

Abstract
Background
After acute myocardial infarction, the recovery of ischemic myocardial blood flow may cause myocardial reperfusion injury, which reduces the efficacy of myocardial reperfusion. Ways to reduce and prevent myocardial ischemia/reperfusion (I/R) injury are of great clinical significance in the treatment of patients with acute myocardial infarction. TRAF1 (tumor necrosis factor receptor–associated factor 1) is an important adapter protein that is implicated in molecular events regulating immunity, inflammation, and cell death. Little is known about the role and impact of TRAF1 in myocardial I/R injury.
Methods and Results
TRAF1 expression is markedly induced in wild‐type mice and cardiomyocytes after I/R or hypoxia/reoxygenation stimulation. I/R models were established in TRAF1 knockout mice and wild type mice (n=10 per group). We demonstrated that TRAF1 deficiency protects against myocardial I/R–induced loss of heat function, inflammation, and cardiomyocyte death. In addition, overexpression of TRAF1 in primary cardiomyocytes promotes hypoxia/reoxygenation‐induced inflammation and apoptosis in vitro. Mechanistically, TRAF1 promotes myocardial I/R injury through regulating ASK1 (apoptosis signal‐regulating kinase 1)–mediated JNK/p38 (c‐Jun N‐terminal kinase/p38) MAPK (mitogen‐activated protein kinase) cascades.
Conclusions
Our results indicated that TRAF1 aggravates the development of myocardial I/R injury by enhancing the activation of ASK1‐mediated JNK/p38 cascades. Targeting the TRAF1–ASK1–JNK/p38 pathway provide feasible therapies for cardiac I/R injury.
Keywords: apoptosis, ASK1, cardiac ischemia reperfusion, inflammation, TRAF1
Subject Categories: Myocardial Infarction, Chronic Ischemic Heart Disease, Heart Failure
Clinical Perspective
What Is New?
The expression of TRAF1 (tumor necrosis factor receptor–associated factor 1) is increased in hearts with ischemia/reperfusion (I/R) and hypoxia/reoxygenation‐induced neonatal rat cardiomyocytes.
TRAF1 promotes I/R or hypoxia/reoxygenation‐induced heart or cardiomyocyte injury, inflammatory response, and apoptosis.
TRAF1 exerts its effect by promoting the ASK1 (apoptosis signal–regulating kinase 1)–mediated JNK/p38 (c‐Jun N‐terminal kinase/p38) MAPK (mitogen‐activated protein kinase) signaling pathway.
What Are the Clinical Implications?
This study revealed a novel function of TRAF1 in cardiac I/R injury and proved that inhibiting the expression of TRAF1 can inhibit the process of cardiac I/R injury.
This finding provides a new therapeutic target for cardiac I/R injury and acute myocardial infarction.
This article is instructive for cardiologists who want to better understand the occurrence of cardiac I/R injury.
Introduction
Acute myocardial infarction is one of the diseases with the highest morbidity and mortality in the world. Rapid recovery of blood flow through the occluded coronary artery by mechanical or pharmacological intervention is the most effective treatment for limiting infarct size and improving clinical outcome after acute myocardial infarction.1 However, reperfusion can also cause harmful effects in cardiomyocytes, limiting the treatment of diseases and the prognosis of cardiac surgery.2 The pathogenesis of cardiac ischemia/reperfusion (I/R) injury is complex. Disorders of energy metabolism, massive production of oxygen free radicals,3 Ca2+ overload,4 myocardial cell apoptosis,5 inflammatory response,6 and vascular endothelial cell dysfunction7 are all thought to play important roles in the occurrence of cardiac I/R. It is widely accepted that inhibition of inflammatory response and cardiomyocyte apoptosis are important for inhibiting the development of cardiac I/R injury.8, 9, 10
TRAF1 (tumor necrosis factor receptor–associated factor 1) is one of the 7 members of the TRAF family. TRAF proteins can function as intracellular signaling adapters by binding to TNF (tumor necrosis factor) receptor and IL‐1 receptor superfamilies and causing activation of NF‐κB (nuclear factor κB) and JNK (c‐Jun N‐terminal kinase).11 TRAF1 is quite different from other members of the TRAF family in structure. It lacks the really interesting new gene (RING) finger domain in the N‐terminal region.12, 13 Studies have shown that TRAF1 can inhibit apoptosis induced by activation of the TNF receptor or the T‐cell receptor.14 Nevertheless, in contrast with these studies, TRAF1 can also initiate apoptosis by recruiting FADD (Fas‐associated protein with death domain).15 An investigation of renal cell carcinoma showed that inhibition of TRAF1 resulted in a decreased apoptotic rate in tumor cells.16 In addition, TRAF1 can activate the JNK prodeath pathway and inhibit the AKT cell survival pathway, promoting the apoptosis of neurons during cerebral I/R.17 TRAF1 can also activate the ASK1 (apoptosis signal‐regulating kinase 1)–JNK pathway and promote hepatic injury. In hepatocytes, TRAF1 deficiency inhibits both the inflammatory responses mediated by NF‐κB and cell death controlled by the ASK/JNK pathway.18 However, the role of TRAF1 in the regulation of myocardial I/R injury remains poorly understood.
