

Summary
Metabolic pathways dynamically regulate tissue development and maintenance. However, the mechanisms that govern the metabolic adaptation of stem or progenitor cells to their local niche are poorly understood. Here, we define the transcription factor PRDM16 as a region-specific regulator of intestinal metabolism and epithelial renewal. PRDM16 is selectively expressed in the upper intestine, with enrichment in crypt-resident progenitor cells. Acute Prdm16 deletion in mice triggered progenitor apoptosis, leading to diminished epithelial differentiation and severe intestinal atrophy. Genomic and metabolic analyses showed that PRDM16 transcriptionally controls fatty acid oxidation (FAO) in crypts. Expression of this PRDM16-driven FAO program was highest in the upper small intestine and declined distally. Accordingly, deletion of Prdm16 or inhibition of FAO selectively impaired the development and maintenance of upper intestinal enteroids, and these effects were rescued by acetate treatment. Collectively, these data reveal that regionally specified metabolic programs regulate intestinal maintenance.
Graphical Abstract
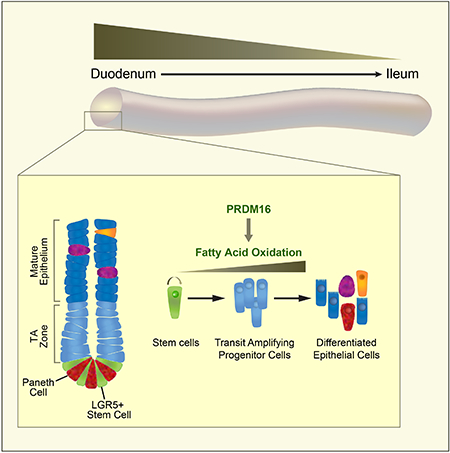
Introduction
The small intestinal epithelium is organized into repeating crypt-villus units. The villi are finger-shaped projections that are specialized for nutrient absorption. The crypt, located at the base of each villus, contains proliferative stem cells responsible for epithelium maintenance (Barker et al., 2007). Stem cells produce transit-amplifying (TA) progenitor cells that divide and migrate out of the crypt and up the villus as they differentiate into absorptive or secretory epithelial cells.
The villus epithelium undergoes rapid renewal by stem cells every few days. The constitutively high levels of intestinal stem and progenitor cell proliferation is supported by specific metabolic pathways (Folmes et al., 2012; Potten and Loeffler, 1990; Wang et al., 2018; Wei et al., 2018). In addition to providing energy and building blocks, cellular metabolism influences chromatin structure and gene expression through a variety of mechanisms (Ito and Ito, 2016). For example, nutritional status and metabolic pathway activity regulate the availability of substrates for DNA and histone modifications (Folmes et al., 2012).
Recent studies have begun to elucidate the importance of metabolism in regulating intestinal stem cells. In particular, pyruvate oxidation in stem cells drives pro-differentiation signaling (Rodriguez-Colman et al., 2017; Schell et al., 2017). Additionally, fatty acid oxidation (FAO) is required for proper stem cell function (Mihaylova et al., 2018). The metabolic behavior of intestinal stem and progenitor cells is modulated by nutritional status, including fasting and high fat diet, accompanied by changes in cell survival, rates of proliferation and tumor-forming capacity (Beyaz et al., 2016; Mihaylova et al., 2018; Wang et al., 2018). However, relatively little is known about the transcriptional pathways that specify the metabolic features of intestinal stem and progenitor cells.
PRDM16 is a transcription factor that drives high levels of oxidative metabolism and mitochondrial respiration in brown fat cells (Cohen et al., 2014; Ohno et al., 2012; Seale et al., 2008; Seale et al., 2011; Wang et al., 2019). PRDM16 enhances the transcriptional function of Peroxisome proliferator-activated receptors (PPARs), including PPARα and PPARγ, to promote metabolic gene transcription in fat cells (Seale et al., 2008). Intriguingly, PRDM16 also regulates the activity of certain stem cell populations, including hematopoietic and neural stem cells (Aguilo et al., 2011; Chuikov et al., 2010; Cohen et al., 2014; Inoue et al., 2017; Shimada et al., 2017).
Here we show that PRDM16 is essential for the maintenance of the small intestinal epithelium. PRDM16 expression is enriched in crypt-resident progenitor cells, selectively in the upper small intestine. Deletion of Prdm16 causes apoptosis in progenitor cells and leads to a diminished production of differentiated progeny. PRDM16 regulates intestinal renewal, at least in part, by stimulating FAO. Notably, the PRDM16-driven FAO program is highest in the proximal small intestine and declines distally. The development and maintenance of enteroids generated from upper but not lower regions of the small intestine rely on PRDM16 and FAO. Together, these results demonstrate that upper intestinal renewal depends on a regionally-specified program of FAO.
Results
Prdm16 is required for small intestinal maintenance
PRDM16 controls brown fat development and regulates hematopoietic and neural stem cell activity (Aguilo et al., 2011; Chuikov et al., 2010; Seale et al., 2008). However, the role of PRDM16 in other adult tissues is largely unknown. We assessed the effects of Prdm16 deletion in 6-week-old mice using a tamoxifen (tmx)-inducible Cre (R26RCre-ERT2; Prdm16loxP/loxP). Deletion of Prdm16 induced a rapid-onset wasting phenotype, including weight loss and diarrhea, with mutant mice dying in ~7-10 days (Fig. 1A). Necropsy revealed gross changes in the small intestine of mutant animals, which were noticeably pale, distended and filled with watery, bile colored feces (Fig. 1B). Prdm16 mRNA was decreased by >90% and PRDM16 protein was undetectable in the duodenum by 3 days post tmx treatment (Fig. S1A–B). By day 7, the small intestines of Prdm16-deficient animals were approximately two-thirds the length of sibling controls (Fig. 1C). H&E staining showed that the duodenum of Prdm16-mutant mice contained highly disorganized columnar epithelial cells and underwent progressive villus shortening (Fig. 1D, S1C).
Figure 1: Prdm16 is required for small intestine maintenance.
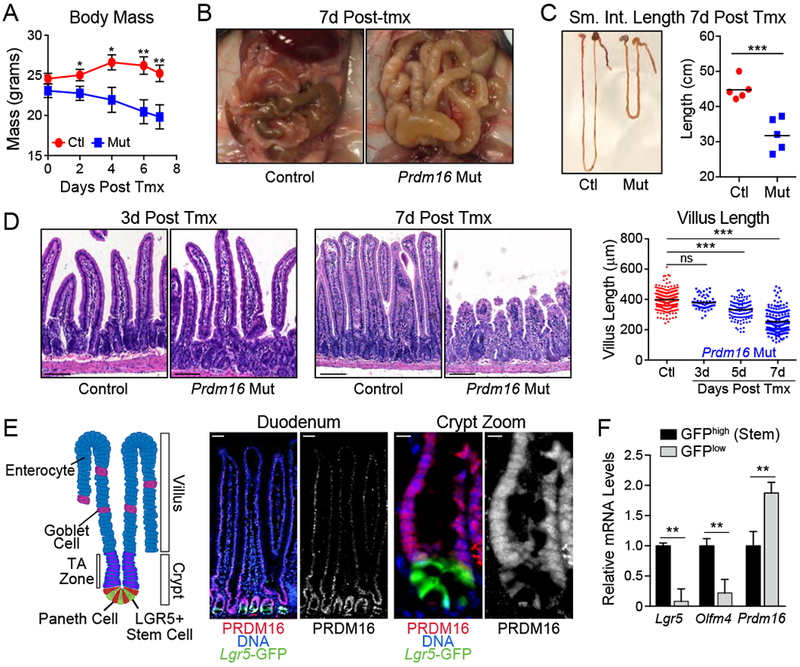
A-D) Phenotypes of Control (Prdm16loxP/loxP) and Prdm16 mutant (R26RCreERT2; Prdm16loxP/loxP) animals after injection with tmx. A) Body mass following tmx injection. n=4 mice/group. B) Gross appearance of small intestines at 7d post tmx. C) Small intestine length at 7d post tmx. D) Hematoxylin/eosin histology of duodenum at 3d and 7d post tmx (left) with quantification of villus length at 3d, 5d, and 7d (right). n=4 mice/group.
E) (left) Schematic of intestinal villus/crypt structure and cell identities. TA Zone=Transit Amplifying Zone. (center, right) Duodenal sections stained for PRDM16 (red) and Lgr5-GFP (green). full villus (center), crypt compartment (right). DNA: DAPI, blue. Sub-panels: [left, overlay] [right] PRDM16 alone (white).
F) mRNA levels of Prdm16 and stem cell markers in FACS isolated Lgr5GFP-low or -high epithelial cells from duodenal crypts. n=3 mice.
All panels show mean ± SEM, *p<0.05, **p<0.01, ***p<0.001. Scale Bars: 100 μm (D,E Left), 10 μm (E Right).
We assessed barrier function by gavaging animals with FITC-dextran and measuring its appearance in the serum. Prdm16-deficient mice had markedly increased blood FITC levels, along with increased colony forming units when whole blood was cultured (Fig. S1D,E). Prdm16-deficient mice also had elevated levels of blood cytokines (IL-10, TNFα, Eotaxin), associated with the onset of sepsis (Fig. S1F) (Shrum et al., 2014).
Prdm16 is highly expressed in small intestinal crypts
Analysis of Prdm16 expression across a panel of adult mouse tissues revealed the highest expression levels in the small intestine and stomach (Fig. S1G). PRDM16 mRNA levels were also at their highest in human duodenum and stomach (Fig. S1H) (https://www.proteinatlas.org) (Uhlen et al., 2015). Immunofluorescence staining of mouse duodenum showed that PRDM16 protein was present at higher levels in crypts relative to villi, with enrichment in TA zone cells, localized directly above Lgr5-GFP+ stem cells at the crypt base (Fig. 1E). This staining was lost in Prdm16-mutant crypts, validating the antibody specificity (Fig. S1I). Consistent with the immunostaining results, Prdm16 mRNA levels were higher in flow-sorted Lgr5-GFPlow cells than in Lgr5-GFPhigh stem cells from crypts (Fig. 1F).
Prdm16 promotes the survival and proliferation of crypt-resident progenitor cells
We next examined the effect of Prdm16-deletion on crypt cell fate. At 5 days post tmx, Prdm16 mutant duodenal crypts contained reduced numbers of proliferating cells, marked by Ki67 expression (Fig. 2A). Mutant crypts also displayed a prominent increase in the number of Cleaved Caspase-3+ apoptotic cells (Fig. 2B). Most apoptotic cells were located in the TA zone and did not correspond to Lgr5-GFPhigh stem cells at the crypt base (Fig. 2B). Crypts from control and mutant mice contained equivalent numbers of Lgr5-GFPhigh stem cells at 5 days post tmx treatment, a time point when villus architecture is disrupted and epithelial cell number is decreased (Fig. 2C,D). Flow cytometry analysis also showed that control and Prdm16-mutant crypts contained similar proportions of Lgr5-GFP+ stem cells (Fig. S2A). Control and mutant crypts also expressed equivalent mRNA levels of Lgr5 and Ascl2, markers of crypt base columnar (CBC) stem cells (Fig. 2E). By contrast, expression of the stem cell marker Olfm4 was downregulated by Prdm16-deletion (Fig. 2E). Marker genes of the reserve or “+4” stem cells, Bmi1 and Hopx, were upregulated in Prdm16-mutant crypts (Fig. 2E) (Takeda et al., 2011; Yan et al., 2012).
Figure 2: Prdm16 deletion induces crypt apoptosis and disrupts cell type composition.
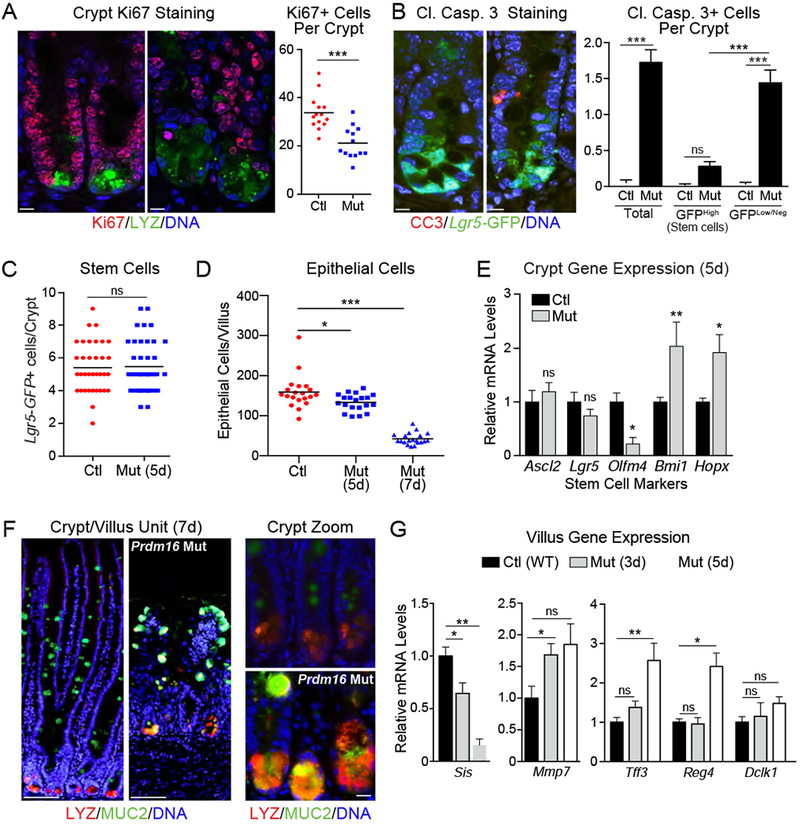
A) (left) Immunofluorescence staining of Ki67+ (red) proliferative cells in crypts of Control (Prdm16loxP/loxP) and Prdm16 mutant (R26RCreERT2; Prdm16loxP/loxP) mice 5d after tmx injection. Lysozyme (LYZ; green), DNA (DAPI, blue). (right) Quantification of Ki67+ cells/crypt. n=13 to 14 crypts, 3 mice per group.
B) (left) Immunofluorescence staining of Cleaved Caspase 3 (CC3; red) in duodenal crypts from Control (corn oil-treated) and Prdm16 mutant (Mut; tmx-treated) R26RCreERT2; Lgr5GFP/CreERT2; Prdm16loxP/loxP mice (at 5d). Lgr5-GFP (green), DNA (DAPI, blue). (right) Quantification of CC3+ stem (GFPhi) or TA (GFPlow/neg) cells. n=88-91 crypts from 3 mice.
C) Quantification of Lgr5-GFP+ stem cells in mice from (B). n=35-41 crypts from 3 mice.
D-G) Control (Prdm16loxP/loxP) and Prdm16 mutant (R26RCreERT2; Prdm16loxP/loxP) (Mut) mice were injected with tmx. D) Counts of epithelial cells/villus at 5d and 7d post tmx. n=20 villi from 3 mice. E) mRNA levels of stem cell markers in isolated crypts 5d post tmx. n=5-7 mice. F) Immunofluorescence in villi and crypts at 7d post tmx. Full villus (left), magnified crypt (right). Goblet cell marker (Mucin 2; MUC2, green), Paneth cell marker (Lysozyme; LYZ, red), DNA (DAPI, blue). arrowheads = MUC2/LYZ double-positive cells. G) mRNA levels of absorptive (left) and secretory cell (right) markers in isolated villi or crypts (for Mmp7) at 3d and 5d post tmx. n= 4-7 mice.
