 | 184 | 95 | 1 | 39 | 1294 | 1014/1294=78.3% | 319/280 | 72 |
| 12 | 932 | 64 | 14 | 0 | 23 | 1010 | 932/1010=92.3% | 119/78 | 52 |
| 13 | 297 | 20 | 4 | 1 | 12 | 332 | 297/332=89.5% | 43/25 | 27 |
| 14 | 613 | 40 | 56 | 4 | 14 | 713 | 613/713=90.0% | 93/100 | 34 |
| 15 | 513 | 43 | 17 | 1 | 30 | 574 | 513/574=89.4% | 73/61 | 40 |
| 16 | 745 | 58 | 10 | 2 | 24 | 815 | 745/815=87.7% | 99/70 | 47 |
| 17 | 1049 | 82 | 13 | 3 | 22 | 1147 | 1049/1147=91.5% | 148/98 | 65 |
| 18 | 250 | 13 | 1 | 2 | 10 | 266 | 250/266=94.0% | 24/16 | 10 |
| 19 | 1271 | 104 | 25 | 2 | 32 | 1402 | 1271/1402=90.7% | 261/131 | 77 |
| 20 | 488 | 43 | 3 | 5 | 12 | 539 | 488/539=90.5% | 82/51 | 44 |
| 21 | 186 | 28 | 18 | 1 | 23 | 233 | 186/233=79.8% | 49/47 | 9 |
| 22 | 432 | 39 | 5 | 1 | 21 | 477 | 432/477=90.6% | 64/45 | 30 |
| X | 720 | 76 | 18 | 5 | 29 | 819 | 720/819=87.9% | 145/99 | 102 |
| Y | 26 | 11 | 3 | 0 | 7 | 40 | 26/40=65% | 16/14 | 1 |
| Mito | 15 | 0 | 0 | 0 | 0 | 15 | 15/15=100% | 0/0 | |
| Unk | 2 | 2 | 0 | 0 | 0 | 4 | 2/4=50% | 2/2 | |
| ALL | 17 694 | 1548 | 510 | 71 | 576 | 19 823 | 17694/19823=89.3% | 2949/2129 | 1254 |
| Sums | 17 699 | 1549 | 510 | 71 | 576 | 19 829 | 17699/19829=89.3% | 2950/2130 | 1254 |
NOTE: There are discrepancies between the true total numbers of proteins in each PE category (ALL) and the Sums because six proteins are derived from two genes on two different chromosomes, and thus appear twice under the per-chromosome table values.
PROGRESS FROM THE CHROMOSOME-CENTRIC C-HPP
The C-HPP initiated a neXt-MP50 challenge at the Sun Moon Lake Workshop following the Taipei HUPO Congress in 2016. The aim was to galvanize the 24 chromosome teams to each find 50 missing proteins as a step toward completing the human proteome parts list. As shown in Table 2, that 50 MP target has been reached for Chromosomes 19, 1, 5, and 17. Chromosomes 2, 3, 10, and X have 43 to 47 net reduction of MPs. The Chr 17 team has published detailed analyses of the paths to reduce PE2,3,4 and increase PE1.14,15 As depicted for the whole proteome in Figure 2 flowcharts, the dynamics of promoting, demoting, gaining, and deleting protein entries in neXtProt is quite complex. More detailed chromosome-by-chromosome analyses are planned for the coming year.
C-HPP investigators generated 10 articles for the sixth annual HPP Special Issue of Journal of Proteome Research (2018) that reported evidence for detection and characterization of a total of 104 MPs:16 13 from cerebrospinal fluid,17 1 from mesenchymal stem cells,18 5 from olfactory epithelium,19 1 from HeLa cells,20 3 from mitochondria,21 14 with the use of multiple proteases for digestion,22 2 using LysargiNase11 which mirrors trypsin specificity,23 26 from embryonic stem cells,24 30 membrane proteins,25 and 9 from other unusual/rare/stressed tissues.26 In addition, evidence for detection of 107 MPs was put forward from reanalyses by MassIVE,27 many of which are included in the 2019−01 PeptideAtlas build.
The limited numbers of MPs found each year by the C-HPP teams and the global proteomics community reflect the increasing difficulty in devising and executing deep discovery of MPs in the human proteome. More sensitive methods (including enrichment techniques) and targeted analyses of understudied tissues and cells are needed. Examples include organ-specific endothelium, hard connective tissues with cartilaginous structures and cortical versus trabecular versus membranous bones,28 dental tissues like dentin and odontoblasts,29 and brain regions. In embryonic tissues, limited bursts of transcription factor expression and specific hydrolases likely account for many differentiating aspects of each cell and tissue in the body. Identifying such temporally or spatially rare proteins will require dedicated searches at precise developmental windows—an ethical and practical challenge. Likewise, tissue responses to bacterial, viral, and parasitic infections, or after injury may harbor MPs key for regeneration or repair of these specific tissues. Integration with the B/D-HPP teams in this task is highly desirable.
Meanwhile, the C-HPP initiated the neXt-CP50 Challenge for the functional characterization of the dark proteome, that is, human proteins with no known or inferred function. These proteins often have antibody-based tissue distribution and intracellular localization in Human Protein Atlas, mass spectrometry-identified peptides in PeptideAtlas, and other information. At the March 2018 launch of the neXt-CP50 Challenge there were 1937 PE1,2,3,4 proteins with no known function, of which 1260 were uPE1 proteins13 (1254 in Table 2 as of 2019−01).