Based on the regulatory effect of TRAF1 on apoptosis and inflammatory response, we hypothesized that TRAF1 may play an important role in the pathogenesis of myocardial I/R injury by regulating inflammation and apoptosis. In this study, we discovered that the mRNA and protein expression levels of TRAF1 were upregulated in heart tissues during myocardial I/R stimulation. TRAF1 depletion ameliorates myocardial infarction, inflammation, and apoptosis. Furthermore, we demonstrated that TRAF1 overexpression can significantly promote hypoxia/reoxygenation (H/R)–induced inflammation and apoptosis in vitro. Moreover, the regulatory effect of TRAF1 on myocardial I/R injury is dependent on promoting the activation of the ASK1–JNK/p38 signaling pathway.
Methods
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure. The data are available from the authors on request.
Reagents
Antibodies against Ikkβ (inhibitor of NF‐κB kinase subunit β), phosphorylated p65 (p‐p65), p65, p‐IkBα (phosphorylated NF‐κB inhibitor α), IkBα, Bax (BCL2 associated X, apoptosis regulator), Bcl2 (BCL2 apoptosis regulator), cleaved Casp3 (caspase 3), p‐ERK (phosphorylated extracellular signal–regulated kinase), ERK, p‐JNK, JNK, p‐p38, p38, p‐ASK1, and GAPDH were purchased from Cell Signaling Technology. Antibodies against p‐Ikkβ and TRAF1 were purchased from Abcam. Antibody against ASK1 was purchased from GeneTex. Goat antimouse and goat antirabbit secondary antibodies were purchased from Jackson Laboratory. FCS was obtained from HyClone. Cell culture reagents and all other reagents were obtained from Sigma‐Aldrich.
Experimental Animals and Mouse Model
The animal protocol was approved by the animal care and use committee of the Center for Animal Experiments at Wuhan University (China). We conducted experiments following the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All animals were housed in an environment with filtered air, uniform temperature (22–24°C), and humidity between 40% and 70%, in a light‐controlled room with food and water available ad libitum.
Myocardial I/R Model
Mice aged 8 to 10 weeks with body weight of 24 to 27 g were anesthetized with sodium pentobarbital (50 mg/kg) via intraperitoneal injections. When the toe pinch reflex was absent, the anesthetized mice were tracheally intubated with a PE‐90 catheter, and a respirator was used to mimic artificial respiration and maintain normal po 2, pco 2, and pH. Body temperature was maintained at 37±0.5°C using a homeothermic blanket. A median sternotomy was performed, and the left coronary artery (LCA) was identified. The LCA was ligated with a 7‐0 silk suture passed with a tapered needle, using blunt dissection. A piece of PE‐10 tubing was placed between the LCA and the 7‐0 silk suture to minimize coronary artery trauma induced by occlusion and to facilitate reperfusion. After 1 hour of ischemia, the ligation line was released and the PE‐10 tubing was removed, and the reperfusion was initiated. The thoracic cavity was closed, and the animal was extubated after recovering consciousness. At 12 or 24 hours after reperfusion, the hearts were harvested (n=10 per group). Sham‐operated animals (n=10 per group) underwent the same surgical procedure without LCA occlusion and reperfusion.
Echocardiography and Hemodynamic Evaluation
Echocardiography and hemodynamic evaluations were performed after the mice (n=10 per group) were anesthetized by inhalation of isoflurane at a concentration of 1.5% to 2.0%. A MyLab30CV system (Biosound Esaote) with a 15‐MHz probe was used for echocardiography. M‐mode tracings derived from the short axis of the left ventricle at the level of the papillary muscles were recorded. Percentage of ejection fraction, left ventricular (LV) end‐diastolic diameter, LV end‐systolic diameter, and LV wall thickness were obtained.