All panels show mean ± SEM, *p<0.05, **p<0.01, ***p<0.001. Scale bars: 10 μm (A,B, F right); 100 μm (F left).
Prdm16-deletion skews intestinal cell type composition
Immunostaining revealed that Prdm16 mutant crypts contained anomalous granular cells co-expressing the Paneth cell marker Lysozyme (LYZ) and the goblet cell marker Mucin 2 (MUC2) (Fig. 2F). The induction of cells co-expressing goblet and Paneth cell markers has been linked to inhibition of the Notch pathway (VanDussen et al., 2012). However, Prdm16-deletion did not reduce expression of the Notch intracellular domain (NICD) or canonical Notch pathway components in crypts (Fig. S2B, C).
Intestinal crypt progenitors normally migrate up the villi, giving rise to differentiated epithelium, including absorptive enterocytes and secretory cells. The Prdm16-mutant villi and crypts expressed higher levels of many secretory cell marker genes, including Tff3 (goblet cells), Mmp7 (Paneth cells) and Reg4 (enteroendocrine cells) along with dramatically reduced levels of absorptive enterocyte markers Sis and Lct (Fig. 2G, Fig. S2D). Prdm16-deletion did not significantly affect expression of tuft cell markers, including Dclk1 (Fig. 2G, S2D). Consistent with these mRNA expression changes, immunofluorescence studies showed that Prdm16 mutant villi contained more CHGA+ enteroendocrine and MUC2+ goblet cells (Fig. S2E,F).
Stem cell-selective Prdm16 deletion reduces the development of mature epithelial cells
To further investigate the role of PRDM16 in regulating intestinal renewal, we generated a stem cell-specific Prdm16 knockout mouse model using the Lgr5GFP/CreERT2 (Prdm16ΔLgr5) driver. This Cre driver has a mosaic expression pattern (Barker et al., 2007; Sato et al., 2009), allowing us to examine the role of PRDM16 in the turnover of intestinal epithelium without the confounding effect of whole organ failure. As anticipated, Prdm16ΔLgr5 mice displayed a less severe intestinal phenotype than global mutant mice (Fig. S3A). We confirmed that PRDM16 protein expression was absent in a subset of crypts from these mice (Fig. 3A). Similar to results in the global mutant animals, Prdm16-deletion in Lgr5+ cells did not affect stem cell number and led to elevated crypt cell apoptosis and the induction of abnormal MUC2+;LYZ+ cells in crypts (Fig. 3B–D). Neither the stem cell specific nor global Prdm16 mutant mice had significantly increased numbers of apoptotic cells in villi (Fig. S3B).
Figure 3: Loss of Prdm16 in stem cells leads to TA cell apoptosis and diminished epithelial differentiation.
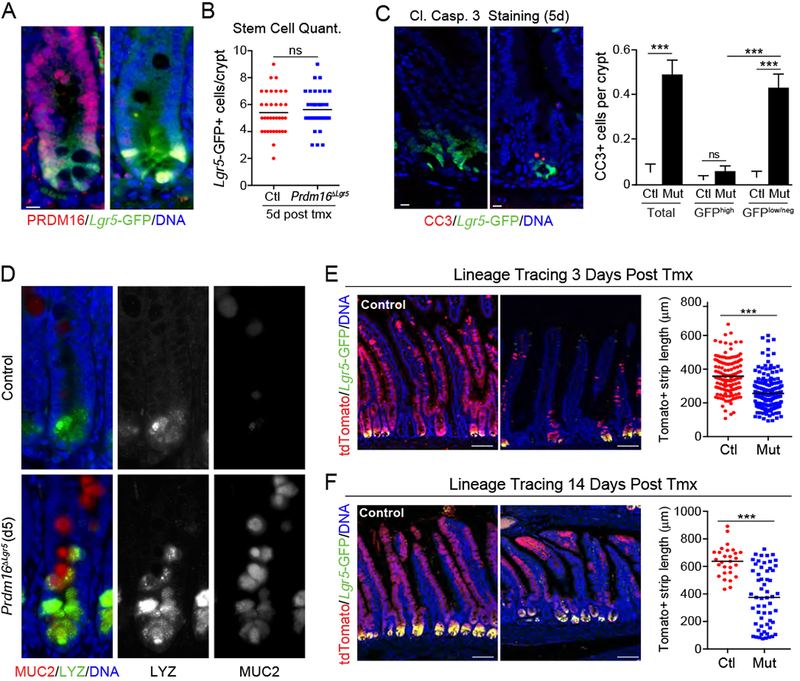
A-D) Lgr5GFP/Cre-ERT2;Prdm16loxP/loxP mice were treated with tmx (mutant; Prdm16ΔLgr5) or vehicle (corn oil; Control). A) Immunostaining of PRDM16 (red) and Lgr5-GFP (green) in duodenal crypts at 7d post tmx. DNA (DAPI, blue). B) Quantification of Lgr5GFP+ stem cells per crypt at 5d. n=35 crypts from 3 mice. C) (left) Duodenal crypts stained for cleaved Caspase 3 (CC3; red) at 5d. (right) Quantification of CC3+ stem (GFPhi) or TA (GFPlow/neg) cells using an intensity threshold (Thermal LUT in Image J). Controls were shared with experiment in Figure 2B. n=91-188 crypts from 3 mice per group. D) Immunostaining of Mucin2 (MUC2; red) and Lysozyme (LYZ; green) in duodenal crypts at 5d. Arrowheads indicate MUC2/LYZ double-positive cells. n=3 mice.
E-F) Lineage tracing of Lgr5+ stem cell progeny in Control (Prdm16loxP/+) and Prdm16 mutant (Prdm16loxP/oxP; Prdm16ΔLgr5) reporter mice (Lgr5GFP/Cre-ERT2;R26Rlox-stop-lox-tdTomato). Marked epithelial strip length was measured for continuous streaks of tdTomato+ (Red) epithelial cells ascending the villi at: E) 3d [n=132-190 villi from 3 mice] or F) 14d [n=28-58 villi from 3 mice] post tmx. Lgr5-GFP (green), DNA (DAPI, blue).
All panels show mean ± SEM, *p<0.05, **p<0.01, ***p<0.001. Scale bars: 10 μm (A, C, ; (D) 100 μm (E, F).
To examine the fate of control and Prdm16-mutant Lgr5+ stem cells, we integrated a Cre-inducible tdTomato reporter gene into the Prdm16ΔLgr5 mice. Tmx induces Cre activity in Lgr5+ cells, driving heritable tdTomato expression and concurrent deletion of Prdm16 in mutant animals. The intestines of control and Prdm16 mutant reporter mice were analyzed 3 and 14 days after tmx treatment. At 3 days, the length of tdTomato-marked epithelial strips along the crypt-villus axis was reduced by ~30% in Prdm16 mutant animals (~380 μM in controls compared to ~270 μM in mutant; Fig. 3E, S3C). At day 14, lineage-traced Prdm16 mutant stem cells (tdTomato+;Lgr5-GFP+) were still present at the base of the crypts (Fig. 3F). Similar to the results at the earlier time point, mutant cells generated reduced numbers of differentiated epithelial progeny in villi, as evidenced by shorter strips of tdTomato+ cells (~625 μM in control vs ~400 μM in mutant; Fig. 3F).
Prdm16 is required for intestinal enteroid differentiation and maintenance
Intestinal enteroids, generated from whole crypts or Lgr5+ stem cells, are an established model system for studying intestinal development and maintenance (Sato et al., 2009). When cultured with growth factors that support a balance of stem cell maintenance and differentiation (EGF + R-Spondin + Noggin = ENR media), stem cells and descendent progenitor cells divide and give rise to mature epithelial cell populations, forming buds with stem cells located away from the enteroid centers.
We generated enteroids from crypts of control (Prdm16loxP/loxP) and mutant (R26RCre-ERT2; Prdm16loxP/loxP) mice in ENR medium. Once established, the enteroids were treated with 4-hydroxytamoxifen (4-OHT) to delete Prdm16. Control enteroids continued to grow in size and underwent extensive budding. By contrast, Prdm16-deletion led to the formation of small, cystic enteroids that failed to bud (Fig. 4A). ShRNA-mediated knockdown of Prdm16 also diminished the recovery of viable enteroids (Fig. S4A).
Figure 4: Prdm16 is required for the development and maintenance of duodenal enteroids.
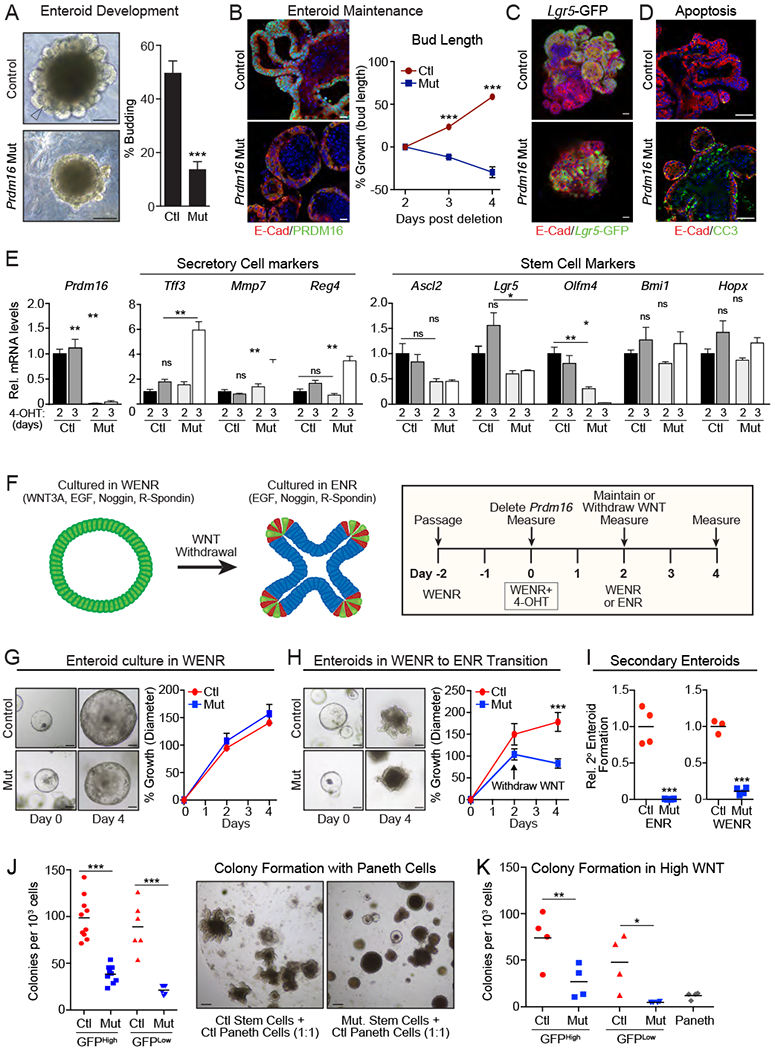
(A) Images and quantification of budding (defined as one or more bud) in Control (Prdm16loxP/loxP) and Prdm16 mutant (R26RCre-ERT2;Prdm16loxP/loxP) duodenal enteroids grown in ENR, 3d following 4-OHT treatment. Arrowhead shows bud. n=410-554 enteroids.
B) Growth or regression of buds following 4-OHT treatment of mature budded enteroids. % growth = change in bud length / initial bud length for n=7-8 enteroids. PRDM16 (green), E-Cadherin (red), DNA (DAPI, blue).
C) Immunostaining of Lgr5-GFP+ (green) stem cells in control and Prdm16-mutant enteroids, 4d after inducing deletion. Enteroids were from Lgr5GPP/CreERT2; Prdm16loxP/loxP (control) or R26RCreERT2; Lgr5GFP/CreERT2; Prdm16loxP/loxP (Mut) mice and treated with vehicle (control) or 4-OHT (Mut). E-Cadherin (red), DNA (DAPI, blue).
D) Cleaved Caspase 3 (CC3+) apoptotic cells (green) in control (Prdm16loxP/loxP) and Prdm16 mutant (R26RCre-ERT2;Prdm16loxP/loxP) duodenal enteroids 3d after treatment with 4-OHT. E-Cadherin (red), DNA (DAPI, blue).
E) mRNA levels of cell type-specific marker genes in control and Prdm16 mutant enteroids at 2d and 3d post 4-OHT treatment. n=4.
F-I) Control and Prdm16 mutant enteroids were established in Wnthigh medium (WENR), prior to treatment with 4-OHT to delete Prdm16. F) Schematic of experimental protocol. G) Enteroid growth in WENR. n=11-13 enteroids. % growth = mean change in diameter / initial diameter. H) Enteroid growth during transfer from WENR to ENR medium. n=16-23 enteroids. I) Secondary enteroids formed per well from passaged fragments of Control or Prdm16 mutant enteroids cultured in WENR or ENR medium. n=3-4 wells.
J-K) Colony formation efficiency of GFPhi and GFPlow cells isolated by FACS from control (Lgr5GFP/Cre-ERT2; Prdm16loxP/+) or Prdm16 mutant (R26RCreERT2; Lgr5GFP/CreERT2; Prdm16loxP/loxP) mice 3d after in vivo tamoxifen injection, when co-cultured with control Paneth Cells (J; n=10 wells) or in high WNT conditions (K; n=4 wells).
All panels show mean ± SEM, *p<0.05, **p<0.01, ***p<0.001. Scale bars: 100 μm (A, G, H, J); 20 μm (B); 50 μm (C,D).
Deletion of Prdm16 in fully mature enteroids induced bud regression with mutant enteroids assuming a stunted and circular morphology (Fig. 4B; S4B). By contrast, control enteroids continued to grow in size and complexity, producing more and larger buds (Fig. 4B). Prdm16-mutant enteroids retained Lgr5-GFP+ stem cells during culture (Fig. 4C). As observed in mouse intestine, Prdm16-deletion in enteroids triggered apoptosis and decreased the expression of Ki67, a proliferation marker (Fig. 4D, S4C). Prdm16-mutant enteroids also expressed elevated levels of many secretory cell genes, including Tff3 (goblet cells), Mmp7 (Paneth cells) and Reg4 (enteroendocrine cells) (Fig. 4E, S4D–G). The mRNA levels of absorptive cell markers were not decreased in Prdm16 mutant enteroids (Fig. S4G). The expression of CBC stem cell markers Ascl2 and Lgr5 were decreased by ~2-fold in Prdm16-deleted enteroids, though these effects were not statistically significant at most time points (Fig. 4E).
Enteroids can be maintained and propagated in an undifferentiated state by adding high amounts of WNT3A to the culture medium (WntHigh + EGF + Noggin + R-Spondin = WENR media) (Fig. 4F) (Rodriguez-Colman et al., 2017; Sato et al., 2009). Spheroids formed under these conditions lack buds and contain homogenously proliferating cells with elevated expression of embryonic markers (Rodriguez-Colman et al., 2017). When cultured in Wnthigh (WENR) conditions, Prdm16-deficient enteroids appeared grossly normal and grew in size at a similar rate as control enteroids (Fig. 4G, S4H). Subsequent removal of WNT3A to induce differentiation caused Prdm16-deficient enteroids to shrink and accumulate dark lumenal debris, while control enteroids continued to grow in size and underwent robust budding (Fig. 4H, S4H).