An interesting example of functional annotation and unusual cell types is MALT1 in B and T cells. MALT1 deficiency causes a rare immunodeficiency with ~50% reduced NFkB activation. The function of this mutant protease was uncovered by TAILS N-terminal positional proteomics. A highly selective molecular connector therapeutic compound was able to graft together two domains of the mutant MALT1 to restore molecular stability in the patient’s B and T cells, rescue NFkB activation, and provide MALT1 cleavage activity in B and T cell antigen receptor activation.30 Meanwhile, a computational approach to predict functions of uPE1 proteins using I-TASSER and COFACTOR was introduced by the Chromosome 17 team31 and subjected to blinded analyses of newly annotated proteins in CAFA3 and in neXtProt-2019.32
NEWLY IDENTIFIED CANONICAL PROTEINS IN PEPTIDE ATLAS
PeptideAtlas33,34 reprocessed and incorporated data from 120 new human sample data sets between the 2018−01 and 2019−01 builds. Not all newly deposited and released MS data sets are automatically captured and incorporated into PeptideAtlas, because, with each additional data set, the stringency applied to all data must be increased to maintain a constant false discovery rate (FDR). Thus, only data sets that are sufficiently novel are subjected to TPP reanalysis and are incorporated. These additional MS data resulted in an increase in the number of PeptideAtlas proteins for which there are at least two uniquely mapping, non-nested 9+ amino acid peptides by 495 to a 2019−01 count of 16 293 (Table 1). Figure 3 depicts the contributions of the top 10 data sets to this increase in the number of proteins, accounting for 292 (59%) of the 495. The three data sets contributing the most canonical proteins (a total of 161) were PXD009737,22 PXD010630,25 and PXD009840,24 all resulting from the sixth HPP special issue in this journal (2018). It is impressive that major progress has been made with membrane proteins. The PeptideAtlas reanalysis of data from the use of multiple proteases by Sun et al.22 yielded 73 new canonical proteins, far more than the 14 new PE1 proteins validated by synthetic peptides and proposed in their original article. Note the important distinction between new PE1 proteins and new canonical proteins in PeptideAtlas (which may already be PE1 via other means). Neither neXtProt nor PeptideAtlas requires confirmatory synthetic peptide MS results for their annual reanalyses of community MS data for the HPP. Also, it is notable that the raw data for PXD009840 were accessed at jPOST35 along with one other new canonical protein from a second jPOST data set. No MS data sets from iProX36 have been included in the current PeptideAtlas or neXtProt releases, but there will be in the next release (see B/D-HPP, below). Also, PeptideAtlas does not yet incorporate SRM results.
From the point of view of PeptideAtlas, the progress in pursuit of a complete human proteome is depicted in Figure 4, using data from Table 1 since 2016, when the HPP Guidelines 2.0 had been applied. In green on the left are the PeptideAtlas canonical proteins, progressing to 16 293 in 2019. These proteins have at least two uniquely mapping, non-nested, peptides of 9 or more amino acids in PeptideAtlas. In yellow in the middle are the proteins classified as PE1 in neXtProt based on evidence from other techniques (see Figure 1 and associated text); this number has been steadily decreasing over the past four years. In red on the right are the PE2,3,4 MPs. The total has been increasing at a small rate as understanding and annotation of the human proteome improves each year. A simple extrapolation of increases in green and decreases in yellow and red (based on 2016 to 2019 data) yields a convergence of all three on the year 2026. Clearly, additional strategies need to be developed to accelerate progress to reach near-complete human proteome coverage at a stringency required by the HPP Mass Spectrometry Data Interpretation Guidelines 3.0 (see below).
Figure 4.

Summary of the evolution since 2016 of the proteome categories of PE1 proteins based on PeptideAtlas (PA) MS evidence (green), PE1 proteins based on non-MS evidence (yellow), and PE2,3,4 MPs (red). Progress has been remarkably linear since 2016.
HPP MASS SPECTROMETRY DATA INTERPRETATION GUIDELINES 3.0
At a full-day workshop prior to the Saint-Malo 21st C-HPP Workshop in May 2019, the HPP leadership gathered for an extended discussion of 25 open questions from the HPP Knowledgebase Resource Pillar and other HPP teams relating to updates of the HPP Mass Spectrometry Data Interpretation Guidelines 2.111 and implementation of the workflow by which the HPP confirms the translation of potential coding genes. Each of these questions was debated, potential solutions listed, and consensus decisions achieved by the participants. The result will be an update of the Guidelines from version 2.1 to version 3.0, with a manuscript44 describing the set of questions, potential solutions, proposed consensus decisions, and the logic behind those proposals. For the checklist required for submitted manuscripts, numbered items will be refactored into logical subgroups and authors will be required to provide page numbers on the checklist so reviewers can see where specific guidelines are addressed in the manuscripts. The term “extraordinary detection claims,” which appears to have caused some confusion, will be replaced with “new PE1 protein detection claims”. The revised guidelines will refine how peptide nesting is defined and specify how sequence-identical protein entries should be handled.
Two new guidelines will be added: (1) the provision of Universal Spectrum Identifiers (USI; http://psidev.info/USI), a feature developed by the HUPO Proteomics Standards Initiative (PSI) that enables the unique identification of a particular spectrum being held up as evidence for a new PE1 protein detection claim across proteomics repositories, suitable for searching; and (2) a guideline for handling HPP data sets that use data-independent acquisition (DIA) workflows including SWATH-MS.45 These guideline changes are expected to take effect for contributions to the 2020 JPR HPP Special Issue.
At the workshop, several changes to the overall pipeline for tracking detections of MPs were considered. The current pipeline begins with deposition to ProteomeXchange, reprocessing of data sets by PeptideAtlas, and final mapping and incorporation by neXtProt. It was agreed that there would be no substantial change to the basic set of guidelines for calling a protein successfully identified by mass spectrometry. However, the meaning of the terms non-nested and uniquely mapping has been interpreted and implemented in slightly different ways among the different components of the pipeline. A consensus interpretation was clarified and will be documented.
For proteins that come close to meeting the guidelines, but do not meet them due to their extreme sequence composition (e.g., very short or very hydrophobic), it was decided that complex exception rules are not warranted. Rather, a panel of researchers including neXtProt curators will be established to review evidence for special cases and classify proteins as PE1 if the available evidence is compelling but falls short of the Guidelines 3.0 due to valid physiochemical reasons. No proteins would be declared too difficult to detect yet, although there was substantial discussion about the extreme difficulties of detecting olfactory receptors and other categories of membrane-bound proteins, let alone those proteins predicted to come from purported genes lacking measurable transcripts. Discussion sections of previous JPR HPP Special Issue Metrics papers have addressed these challenges.7 Finally, a plan was initiated for incorporating the data set reprocessing results of MassIVE-KB ProteinExplorer,27,46 including BioPlex data (while excluding bait) through the PeptideAtlas TPP to feed into the 2020 neXtProt HPP release. Refinements to the overall HPP pipeline for tracking high stringency identification of MPs should facilitate confident completion of the attainable protein parts list over the next several years.