For hemodynamic analysis, cardiac catheterization was conducted using a catheter conductor. Under the condition of pressure control, the catheter was inserted into the LV cavity via the right carotid artery. An Aria pressure‐volume conductance system (MPVS‐300 Signal Conditioner; Millar Instruments) coupled with a PowerLab/4SP A/D converter were used to recorded pressure signals and decreased rate of pressure increase.
Myocardial Infarct Size Determination
After reperfusion, mice (n=5 per group) were anesthetized with sodium pentobarbital, a median sternotomy was performed, and the LCA was religated at the same location. Evans blue dye (2.5% solution, 2 mL) was injected into the jugular vein to expose the nonischemic zone. The hearts were excised, rinsed with saline, and frozen at −20°C for 5 minutes. Frozen ventricles were cut transversely into 5 slices, which were further incubated with 2% TTC for 15 minutes at 37°C. The viable myocardium was stained in red, and the nonviable myocardium could not be stained and appeared white. The sections were imaged, and the area at risk and the infarct area were quantified with Image‐Pro Plus software (v6.0; Media Cybernetics).
Analysis of Myocardial Injury
Serum levels of CPK (creatine phosphokinase) and LDH (lactate dehydrogenase) were assessed with commercial kits (K777‐100 and K726‐500, respectively; BioVision), according to the manufacturer's instructions (n=10 per group).
Immunofluorescence Staining
Heart samples (n=3 per group) were embedded in paraffin for sectioning (5 μm). The sections were dried, dewaxed, hydrated, repaired, washed, and sealed with 10% goat serum for 1 hour. Subsequently, they were incubated overnight at 4°C with the following primary antibodies: anti‐LY6G (anti–lymphocyte antigen 6G) and anti‐CD11b. Next, the sections were washed with PBS and incubated with the corresponding secondary antibodies (for LY6G staining, Alexa Fluor 555 antirat IgG; for CD11b staining, Alexa Fluor 568 antirabbit IgG) for 1 hour. The nuclei were stained with DAPI (4′,6‐diamidino‐2‐phenylindole). Images were collected with a fluorescence microscope (Olympus DX51).
TUNEL Staining
An apoptosis detection kit (ApopTag Plus In Situ Apoptosis Fluorescein Detection Kit; Millipore) was used for TUNEL (terminal deoxynucleotidyl transferase dUTP nick‐end labeling) staining. TUNEL assays were performed according to the manufacturer's instructions (n=3 per group). Images were collected with a fluorescence microscope (Olympus DX51).
Western Blotting and Quantitative Real‐Time Polymerase Chain Reaction
Proteins were extracted from heart tissues and primary cardiomyocytes according to standard protocols. Tissue or cell samples were lysed in RIPA lysis buffer containing the complete Protease Inhibitor (Roche) and PhosStop phosphatase inhibitor (Roche). Protein concentrations were determined using a Pierce BCA Protein Assay Kit. The protein samples were subjected to SDS‐PAGE and transferred to polyvinylidene difluoride membranes. The polyvinylidene difluoride membranes were blocked using 5% nonfat dry milk in a Tris‐buffered saline with Tween 20 buffer and incubated with the corresponding primary antibodies overnight at 4°C. After that, the membranes were rinsed with Tris‐buffered saline with Tween 20 buffer and incubated with horseradish peroxidase–conjugated secondary antibodies. The bands were visualized using Bio‐Rad ChemiDoc XRS. Protein expression levels were normalized to corresponding GAPDH levels.
TRIzol reagent (Invitrogen) was used to extract total mRNA. The mRNA was converted to cDNA using oligo (dT) primers with a Transcriptor First Strand cDNA Synthesis Kit (Roche). Quantitative real‐time PCR (RT‐PCR) amplification of the indicated genes was performed using SYBR Green (Roche). Target gene expression was normalized to Gapdh gene expression. The primers for RT‐PCR are shown in Table.
Table 1.