We tested whether PRDM16 was required for the formation of secondary enteroids, reflecting the renewal capacity of enteroid-forming (stem or progenitor) cells. Prdm16-deficient enteroids had no capacity to produce secondary enteroids when passaged in ENR conditions (Fig. 4I). In WntHigh (WENR) conditions, Prdm16-deficient enteroids generated secondary enteroids, albeit at lower rates than control enteroids (Fig. 4I). We also compared the enteroid-forming capacity of control and Prdm16 mutant stem and progenitor cells. Lgr5-GFPhigh stem cells and Lgr5-GFPlow cells (Lgr5+ descendants) were isolated by FACS from control and Prdm16 mutant intestines three days after tmx treatment. The sorted cells were combined with wildtype Paneth cells (CD24hi, c-KIThi, GFPneg) at a 1:1 ratio prior to embedding in Matrigel. Lgr5-GFPhigh and Lgr5-GFPlow cells from control mice displayed robust colony forming activity (Fig. 4J). Prdm16-deletion reduced colony formation in GFPhigh and GFPlow cells by 61% and 72%, respectively, relative to corresponding controls (Fig. 4J). At a morphological level, control stem cells formed highly budded enteroids over the course of 14 days, while mutant stem cells generated cystic colonies that failed to bud (Fig. 4J). We also quantified colony formation of Lgr5-GFPhigh and Lgr5-GFPlow cells cultured in Wnthigh (WENR) medium. Again, Prdm16 mutant cells generated fewer colonies than control (wildtype) cells (Fig. 4K). In particular, the colony forming activity of Prdm16-deficient Lgr5-GFPlow (Lgr5+ descendant) cells was reduced by >90%. These data demonstrate that PRDM16 regulates the renewal capacity of enteroid-forming cells.
Prdm16 deletion induces type I interferon and p53 gene signatures
To identify PRDM16-target pathways in intestinal crypts, we compared the gene expression profiles of control and Prdm16-mutant duodenal crypts 3 days post tmx injection. At this time point, PRDM16 protein was depleted in mutant crypts, but changes in intestinal architecture had not yet manifested (Fig. 1D, S1B). The upregulated genes in Prdm16-mutant vs. control crypts were highly enriched for p53 and type I interferon pathway components (Fig. S5A,B, Table S1A,B) (Porta et al., 2005). Crossing Prdm16-mutant mice onto a conditional p53 mutant background prevented the P53 target gene response in crypts, but did not rescue their intestinal renewal deficit (Fig. S5C–F). Enteroids generated from Prdm16/p53-mutant mice displayed severe growth and maturation defects, though expression of Olfm4 was restored back to control levels (Fig. S5G–H). Additionally, blocking type I interferon signaling and target gene expression using a neutralizing antibody did not normalize the phenotype of Prdm16-mutant enteroids (Fig. S5I–K). These results indicate that neither elevated p53 activity nor interferon signaling are sufficient to cause the intestinal deficits caused by Prdm16-deletion.
PRDM16 regulates metabolic genes/pathways in small intestinal crypts
Pathway analysis of downregulated genes in Prdm16-mutant vs. control crypts revealed a striking enrichment in metabolic pathways, including biological oxidation, mitochondrial fatty acid beta-oxidation, retinoic acid biosynthesis and fatty acid, triacylglycerol and ketone body degradation (Fig. 5A, Table S1C,D). In particular, Prdm16-mutant crypts expressed lower levels of genes involved in multiple steps of fatty acid utilization, including fatty acid uptake and acylation (Cd36, Slc27a2, Acsl1), acyl-carnitine mitochondrial import (Cpt1a and Cpt2), mitochondrial fatty acid oxidation (FAO; Acadm, Acaa2, Hadh), and mono and polyunsaturated FAO (Eci1, Eci2, Decr1) (Fig. 5B,C). Many of these genes were further downregulated at later time points as the mutant phenotype progressed (Fig. 5D). FAO gene levels were also reduced, albeit to a lesser extent, in mutant compared to control villi (Fig. S5L). In addition to decreasing FAO gene levels, loss of Prdm16 reduced the expression of genes involved in the TCA cycle, amino acid catabolism and retinol metabolism (Fig. S5M).
Figure 5: PRDM16 binds and promotes the expression of fatty acid oxidation genes.
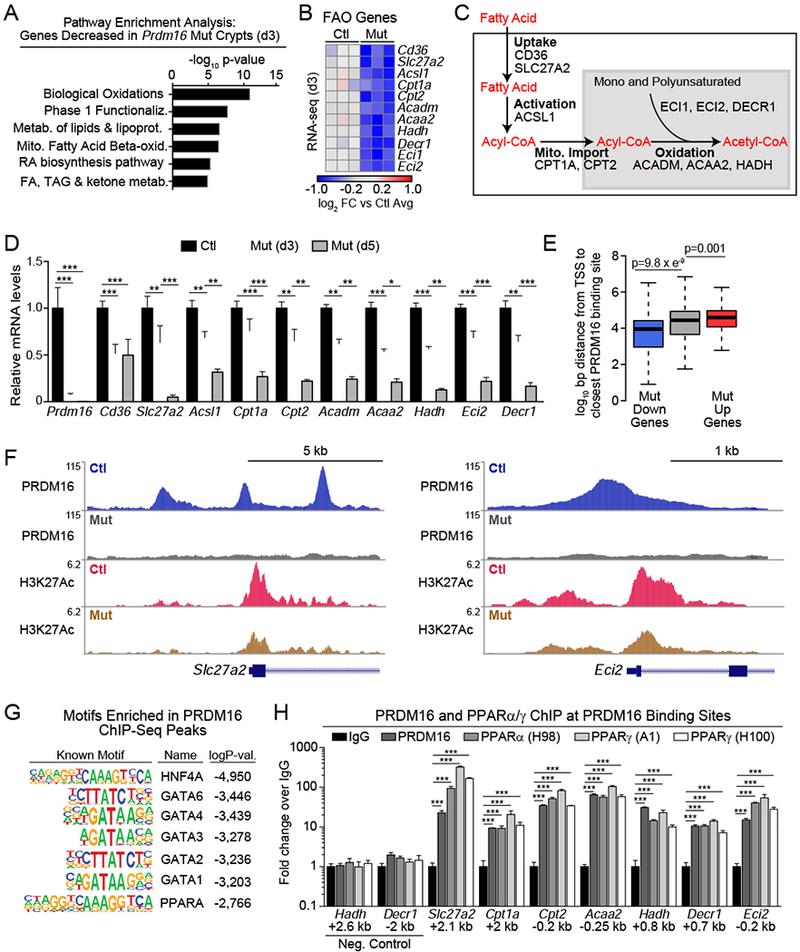
A-B) RNA-seq analysis of isolated duodenal crypts from Prdm16loxP/loxP (control) or R26RCre-ERT2;Prdm16loxP/loxP (mutant) animals at 3d after tmx injection. A) Pathway analysis of down-regulated genes (FDR<0.05) in mutant crypts. B) Downregulated FAO genes meeting a threshold of FDR<0.1 are displayed in a heat map of log2 fold-change (FC) relative to mean expression in controls. n=3 mice per group.
C) Schematic of key steps of FAO to acetyl-CoA generation with associated enzymes.
D) mRNA levels of FAO genes in control and Prdm16 mutant crypts at 3d and 5d after tmx injection. n=4 (control/mutant d3), n=5 (mutant d5).
E) Distance of PRDM16 ChIP-Seq peaks from the transcriptional start site (TSS) of downregulated (blue), unchanged (gray), or upregulated (red) genes in Prdm16-mutant crypts by RNA-seq at 3d post tmx.
F) ChIP-seq tracks for PRDM16 and H3K27Ac at FAO genes Slc27a2 and Eci2 in control and mutant duodenal crypts.
G) Transcription factor binding motifs enriched at PRDM16 ChIP-seq peaks.
H) CHIP-qPCR analysis of PRDM16, PPARα and PPARγ (two antibodies) binding at PRDM16-ChIP-seq binding regions near FAO gene promoters in duodenal crypts. Negative control sites are ~3 kb from PRDM16 peaks. Distance from TSS is shown in kilobases (kb), fold-change shown relative to normal-IgG control ChIP. n=3 biological ChIP replicates.
Panels (D,H) show mean ± SEM, *p<0.05, **p<0.01, ***p<0.001.
To investigate the mechanism by which PRDM16 regulates FAO genes, we performed ChIP-seq for PRDM16 and the active enhancer mark H3K27Ac in control and Prdm16-deficient crypts. PRDM16 binding sites were located closer to genes whose expression was downregulated in Prdm16-mutants, compared to unchanged or increased genes, suggesting that PRDM16 largely functions as a transcriptional activator in crypts (Fig. 5E). Prominent PRDM16 binding sites were identified at several FAO gene loci, including Slc27a2, Acsl1, Cpt2, Acaa2, Hadh, Decr1 and Eci2, with many of these sites showing decreased H3K27Ac levels in mutant vs. control crypts (Fig. 5F, S5N).
PRDM16 binding sites were enriched with motifs for several transcription factors, including PPARs, which are known to regulate FAO (Fig. 5G, Table S1E) (Ito et al., 2012). Motif analysis of regions with reduced H3K27Ac levels in mutant vs. control crypts identified a similar set of motifs (Fig. S5O, Table S1F). ChIP-qPCR analysis of intestinal crypts showed that PPARα and PPARγ bind to the same sites as PRDM16 at many FAO genes, including Slc27a2, Cpt1a, Cpt2, Acaa2, Hadh, Decr1 and Eci2 (Fig. 5H). Previous studies have shown that PRDM16 physically interacts with PPARα and PPARγ, and enhances their transcriptional function (Hondares et al., 2011; Seale et al., 2008). Treatment of wild type duodenal enteroids with antagonists of PPARγ and PPARα, either separately or in combination, led to severe developmental defects. Antagonism of PPARγ blocked enteroid budding and induced apoptosis, while inhibition of PPARα caused a complete loss of viability (Fig S5P,Q). Thus, these data provide evidence that PRDM16, along with PPAR proteins, directly activate the expression of FAO genes in intestinal crypts.
Prdm16 regulates FAO in small intestinal crypts
To assess the role of PRDM16 in regulating FAO, we performed stable isotope tracing in control and Prdm16-deficient duodenal crypts. Crypts isolated from control and mutant mice 3 days after tmx injection were incubated in medium containing uniformly labeled 13C16-palmitate (Fig. 6A, S6A). We then measured 13C integration into the TCA cycle intermediates citrate and α-ketoglutarate (Fig. 6B). Labeling was assessed by calculating the percentage of 13C labeled carbons in each metabolite pool (Fendt et al., 2013). 13C16-palmitate was efficiently incorporated into citrate and α-ketoglutarate in control crypts, with over 20 percent of carbons labeled with 13C (Fig. S6B). Prdm16 deficient crypts displayed a ~30% reduction in relative 13C labeling of both citrate and α-ketoglutarate (Fig. 6B, S6B). At this time point, the expression of FAO genes had begun to decrease in Prdm16-deficient crypts and continued to drop over subsequent days (Fig. 5D). Of note, the FAO regulators Ppara and Pparg as well as the mitochondrial acyl-carnitine importers Cpt1a and Cpt2 were expressed at higher levels in Lgr5-GFPlow (Lgr5+ descendant) cells compared to Lgr5-GFPHigh stem cells, following the same pattern as Prdm16 (Fig. 6C, 1F).
Figure 6: PRDM16 regulates intestinal growth and maintenance by promoting FAO.

A-B) Mass spectrometry analysis of 13C16-palmitate incorporation into TCA cycle metabolites. A) schematic. B) Relative 13C-labeling of the citrate/isocitrate and α-ketoglutarate pools in control (Prdm16loxP/loxP) vs. Prdm16 mutant (R26RCreERT2; Prdm16loxP/loxP) crypts isolated 3d after tmx injection. 13C-labeling = Σ (% of isotopomer multiplied by labeled carbons in isotopomer) divided by the number of carbons in molecule. n=4-5 mice per group.
C) mRNA levels of indicated FAO genes in FACS-isolated Lgr5-GFPHi or Lgr5-GFPlow epithelial cells from the duodenum of Lgr5GPP/Cre-ERT2 mice. n=3 mice.
D-G) Wildtype enteroids (in ENR medium) were treated with: (1) vehicle or etomoxir, and (2) sodium chloride (No Acetate) or Acetate (Ac). D) Brightfield images with magnified inset. E) Quantification of enteroid growth. n=32-41 enteroids. F) Quantification of enteroid budding (one or more buds). n=4 wells. G) Secondary enteroid formation from fragments of passaged enteroids. n=4 wells. Etomoxir (Eto), 50 μM in water; NaAc/NaCl, 5 mM in water. Enteroids were treated with NaAc / NaCl for 2d before addition of Eto treatment.
H-N) Control or Prdm16 mutant enteroids (in ENR medium) were treated with 5mM sodium acetate (Ac) or 5mM sodium chloride (No Acetate). H) Brightfield images with magnified insets. I) Enteroid growth. n=16-21 enteroids. J) Immunostaining of Lysozyme-positive (green) Paneth cells in control and Prdm16-mutant enteroids, 4d after inducing deletion. Enteroids were from Prdm16loxP/loxP (control) or R26RCreERT2; Prdm16loxP/loxP (Mut) mice and treated with 4-OHT with additional treatment of acetate or no acetate treatment (NaCl, 5mM). E-Cadherin (red), DNA (DAPI, blue). Percentage of enteroid cells positive for Lysozyme staining are quantified in the graph. K) mRNA levels of cell type-specific marker genes in control and Prdm16 mutant enteroids at 4days post 4-OHT treatment with or without acetate treatment. n=4. L) Secondary enteroid formation from fragments of passaged enteroids. M) Enteroid budding (defined as one or more bud). n=4-6 wells. n=4 wells; NaAc/NaCl, 5 mM in water. Enteroids were pretreated with NaAc / NaCl for 2d after plating but before treatment with 4-OHT.
N) Lineage tracing of stem cell progeny in Prdm16loxP/+ (control) or Prdm16loxP/loxP (mutant) tdTomato reporter mice. Animals received drinking water containing either 150 mM NaAc (Ac) or NaCl (No Acetate) 2d before tmx treatment. Length of marked villus epithelial cell strips was measured for continuous strips of Tomato+ (red) epithelial cells ascending the villi at 4d post tmx treatment. n=200-291 villi measured from >=4 mice per group. tdTomato (red), DNA (DAPI, blue). O) Quantification of Cleaved caspase-3 positive cells per tdTomato positive crypt relative to the number of cleaved caspase-3 positive cells in tdTomato negative crypts. N=303-423 crypts from 3 or more mice per group.
All panels show mean ± SEM, *p<0.05, **p<0.01, ***p<0.001. Scale bars: 100 μm (D, H, J); 50 μm (N).
We also assessed glucose metabolism in control and Prdm16-mutant enteroids using 13C6-glucose stable isotope tracing. Prdm16 mutant enteroids displayed a small but significant increase in the incorporation of 13C labeled carbons (from glucose) into citrate and α-ketoglutarate (Fig. S6C). This result demonstrates that loss of Prdm16 does not disrupt general mitochondrial oxidative activity and, if anything, increases glucose utilization. While we did not observe widespread expression changes in the levels of glycolytic genes in mutant crypts, the glucose transporter Glut1/Slc2a1 and hexokinases Hk1 and Hk2 were upregulated (Fig. S6D).