PROTEOMEXCHANGE (PX)
The ProteomeXchange Consortium47 was founded a decade ago by the European Bioinformatics Institute (EBI) and HUPO based on PRIDE and PeptideAtlas. As of 23 May 2019, ProteomeXchange (PX) contained 8134 publicly released data submissions, of which 3395 are human data sets. PRIDE48,49 (EMBL-EBI, Cambridge, UK) and PeptideAtlas (ISB, Seattle, WA, USA) are the founding members, joined now by MassIVE (UCSD, San Diego, CA, USA), jPOST (various institutions, Japan), iProX (Phoenix National Center for Protein Sciences, Beijing, China), and Panorama Public50 (targeted proteomic data sets from Skyline, University of Washington, Seattle, USA). Each assigns ProteomeXchange identifiers (PXD) to its data sets. The PRIDE (PRoteomics IDEntification) database had 12 585 projects [https://www.ebi.ac.uk/pride/archive/]. Peptide Atlas had 1547 human data sets (120 new in 2018) with 16 303 canonical PE1−4 proteins and 2949 uncertain or redundant protein entries [www.peptideatlas.org/hupo/chpp/]. MassIVE-KB [Mass Spectrometry Interactive Virtual Environment; https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp] comprised 9286 public data sets, with 20 116 proteins, 11 M peptide variants, and 505 modifications. iProX [www.iprox.org] contained 553 projects (247 public), and 112 608 data files (14 404 added in the past year). The large data set for the 2019 early stage hepatocellular carcinoma paper from Jiang et al.51 will be included in 2020 HPP updates; this data set was uploaded via ProteomeXchange to PRIDE as well as to iProX. JPOSTdb [https://globe.jpostdb.org/] held 66 human, 25 mouse, and 12 bacterial data sets.
HIGHLIGHTS OF PROGRESS FROM THE B/D-HPP
The Biology and Disease-driven Human Proteome Project (B/D-HPP) currently encompasses the efforts of 19 independent initiatives (https://www.hupo.org/B/D-HPP). The main aims are to elucidate the molecular basis of human biology, to uncover protein drivers of human disease, and to promote the development of novel proteomics-based tools to improve the clinical management of patients. Collaborative research is conducted in close interaction with specific biomedical and clinical communities. Overall, the research work done in 2018 by B/D-HPP teams resulted in 160 published papers and participation in 82 international congresses, among which 40% were organized by scientific and clinical societies focused on specific topics, including cardiovascular, cancer, infectious diseases, gastroenterology, and nutrition. Also, there were 28 educational activities, including the first Summer School in Immunopeptidomics (https://hupo.org/B/D-HPP/).
Web tools for computational bibliometric analyses provide researchers prioritized lists of proteins in relation to particular organ systems52 or disease categories,53 and metabolites or chemicals.54 In the annual survey of B/D teams, 13 responded that they are using (11) or planning to use (2) such popular proteins methodology and databases in their research.
B/D-HPP teams have made substantial contributions to the elucidation of molecular mechanisms driving human disorders, moving toward “precision medicine”. For example, pathological conformations of TDP-43, a nucleic acid binding protein that regulates splicing and expression of CFTR, can distinguish four pathological subtypes of frontotemporal lobar degeneration.55 The human arterial proteome has been extensively analyzed for proteins associated with early atherosclerosis; a subset of plasma proteins emerged as efficient predictors of angiographically defined coronary disease.56 Biomarker identification has been pursued in urine of diabetic patients,57 peptides from mucins, fibrinogen, and collagen fragments in ~1000 cancer patients,58 and coregulated groups of circulating proteins correlating to past, current and future disease states in 5500 individuals in Iceland.59 Studies from the Beijing Proteome Research Center addressed human HBV-associated HCC60 and a mouse model of metabolic syndrome and fatty liver.61 A major analysis of patients with early stage hepatocellular cancers delineated three subtypes, notably S−III with high expression of sterol-O-acyl transferase (SOAT1), which is well-suited for targeted therapy, as demonstrated in xenograft models.51
PTMs represent a functional regulatory level that is central to understanding progression of diseases and to define novel therapeutic targets. Cross-talk across different PTMs in the context of cardiovascular physiopathology has been addressed.62 A total of 1655 proteins with 3324 oxidized cysteine sites were identified in a mouse cardiac hypertrophy model.63 Changes in protein phosphorylation patterns have been associated with HCC64 and ovarian tumors.65 Protein glycosylation profiles have been explored in plasma from 300 cancer patients combining N-glycosite enrichment and SWATH; some glycoproteins, notably those related to blood platelets, are common changes across several cancers, while others are highly cancer-type specific.66 Finally, Murray et al. have demonstrated how dynamic changes in protein acetylation participate in the herpes virus human cytomegalovirus replication and in the host cell defense.67
Many efforts have been devoted to development and standardization of new proteomics-based applications. MALDI-TOF profiling has been used to discriminate different infant milk formula in pediatric clinical settings.68 Piazza et al. have defined protein-metabolite interaction networks and identified 1700 interactions and 7000 interaction sites, revealing principles of chemical communication, mechanisms of enzyme promiscuity, and estimates of metabolite binding at proteome-wide scale.69 Reference proteomes have been reported for clinical studies of cerebrospinal fluid70 and the NCI-7 cell line panel.71 Gut microbiota have been investigated increasingly as a complex community that influences many aspects of human physiology and disease; the platform iMetaLab (http://imetalab.ca) facilitates functional studies of microbiota.72 A key need in proteomic workflows is format and data analysis standardization; an example is the Minimal Information About an Immuno-Peptidomics Experiment (MIAIPE) guidelines.73 Finally, an interlaboratory study to assess the performance of glycoproteomics software for automated intact N- and O-glycopeptide identification from high resolution MS/MS spectral data is ongoing.
The functional annotation of the human proteome requires a collaborative effort. Fertilization of interactions across C-HPP and B/D-HPP teams21,74 to share resources and knowledge will help decipher the code of life and set the basis for future molecular precision medicine.
HIGHLIGHTS FROM THE HUMAN PROTEIN ATLAS
During 2018, the Human Protein Atlas (HPA)75 released its version 18.1 that summarizes data from 26 000 antibodies targeting proteins from 17 000 human protein-coding genes. There is continuously growing global interest with 150 000 web visitors per month. The latest update included interactive survival scatter plots as part of the Pathology Atlas.76 The Cell Atlas77 is being recognized in other international projects, such as the Human Cell Atlas,78 as well as deep learning and citizen science projects.79 HPA continues to enhance its work on antibody validation80 and on annotation of protein expression in currently under-explored tissues.81 Fredolini et al. have applied a magnetic bead-assisted workflow and immunoprecipitation-MS/MS to assess antibody selectivity for enrichment and detection of 120 proteins in human EDTA-plasma; most of the antibodies coenriched other proteins besides the intended target, due to sequence homology.82
During 2019, the HPA plans to release three new components focusing on blood, the brain, and metabolism. In addition, HPA is combining mRNA expression data obtained from human tissues that are currently derived from different types of sequencing data; the harmonized RNA expression data will be a valuable reference resource for neXtProt Protein Evidence curation. A quantitative paired analysis of the proteome and transcriptome abundance for 29 healthy human tissues identified 13 640 proteins, but only 37 without prior protein level evidence. Hundreds of proteins were not detected even when the corresponding mRNAs were highly expressed, particularly in testis (PXD010154).83 These findings, and many others, will be captured for standardized reanalysis with the PeptideAtlas TransProteomicPipeline to be included in PeptideAtlas 2020−01 and incorporated into neXtProt release 2020−01.