Primer Sequences for Reverse Transcription Polymerase Chain Reaction
| Gene | Forward | Reverse |
|---|---|---|
| TRAF1 | ATACGATGCTCTCCTGCCCT | CATCTTTGACGTAGGCGTGC |
| TNF‐α | CATCTTCTCAAAATTCGAGTGACAA | TGGGAGTAGACAAGGTACAACCC |
| IFN‐γ | TGCCAAGTTTGAGGTCAACAACCCA | ACCCCGAATCAGCAGCGACT |
| IL‐1β | CCGTGGACCTTCCAGGATGA | GGGAACGTCACACACCAGCA |
| IL‐6 | AGTTGCCTTCTTGGGACTGA | TCCACGATTTCCCAGAGAAC |
| IL‐10 | TGAATTCCCTGGGTGAGAAG | CTCTTCACCTGCTCCACTGC |
| Ccl2 | TGGCTCAGCCAGATGCAGT | CCAGCCTACTCATTGGGATCA |
| Bax | TGAGCGAGTGTCTCCGGCGAAT | GCACTTTAGTGCACAGGGCCTTG |
| Bcl2 | TGGTGGACAACATCGCCCTGTG | GGTCGCATGCTGGGGCCATATA |
| Bad | CCAGAGTTTGAGCCGAGTGAGCA | ATAGCCCCTGCGCCTCCATGAT |
| ICAM‐1 | GGAGCCTCCGGACTTTCGATCT | AGCGGCAGGGTTCTGTCGAA |
| IRF1 | ATGCCAATCACTCGAATGCG | TTGTATCGGCCTGTGTGAATG |
| HIF‐1α | ACCTTCATCGGAAACTCCAAAG | CTGTTAGGCTGGGAAAAGTTAGG |
| PGE2 | GGAGGACTGCAAGAGTCGTC | GCGATGAGATTCCCCAGAACC |
| CD38 | TCTCTAGGAAAGCCCAGATCG | GTCCACACCAGGAGTGAGC |
| Gpr18 | CACCCTGAGCAATCACAACCA | AGTGACATTAACAAACAGCCCA |
| Fpr2 | GAGCCTGGCTAGGAAGGTG | TGCTGAAACCAATAAGGAACCTG |
| Egr2 | GCCAAGGCCGTAGACAAAATC | CCACTCCGTTCATCTGGTCA |
| c‐Myc | ATGCCCCTCAACGTGAACTTC | CGCAACATAGGATGGAGAGCA |
| GAPDH | ACTCCACTCACGGCAAATTC | TCTCCATGGTGGTGAAGACA |
Bad indicates BCL2 associated agonist of cell death; Bax, BCL2 associated X, apoptosis regulator; Ccl2, C‐C motif chemokine ligand 2; Egr2, early growth response 2; Fpr2, formyl peptide receptor 2; Gpr18, G protein‐coupled receptor 18; HIF‐1α, hypoxia inducible factor 1 subunit α; ICAM1, intercellular adhesion molecule 1; IFN‐γ, interferon γ; IL, interleukin; IRF1, interferon regulatory factor 1; NF‐κB, nuclear factor κB; PGE2, prostaglandin E2; TNF‐α, tumor necrosis factor α; TRAF1, tumor necrosis factor receptor–associated factor 1.
Neonatal Rat Cardiomyocyte Isolate and Culture
Neonatal rat cardiomyocytes (NRCMs) were isolated and cultured, as described previously.19 In brief, the 1‐ to 2‐day‐old Sprague‐Dawley rats were used for isolating primary cardiomyocytes. The hearts were excised and digested with PBS containing 0.03% trypsin and 0.04% collagenase type II to isolate the NRCMs from the fibroblasts. NRCMs were enriched and seeded at a density of 1×106 cells per well in 6‐well culture plates. The NRCMs were cultured with DMEM/F12 medium. For cardiomyocytes treated with the reactive oxygen species (ROS) scavenger or ASK1 inhibitor NAC (N‐acetyl‐L‐cysteine; 10 mmol/L), GS‐4997 (selonsertib; 80 μmol/L) was added to the media.
NRCMs H/R Stimulated
After 24 hours of normal culture, the medium was replaced with F10 medium containing 0.1% FCS, and 0.1 mmol/L BrdU (bromodeoxyuridine; to inhibit the proliferation of fibroblasts) to induce hypoxia. The cultured cardiomyocytes were exposed to hypoxia stimulation in a BioSpherix C‐Chamber (model C‐274; BioSpherix) with a standard cell culture chamber. A concentration of 5% O2 and 5% CO2 was maintained inside the C‐Chamber by injecting N2 and CO2 using a ProOx 110 oxygen controller and a ProCO2 CO2 controller (BioSpherix). For the control group, the C‐Chamber was maintained at 37°C and filled with 5% CO2 and 95% air. After 60 minutes of hypoxia, the cells were then incubated under normal conditions for 3 hours.