To determine if blocking FAO mimics the phenotypic effect of Prdm16-deficiency, we treated duodenal enteroids with the CPT1 inhibitor etomoxir. Etomoxir treatment of enteroids reduced their growth, budding and capacity to generate secondary enteroids (Fig. 6D–G). Acetate, by providing an alternate source of acetyl-CoA rescues genetic or etomoxir-mediated inhibition of FAO (Schug et al., 2016; Wong et al., 2017). While etomoxir has off-target effects in some tissues, these effects are not reversed by acetate (Yao et al., 2018). We found that acetate completely rescued the growth and budding defects and significantly increased secondary enteroid formation in etomoxir-treated enteroids (Fig. 6E–G). Acetate did not augment the growth, budding or passaging capacity of control enteroids.
These results led us to ask whether acetate could substitute for FAO and rescue the intestinal defects caused by Prdm16-deficiency. Strikingly, acetate supplementation fully rescued the altered morphology and growth deficit of Prdm16-deficient enteroids, while also suppressing the induction of Cleaved Caspase-3+ (apoptotic) cells (Fig. 6H, I, S6E). Acetate also prevented the increased formation of Lysozyme+ cells in Prdm16-deleted enteroids and normalized the expression of secretory cell marker genes (Tff3, Mmp7, Chga) (Fig. 6J,K). The stem cell genes Ascl2 and Lgr5 were moderately downregulated by acetate in control enteroids. However, acetate prevented the downregulation of these genes upon Prdm16-deletion (Fig. 6K). As shown above, Prdm16-deletion severely impaired secondary enteroid formation in ENR medium (Fig. 6L). However, in acetate-supplemented medium, control and mutant enteroids generated comparable numbers of secondary enteroids, similar to that of control (no acetate) enteroids (Fig. 6L, images in S6F). Finally, acetate significantly increased budding frequency in Prdm16-mutant enteroids (Fig. 6M).
We next tested if acetate could rescue the impaired development of villus epithelium by Prdm16-deficient stem cells in vivo. To do this, control and Prdm16-mutant Lgr5CreERT; R26R-tdTomato reporter mice were given drinking water containing either 150 μM sodium acetate or 150 mM sodium chloride (as control) beginning two days before tmx injection and continuing throughout the course of the experiment. As observed before, the development of tdTomato+ villus epithelial cells was severely reduced in Prdm16 mutant mice under control conditions (Fig. 6N). Acetate-treatment had very little effect on epithelial cell development in control mice. By contrast, acetate supplementation markedly increased the development of tdTomato+ villus epithelial cells in Prdm16-mutant mice, almost reaching the levels observed in controls (Fig. 6N). In line with this, acetate blocked the induction of apoptotic cells and Lysozyme+ cells and blunted the induction of Mucin2+ cells in Prdm16-mutant mice (Fig. 6O, S6G–I). Thus, acetate treatment rescues many of the phenotypic consequences of Prdm16-deficiency in intestinal stem cells and further implicates a crucial role for PRDM16-driven FAO in promoting intestinal renewal.
PRDM16 is a region-specific regulator of small intestinal metabolism and renewal
We noted that the jejunum and ileum had milder phenotypes than the duodenum in Prdm16-mutant mice (Fig. S7A). This led us to examine PRDM16 expression along the length of the intestine. Immunostaining of a full length, small intestinal “Swiss roll” showed high PRDM16 protein expression in the duodenum, with gradually decreasing expression toward the ileum (Fig. 7A). This PRDM16 expression pattern was confirmed by western blot analyses of crypt samples from different intestinal regions (Fig. 7B). Prdm16 mRNA levels were also highest in duodenal crypts compared to crypts isolated from more distal regions (Fig. 7C).
Figure 7: PRDM16-driven FAO selectively regulates the development and maintenance of upper intestinal enteroids.
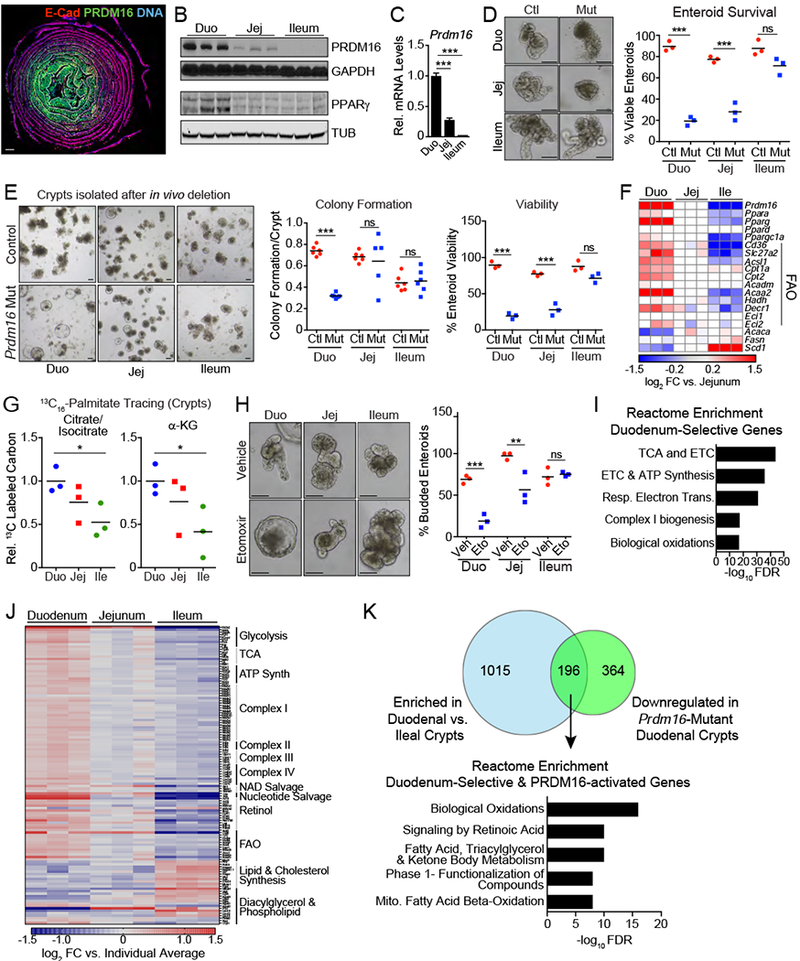
A) “Swiss roll” immunostaining of PRDM16 (green) in small intestine, with duodenum at the center and ileum at the periphery. E-cadherin (red), DNA (DAPI, blue). Panel is stitched composite of 10X images.
B) Western blot analysis of PRDM16 and PPARγ expression in duodenal, jejunal, and ileal crypts. Loading controls are GAPDH and α-Tubulin (TUB). n=3 mice.
C) mRNA levels of Prdm16 in duodenal, jejunal, and ileal crypts. n=3 mice.
D) (left) Control (Prdm16loxP/loxP) and Prdm16 mutant (R26RCreERT2; Prdm16loxP/loxP) enteroids derived from duodenal, jejunal, and ileal crypts. Enteroids were analyzed 7d post 4-OHT treatment. (right) Quantification of viable enteroids. n=54-121 enteroids, n=3 wells.
E) Enteroid formation from duodenal, ileal, and jejunal crypts of control (Prdm16loxP/loxP) and Prdm16 mutant (R26RCreERT2; Prdm16loxP/loxP) mice 3d after tmx injection. Colony formation and viability were assessed 2d and 7d after plating, respectively.
F) Expression heat map of Prdm16, Ppar isoforms, FAO, and fatty acid synthesis genes, as measured by qRT-PCR. n=3 mice.
G) Mass spectrometry analysis of relative 13C-labeling of the citrate/isocitrate and α-ketoglutarate pools in duodenal, jejunal, and ileal crypts after a 90-minute incubation in 13C16-palmitate (relative to duodenum). 13C-labeling = Σ (% of isotopomer multiplied by labeled carbons in isotopomer) divided by the number of carbons in molecule. n=3 mice.
H) Quantification of budding (one or more buds) in duodenal, jejunal, and ileal enteroids following treatment with vehicle or etomoxir (Eto; 50 μM). n=79-239 from 4 wells.
I-K) RNA-seq analysis of wildtype crypts from duodenum, jejunum, and ileum. n=3. I) Reactome enrichment analysis of genes with selective expression in duodenal vs. ileal crypts. J) Expression heat map of a curated set of metabolism genes showing relative regional expression. Scale is log2 fold change versus average expression across crypts for each individual replicate. K) Venn diagram of overlap between duodenal crypt-enriched genes and genes downregulated in Prdm16 mutant crypts (top). Intersecting gene set was used for Reactome enrichment analysis (bottom).
All panels show mean ± SEM, *p<0.05, **p<0.01, ***p<0.001. Scale bars: 1 mm (A), 100 μm (D, E, H). Duo= duodenal, Jej=jejunal, Ile=ileal. Ctl= Control. Veh=vehicle.
To determine if PRDM16 is required in a region-specific manner, we studied the effect of Prdm16-deletion in enteroids established from duodenal, jejunal and ileal crypts. The cultured enteroids expressed region-selective signature genes (Middendorp et al., 2014; Thompson et al., 2017), indicating that enteroid identity was maintained ex vivo (Fig. S7B). Enteroids were grown under differentiation-permissive ENR conditions. Upon Prdm16 ablation, duodenal enteroids displayed the most severe phenotype (Fig. 7D, S7C). By contrast, ileal enteroids lacking Prdm16 underwent robust budding similar to that of their controls and appeared unaffected overall, with no decrease in survival (Fig. 7D, S7C). Additionally, Prdm16-deficient duodenal enteroids were unable to form secondary enteroids, whereas mutant ileal enteroids could be readily passaged, though at a reduced efficiency compared to control (Fig. S7D). Consistent with a reduced role of PRDM16 in the ileal epithelium, the length of epithelial tdTomato+ cell strips was slightly but significantly decreased in the ileum of Prdm16-mutant Lgr5CreERT; tdTomato reporter mice (Fig. S7E).
To determine the effect of in vivo Prdm16 deletion on enteroid formation from different regions, we isolated duodenal, jejunal or ileal crypts 3 days after administration of tmx. In these experiments, mutant duodenal crypts generated few viable enteroids, and those that survived had a cystic morphology. On the other hand, there was no detectable difference in enteroid formation or viability between control and Prdm16 mutant ileal crypts (Fig. 7E).
We next sought to determine if tapering PRDM16 expression corresponded to changes in crypt metabolism along the small intestine. Indeed, a large fraction of the FAO genes regulated by PRDM16 (e.g. Slc27a2, Acsl1, Cpt1a, Cpt2, Acaa2, Hadh, Decr1), were expressed at high levels in crypts of the duodenum, intermediate levels in jejunum and low levels in the ileum (Fig. 7F). Interestingly, Ppara and Pparg were also expressed in a similar graded fashion from duodenum to ileum (Fig. 7F). PPARγ, in particular, displayed a striking region-specific expression pattern at both the mRNA and protein level (Fig. 7B,F). Stable isotope tracing showed that palmitate gave the highest contribution to TCA cycle intermediates in duodenal crypts, with lowest contribution in ileal crypts (Fig. 7G, S7F).
These results raised the notion that stem and/or progenitor cells in different intestinal regions may exhibit differential reliance on FAO. To test this, enteroids generated from different regions were treated with etomoxir to block FAO. As above, etomoxir severely impaired the budding of duodenal enteroids (Fig. 7H). Jejunal enteroids were much less affected, while ileal enteroids budded robustly in the presence of etomoxir (Fig. 7H), indicating that intestinal region-specific stem and/or progenitor cells possess different metabolic requirements.
Finally, we performed RNA-seq expression profiling of duodenal, jejunal and ileal crypts to identify region-selective metabolic programs in an unbiased manner. Among genes with higher expression in duodenal (vs. Ileal or Jejunal) crypts, pathway analysis identified a strong enrichment of genes involved in oxidative metabolism, including TCA cycle, electron transport chain, ATP synthase complex and FAO (Fig 7I–J, Table S2A,B). Conversely, ileal crypts showed relatively higher expression of genes involved in acyl chain metabolism and cholesterol biosynthesis (Fig. 7J, S7G, Table S2C). At a global level, a substantial proportion (35%) of the genes that were downregulated by Prdm16 deletion were also enriched in duodenal vs. ileal crypts (Fig. 7K). The top enriched pathways for these genes were metabolic processes, including “Fatty acid, triacylglycerol, and ketone body metabolism” and “Mitochondrial Fatty Acid Beta-Oxidation” (Fig. 7K, Table S2D,E). Overall, these results demonstrate that crypts from different intestinal regions express divergent metabolic programs and that PRDM16 is a major regulator of the duodenal-specific profile.
Discussion
This study identifies PRDM16 as a critical transcriptional regulator of metabolism and differentiation in the small intestinal crypt. PRDM16 controls the expression of FAO pathway genes and is required for the survival of Lgr5+ descendant progenitor cells and epithelial renewal, selectively in the upper region of the small intestine.
PRDM16 and its binding partners PPARα and PPARγ are expressed at high levels in the upper intestine (duodenum and proximal jejunum) and decline distally. FAO genes also follow this expression pattern, accompanied by a higher contribution of FAO to TCA cycle metabolites in upper compared to lower small intestine. This pattern is physiologically rational, as the upper intestine is exposed to the highest level of dietary fat, which is broken down into fatty acids by pancreatic lipases secreted into the duodenum (Iqbal and Hussain, 2009). The specialization of upper intestinal crypt cells for fatty acid catabolism presumably allows these cells to function more efficiently in this metabolic milieu.
A reliance on fatty acid utilization sensitizes duodenal stem and progenitor cells to disruptions in the FAO pathway, as revealed upon Prdm16-deletion. Intestinal loss of the FAO enzyme CPT1A causes similar, though less severe phenotypes, including reduced development of mature villus epithelial cells, elevated crypt apoptosis and reduced stem cell function (Mihaylova et al., 2018). The ability of acetate to rescue many of the phenotypic effects of Prdm16-deficiency suggests that FAO plays a major role in mediating PRDM16-function. In particular, acetate robustly rescued the growth, survival and renewal capacity of Prdm16-mutant duodenal enteroids. However, acetate only partially rescued the budding deficit of Prdm16-mutant enteroids, suggesting that PRDM16 controls budding through other mechanisms. In this regard, we noted that PRDM16 regulates other metabolic pathways (Fig. S5M), in addition to FAO, that could contribute to the budding defect of Prdm16-mutant enteroids. Presumably, PRDM16 also regulates non-metabolic genes that influence intestinal development.
Our results suggest that PRDM16 plays a critical role in regulating the function of Lgr5+ descendant progenitor cells in the upper intestine. PRDM16 is expressed at higher levels in the TA zone of crypts relative to that of Lgr5+ stem cells, localized at the crypt base. Moreover, loss of PRDM16 reduced proliferation and triggered apoptosis, specifically in the TA zone, supporting the notion that PRDM16 directly regulates progenitor cell function. Although our most striking phenotypes manifest in the progenitor cell compartment, stem cells lacking Prdm16 show defects in enteroid forming capacity, and we cannot rule out a proliferation defect in the stem cell compartment. It is therefore possible that Prdm16 deletion compromises the function of Lgr5+ stem cells, which could contribute to the loss of TA progenitors and deficit in epithelial renewal. Additionally, a recent study focusing on the enteroendocrine lineage hierarchy identified Prdm16 as an enriched transcription factor in enterochromaffin precursors (Gehart et al., 2019). Thus, in addition to broadly regulating metabolic pathways and epithelial renewal in the duodenum, PRDM16 may play other roles in regulating enteroendocrine differentiation.