CONCLUSIONS
The Human Proteome Project continues to provide a quality-assured framework for capturing sustained progress on the protein parts list and the characterization of the features and functions of human proteins. PeptideAtlas identified 495 new canonical proteins during the past year, including many neXtProt PE1 proteins that previously were classified based on non-MS data. NeXtProt had a net increase of 224 PE1 proteins. As of the baseline for the 2019 studies (neXtProt release 2019−01), there were still 2129 “Missing Proteins” (PE2,3,4) awaiting the application of advanced techniques and analysis of understudied specimen types. Since 2016 there has been a surprisingly linear decrease in the number of missing proteins, which is likely to continue, though a hard core of perhaps 1000 predicted proteins may be so low in abundance, so unusual in the sites or conditions of expression, or so unsuited to detection that they will be “out of reach.” We suspected that we were approaching the limit 2−3 years ago, but the pace of progress has not diminished. Over the next few years we will clarify that question while enhancing our knowledge of the proteins and stimulating the broader and broader use of proteomics in biomedical research.
ACKNOWLEDGMENTS
We appreciate the guidance from the HPP Executive Committee and the participation of all HPP investigators. We thank the UniProt groups at SIB, EBI, and PIR for providing high-quality annotations for the human proteins in UniProtKB/Swiss-Prot. The neXtProt server is hosted by VitalIT in Switzerland. The PeptideAtlas server is hosted at the Institute for Systems Biology in Seattle. G.S.O. acknowledges grant support from National Institutes of Health grants P30ES017885-01A1 and U24CA210967; E.W.D. from National Institutes of Health grants, R01GM087221, R24GM127667, U54EB020406, and the U19AG023122; L.L. and neXtProt from the SIB Swiss Institute of Bioinformatics; C.M.O. by a Canadian Institutes of Health Research Foundation Grant and a Canada Research Chair in Protease Proteomics and Systems Biology; M.S.B. by NHMRC Project Grant APP1010303; J.M.S. by the Knut and Alice Wallenberg Foundation for the Human Protein Atlas; and Y.-K.P. by grants from the Korean Ministry of Health and Welfare HI13C22098 and HI16C0257.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Legrain P; Aebersold R; Archakov A; Bairoch A; Bala K; Beretta L; Bergeron J; Borchers CH; Corthals GL; Costello CE; Deutsch EW; Domon B; Hancock W; He F; Hochstrasser D; Marko-Varga G; Salekdeh GH; Sechi S; Snyder M; Srivastava S; Uhlen M; Wu CH; Yamamoto T; Paik YK; Omenn GS The Human Proteome Project: current state and future direction. Mol. Cell. Proteomics 2011, 10 (7), M111.009993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Marko-Varga G; Omenn GS; Paik YK; Hancock WS A first step toward completion of a genome-wide characterization of the human proteome. J. Proteome Res 2013, 12 (1), 1–5. [DOI] [PubMed] [Google Scholar]
- (3).Lane L; Bairoch A; Beavis RC; Deutsch EW; Gaudet P; Lundberg E; Omenn GS Metrics for the human proteome project 2013−2014 and strategies for finding missing proteins. J. Proteome Res 2014, 13 (1), 15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Omenn GS; Lane L; Lundberg EK; Beavis RC; Nesvizhskii AI; Deutsch EW Metrics for the Human Proteome Project 2015: progress on the human proteome and guidelines for high-confidence protein identification. J. Proteome Res 2015, 14 (9), 3452–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Omenn GS; Lane L; Lundberg EK; Beavis RC; Overall CM; Deutsch EW Metrics for the human proteome project 2016: Progress on identifying and characterizing the human proteome, including post-translational modifications. J. Proteome Res 2016, 15 (11), 3951–3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Omenn GS; Lane L; Lundberg EK; Overall CM; Deutsch EW Progress on the HUPO draft human proteome: 2017 metrics of the Human Proteome Project. J. Proteome Res 2017, 16 (12), 4281–4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Omenn GS; Lane L; Overall CM; Corrales FJ; Schwenk JM; Paik YK; Van Eyk JE; Liu S; Snyder M; Baker MS; Deutsch EW Progress on identifying and characterizing the human proteome: 2018 metrics from the HUPO Human Proteome Project. J. Proteome Res 2018, 17 (12), 4031–4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Gaudet P; Michel PA; Zahn-Zabal M; Britan A; Cusin I; Domagalski M; Duek PD; Gateau A; Gleizes A; Hinard V; Rech de Laval V; Lin J; Nikitin F; Schaeffer M; Teixeira D; Lane L; Bairoch A The neXtProt knowledgebase on human proteins: 2017 update. Nucleic Acids Res. 2017, 45 (D1), D177–d182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Keller A; Eng J; Zhang N; Li XJ; Aebersold R A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol. Syst. Biol 2005, 1, 0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Deutsch EW; Mendoza L; Shteynberg D; Slagel J; Sun Z; Moritz RL Trans-proteomic pipeline, a standardized data processing pipeline for large-scale reproducible proteomics informatics. Proteomics: Clin. Appl 2015, 9 (7−8), 745–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Deutsch EW; Overall CM; Van Eyk JE; Baker MS; Paik YK; Weintraub ST; Lane L; Martens L; Vandenbrouck Y; Kusebauch U; Hancock WS; Hermjakob H; Aebersold R; Moritz RL; Omenn GS Human Proteome Project mass spectrometry data interpretation guidelines 2.1. J. Proteome Res 2016, 15 (11), 3961–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Duek P; Gateau A; Bairoch A; Lane L Exploring the uncharacterized human proteome using neXtProt. J. Proteome Res 2018, 17 (12), 4211–4226. [DOI] [PubMed] [Google Scholar]