Plasmid Construct and Transfection
To overexpress TRAF1, the entire coding region of the rat TRAF1 gene was cloned into the replication‐defective adenoviral vector under the control of the cytomegalovirus promoter (pHBAd 3?flag MCS vector, Hanbio Biotechnology Co., Ltd., Shanghai, China) to obtain the overexpression recombinant adenovirus plasmid. The primer sequences were as follows: forward, 5′‐TCCAGGTACCATTACTAGTATGGCCTCCAGCTCAGCC‐3′; reverse, 5′‐ GTGTTTAAACCTGACTAGTCTAAGCACTGGTGTCCACAATG‐3′. Recombinant constructs were inserted into a replication‐defective adenoviral vector. An adenoviral vector encoding the GFP (green fluorescent protein) gene was used as a control. NRCMs were infected with AdGFP and AdTRAF1 in diluted media at a multiplicity of infection of 10 for 24 hours.
Statistical Analysis
The data are presented as mean±SD. SPSS 19.0 software (IBM Corp) was used for all statistical analyses. A normal distribution test was carried out before subsequent statistical analyses. For data that showed a normal distribution and homogeneity of variance, a 1‐way ANOVA or 2‐tailed Student t test was performed. Statistical differences among >2 groups were compared using 1‐way ANOVA, followed by Bonferroni analysis (for data meeting homogeneity of variance) or Tamhane T2 analysis (for data demonstrating heteroscedasticity). Statistical differences between 2 groups were compared with a 2‐tailed Student t test. P<0.05 was considered significant.
Results
TRAF1 Expression Is Induced by ROS During Myocardial I/R Injury
To investigate the changes of TRAF1 expression during I/R injury, a mouse model of myocardial I/R injury was constructed and the mRNA levels of Traf1 were detected by RT‐PCR. The results showed that the mRNA expression levels of Traf1 were gradually upregulated after I/R injury (Figure 1A). Western blot results showed that the protein level of TRAF1 was significantly increased at 24 hours after reperfusion, but no difference was noted between the sham and ischemia surgery groups (Figure 1B). Consistent with the in vivo results, the protein levels of TRAF1 were also significantly increased after H/R treatment for 3 hours in the primary NRCMs (Figure 1C). These results indicated that the expression of TRAF1 was remarkably increased in mouse heart after I/R injury in vivo and in H/R stimulation in vitro. Next, we investigated the reason for the significant TRAF1 increase during myocardial I/R injury. Among many factors contributing to myocardial I/R injury, oxidative stress and ROS played important roles.20, 21 To evaluate whether ROS mediates the increase expression of TRAF1 during myocardial I/R injury, we subjected NRCMs to H2O2 stimulation and found that TRAF1 protein expression was significantly enhanced (Figure 1D). More important, upregulation of TRAF1 mRNA and protein expression induced by H/R stimulation were blocked by treating with ROS scavenger NAC (Figure 1E and 1F). Collectively, these results demonstrate that TRAF1 expression is induced by H/R stimulation through ROS.
Figure 1.