Both PRDM16 and FAO were dispensable for the formation, growth and renewal of enteroids derived from distal regions of the small intestine. Presumably, progenitor cells in these regions have a higher capacity to utilize other fuel sources or may depend less on oxidative metabolism. Interestingly, ileal crypts selectively expressed genes involved in lipid metabolism and cholesterol biosynthesis, pathways that were recently implicated in regulating intestinal stem cell activity and tumorigenesis (Wang et al., 2018). Overall, our findings highlight the importance of studying intestinal stem cell metabolism in the context of its regional origin rather than in aggregate or from a single region.
An important outstanding question is why duodenal enteroids rely on FAO, considering the availability of other nutrients in the medium. FAO is required in other stem cell niches (Ito et al., 2012; Knobloch et al., 2017), though the reason for this requirement over other pathways is poorly understood. FAO may generate a specific metabolite that is not adequately produced by other pathways and/or affect the sub-cellular distribution of certain metabolites. Metabolism could also influence progenitor function through many epigenetic pathways (Carey et al., 2015; TeSlaa et al., 2016; Wei et al., 2018; Wellen et al., 2009). In this regard, fatty acids can serve as a dominant source of acetyl-CoA for histone modifications (McDonnell et al., 2016). Future studies examining the fate of fatty acid-derived acetyl-CoA in intestinal progenitors will address this question.
This study expands the role of PRDM16 as a metabolic regulator outside of its heavily studied function in brown fat. Many of the PRDM16 binding sites at FAO genes are common to intestine and brown fat, suggesting that PRDM16 has conserved core functions in multiple cell types (Harms et al., 2015; Shapira et al., 2017). PRDM16 has also been linked to metabolism in other stem cell niches, including hematopoietic and neuronal stem cells (Aguilo et al., 2011; Chuikov et al., 2010). Prdm16-deficient hematopoietic stem cells show increased respiration and an upregulation of electron transport genes (Corrigan et al., 2018), effects that are not evident in intestine or brown fat. In neuronal progenitors, PRDM16 regulates the expression of Hgf to reduce ROS generation (Chuikov et al., 2010). Taken together, these studies highlight multiple pathways through which PRDM16 can regulate stem and progenitor cell activity.
Our studies also suggest an important role for PPARγ in establishing region-specific intestinal metabolism. Notably, PPARγ has a protective effect on the small intestine during injury and in the context of Crohn’s disease (Baregamian et al., 2009; Sugawara et al., 2005). PPARγ also modulates microbiota/host interaction in the small intestine following high fat diet (Tomas et al., 2016). PPARγ and its related family members PPARα and PPARδ are nuclear receptors whose activity can be modulated by a variety of synthetic and proposed natural agonists, including many fatty acids. This property allows PPAR proteins to function as nutritional sensors for integrating environmental changes in fatty acid levels to adaptive changes in metabolic programs. Related to this, PPARδ, whose mRNA is expressed at similar levels as Ppara in duodenal crypts, can sense changes in dietary fatty acids as a result of high fat diet and enhance the activity of stem cells and transit-amplifying cells (Beyaz et al., 2016; Mihaylova et al., 2018).
In summary, our studies define PRDM16 as a critical regulator of small intestinal maintenance and uncover an unanticipated role for region-specific metabolic pathways in regulating intestinal epithelial renewal. It will now be important to identify the mechanisms by which specific metabolic programs and metabolites control intestinal stem and progenitor cell activity and to determine if dysregulation of these pathways underlie intestinal disease or dysfunction.
Star Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Patrick Seale (sealep@pennmedicine.upenn.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
The following mouse strains were purchased from the Jackson Laboratory: Rosa26CreERT2 (B6;129-Gt(ROSA)26Sortm1(cre/ERT)Nat/J, RRID: IMSR_JAX:004847); Lgr5EGFP-iREs-creERT2 (B6.129P2-Lgr5tm1(cre/ERT2)Cle/J, RRID :IMSR_JAX:008875); Rosa26LoxP-Stop-LoxP-tdTomato (B6.Cg-Gt(ROSA)26Sortm14 (CAG-tdTomato) Hze/J, RRID: IMSR_JAX:007914); and p53loxP/loxP (B6.129P2-Trp53tm1Brn/J, RRID: IMSR_JAX:008462). The Prdm16 conditional knockout mouse line was generated by Patrick Seale and Bruce Spiegelman and is available from the Jackson Laboratory: Prdm16loxP/loxP (B6.129-Prdm16tm1.1Brsp/J, RRID: IMSR_JAX:024992). All animal work was approved by the University of Pennsylvania’s Institutional Animal Care and Use Committee. Mice were housed under the care of University of Pennsylvania University Laboratory Animal Resources (ULAR), which provides both basic husbandry and veterinary care. Animals were specific-pathogen free (SPF) and raised at room temperature on standard chow with a 12-hour light/dark cycle. Both sexes were used for experiments. All experiments were done on adult animals between the ages of 6 to 10 weeks at the onset of the experiment.
METHOD DETAILS
In Vivo Treatments
Tamoxifen (tmx) was used to activate CreER in conditional knockout and lineage-tracing models. For gene knockout, 6-8 week-old-mice (~18-25g) were intraperitoneally injected with tmx (Sigma, stock 20 mg/ml in corn oil) at a dose of 3 mg/d for three consecutive days. For lineage tracing experiments, mice were injected with a single 3 mg dose of tmx. For in vivo acetate treatment, drinking water was supplemented with either 150 mM NaCl (Sigma) as control or 150 mM NaAcetate (Sigma). Sibling- or age-matched experimental and control animals were randomly allocated into experimental groups based only on genotype and date of birth. Both female and male mice were analyzed separately for each experiment. All results were consistent between sexes and combined when appropriate. All animal experiments were approved by the University of Pennsylvania’s Institutional Animal Care and Use Committee.
Analysis of intestinal barrier function and serum cytokines
For barrier function analysis, mice were gavaged orally with 150 μl of 80 mg/ml fluoroscein isothiocyanate (FITC) dextran (Sigma) dissolved in sterile PBS (6d after tmx injection). Four hours after gavage, mice were sacrificed, and blood was collected by cardiac puncture. Serum was isolated by centrifugation, diluted 1:10 in PBS and loaded onto a black, opaque bottomed microplate for measurement using a Synergy HT plate reader (BioTek) excited at 485 nm and read at 530 nm. Concentration was determined using a FITC-dextran standard curve. To determine colony formation, whole blood samples were diluted 1:2 or 1:10 in sterile PBS and 100 μl was plated on blood agar plates (Fisher). Plates were incubated at 37°C for 20 hours and colonies were quantified. For cytokine analysis, serum was analyzed by MILLIPLEX Mouse Cytokine/Chemokine Magnetic Bead Immunology Multiplex Assay (Millipore) using a Luminex Flexmap 3D machine in conjunction with the UPenn Human Immunology Core.
Histology and Immunofluorescence
Tissues were fixed in 10% buffered formalin overnight, washed in PBS, dehydrated in ethanol, paraffin-embedded and sectioned. Following deparaffinization, slides were subjected to heat antigen retrieval in a pressure cooker with Bulls Eye Decloaking buffer (Biocare), unless otherwise noted. Slides were incubated in primary antibody overnight and secondary antibody conjugated to peroxidase and then developed using Tyramide Signal Amplification (TSA, Akoya Biosciences). Primary antibodies used for staining were: rabbit anti-PRDM16 (Seale et al., 2011) 1:200, goat anti-Lysozyme C (W-20) (1:50, Santa Cruz Cat# sc-27956, RRID:AB_2138793 (Disc)), rabbit anti-MUC2 (H-300) (1:50, Santa Cruz, Cat#sc-15334, RRID:AB_2146667 no TSA amplification), rabbit anti-MUC2 (1:500, Cloud Clone, Cat#PAA705MU02, no TSA amplification, goat anti-rabbit Alexa 488 secondary), mouse anti-E-cadherin, (1:200, BD Biosciences, Cat#610181, RRID:AB_397580), rabbit anti-KI67 (Abcam Cat#ab16667, RRID:AB_302459, no TSA amplification, goat anti rabbit Alexa 555 secondary), rabbit anti-cleaved NOTCH1 (Val1744) (1:50, Cell Signaling, Cat#4147, RRID:AB_2153348), rabbit anti-cleaved Caspase-3 (1:500, Cell Signaling Cat#9664, RRID: AB_2070042, Borg decloaker), goat anti-GFP (1:500, Abcam, Cat#ab6673, RRID:AB_305643), rabbit anti-RFP (1:250, Rockland Cat#RL600-401-379, RRID:AB_2209751), rabbit anti Chromogranin A (1:500, Abcam, Cat#Ab15160, RRID:AB_301704), rabbit anti-Lysozyme C (1:500, Agilent Cat#A0099, RRID:AB_2341230) and DAPI (1:1000 Roche). Images were captured on a Leica TCS SP8 confocal microscope or Keyence BZ-X700 fluorescent light microscope. Villi length was measured from the top of the crypt to the tip of the villi using the measurement tool in photoshop and pixel length was converted to microns. For lineage tracing experiments, full length small intestines were arranged into tight swiss rolls to maximize cross sectional villus area. Complete swiss rolls were imaged using the multi-image stitching setting of the Keyence BZ-X700 microscope with the 10X objective. Lineage traced experiments were scored blinded.
For enteroid staining, whole enteroids were collected in ice-cold PBS to dissolve Matrigel (Corning) and fixed in 1.5 ml Eppendorf tubes with 4% PFA (EMS) for 15 minutes. Enteroids were washed and blocked with normal donkey serum (Sigma). Primary antibody was incubated overnight, enteroids were washed and secondary antibody was incubated for 1 hour. Primary antibodies were used as listed above. Secondary antibodies were donkey anti-goat IgG, Alexa 488 (1:400 ThermoFisher, Cat#A11055, RRID:AB_2534102), donkey anti-mouse IgG, Alexa 555 (1:200 ThermoFisher, Cat#A31570, RRID:AB_2536180), donkey anti-rabbit IgG, Alexa 647 (1:200 ThermoFisher, Cat#A31573, RRID:AB_2536183), goat anti-rabbit IgG, Alexa 488 (1:400 ThermoFisher, Cat#A11034, RRID:AB_2536180) and DAPI (1:1000, Roche). Enteroids were then washed 3 times and, after the final wash, mounted onto slides in Vectashield (Vector Biolabs) for imaging using a Leica TCS SP8 confocal microscope. All washes were with PBS + 0.1% T riton X with the exception of a final wash in PBS.
Crypt Isolation and culture
Intestinal crypts were isolated according to published methods (Chatterji et al., 2018; Sato et al., 2009). Briefly, 8-10 cm of a specified region of the small intestine were dissected and cleaned with PBS. For duodenum samples, 8 cm was isolated immediately adjacent to the stomach. For jejunum samples, 10 cm was isolated at the midpoint of the small intestine. For ileum samples, 10 cm was isolated immediately adjacent to the cecum. All tissues were kept on ice in Ca2+ and Mg2+-free Hank’s buffered salt solution (CMF-HBSS, Thermo Fisher) supplemented with 1 mM N-acetyl cysteine (NAC, Sigma). Samples were then transferred to CMF-HBSS containing 10 mM EDTA (Fisher) and 1 mM NAC and rotated for 45 min at 4°C. To dissociate villi and crypts from underlying mesenchyme, tubes were vortexed for 30s following a 30s rest on ice four times (six times for ileal samples). Samples were passed over a 70-μm filter to remove villi, and crypts were gently pelleted. Crypts were resuspended in Basal+ medium (advanced DMEM/F12 medium containing 2 mM Glutamax, 10 mM HEPES, 1X penicillin/streptomycin, 1X N2 supplement, and 1X B27 supplement (all Thermo Fisher), with 5 μM CHIR99021 (Biovision) and 1 mM NAC. Crypts were then counted, pelleted and resuspended at a concentration of 15 crypts/μl in an 80/20 mixture of Matrigel matrix (Corning) and ENR (Basal+ medium also containing 50 ng/mL mouse epidermal growth factor [Preprotech], 60 ng/ml Noggin [Preprotech] and 0.5 μg/ml R-Spondin [Preprotech]) or Intesticult (Stem Cell Technologies) medium. Crypt/matrix mixtures were then plated as droplets into 24- or 96-well plates. After solidification of the matrix, crypt-containing droplets were overlaid with ENR medium. Medium was changed every 48-72 hours. Enteroids were passaged using mechanical dissociation or 2-minute TrypLE Express (Invitrogen) digestion (for Wnthigh experiments) every 6-8 days. For experiments involving Wnthigh conditions, WENR media was made by supplementing ENR media with 100 ng/ml Wnt3A (R&D Systems) and 10 μM Nicotinamide (Sigma), with 10 μM Y-27632 (Cell Signaling) added for the first 24 hours after passage.
For ex vivo Prdm16 deletion, 1 μM 4-OHT (Sigma) or vehicle (ethanol, 1:10,000) was added directly to the media for 24 hours. For IFNAR neutralizing antibody experiments, anti-IFNAR antibody (Leinco, Cat#I-401, RRID:AB_2491621) was added directly to enteroid media at a concentration of 1ug/ml immediately after plating. For etomoxir experiments, 50 μM Eto (Sigma) was added directly to the media, and media was refreshed every 48 hours. For acetate rescue experiments, filter sterilized 1M sodium acetate solution was added directly to culture medium to a final concentration of 5 mM. For PPARA and PPARG antagonist experiments, 5uM concentrations of GW9662 or GW6271 (both from Sigma) were added directly to the media. Antagonist treated media was also used to create the 80/20 Matrigel/media mixture. Media was changed every 2 days. DMSO was used as a vehicle control.