- (13).Paik YK; Lane L; Overall CM Launching the C-HPP neXtCP50 pilot project for functional characterization of identified proteins with no known function. J. Proteome Res 2018, 17, 4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Siddiqui O; Zhang H; Guan Y; Omenn GS Chromosome 17 missing proteins: Recent progress and future directions as part of the neXt-MP50 challenge. J. Proteome Res 2018, 17 (12), 4061–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zhang H; Siddiqui O; Guan Y; Omenn GS Chromosome 17: neXt-MP50 challenge completed. J. Proteome Res 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Paik Y-K; Overall CM; Corrales F; Deutsch EW; Lane L; Omenn GS Toward completion of the human proteome parts list: Progress uncovering proteins that are missing or have unknown function and developing analytical methods. J. Proteome Res 2018, 17 (12), 4023–4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Macron C; Lane L; Núñez Galindo A; Dayon L Deep dive on the proteome of human cerebrospinal fluid: A valuable data resource for biomarker discovery and missing protein identification. J. Proteome Res 2018, 17 (12), 4113–4126. [DOI] [PubMed] [Google Scholar]
- (18).Clemente LF; Hernáez ML; Ramos-Fernández A; Ligero G; Gil C; Corrales FJ; Marcilla M Identification of the missing protein hyaluronan synthase 1 in human mesenchymal stem cells derived from adipose tissue or umbilical cord. J. Proteome Res 2018, 17 (12), 4325–4328. [DOI] [PubMed] [Google Scholar]
- (19).Hwang H; Jeong JE; Lee HK; Yun KN; An HJ; Lee B; Paik Y-K; Jeong TS; Yee GT; Kim JY; Yoo JS Identification of missing proteins in human olfactory epithelial tissue by liquid chromatography−tandem mass spectrometry. J. Proteome Res 2018, 17 (12), 4320–4324. [DOI] [PubMed] [Google Scholar]
- (20).Robin T; Bairoch A; Müller M; Lisacek F; Lane L Largescale reanalysis of publicly available HeLa cell proteomics data in the context of the human proteome project. J. Proteome Res 2018, 17 (12), 4160–4170. [DOI] [PubMed] [Google Scholar]
- (21).Ronci M; Pieroni L; Greco V; Scotti L; Marini F; Carregari VC; Cunsolo V; Foti S; Aceto A; Urbani A Sequential fractionation strategy identifies three missing proteins in the mitochondrial proteome of commonly used cell lines. J. Proteome Res 2018, 17 (12), 4307–4314. [DOI] [PubMed] [Google Scholar]
- (22).Sun J; Shi J; Wang Y; Chen Y; Li Y; Kong D; Chang L; Liu F; Lv Z; Zhou Y; He F; Zhang Y; Xu P Multiproteases combined with high-pH reverse-phase separation strategy verified fourteen missing proteins in human testis tissue. J. Proteome Res 2018, 17 (12), 4171–4177. [DOI] [PubMed] [Google Scholar]
- (23).Huesgen PF; Lange PF; Rogers LD; Solis N; Eckhard U; Kleifeld O; Goulas T; Gomis-Ruth FX; Overall CM LysargiNase mirrors trypsin for protein C-terminal and methylation-site identification. Nat. Methods 2015, 12 (1), 55–8. [DOI] [PubMed] [Google Scholar]
- (24).Weldemariam MM; Han C-L; Shekari F; Kitata RB; Chuang C-Y; Hsu W-T; Kuo H-C; Choong W-K; Sung T-Y; He F-C; Chung MCM; Salekdeh GH; Chen Y-J Subcellular proteome landscape of human embryonic stem cells revealed missing membrane proteins. J. Proteome Res 2018, 17 (12), 4138–4151. [DOI] [PubMed] [Google Scholar]
- (25).Zhang Y; Lin Z; Hao P; Hou K; Sui Y; Zhang K; He Y; Li H; Yang H; Liu S; Ren Y Improvement of peptide separation for exploring the missing proteins localized on membranes. J. Proteome Res 2018, 17 (12), 4152–4159. [DOI] [PubMed] [Google Scholar]
- (26).Sjöstedt E; Sivertsson Å; Hikmet Noraddin F; Katona B; Näsström Å; Vuu J; Kesti D; Oksvold P; Edqvist P-H; Olsson I; Uhlén M; Lindskog C Integration of transcriptomics and antibody-based proteomics for exploration of proteins expressed in specialized tissues. J. Proteome Res 2018, 17 (12), 4127–4137. [DOI] [PubMed] [Google Scholar]
- (27).Pullman BS; Wertz J; Carver J; Bandeira N ProteinExplorer: A repository-scale resource for exploration of protein detection in public mass spectrometry data sets. J. Proteome Res 2018, 17 (12), 4227–4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Bell P; Solis N; Kizhakkedathu J; Matthew I; Overduin B Proteomic and N-terminomic TAILS analyses of human alveolar bone proteins: Improved protein extraction methodology and LysargiNase digestion increases proteome coverage missing protein identification. J. Proteome Res 2019. [DOI] [PubMed] [Google Scholar]
- (29).Abbey SR; Eckhard U; Solis N; Marino G; Matthew I; Overall CM The human odontoblast cell layer and dental pulp proteomes and N-terminomes. J. Dent. Res 2018, 97 (3), 338–346. [DOI] [PubMed] [Google Scholar]
- (30).Quancard J; Klein T; Fung SY; Renatus M; Hughes N; Israel L; Priatel JJ; Kang S; Blank MA; Viner RI; Blank J; Schlapbach A; Erbel P; Kizhakkedathu J; Villard F; Hersperger R; Turvey SE; Eder J; Bornancin F; Overall CM An allosteric MALT1 inhibitor is a molecular corrector rescuing function in an immunodeficient patient. Nat. Chem. Biol 2019, 15 (3), 304–313. [DOI] [PubMed] [Google Scholar]