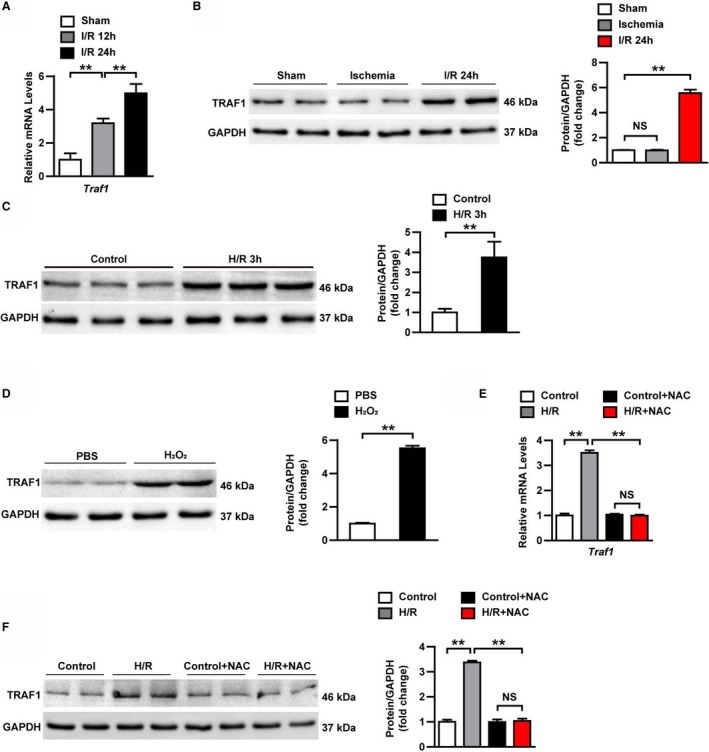
TRAF1 expression is increased by reactive oxygen species during myocardial I/R. A, mRNA expression level of Traf 1 in heart samples of mice at indicated points after I/R (n=3 per group). B, TRAF1 protein expression level in heart tissues in the indicated groups (n=4 per group). C, TRAF1 protein expression level in neonatal rat primary cardiomyocytes exposed to H/R. D, TRAF1 protein expression level in neonatal rat primary cardiomyocytes exposed to H2O2. E, mRNA level of Traf 1 in neonatal rat primary cardiomyocytes in the indicated groups. F, TRAF1 protein expression level in neonatal rat primary cardiomyocytes in the indicated groups. Results shown are representative of 3 blots. For panels (B, C, D, and F), GAPDH served as loading control. For statistical analysis, 1‐way ANOVA was used for panels (A, B, D, and E); a 2‐tailed Student t test was used for panels (C and D). **P<0.01. H/R indicates hypoxia/reoxygenation; I/R, ischemia/reperfusion; NAC, N‐acetyl‐L‐cysteine; NS, not significant; TRAF1, tumor necrosis factor receptor–associated factor 1.
TRAF1 Deficiency Protects Against Myocardial I/R Injury
Elevated TRAF1 expression in mouse hearts after myocardial I/R injury promotes us to investigate the potential functions of TRAF1 in myocardial I/R injury. TRAF1 gene knockout (TRAF1‐KO) mice and wild‐type (WT) mice were used to construct myocardial I/R models (Figure 2A). Serial echocardiography and hemodynamic measurements were used to detect cardiac function at 24 hours after myocardial I/R injury. The echocardiography results showed that absence of TRAF1 significantly inhibited the increase of LV end‐diastolic diameter and LV end‐systolic diameter induced by myocardial I/R injury (Figure 2B). In addition, myocardial I/R injury–induced cardiac dysfunction was also strikingly reduced by TRAF1 deficiency, as indicated by higher LV ejection fraction percentage, and decreased maximum and minimum rates of pressure increase in the TRAF1‐KO group compared with the WT group at 24 hours after I/R injury (Figure 2C and 2D; Figure S1). Consistently, TTC staining exhibited that the ratio of area at risk/LV was similar between the WT and TRAF1‐KO groups at 24 hours after I/R injury, whereas the ratio of infarction size/area at risk was significantly reduced in the TRAF1‐KO group compared with WT after I/R injury (Figure 2E). Moreover, serum levels of CPK and LDH in the TRAF1‐KO I/R group were lower than those in the WT I/R group (Figure 2F and 2G). These data demonstrate that TRAF1 deficiency protects against myocardial I/R injury.
Figure 2.