For shRNA experiments, PLKO.1-shRNA viral plasmids (see Key Resources Table) were transfected into HEK293T cells along with packaging plasmids pMD2.G and psPAX2 (Addgene #12259 and #12260, a gift from Didier Trono) using Lipofectamine 2000 (Invitrogen). After media change, virus was collected for 48 hours following transfection and concentrated using PEG-It (System Bioscience). Enteroids were infected with concentrated lentivirus using published methods (Andersson-Rolf et al., 2014). Wildtype (B6) enteroids were grown in WENR with 10 μM Nicotinamide (Sigma), with 10 μM Y-27632 (Cell Signaling) to achieve cystic morphology and then trypsinized, split into groups and infected with concentrated control (pLKO-shScr) or shPrdm16 (pLKO-shPrdm16) lentivirus in the same medium plus 8 mg/ml polybrene. Enteroid fragments were spinoculated in a 48 well plate at 600 x g for 1 hour at 32° C and then incubated at 37° C for 5 hours. Enteroid fragments were spun down, plated in Matrigel and incubated in WENR media for 4 days, followed by 4d treatment with 1 μg/ml Puromycin to select for transduced cells. Enteroids were then transferred to ENR medium to stimulate budding. For in vivo crypt knockout experiments, mice were injected with tmx. 3 days later, crypts were isolated and plated (150/per well) in 24 well plates. Enteroid formation was scored on d7.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rb anti Cleaved Notch1 (Val1744) (D3B8) | Cell Signaling | RRID:AB_2153348 |
| Rb anti Cleaved Caspase 3 (Asp175) (5A1E) | Cell Signaling | RRID: AB_2070042 |
| Ms anti E-Cadherin | BD Transduction | RRID:AB_397580 |
| Rb anti KI67 (SP6) | Abcam | RRID:AB_302459 |
| Gt anti Lysozyme C Antibody (W-20) | Santa Cruz | RRID:AB_2138793 (Disc) |
| Rb anti Muc2 (H-300) | Santa Cruz | RRID:AB_2146667 (Disc) |
| Ms anti-p53 (1C12) | Cell Signaling | RRID:AB_331743 |
| Gt anti GFP | Abcam | RRID:AB_305643 |
| Rb anti Prdm16 | P. Seale | Made in house |
| Rb anti Prdm16 | R and D | RRID:AB_10717965 |
| Ms anti GAPDH (GA1R) | Thermo | RRID:AB_10977387 |
| Ms anti Tubulin (DM1A) | Sigma | RRID:AB_477583 |
| Rb anti Pparg (H-100) | Santa Cruz | RRID:AB_654710 |
| Rb anti Pparg (81B8) | Cell Signaling | RRID:AB_823598 |
| Rb anti Ppara (H-98) | Santa Cruz | RRID:AB_2165737 |
| Rb anti Lysozyme C | Agilent | RRID:AB_2341230 |
| Rb anti Chromogranin A | Abcam | RRID:AB_301704 |
| Rb anti MUC2 | Cloud Clone | RRID:AB_2811028 |
| Rb anti RFP | Rockland | RRID:AB_2209751 |
| Ms anti IFNAR1 (MAR15A3) | Leinco Tech. Inc | RRID:AB_2491621 |
| anti-rabbit IgG, Alexa 647 donkey | Thermo Fisher | RRID:AB_2536183 |
| anti-goat IgG, Alexa 488 donkey | Thermo Fisher | RRID:AB_2534102 |
| anti-mouse IgG, Alexa 555 donkey | Thermo Fisher | RRID:AB_2536180 |
| anti-rabbit Alexa 488 goat | Thermo Fisher | RRID:AB_2536180 |
| DAPI | Roche | Cat#10236276001 |
| anti- CD117 (c-Kit) APC/Cy7 (2B8) | Biolegend | RRID:AB_1626280 |
| anti-CD24 Pacific Blue (MI/69) | Biolegend | RRID:AB_572011 |
| anti-CD31 PE (390) | Biolegend | RRID:AB_312902 |
| anti-CD45 PE (30-F11) | Biolegend | RRID:AB_312971 |
| anti-Ter119 PE (Ly 76) | Biolegend | RRID:AB_313708 |
| anti-CD326 (EpCAM) APC (G8.8) | EBioscience | RRID:AB_2734965 |
| Anti-mouse IgG IR Dye 680RD | Li-Cor | RRID:AB_10953628 |
| and anti-rabbit IgG IRDye 800CW | Li-Cor | RRID:AB_621848 |
| anti-rabbit-IgG-HRP | Cell Signaling | RRID:AB_2099233 |
| anti-mouse IgG (H/L)-HRP | Cell Signaling | RRID:AB_330924 |
| Anti-H3K27ac | Active Motif | RRID:AB_2561016 |
| Normal Rabbit IgG | Cell Signaling | RRID:AB_1031062 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Intesticult | Stem Cell Tech. | Cat#06005 |
| Murine EGF, Recombinant | Preprotech | Cat#315-09 |
| Murine Noggin, Recombinant | Preprotech | Cat#250-38 |
| Human R-Spondin-1, Recombinant | Preprotech | Cat#120-38 |
| Human Wnt3a, Recombinant | R&D Systems | Cat#5036-WN-010 |
| HBSS, No Ca+, No Mg+ | Thermo Fisher | Cat#14175079 |
| EDTA (Disodium Salt) | Fisher | Cat#BP 120-500 |
| CHIR 99021 | Biovision | Cat#1677-5 |
| N-Acetyl-L-cysteine | Sigma | Cat#A9165 |
| Y-27632 | Cell Signaling | Cat#13624S |
| N2 Supplement | Thermo Fisher | Cat#17502048 |
| B-27 Supplement | Thermo Fisher | Cat#17504044 |
| Advanced DMEM F12 Medium | Thermo Fisher | Cat#12634010 |
| Matrigel | Corning | Cat#356231 |
| Nicotinamide | Sigma | Cat#N0636 |
| RPMI 1640 | Invitrogen | Cat#11835030 |
| L-Carnitine | Sigma | Cat#C0283 |
| L-Glutathione, Reduced | Sigma | Cat#G4251 |
| Dulbecco’s MEM (DMEM) F-12, w/o Amino Acids, L-Glutamine, Glucose, Sodium Pyruvate | US Biologicals | Cat#D9807-11 |
| Glutamax 100x | Thermo Fisher | Cat#35050061 |
| Hepes 1M | Thermo Fisher | Cat#15630080 |
| MEM non-essential amino acids, 100x | Thermo Fisher | Cat#11140050 |
| MEM Amino Acids Solution, 50X | Thermo Fisher | Cat#11130051 |
| D-Glucose (U-13C6, 99%) | Cambridge Isotopes | Cat#CLM-1396 |
| Palmitic Acid (13C16, 99%) | Cambridge Isotopes | Cat# CLM-3957 |
| Palmitic Acid | Sigma | Cat#76119 |
| TrypLE Express | Invitrogen | Cat#12605010 |
| Tamoxifen (Free Base) | Sigma | Cat#T5648 |
| 4-hydroxy-tamoxifen | Sigma | Cat#H6278 |
| Etomoxir | Sigma | Cat#E1905 |
| Corn Oil | Sigma | Cat#C8267 |
| 16% Paraformaldehyde | EMS | Cat#15710 |
| Normal Donkey Serum | Sigma | Cat#D9663 |
| Vectashield | Vector Biolabs | Cat#H-1000 |
| Protein-A Sepharose CL-4B Beads | GE Healthcare | Cat#17-0780-01 |
| Acetonitrile (HPLC) | Fisher | Cat#A998-1 |
| Methanol (HPLC) | VWR | Cat#MX0475-1 |
| Water (HPLC) | Fisher Chemical | Cat#W5-1 |
| Formic Acid LC/MS Grade | Pierce | Cat#28905 |
| Fluoroscein isothiocyanate (FITC) dextran | Sigma | Cat#FD4 |
| Blood Agar (TSA w/5% Sheep Blood) | Fisher | Cat#R01201 |
| TruSeq RNA Sample Prep Kit v2 set A | Illumina | Cat#RS-122-2001 |
| PCR Master Mix, Power SYBR Green | Applied Biosystems | Cat#4367659 |
| MILLIPLEX Mouse Cytokine/Chemokine Magnetic Bead Immunology Multiplex Assay | Millipore | Cat#MCYTMAG-70K-PX32 |
| Lipofectamine 2000 | Invitrogen | Cat#11668027 |
| PEG-It | System Bioscience | Cat#LV810A-1 |
| NEBNext Ultra II DNA Library Prep with Sample Purification Beads | NEB | Cat#E7103 |
| NEB Next Ultra RNA Library Prep Kit | NEB | Cat#E7530 |
| Sodium Acetate | Sigma | Cat#S5636 |
| Sodium Chloride | Sigma | Cat#S9888 |
| DNase I, RNase Free, Recombinant | Roche | Cat#04716728001 |
| DMEM | MediaTech | Cat#MT10-017-CV |
| DTT | Roche | Cat#3117006007 |
| Red Blood Cell Lysis Buffer | Biolegend | Cat#420301 |
| Dispase II | Roche | Cat#4942078001 |
| Fetal Bovine Serum (Lot 401714) | Omega Scientific | Cat#FB-11 |
| Fluorobrite DMEM | Gibco | Cat#A18967 |
| AbC Total Compensation Beads | Molecular Probes | Cat#A10497 |
| 7-AAD Viability Dye | Biolegend | Cat#420404 |
| Arcturus PicoPure RNA Isolation Kit | ThermoFisher | Cat#KIT0204 |
| TRIzol | Invitrogen | Cat#15596018 |
| Purelink RNA Mini columns | Invitrogen | Cat#LT-12183018 |
| SuperSignal West Dura ECL | Fisher | Cat#PIA34075 |
| HyBlot CL Autoradiography Film | Denville | Cat#E3012 |
| ABI High-Capacity cDNA Synthesis kit | Applied Biosystems | Cat#4368813 |
| Borg Decloaker RTU | Biocare | Cat#BD1000 |
| TSA TMR Tyramide Reagent Pack | Akoya Biosciences | Cat#NEL742001KT |
| TSA Fluorescein Tyramide Reagent Pack | Akoya Biosciences | Cat#NEL741001KT |
| Bulls Eye Decloaking Buffer | Biocare | Cat#BULL1000 MX |
| GW9662 | Sigma | Cat#M6191 |
| GW6471 | Sigma | Cat#G5045 |
| Deposited Data | ||
| RNA sequencing data | GEO repository | GSE121014 |
| H3K27Ac ChIP Sequencing data | GEO repository | GSE121014 |
| PRDM16 ChIP Sequencing | GEO repository | GSE121014 |
| Experimental Models: Organisms/Strains | ||
| R26CreER | The Jackson Laboratory | RRID:IMSR_JAX:004847 |
| Lgr5-EGFP-IRES-creERT2 | The Jackson Laboratory | RRID:IMSR_JAX:008875 |
| Prdm16 loxP/loxP | Dr. Patrick Seale | RRID:IMSR_JAX:024992 |
| Rosa26 loxp-stop-loxp tdTomato Reporter | The Jackson Laboratory | RRID:IMSR_JAX:007914 |
| p53 loxP/loxP | The Jackson Laboratory | RRID:IMSR_JAX:008462 |
| Oligonucleotides | ||
| See Table S3 for Primers for Real-time PCR | Integrated DNA Technologies (IDT) | NA |
| Recombinant DNA | ||
| PLKO.1-shRNA-Scramble | Addgene | RRID:Addgene_1864 |
| PLKO.1-shRNA-Prdm16 2 (Sequence in table) | The RNAi Consortium (TRC)- Broad Institute via UPenn High-Throughput Screening Core | TRCN0000075460 |
| PLKO.1-shRNA-Prdm16 1 (Sequence in table) | The RNAi Consortium (TRC)- Broad Institute via UPenn High-Throughput Screening Core | TRCN0000075458 |
| pMD2.G | Addgene (D. Trono) | RRID:Addgene_12259 |
| psPAX2 | Addgene (D. Trono) | RRID:Addgene_12260 |
| Software and Algorithms | ||
| GraphPad Prism 7 | Graphpad (https://www.graphpad.com/scientific-software/prism/) | RRID:SCR_002798 |
| UCSC Genome Browser | UCSC (http://genome.ucsc.edu/) | RRID:SCR_005780 |
| Human Protein Atlas (Human Prdm16 expression) | www.proteinatlas.org | RRID:SCR_006710 |
| Morpheus (Heat Maps) by the Broad Institute | https://software.broadinstitute.org/morpheus/ | RRID:SCR_017386 |
| Homer | http://homer.ucsd.edu/ | RRID:SCR_010881 |
| Star Aligner | https://github.com/alexdobin/STAR | RRID:SCR_015899 |
| ToppGene | http://toppgene.cchmc.org/ | RRID:SCR_005726 |
For passaging experiments, 1 well of enteroids was trypsinized for 2 minutes in TrypLE Express (Invitrogen) to form fragments of ~10-20 cells and plated into 4 wells. The number of enteroids recovered from each well was quantified. Enteroids were removed from Matrigel and resuspended in PBS droplets for counting if numbers were high or accurate counts could not be obtained within the Matrigel droplet. The 4 wells plated from each parent well were averaged and represent an N of 1.
All quantifications were performed by taking images of at least 3 wells at 10× magnification using Leica DM IL LED inverted light microscope with a Canon EOS Rebel T1i 15.1 MP CMOS Digital SLR Camera. For enteroid/bud growth, enteroids were photographed over time and the same enteroids were measured using the Photoshop measuring tool. Enteroid fields to photograph were chosen randomly by marking the bottom of culture plate well with 3 dots. All images over the timecourse were centered around the dots and taken in multiple focal planes to allow for tracking of enteroids over time. For buds, measurements were from tip to enteroid body as shown in example images in Fig. S4B. For spheroids/enteroids, measurements made of widest diameters, as shown in example images in Fig. S4G. All replicate measures were converted to percent growth of single enteroids over time, aggregated and graphed. Viable enteroids were defined as having a clear and unbroken epithelial cell border. Non-viable enteroids were counted if they could be identified as viable at earlier timepoints. “Non-budding” or spheroid enteroids were defined as have a round or oval shape with no protrusions of the epithelium. “Budding” was defined as an epithelial protrusion representing a crypt emerging from the spheroid body.
RNA Extraction and qRT-PCR.
Total RNA was extracted from isolated crypts or enteroids (following one ice cold PBS wash to dissolve Matrigel) using TRIzol (Invitrogen) combined with Purelink RNA Mini columns (Fisher) and then reverse transcribed into cDNA using the ABI High-Capacity cDNA Synthesis kit (ABI). Real-time PCR was performed on an ABI7900HT PCR machine using SYBR green fluorescent dye (Applied Biosystems). Fold changes were calculated using the delta delta CT method, with Tata Binding Protein (TBP) serving as a normalization control. Primer sequences can be found in Table S3.
Flow Cytometry
Intestines were opened and cleaned in PBS, followed by soaking in HBSS with 30mM EDTA and 1.5mM DTT (Roche) on ice for 20 minutes. Intestines were shifted to HBSS with 30mM EDTA and rotated at 37C for 8 minutes, with shaking every 2 minutes. Samples were strained through a 70 μm filter and pelleted at 300g for 5 minutes at 4°C. Pellets were resuspended in 5 ml DMEM with 0.3U/ml Dispase II (Roche) and rotated at 37°C for 10 min with shaking every 2 min and then quenched with FBS + 10 U/ml DNase (Roche), followed by filtration through a 40 μM filter. Samples were again pelleted at 500g for 5 min at 4°C. Pellets were suspended in 5 ml room temperature Red Blood Cell Lysis Buffer (Biolegend) for 4 min and quenched with DMEM +10% FBS (Omega). Samples were pelleted at 500g for 5 minutes at 4°C and resuspended in FACS medium (FluoroBrite DMEM (Gibco) with 3% FBS, 10U/ml DNase, 10uM Y27632). Samples were stained with: anti-CD45 PE (1:100, Biolegend, Cat#103106, RRID:AB_312971), anti-Ter119 PE (1:100, Biolegend, Cat#116207, RRID:AB_313708), anti- CD31 PE (1:100, Biolegend, Cat#102407, RRID:AB_312902), anti- Epcam APC (1:100, EBiosciences, Cat#17-5791-80, RRID:AB_2734965), anti- CD24 Pacific Blue (1:100, Biolegend, Cat#101820, RRID:AB_572011), anti- c-kit APC-Cy7 (1:100, Biolegend, Cat#105825, RRID:AB_1626280) and 7-AAD viability stain (1:10, Biolegend) for 30 minutes at 4C with rocking. Cells were sorted on an BD FACs Aria II with compensation being performed at the time of acquisition in Diva software by using AbC Total Compensation beads (Molecular Probes) for single-color staining and isolated crypt cells for negative staining and fluorescence (7-AAD and GFP). GFPhi stem cells, GFPlow progenitor cells and Paneth cells were isolated according to the sorting strategy described by (Beyaz et al., 2016). Stem cells were identified as Lgr5-GFPhi, Epcampos, CD24low, C-KITneg/ CD31neg, Ter119neg,CD45neg,7-AADneg. GFPlow progenitors were identified as GFPlow, Epcampos, CD24low, C-KITneg /CD31neg, Ter119neg, CD45neg, 7-AADneg. Paneth cells were isolated as CD24hi, C-KIThi, SSChi, GFPneg, CD31neg,Ter119neg,CD45neg,7-AADneg. Cells were collected in basal crypt culture medium supplemented with 10% FBS, 10 U/ml DNase and 10 μM Y27632. For RNA isolation after sorting, FACS isolated cells were pelleted at 500g and RNA was isolated using Trizol extraction and an Arcturus PicoPure RNA Isolation Kit. cDNA synthesis was performed as above.