- (31).Zhang C; Wei X; Omenn GS; Zhang Y Structure and protein interaction-based gene ontology annotations reveal likely functions of uncharacterized proteins of human chromosome 17. J. Proteome Res 2018, 17 (12), 4186–4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Zhang C; Lane L; Omenn GS; Zhang Y A blinded testing of function annotation for uPE1 proteins by the I-TASSER/COFACTOR pipeline using the 2018−2019 additions to neXtProt and CAFA3 challenge. J. Proteome Res 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Desiere F; Deutsch EW; Nesvizhskii AI; Mallick P; King NL; Eng JK; Aderem A; Boyle R; Brunner E; Donohoe S; Fausto N; Hafen E; Hood L; Katze MG; Kennedy KA; Kregenow F; Lee H; Lin B; Martin D; Ranish JA; Rawlings DJ; Samelson LE; Shiio Y; Watts JD; Wollscheid B; Wright ME; Yan W; Yang L; Yi EC; Zhang H; Aebersold R Integration with the human genome of peptide sequences obtained by high-throughput mass spectrometry. Genome Biol. 2004, 6 (1), R9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Deutsch EW; Sun Z; Campbell D; Kusebauch U; Chu CS; Mendoza L; Shteynberg D; Omenn GS; Moritz RL State of the human proteome in 2014/2015 as viewed through PeptideAtlas: enhancing accuracy and coverage through the AtlasProphet. J. Proteome Res 2015, 14 (9), 3461–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Moriya Y; Kawano S; Okuda S; Watanabe Y; Matsumoto M; Takami T; Kobayashi D; Yamanouchi Y; Araki N; Yoshizawa AC; Tabata T; Iwasaki M; Sugiyama N; Tanaka S; Goto S; Ishihama Y The jPOST environment: an integrated proteomics data repository and database. Nucleic Acids Res. 2019, 47 (D1), D1218–d1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Ma J; Chen T; Wu S; Yang C; Bai M; Shu K; Li K; Zhang G; Jin Z; He F; Hermjakob H; Zhu Y iProX: an integrated proteome resource. Nucleic Acids Res. 2019, 47 (D1), D1211–d1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Muller MM; Lehmann R; Klassert TE; Reifenstein S; Conrad T; Moore C; Kuhn A; Behnert A; Guthke R; Driesch D; Slevogt H Global analysis of glycoproteins identifies markers of endotoxin tolerant monocytes and GPR84 as a modulator of TNFalpha expression. Sci. Rep 2017, 7 (1), 838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Deeb SJ; Cox J; Schmidt-Supprian M; Mann M N-linked glycosylation enrichment for in-depth cell surface proteomics of diffuse large B-cell lymphoma subtypes. Mol. Cell. Proteomics 2014, 13 (1), 240–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Bassani-Sternberg M; Pletscher-Frankild S; Jensen LJ; Mann M Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol. Cell. Proteomics 2015, 14 (3), 658–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Bassani-Sternberg M; Braunlein E; Klar R; Engleitner T; Sinitcyn P; Audehm S; Straub M; Weber J; Slotta-Huspenina J; Specht K; Martignoni ME; Werner A; Hein R; D, H. B.; Peschel C; Rad R; Cox J; Mann M; Krackhardt AM Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun 2016, 7, 13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Kulak NA; Geyer PE; Mann M Loss-less nano-fractionator for high sensitivity, high coverage proteomics. Mol. Cell. Proteomics 2017, 16 (4), 694–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Klaeger S; Heinzlmeir S; Wilhelm M; Polzer H; Vick B; Koenig PA; Reinecke M; Ruprecht B; Petzoldt S; Meng C; Zecha J; Reiter K; Qiao H; Helm D; Koch H; Schoof M; Canevari G; Casale E; Depaolini SR; Feuchtinger A; Wu Z; Schmidt T; Rueckert L; Becker W; Huenges J; Garz AK; Gohlke BO; Zolg DP; Kayser G; Vooder T; Preissner R; Hahne H; Tonisson N; Kramer K; Gotze K; Bassermann F; Schlegl J; Ehrlich HC; Aiche S; Walch A; Greif PA; Schneider S; Felder ER; Ruland J; Medard G; Jeremias I; Spiekermann K; Kuster B The target landscape of clinical kinase drugs. Science 2017, 358 (6367), eaan4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Doll S; Dressen M; Geyer PE; Itzhak DN; Braun C; Doppler SA; Meier F; Deutsch MA; Lahm H; Lange R; Krane M; Mann M Region and cell-type resolved quantitative proteomic map of the human heart. Nat. Commun 2017, 8 (1), 1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Deutsch EW; Lane L; Overall CM; Baker MS; Pineau C; Mortiz RL; Bandeira N; Corrales F; Orchard S; Van Eyk J; Paik Y-K; Weintraub ST; Vandenbrouck Y; Omenn GS Human Proteome Project Mass Spectrometry Data Interpretation Guidelines 3.0. J. Proteome Res 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Gillet LC; Navarro P; Tate S; Rost H; Selevsek N; Reiter L; Bonner R; Aebersold R Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics 2012, 11 (6), O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Deutsch EW; Csordas A; Sun Z; Jarnuczak A; Perez-Riverol Y; Ternent T; Campbell DS; Bernal-Llinares M; Okuda S; Kawano S; Moritz RL; Carver JJ; Wang M; Ishihama Y; Bandeira N; Hermjakob H; Vizcaino JA The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017, 45 (D1), D1100–d1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Vizcaino JA; Deutsch EW; Wang R; Csordas A; Reisinger F; Rios D; Dianes JA; Sun Z; Farrah T; Bandeira N; Binz PA; Xenarios I; Eisenacher M; Mayer G; Gatto L; Campos A; Chalkley RJ; Kraus HJ; Albar JP; Martinez-Bartolome S; Apweiler R; Omenn GS; Martens L; Jones AR; Hermjakob H ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol 2014, 32 (3), 223–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Martens L; Hermjakob H; Jones P; Adamski M; Taylor C; States D; Gevaert K; Vandekerckhove J; Apweiler R PRIDE: the proteomics identifications database. Proteomics 2005, 5 (13), 3537–45. [DOI] [PubMed] [Google Scholar]
- (49).Perez-Riverol Y; Csordas A; Bai J; Bernal-Llinares M; Hewapathirana S; Kundu DJ; Inuganti A; Griss J; Mayer G; Eisenacher M; Perez E; Uszkoreit J; Pfeuffer J; Sachsenberg T; Yilmaz S; Tiwary S; Cox J; Audain E; Walzer M; Jarnuczak AF; Ternent T; Brazma A; Vizcaino JA The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019, 47 (D1), D442–d450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Sharma V; Eckels J; Schilling B; Ludwig C; Jaffe JD; MacCoss MJ; MacLean B Panorama public: A public repository for quantitative data sets processed in Skyline. Mol. Cell. Proteomics 2018, 17 (6), 1239–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Jiang Y; Sun A; Zhao Y; Ying W; Sun H; Yang X; Xing B; Sun W; Ren L; Hu B; Li C; Zhang L; Qin G; Zhang M; Chen N; Zhang M; Huang Y; Zhou J; Zhao Y; Liu M; Zhu X; Qiu Y; Sun Y; Huang C; Yan M; Wang M; Liu W; Tian F; Xu H; Zhou J; Wu Z; Shi T; Zhu W; Qin J; Xie L; Fan J; Qian X; He F Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature 2019, 567 (7747), 257–261. [DOI] [PubMed] [Google Scholar]