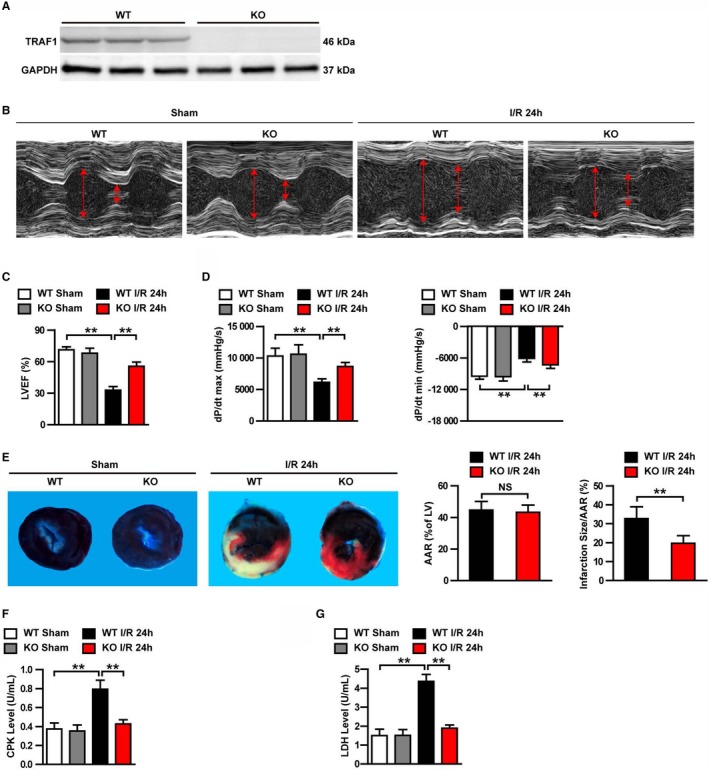
TRAF1 deficiency protected mouse hearts against I/R injury. A, Western blot of TRAF1 expression in KO and WT mice (n=3 per group). GAPDH served as loading control. B, Representative images of echocardiography in WT and TRAF1‐KO mice before (sham) and 24 hours after I/R. C and D, Echocardiographic (C) and hemodynamic (D) assessment of cardiac function in sham and I/R mice (n=10 per group). E, Images of myocardial tissues (Evans blue combination with TTC staining) from WT and TRAF1‐KO mice before (sham) and 24 hours after I/R. The ratios of AAR to left ventricle and infarction size to AAR were quantified by Image‐Pro Plus 6.0 (Media Cybernetics) in the WT and KO groups (n=5 per group). F and G, Quantitative results of serum CKP (F) and LDH (G) before (sham) and 24 hours after I/R (n=10 per group). For statistical analysis, 1‐way ANOVA was used for panels B, D, F, and G; a 2‐tailed Student t test was used for panel (E). **P<0.01. Red arrows represent the left ventricular end‐diastolic and end‐systolic dimension. AAR indicates area at risk; CPK, creatine phosphokinase; dP/dt max, decreased maximum rate of pressure increase; dP/dt min, decreased minimum rate of pressure increase; I/R, ischemia/reperfusion; KO, knockout; LDH, lactate dehydrogenase; LV, left ventricle; LVEF, left ventricular ejection fraction; NS, not significant; TRAF1, tumor necrosis factor receptor–associated factor 1; WT, wild type.
TRAF1 Deficiency Suppresses Inflammatory Response Induced by Myocardial I/R Injury
Because inflammatory response is a key component of myocardial I/R injury,22 we examined the effects of TRAF1 deletion on inflammatory cell infiltration, expression of inflammatory cytokines, and regulation of inflammatory signaling pathways. As shown in Figure 3A, CD11b‐ and LY6G‐positive inflammatory cells accumulated in the hearts of WT mice after myocardial I/R injury, whereas the infiltration of these inflammatory cells was significantly reduced in TRAF1‐KO group (Figure 3A). RT‐PCR results further showed that type 1 macrophage markers (CD38, Gpr18 [G protein‐coupled receptor 18], and Fpr2 [formyl peptide receptor 2]) were significantly decreased, whereas type 2 macrophage markers (Efr2 [early growth response 2] and c‐Myc) were significantly elevated in the cardiac tissue of the TRAF1‐KO group, indicated TRAF1 inhibiting macrophage polarization to M1 (ie, classically activated macrophages; Figure S2). In addition, the mRNA levels of cytokines (TNF‐α, IFN‐γ [interferon γ], IL‐1β [interleukin 1β]) and chemokine (Ccl2 [C‐C motif chemokine ligand 2]) as well as downstream of IL‐1β (HIF‐1α [hypoxia inducible factor 1 subunit α], PGE2 [prostaglandin E2]) and downstream of IFN‐γ (ICAM1 [intercellular adhesion molecule 1], IRF1 [interferon regulatory factor 1]) were markedly attenuated in the hearts of the TRAF1‐KO I/R group compared with the WT I/R group (Figure 3B). NF‐κB signaling is one of the most important pathways in regulating inflammatory response.23 Therefore, to explore the mechanism of TRAF1 deletion in inhibiting immune cell infiltration and inflammatory factor expression, we examined the regulation of TRAF1 on NF‐κB pathways. Western blot results showed that the protein levels of phosphorylated Ikkβ, IkBα, and p65 were significantly decreased in the TRAF1‐KO I/R group compared with the WT I/R group (Figure 3C). Taken together, these results demonstrate that TRAF1 deficiency suppresses inflammatory response induced by myocardial I/R injury.
Figure 3.