Colony Formation Assays
Sorted cells were gently pelleted (500g) and resuspended in complete medium at a concentration of 100 cells/μl. For Stem cell and Paneth cell mixing experiments, cells were mixed at a 1:1 ratio, pelleted again and resuspended at a concentration of 1000 stem cells/10 μl Matrigel with 20% complete medium. Matrigel was solidified for 10 min and overlaid with medium. For stem cell assays without Paneth cells, 1000 cells/10 μl Matrigel were plated with WENR media. Enteroid formation was quantified after 4d and 14d. All colony formation experiments reported were repeated 3 or more times.
RNA-seq analysis
RNA-seq libraries were generated using a Truseq RNA Sample Preparation kit (Version 2, Illumina). RNA-seq was performed by the University of Pennsylvania Next Gen Sequencing Core on a HiSeq 2500 high output sequencer using 100 base pair single-end reads. RNA-seq reads were aligned to UCSC mm9 using STAR pipeline (Dobin et al., 2013) allowing unique mapping only. All samples had uniquely mapped read percentages between 81% and 89% (Table S2F). Tags were counted for each gene using featureCounts (Liao et al., 2014). Differential gene expression was done using edgeR (Robinson et al., 2010). Differential expression was determined using the EdgeR software package using quantile normalized log2 fold change of mutant divided by control. 0-1 False Discovery Rates were calculated from the p-value using a Benjamini-Hochberg correction. Upregulated genes with a FDR <0.05 or downregulated genes with an FDR <0.05 were analyzed using ToppFun Reactome Pathways tool (https://toppgene.cchmc.org/enrichment.jsp) (Chen et al., 2009); FDR<0.1, between 1 and 2000 genes using RefSeq ID entry. Heat maps were created using Morpheus (https://software.broadinstitute.org/morpheus). RNA-seq data sets have been deposited in Gene Expression Omnibus (GEO) with the following accession number: GSE121009. For regional crypt RNA-seq, total RNA was isolated from purified crypts of the duodenum (top 8cm), jejunum (10 cm around small intestinal midpoint) and ileum (bottom 10 cm) in C57/B6J mice. Total RNA was sent to Novogene for library preparation and sequencing. Libraries were prepared using the NEB Next Ultra RNA Library Prep Kit and sequenced on a HiSeqX Ten (150 base pair, paired-end reads). FASTQ files were aligned to mm10 by using STAR-2.5.2a, and all samples had uniquely mapped read percentages between 81% and 87%. Differential gene expression was analyzed by using the R package DESeq2. For comparison of overlap in Prdm16 control vs Mut and Duodenum vs Ileum differentially expressed gene sets, FASTQ files for control vs Prdm16 KO animals were aligned to mm10 by using STAR-2.5.2a, and differential gene expression was analyzed by using the R package DESeq2. For the control vs Mut comparison, gene lists were generated with padj<0.2 and log2 Fold Change > 0.5 for upregulated genes or <−0.5 for downregulated genes (in the control compared to the KO). For the Duodenum vs Jejunum comparison, gene lists were generated with padj<0.1 and log2 Fold Change > 0.5 for upregulated genes or <−0.5 for downregulated genes (in the Duodenum compared to the Ileum). Enrichment analysis was performed using the Reactome tool (https://toppgene.cchmc.org/enrichment.jsp) analysis on the genes that were upregulated (or downregulated) in both the control vs mutant and Duodenum vs Ileum comparison (Chen et al., 2013; Kuleshov et al., 2016). All RNAseq data sets includes 3 biological replicates.
13C-Glucose and 13C-Palmitate Metabolomics Labeling and LC/MS
For U-13C-Palmitate labeling, crypts were isolated from the specified small intestine regions. Control (Prdm16loxP/loxP) or global Prdm16 mutant (Rosa26CreERT2; Prdm16loxP/loxP) mice were sacrificed 72h following initial tmx injection. Crypts were counted, and 2,000 crypts were seeded per well of a 24 well plate coated in a 10% Matrigel/90% RPMI mixture. Crypts were overlaid with 500 μl RPMI media containing growth factors (EGF, Noggin, R-spondin), 5 μM CHIR99021, 1 mM NAC, 1 μg/ml L-carnitine (Sigma) and 150 μM U-13C16-Palmitatic-Acid (Cambridge) conjugated to fatty acid-free BSA. Crypts were incubated for 90 min and then 3 wells were combined and spun down in ice cold labeling medium to remove Matrigel and resuspended in 800 μl of extraction buffer (40% methanol (VWR), 40% Acetonitrile (Fisher), 20% water (Fisher), all LC/MS grade with 0.5% formic acid). Samples were vortexed and placed on ice. Within 5 min, samples were neutralized by adding 15% ammonium bicarbonate (Sigma, amount determined before each experiment), vortexed again and returned to ice for 10 min. Samples were cleared in a cold centrifuge at max speed for 10 min. Supernatants were collected and then dried in a vacuum dryer or under a stream of nitrogen, followed by resuspension in 100 μl LC/MS grade water for mass spec analysis.
For glucose labeling, enteroids were grown in 50% Matrigel, 50% media and treated with 1 μM 4-OHT for 48 hours prior to exposure to labeled glucose. To make labeling media, Dulbecco’s MEM (DMEM) F-12, lacking Amino Acids, L-Glutamine, Glucose and Sodium Pyruvate (US Biologicals) was prepared and supplemented with 1X essential and non-essential amino acids (ThermoFisher), 1 μg/ml glutathione (Sigma) and 29 mM sodium bicarbonate (Sigma) and 17.5 mM 13C6 Glucose (Cambridge). This media was used as a base for complete enteroid media (substituted for Advanced DMEM/F12), which was prepared as described above. Enteroids were incubated for 4h in labeled glucose. Enteroids were then recovered by dissolving Matrigel in ice cold medium, spinning briefly and resuspending in extraction buffer and processing as described above.
Cell extracts were analyzed by LC-MS analysis on a Q Exactive PLUS hybrid quadrupole-orbitrap mass spectrometer (Thermo Scientific) coupled to hydrophilic interaction chromatography (HILIC) via electrospray ionization. The LC separation was performed on a XBridge BEH Amide column (150 mm χ 2.1 mm, 2.5 μM particle size, Waters, Milford, MA) using a gradient of solvent A (95%:5% H2O:acetonitrile with 20 mM ammonium acetate, 20 mM ammonium hydroxide, pH 9.4), and solvent B (100% acetonitrile). The gradient was 0 min, 85% B; 2 min, 85% B; 3 min, 80% B; 5 min, 80% B; 6 min, 75% B; 7 min, 75% B; 8 min, 70% B; 9 min, 70% B; 10 min, 50% B; 12 min, 50% B; 13 min, 25% B; 16 min, 25% B; 18 min, 0% B; 23 min, 0% B; 24 min, 85% B; 30 min, 85% B. The flow rate was 150 μL min–1 . Injection volume was 10 μL and autosampler and column temperature were 4 °C; and 25 °C, respectively. The MS scans were in both positive and negative ion mode with a resolution of 70,000 at m/z 200. The automatic gain control (AGC) target was 5 χ 105 and the scan range was 75–1000 at 1Hz. Data were analyzed using MAVEN software (Melamud et al., 2010). Isotope labeling was corrected for natural abundance of 13C using an in-house correction code in R (Su et al., 2017). Percent carbon with 13C label was calculated in Excel using the equation [(%M+1) + 2* (%M+2) + 3*(%M+3) + 4*(%M+4) + 5*(%M+5) + 6*(%M+6)]/6 *100 (for citrate/isocitrate) and [(%M+1) + 2* (%M+2) + 3*(%M+3) + 4*(%M+4) + 5*(%M+5) /5*100] (for α-ketoglutarate) (Fendt et al., 2013).
Human Protein Atlas RNA-seq
Human RNA-seq data from a panel of tissues was accessed via Human Protein Atlas (https://www.proteinatlas.org). Gene level RNA-seq (Uhlen et al., 2015) was downloaded from https://www.proteinatlas.org/download/rna_tissue.tsv.zip.
Chromatin Immunoprecipitation
ChIP analysis was performed as previously described (Shapira et al., 2017). Crypts were isolated and fixed with 1.1% formaldehyde for 15 min at room temperature and then quenched with 2.5 M glycine for 5 mins at room temperature. Samples were washed with PBS, pelleted and resuspended in ChIP lysis buffer (0.6% SDS, 1% Triton X-100, 0.15 M NaCl, 1 mM EDTA, 20 mM Tris at pH 8) with protease inhibitors (Roche) and 1 mM PMSF. Crypts were sonicated to shear chromatin using an Active Motif EpiShear platform (8 minutes active time, 30 sec on, 20 sec off, amplitude 25%). To a achieve a final concentration of 0.1% SDS, chromatin was diluted 1:6 with ChIP dilution buffer (1% Triton X-100, 0.15 M NaCl, 1 mM EDTA, 20 mM Tris at pH 8). Inputs were removed, and primary antibodies were added for overnight incubation at 4°C with rotation. Antibodies used were 3 μg of anti-PRDM16 (P. Seale, made in house), 4 μg of anti-PPARα H98 (Santa Cruz, Cat#sc-9000X, RRID:AB_2165737), 4 μg of anti-PPARγ H100 (Santa Cruz, Cat#sc-7196 X, RRID:AB_654710), 10 μl of anti PPARγ A1 (Cell Signaling, Cat#2443S, RRID:AB_823598), 3 μg anti-H3K27Ac (Active Motif, Cat#39133, RRID:AB_2561016) and 3 μg of normal rabbit IgG (Cell Signaling, RRID:AB_1031062). Protein A Sepharose beads (GE healthcare) were then added for 4 hours at 4°C Samples were washed twice with wash buffer 1 (0.1% SDS, 0.1% NaDOC, 1% Triton X-100, 0.15 M NaCl, 1 mM EDTA, 20 mM Tris at pH 8), once with wash buffer 2 (0.1% SDS, 0.1% NaDOC, 1% Triton X-100, 0.5 M NaCl, 1 mM EDTA, 20 mM Tris at pH 8), once with wash buffer 3 (0.25 M LiCl, 0.5% NaDOC, 0.5% NP-40, 1 mM EDTA, 20 mM Tris at pH 8), and twice with wash buffer 4 (1 mM EDTA, 20 mM Tris at pH 8). Samples were then eluted from beads with warm 100 mM NaHCO3/1% SDS buffer. Samples were reverse cross-linked overnight at 65°C with RN ase A and then treated with proteinase K and column purified (Clontech NucleoSpin). ChIP enrichment was calculated as percent of input. H3K27Ac ChIP control and global Prdm16 mutant samples were prepared as ChIP-seq libraries using the NEBNext ChIP-seq DNA Sample Prep Master Mix Set 1 (NEB) according to manufacturer instructions. PRDM16 ChIP control and global mutant samples were prepared as ChIP-seq libraries using the NEBNext Ultra II DNA Library Prep kit with Sample Purification Beads. Adaptor and primer sequences can be found in the Key Resources table. Libraries were sequenced using a HiSeq 4000 high output sequencer with 100 base pair, single end reads. ChlP-seq data sets have been deposited in Gene Expression Omnibus (GEO). For H3K27Ac, ChIP-seq data was analyzed, using downsampling to match sequencing depth between control and global Prdm16 mutant H3K27Ac samples. All bigwig files for ChIP-seq data were generated using Homer and bedGraphToBigWig command in RPM scale. Sequencing reads were aligned to mm9 using bowtie (Langmead et al., 2009). Peak calling was done by ‘findPeak’ in Homer (Heinz et al., 2010) using an option “-style histone -nfr”. Peaks from control and Prdm16 mutant were pooled to make a master peak set and overlapping peaks were merged if their center distance was <100bp. These signals were further normalized using ‘normalize.loess’ in affy, R package. Differential H3K27ac peaks were defined by 1.5-fold-change cut off. De novo motif search and motif scan were performed using Homer. For PRDM16 ChIP-seq, ChIP-seq reads were aligned to UCSC mm9 using the STAR pipeline. All samples had uniquely mapped read percentages between 74% and 81%. Biological replicates were pooled after deduplicating. Prdm16 peaks were called against KO as a control using Homer (Heinz et al., 2010). To examine the relationship between PRDM16 peaks and gene expression, we measured the distances from transcription start sites of expressed genes to the closest PRDM16 binding sites then compared them by grouping Prdm16 deletion-induced gene expression changes. ChIP-Seq experiments included 2 biological replicates. ChIP qPCR experiments included 3 or more replicates.
Western blot
Protein extracts were prepared by resuspending isolated crypt pellets in RIPA buffer with protease inhibitors (Roche). Samples were sonicated using a Diagenode Bioruptor for 2.5 minutes (30 sec on, 30 sec off, high) and protein was quantified using a standard DC assay (BioRad). Proteins were separated in 4–12% Bis–Tris NuPAGE gels (Invitrogen) and transferred to low-fluorescent PVDF membranes using either a standard wet transfer (90 minutes at 325 amps for PRDM16) or a BioRad Transblot Turbo system (for PPARγ). Antibodies used were as follows: anti-PRDM16, 1:1000 (Seale et al., 2011), anti-PPARγ 1:1000 (Cell Signaling, Cat#2443S, RRID:AB_823598), anti-p53 1:1000 (Cell Signaling, Cat#2524S, RRID:AB_331743), anti-αTUB 1:10,000 (DM1A) (Sigma, Cat#CP06 RRID:AB_477583), and anti-GAPDH 1:10,000 (Thermo, Cat#MA515738, RRID:AB_10977387). Blots were developed using either film or LiCor. For film, blots were incubated with either anti-rabbit-IgG-HRP (Cell Signaling, Cat#7074S, RRID:AB_2099233) or anti-mouse IgG-HRP (Cell Signaling, Cat#7076S, RRID:AB_330924) secondary antibodies, developed using SuperSignal West Dura ECL (Fisher). Alternatively, blots were incubated with the LiCor secondaries anti-mouse IgG IRDye 680RD (Li-Cor, Cat#926-68072, RRID:AB_10953628) and anti-rabbit IgG, donkey, IRDye 800CW (Li-Cor, Cat#926-32213, RRID:AB_621848) and imaged on a LiCOR Odyssey imaging system.