- (52).Lam MP; Venkatraman V; Xing Y; Lau E; Cao Q; Ng DC; Su AI; Ge J; Van Eyk JE; Ping P Data-driven approach to determine popular proteins for targeted proteomics translation of six organ systems. J. Proteome Res 2016, 15 (11), 4126–4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Yu KH; Lee TM; Wang CS; Chen YJ; Ré C; Kou SC; Chiang JH; Kohane IS; Snyder M Systematic protein prioritization for targeted proteomics studies through literature mining. J. Proteome Res 2018, 17 (4), 1383–1396. [DOI] [PubMed] [Google Scholar]
- (54).Yu KH; Lee TM; Chen YJ; Re C; Kou SC; Chiang JH; Snyder M; Kohane IS A cloud-based metabolite and chemical prioritization system for the biology/disease-driven human proteome project. J. Proteome Res 2018, 17 (12), 4345–4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Laferriere F; Maniecka Z; Perez-Berlanga M; Hruska-Plochan M; Gilhespy L; Hock EM; Wagner U; Afroz T; Boersema PJ; Barmettler G; Foti SC; Asi YT; Isaacs AM; Al-Amoudi A; Lewis A; Stahlberg H; Ravits J; De Giorgi F; Ichas F; Bezard E; Picotti P; Lashley T; Polymenidou M TDP-43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting disease progression rates. Nat. Neurosci 2019, 22 (1), 65–77. [DOI] [PubMed] [Google Scholar]
- (56).Herrington DM; Mao C; Parker SJ; Fu Z; Yu G; Chen L; Venkatraman V; Fu Y; Wang Y; Howard TD; Jun G; Zhao CF; Liu Y; Saylor G; Spivia WR; Athas GB; Troxclair D; Hixson JE; Vander Heide RS; Wang Y; Van Eyk JE Proteomic architecture of human coronary and aortic atherosclerosis. Circulation 2018, 137 (25), 2741–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Hirao Y; Saito S; Fujinaka H; Miyazaki S; Xu B; Quadery AF; Elguoshy A; Yamamoto K; Yamamoto T Proteome profiling of diabetic mellitus patient urine for discovery of biomarkers by comprehensive MS-based proteomics. Proteomes 2018, 6 (1), 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Belczacka I; Latosinska A; Siwy J; Metzger J; Merseburger AS; Mischak H; Vlahou A; Frantzi M; Jankowski V Urinary CE-MS peptide marker pattern for detection of solid tumors. Sci. Rep 2018, 8 (1), 5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Emilsson V; Ilkov M; Lamb JR; Finkel N; Gudmundsson EF; Pitts R; Hoover H; Gudmundsdottir V; Horman SR; Aspelund T; Shu L; Trifonov V; Sigurdsson S; Manolescu A; Zhu J; Olafsson O; Jakobsdottir J; Lesley SA; To J; Zhang J; Harris TB; Launer LJ; Zhang B; Eiriksdottir G; Yang X; Orth AP; Jennings LL; Gudnason V Co-regulatory networks of human serum proteins link genetics to disease. Science 2018, 361 (6404), 769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Cao P; Yang A; Wang R; Xia X; Zhai Y; Li Y; Yang F; Cui Y; Xie W; Liu Y; Liu T; Jia W; Jiang Z; Li Z; Han Y; Gao C; Song Q; Xie B; Zhang L; Zhang H; Zhang J; Shen X; Yuan Y; Yu F; Wang Y; Xu J; Ma Y; Mo Z; Yu W; He F; Zhou G Germline duplication of SNORA18L5 increases risk for HBV-related hepatocellular carcinoma by altering localization of ribosomal proteins and decreasing levels of p53. Gastroenterology 2018, 155 (2), 542–556. [DOI] [PubMed] [Google Scholar]
- (61).Wei J; Yuan Y; Chen L; Xu Y; Zhang Y; Wang Y; Yang Y; Peek CB; Diebold L; Yang Y; Gao B; Jin C; Melo-Cardenas J; Chandel NS; Zhang DD; Pan H; Zhang K; Wang J; He F; Fang D ER-associated ubiquitin ligase HRD1 programs liver metabolism by targeting multiple metabolic enzymes. Nat. Commun. 2018, 9 (1), 3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Fert-Bober J; Murray CI; Parker SJ; Van Eyk JE Precision profiling of the cardiovascular post-translationally modified proteome: Where there is a will, there is a way. Circ. Res 2018, 122 (9), 1221–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Wang J; Choi H; Chung NC; Cao Q; Ng DCM; Mirza B; Scruggs SB; Wang D; Garlid AO; Ping P Integrated dissection of cysteine oxidative post-translational modification proteome during cardiac hypertrophy. J. Proteome Res 2018, 17 (12), 4243–4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Ren L; Li C; Wang Y; Teng Y; Sun H; Xing B; Yang X; Jiang Y; He F In vivo phosphoproteome analysis reveals kinome reprogramming in hepatocellular carcinoma. Mol. Cell. Proteomics 2018, 17 (6), 1067–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Song G; Chen L; Zhang B; Song Q; Yu Y; Moore C; Wang TL; Shih IM; Zhang H; Chan DW; Zhang Z; Zhu H Proteome-wide tyrosine phosphorylation analysis reveals dysregulated signaling pathways in ovarian tumors. Mol. Cell. Proteomics 2019, 18 (3), 448–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Sajic T; Liu Y; Arvaniti E; Surinova S; Williams EG; Schiess R; Huttenhain R; Sethi A; Pan S; Brentnall TA; Chen R; Blattmann P; Friedrich B; Nimeus E; Malander S; Omlin A; Gillessen S; Claassen M; Aebersold R Similarities and differences of blood N-Glycoproteins in five solid carcinomas at localized clinical stage analyzed by SWATH-MS. Cell Rep. 2018, 23 (9), 2819–2831. [DOI] [PubMed] [Google Scholar]
- (67).Murray LA; Sheng X; Cristea IM Orchestration of protein acetylation as a toggle for cellular defense and virus replication. Nat. Commun 2018, 9 (1), 4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Di Francesco L; Di Girolamo F; Mennini M; Masotti A; Salvatori G; Rigon G; Signore F; Pietrantoni E; Scapaticci M; Lante I; Goffredo BM; Mazzina O; Elbousify AI; Roncada P; Dotta A; Fiocchi A; Putignani L A MALDI-TOF MS approach for mammalian, human, and formula milks profiling. Nutrients 2018, 10 (9), 1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Piazza I; Kochanowski K; Cappelletti V; Fuhrer T; Noor E; Sauer U; Picotti P A map of protein-metabolite interactions reveals principles of chemical communication. Cell 2018, 172 (1−2), 358–372. [DOI] [PubMed] [Google Scholar]
- (70).Barkovits K; Linden A; Galozzi S; Schilde L; Pacharra S; Mollenhauer B; Stoepel N; Steinbach S; May C; Uszkoreit J; Eisenacher M; Marcus K Characterization of cerebrospinal fluid via data-independent acquisition mass spectrometry. J. Proteome Res 2018, 17 (10), 3418–3430. [DOI] [PubMed] [Google Scholar]
- (71).Clark DJ; Hu Y; Bocik W; Chen L; Schnaubelt M; Roberts R; Shah P; Whiteley G; Zhang H Evaluation of NCI-7 cell line panel as a reference material for clinical proteomics. J. Proteome Res 2018, 17 (6), 2205–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Liao B; Ning Z; Cheng K; Zhang X; Li L; Mayne J; Figeys D iMetaLab 1.0: a web platform for metaproteomics data analysis. Bioinformatics 2018, 34 (22), 3954–3956. [DOI] [PubMed] [Google Scholar]
- (73).Lill JR; van Veelen PA; Tenzer S; Admon A; Caron E; Elias JE; Heck AJR; Marcilla M; Marino F; Muller M; Peters B; Purcell A; Sette A; Sturm T; Ternette N; Vizcaino JA; Bassani-Sternberg M Minimal information about an immunopeptidomics experiment (MIAIPE). Proteomics 2018, 18 (12), No. e1800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Kopylov AT; Ponomarenko EA; Ilgisonis EV; Pyatnitskiy MA; Lisitsa AV; Poverennaya EV; Kiseleva OI; Farafonova TE; Tikhonova OV; Zavialova MG; Novikova SE; Moshkovskii SA; Radko SP; Morukov BV; Grigoriev AI; Paik YK; Salekdeh GH; Urbani A; Zgoda VG; Archakov AI 200+ protein concentrations in healthy human blood plasma: targeted quantitative SRM SIS screening of Chromosomes 18, 13, Y, and the mitochondrial chromosome encoded proteome. J. Proteome Res 2018, 18 (1), 120–129. [DOI] [PubMed] [Google Scholar]
- (75).Uhlen M; Fagerberg L; Hallstrom BM; Lindskog C; Oksvold P; Mardinoglu A; Sivertsson A; Kampf C; Sjostedt E; Asplund A; Olsson I; Edlund K; Lundberg E; Navani S; Szigyarto CA; Odeberg J; Djureinovic D; Takanen JO; Hober S; Alm T; Edqvist PH; Berling H; Tegel H; Mulder J; Rockberg J; Nilsson P; Schwenk JM; Hamsten M; von Feilitzen K; Forsberg M; Persson L; Johansson F; Zwahlen M; von Heijne G; Nielsen J; Ponten F Proteomics. Tissue-based map of the human proteome. Science 2015, 347 (6220), 1260419. [DOI] [PubMed] [Google Scholar]
- (76).Uhlen M; Zhang C; Lee S; Sjostedt E; Fagerberg L; Bidkhori G; Benfeitas R; Arif M; Liu Z; Edfors F; Sanli K; von Feilitzen K; Oksvold P; Lundberg E; Hober S; Nilsson P; Mattsson J; Schwenk JM; Brunnstrom H; Glimelius B; Sjoblom T; Edqvist PH; Djureinovic D; Micke P; Lindskog C; Mardinoglu A; Ponten F A pathology atlas of the human cancer transcriptome. Science 2017, 357 (6352), eaan2507. [DOI] [PubMed] [Google Scholar]
- (77).Thul PJ; Akesson L; Wiking M; Mahdessian D; Geladaki A; Ait Blal H; Alm T; Asplund A; Bjork L; Breckels LM; Backstrom A; Danielsson F; Fagerberg L; Fall J; Gatto L; Gnann C; Hober S; Hjelmare M; Johansson F; Lee S; Lindskog C; Mulder J; Mulvey CM; Nilsson P; Oksvold P; Rockberg J; Schutten R; Schwenk JM; Sivertsson A; Sjostedt E; Skogs M; Stadler C; Sullivan DP; Tegel H; Winsnes C; Zhang C; Zwahlen M; Mardinoglu A; Ponten F; von Feilitzen K; Lilley KS; Uhlen M; Lundberg E A subcellular map of the human proteome. Science 2017, 356 (6340), eaal3321. [DOI] [PubMed] [Google Scholar]
- (78).Regev A; Teichmann SA; Lander ES; Amit I; Benoist C; Birney E; Bodenmiller B; Campbell P; Carninci P; Clatworthy M; Clevers H; Deplancke B; Dunham I; Eberwine J; Eils R; Enard W; Farmer A; Fugger L; Gottgens B; Hacohen N; Haniffa M; Hemberg M; Kim S; Klenerman P; Kriegstein A; Lein E; Linnarsson S; Lundberg E; Lundeberg J; Majumder P; Marioni JC; Merad M; Mhlanga M; Nawijn M; Netea M; Nolan G; Pe’er D; Phillipakis A; Ponting CP; Quake S; Reik W; Rozenblatt-Rosen O; Sanes J; Satija R; Schumacher TN; Shalek A; Shapiro E; Sharma P; Shin JW; Stegle O; Stratton M; Stubbington MJT; Theis FJ; Uhlen M; van Oudenaarden A; Wagner A; Watt F; Weissman J; Wold B; Xavier R; Yosef N The Human Cell Atlas. eLife 2017, DOI: 10.7554/eLife.27041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Sullivan DP; Winsnes CF; Akesson L; Hjelmare M; Wiking M; Schutten R; Campbell L; Leifsson H; Rhodes S; Nordgren A; Smith K; Revaz B; Finnbogason B; Szantner A; Lundberg E Deep learning is combined with massive-scale citizen science to improve large-scale image classification. Nat. Biotechnol 2018, 36 (9), 820–828. [DOI] [PubMed] [Google Scholar]
- (80).Edfors F; Hober A; Linderback K; Maddalo G; Azimi A; Sivertsson A; Tegel H; Hober S; Szigyarto CA; Fagerberg L; von Feilitzen K; Oksvold P; Lindskog C; Forsstrom B; Uhlen M Enhanced validation of antibodies for research applications. Nat. Commun 2018, 9 (1), 4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Sjostedt E; Sivertsson A; Hikmet Noraddin F; Katona B; Nasstrom A; Vuu J; Kesti D; Oksvold P; Edqvist PH; Olsson I; Uhlen M; Lindskog C Integration of transcriptomics and antibody-based proteomics for exploration of proteins expressed in specialized tissues. J. Proteome Res 2018, 17 (12), 4127–4137. [DOI] [PubMed] [Google Scholar]
- (82).Fredolini C; Bystrom S; Sanchez-Rivera L; Ioannou M; Tamburro D; Ponten F; Branca RM; Nilsson P; Lehtio J; Schwenk JM Systematic assessment of antibody selectivity in plasma based on a resource of enrichment profiles. Sci. Rep 2019, 9 (1), 8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Wang D; Eraslan B; Wieland T; Hallstrom B; Hopf T; Zolg DP; Zecha J; Asplund A; Li LH; Meng C; Frejno M; Schmidt T; Schnatbaum K; Wilhelm M; Ponten F; Uhlen M; Gagneur J; Hahne H; Kuster B A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol 2019, 15 (2), No. e8503. [DOI] [PMC free article] [PubMed] [Google Scholar]