Quantification of fluorescent images
For quantification of area on immunofluorescence images (used for tdTomato and lysozyme), channels of interest were opened in ImageJ. A gaussian blur was applied to the image and threshold was set to identify positive pixels. The image was converted to a mask and particles were analyzed to calculate area. Images were acquired on blinded samples.
QUANTIFICATION AND STATISTICAL ANALYSIS
All experiments reported in this study were repeated at least two independent times. Unless otherwise specified in the main text or figure legends, all sample numbers (n) listed in legends represent biological replicates. For murine enteroid assays, 2-4 wells per group with at least 3 different mice were analyzed. All center values shown in graphs refer to the mean. For analysis of the statistical significance of differences between two groups, two-tailed unpaired Student’s t tests were used. For analysis of three or more groups, a one-way Anova was used. For statistical analysis of two outcomes (i.e. budding, viability), a categorical Fisher exact test was used. No samples or animals were excluded from analysis with the exception of one 13C16-palmitate metabolomic sample from the control group, which was incorrectly extracted and quenched during processing. Sample size estimates were not used. Animals were randomly assigned to groups. Lineage tracing measurements were performed blinded.
DATA AND CODE AVAILABILITY
The accession number for the high throughput sequencing data reported in this paper is GEO: GSE121014.
Supplementary Material
Acknowledgements
We thank members of the Seale laboratory for helpful discussions and careful reading of the manuscript. We thank M. Zhou, R. Fadnavis, and C. Ghougasian for technical assistance and Chi Dang for helpful comments. Thanks to the Functional Genomics Core of the Penn Diabetes Center (DK19525) for sequencing, the Human Immunology Core (P30-Ca016520) for Luminex analysis, and the UPenn Histology and Gene Expression Core. This work was supported by: start-up funds provided by the UPenn Institute for Diabetes, Obesity and Metabolism, NIH grants DK10300802 and DK107589 to P.S.; DK10574303 to R.R.S. and a pilot grant from the Center for Molecular Studies in Digestive and Liver Diseases (NIH P30 DK050306).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession
All sequencing data is available at GSE121014.
Declaration of Interests: The authors declare no competing interests.
References
- Aguilo F, Avagyan S, Labar A, Sevilla A, Lee DF, Kumar P, Lemischka IR, Zhou BY, and Snoeck HW (2011). Prdm16 is a physiologic regulator of hematopoietic stem cells. Blood 117, 5057–5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson-Rolf A, Fink J, Mustata RC, and Koo BK (2014). A video protocol of retroviral infection in primary intestinal organoid culture. J Vis Exp, e51765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baregamian N, Mourot JM, Ballard AR, Evers BM, and Chung DH (2009). PPAR-gamma agonist protects against intestinal injury during necrotizing enterocolitis. Biochem Biophys Res Commun 379, 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, et al. (2007). Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449, 1003–1007. [DOI] [PubMed] [Google Scholar]
- Beyaz S, Mana MD, Roper J, Kedrin D, Saadatpour A, Hong SJ, Bauer-Rowe KE, Xifaras ME, Akkad A, Arias E, et al. (2016). High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 531, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey BW, Finley LW, Cross JR, Allis CD, and Thompson CB (2015). Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterji P, Hamilton KE, Liang S, Andres SF, Wijeratne HRS, Mizuno R, Simon LA, Hicks PD, Foley SW, Pitarresi JR, et al. (2018). The LIN28B-IMP1 post-transcriptional regulon has opposing effects on oncogenic signaling in the intestine. Genes Dev 32, 1020–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Bardes EE, Aronow BJ, and Jegga AG (2009). ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 37, W305–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuikov S, Levi BP, Smith ML, and Morrison SJ (2010). Prdm16 promotes stem cell maintenance in multiple tissues, partly by regulating oxidative stress. Nat Cell Biol 12, 999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Levy JD, Zhang Y, Frontini A, Kolodin DP, Svensson KJ, Lo JC, Zeng X, Ye L, Khandekar MJ, et al. (2014). Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell 156, 304–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrigan DJ, Luchsinger LL, Justino de Almeida M, Williams LJ, Strikoudis A, and Snoeck HW (2018). PRDM16 isoforms differentially regulate normal and leukemic hematopoiesis and inflammatory gene signature. J Clin Invest 128, 3250–3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendt SM, Bell EL, Keibler MA, Davidson SM, Wirth GJ, Fiske B, Mayers JR, Schwab M, Bellinger G, Csibi A, et al. (2013). Metformin decreases glucose oxidation and increases the dependency of prostate cancer cells on reductive glutamine metabolism. Cancer Res 73, 4429–4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmes CD, Dzeja PP, Nelson TJ, and Terzic A (2012). Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell 11, 596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehart H, van Es JH, Hamer K, Beumer J, Kretzschmar K, Dekkers JF, Rios A, and Clevers H (2019). Identification of Enteroendocrine Regulators by Real-Time Single-Cell Differentiation Mapping. Cell 176, 1158–1173 e1116. [DOI] [PubMed] [Google Scholar]
- Harms MJ, Lim HW, Ho Y, Shapira SN, Ishibashi J, Rajakumari S, Steger DJ, Lazar MA, Won KJ, and Seale P (2015). PRDM16 binds MED1 and controls chromatin architecture to determine a brown fat transcriptional program. Genes Dev 29, 298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondares E, Rosell M, Diaz-Delfin J, Olmos Y, Monsalve M, Iglesias R, Villarroya F, and Giralt M (2011). Peroxisome proliferator-activated receptor alpha (PPARalpha) induces PPARgamma coactivator 1alpha (PGC-1alpha) gene expression and contributes to thermogenic activation of brown fat: involvement of PRDM16. J Biol Chem 286, 43112–43122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Iwai R, Tabata H, Konno D, Komabayashi-Suzuki M, Watanabe C, Iwanari H, Mochizuki Y, Hamakubo T, Matsuzaki F, et al. (2017). Prdm16 is crucial for progression of the multipolar phase during neural differentiation of the developing neocortex. Development 144, 385–399. [DOI] [PubMed] [Google Scholar]
- Iqbal J, and Hussain MM (2009). Intestinal lipid absorption. Am J Physiol Endocrinol Metab 296, E1183–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE, Schafer ZT, Evans RM, Suda T, Lee CH, et al. (2012). A PML-PPAR-delta pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med 18, 1350–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, and Ito K (2016). Metabolism and the Control of Cell Fate Decisions and Stem Cell Renewal. Annu Rev Cell Dev Biol 32, 399–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura S, Seale P, Tomaru T, Erdjument-Bromage H, Cooper MP, Ruas JL, Chin S, Tempst P, Lazar MA, and Spiegelman BM (2008). Regulation of the brown and white fat gene programs through a PRDM16/CtBP transcriptional complex. Genes Dev 22, 1397–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobloch M, Pilz GA, Ghesquiere B, Kovacs WJ, Wegleiter T, Moore DL, Hruzova M, Zamboni N, Carmeliet P, and Jessberger S (2017). A Fatty Acid Oxidation-Dependent Metabolic Shift Regulates Adult Neural Stem Cell Activity. Cell Rep 20, 2144–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44, W90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- McDonnell E, Crown SB, Fox DB, Kitir B, Ilkayeva OR, Olsen CA, Grimsrud PA, and Hirschey MD (2016). Lipids Reprogram Metabolism to Become a Major Carbon Source for Histone Acetylation. Cell Rep 17, 1463–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamud E, Vastag L, and Rabinowitz JD (2010). Metabolomic analysis and visualization engine for lC-MS data. Anal Chem 82, 9818–9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middendorp S, Schneeberger K, Wiegerinck CL, Mokry M, Akkerman RD, van Wijngaarden S, Clevers H, and Nieuwenhuis EE (2014). Adult stem cells in the small intestine are intrinsically programmed with their location-specific function. Stem Cells 32, 1083–1091. [DOI] [PubMed] [Google Scholar]
- Mihaylova MM, Cheng CW, Cao AQ, Tripathi S, Mana MD, Bauer-Rowe KE, Abu-Remaileh M, Clavain L, Erdemir A, Lewis CA, et al. (2018). Fasting Activates Fatty Acid Oxidation to Enhance Intestinal Stem Cell Function during Homeostasis and Aging. Cell Stem Cell 22, 769–778 e764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno H, Shinoda K, Ohyama K, Sharp LZ, and Kajimura S (2013). EHMT1 controls brown adipose cell fate and thermogenesis through the PRDM16 complex. Nature 504, 163–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno H, Shinoda K, Spiegelman BM, and Kajimura S (2012). PPARgamma agonists induce a white-to-brown fat conversion through stabilization of PRDM16 protein. Cell Metab 15, 395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro I, Margueron R, Shukeir N, Eisold M, Fritzsch C, Richter FM, Mittler G, Genoud C, Goyama S, Kurokawa M, et al. (2012). Prdm3 and Prdm16 are H3K9me1 methyltransferases required for mammalian heterochromatin integrity. Cell 150, 948–960. [DOI] [PubMed] [Google Scholar]
- Porta C, Hadj-Slimane R, Nejmeddine M, Pampin M, Tovey MG, Espert L, Alvarez S, and Chelbi-Alix MK (2005). Interferons alpha and gamma induce p53-dependent and p53-independent apoptosis, respectively. Oncogene 24, 605–615. [DOI] [PubMed] [Google Scholar]
- Potten CS, and Loeffler M (1990). Stem cells: attributes, cycles, spirals, pitfalls and uncertainties. Lessons for and from the crypt. Development 110, 1001–1020. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Colman MJ, Schewe M, Meerlo M, Stigter E, Gerrits J, Pras-Raves M, Sacchetti A, Hornsveld M, Oost KC, Snippert HJ, et al. (2017). Interplay between metabolic identities in the intestinal crypt supports stem cell function. Nature 543, 424–427. [DOI] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, et al. (2009). Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265. [DOI] [PubMed] [Google Scholar]
- Schell JC, Wisidagama DR, Bensard C, Zhao H, Wei P, Tanner J, Flores A, Mohlman J, Sorensen LK, Earl CS, et al. (2017). Control of intestinal stem cell function and proliferation by mitochondrial pyruvate metabolism. Nat Cell Biol 19, 1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug ZT, Vande Voorde J, and Gottlieb E (2016). The metabolic fate of acetate in cancer. Nat Rev Cancer 16, 708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, Scime A, Devarakonda S, Conroe HM, Erdjument-Bromage H, et al. (2008). PRDM16 controls a brown fat/skeletal muscle switch. Nature 454, 961–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P, Conroe HM, Estall J, Kajimura S, Frontini A, Ishibashi J, Cohen P, Cinti S, and Spiegelman BM (2011). Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J Clin Invest 121, 96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira SN, Lim HW, Rajakumari S, Sakers AP, Ishibashi J, Harms MJ, Won KJ, and Seale P (2017). EBF2 transcriptionally regulates brown adipogenesis via the histone reader DPF3 and the BAF chromatin remodeling complex. Genes Dev 31, 660–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada IS, Acar M, Burgess RJ, Zhao Z, and Morrison SJ (2017). Prdm16 is required for the maintenance of neural stem cells in the postnatal forebrain and their differentiation into ependymal cells. Genes Dev 31, 1134–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrum B, Anantha RV, Xu SX, Donnelly M, Haeryfar SM, McCormick JK, and Mele T (2014). A robust scoring system to evaluate sepsis severity in an animal model. BMC Res Notes 7, 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Lu W, and Rabinowitz JD (2017). Metabolite Spectral Accuracy on Orbitraps. Anal Chem 89, 5940–5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara K, Olson TS, Moskaluk CA, Stevens BK, Hoang S, Kozaiwa K, Cominelli F, Ley KF, and McDuffie M (2005). Linkage to peroxisome proliferator-activated receptor-gamma in SAMP1/YitFc mice and in human Crohn’s disease. Gastroenterology 128, 351–360. [DOI] [PubMed] [Google Scholar]
- Takeda N, Jain R, LeBoeuf MR, Wang Q, Lu MM, and Epstein JA (2011). Interconversion between intestinal stem cell populations in distinct niches. Science 334, 1420–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TeSlaa T, Chaikovsky AC, Lipchina I, Escobar SL, Hochedlinger K, Huang J, Graeber TG, Braas D, and Teitell MA (2016). alpha-Ketoglutarate Accelerates the Initial Differentiation of Primed Human Pluripotent Stem Cells. Cell Metab 24, 485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CA, Wojta K, Pulakanti K, Rao S, Dawson P, and Battle MA (2017). GATA4 Is Sufficient to Establish Jejunal Versus Ileal Identity in the Small Intestine. Cell Mol Gastroenterol Hepatol 3, 422–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomas J, Mulet C, Saffarian A, Cavin JB, Ducroc R, Regnault B, Kun Tan C, Duszka K, Burcelin R, Wahli W, et al. (2016). High-fat diet modifies the PPAR-gamma pathway leading to disruption of microbial and physiological ecosystem in murine small intestine. Proc Natl Acad Sci U S A 113, E5934–E5943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- VanDussen KL, Carulli AJ, Keeley TM, Patel SR, Puthoff BJ, Magness ST, Tran IT, Maillard I, Siebel C, Kolterud A, et al. (2012). Notch signaling modulates proliferation and differentiation of intestinal crypt base columnar stem cells. Development 139, 488–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Rong X, Palladino END, Wang J, Fogelman AM, Martin MG, Alrefai WA, Ford DA, and Tontonoz P (2018). Phospholipid Remodeling and Cholesterol Availability Regulate Intestinal Stemness and Tumorigenesis. Cell Stem Cell 22, 206–220 e204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Ishibashi J, Trefely S, Shao M, Cowan AJ, Sakers A, Lim HW, O’Connor S, Doan MT, Cohen P, et al. (2019). A PRDM16-Driven Metabolic Signal from Adipocytes Regulates Precursor Cell Fate. Cell Metab 30, 174–189 e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei P, Dove KK, Bensard C, Schell JC, and Rutter J (2018). The Force Is Strong with This One: Metabolism (Over)powers Stem Cell Fate. Trends Cell Biol 28, 551–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, and Thompson CB (2009). ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong BW, Wang X, Zecchin A, Thienpont B, Cornelissen I, Kalucka J, Garcia-Caballero M, Missiaen R, Huang H, Bruning U, et al. (2017). The role of fatty acid beta-oxidation in lymphangiogenesis. Nature 542, 49–54. [DOI] [PubMed] [Google Scholar]
- Yan KS, Chia LA, Li X, Ootani A, Su J, Lee JY, Su N, Luo Y, Heilshorn SC, Amieva MR, et al. (2012). The intestinal stem cell markers Bmi1 and Lgr5 identify two functionally distinct populations. Proc Natl Acad Sci U S A 109, 466–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao CH, Liu GY, Wang R, Moon SH, Gross RW, and Patti GJ (2018). Identifying off-target effects of etomoxir reveals that carnitine palmitoyltransferase I is essential for cancer cell proliferation independent of beta-oxidation. PLoS Biol 16, e2003782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X, Jedrychowski MP, Chen Y, Serag S, Lavery GG, Gygi SP, and Spiegelman BM (2016). Lysine-specific demethylase 1 promotes brown adipose tissue thermogenesis via repressing glucocorticoid activation. Genes Dev 30, 1822–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the high throughput sequencing data reported in this paper is GEO: GSE121014.