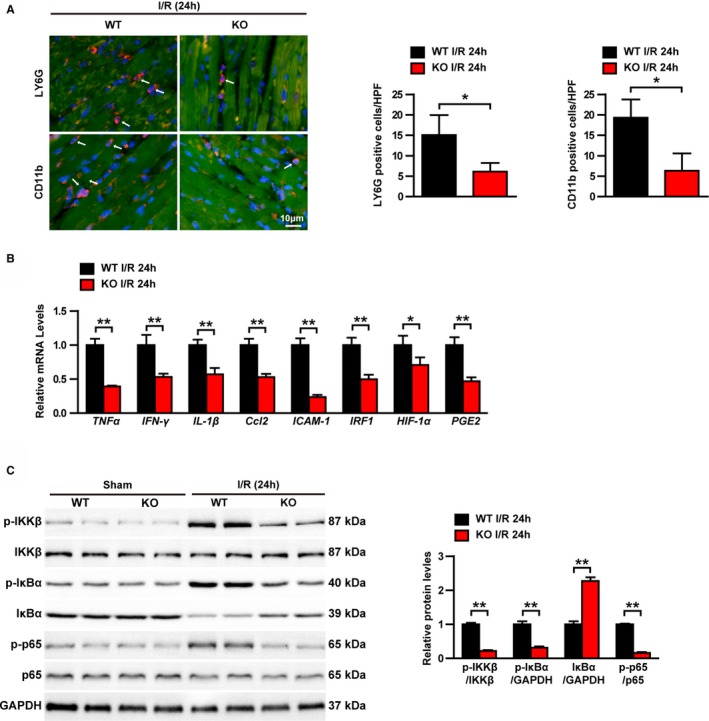
TRAF1 deficiency inhibited the I/R‐induced inflammatory response in heart. A, Immunofluorescence staining and the counted results of positive cells in each high‐power field of CD11b and LY6G in the I/R group (n=3 per group). Blue indicates nuclei; red indicates CD11b‐ and LY6G‐positive staining. B, Relative mRNA levels of cytokines, including TNF‐α, IFN‐γ, IL‐1β, Ccl2, ICAM1, IRF1, HIF‐1α, and PGE2 in heart tissues (n=4 per group). C, Western blot and quantitative results of NF‐κB signaling in sham and I/R groups. Results shown are representative of 3 blots, GAPDH served as loading control. For statistical analysis, a 2‐tailed Student t test was used for panels B and C. *0.01≤ P <0.05, **P<0.01. HPF, high‐power filed. Ccl2 indicates C‐C motif chemokine ligand 2; HIF‐1α, hypoxia inducible factor 1 subunit α; ICAM1, intercellular adhesion molecule 1; IFN‐γ, interferon γ; IL‐1β, interleukin 1β; I/R. ischemia/reperfusion; IRF1, interferon regulatory factor 1; KO, knockout; LY6G, lymphocyte antigen 6G; NF‐κB, nuclear factor κB; p‐, phosphorylated; PGE2, prostaglandin E2; TNF‐α, tumor necrosis factor α; TRAF1, tumor necrosis factor receptor–associated factor 1; WT, wild type.
TRAF1 Deficiency Reduces Apoptosis Induced by Myocardial I/R Injury
Apoptosis is a major contributor to mortality and infarct size after myocardial I/R injury.24 Consequently, we investigated whether TRAF1 affects cardiomyocyte apoptosis. TUNEL staining indicated that myocardial I/R injury–induced cardiomyocyte apoptosis was significantly less in the TRAF1‐KO I/R group compared with the WT I/R group (Figure 4A). Furthermore, the mRNA levels of proapoptotic molecules Bad (BCL2 associated agonist of cell death) and Bax were decreased in the TRAF1‐KO I/R group compared with the WT I/R group, whereas the mRNA levels of antiapoptotic molecule Bcl2 was increased (Figure 4B). Consistent with this result, Western blot results showed that the amount of cleaved Casp3 (caspase 3) and Bax were dramatically increased in the hearts of WT mice after I/R injury but remained significantly lower in the hearts of TRAF1‐KO mice after injury. In contrast, Bcl2 was decreased dramatically in the WT I/R mice but remained at a significantly higher level in the TRAF1‐KO I/R mice (Figure 4C). Collectively, these results indicate that TRAF1 deficiency inhibits cardiomyocyte apoptosis induced by myocardial I/R injury.
Figure 4.
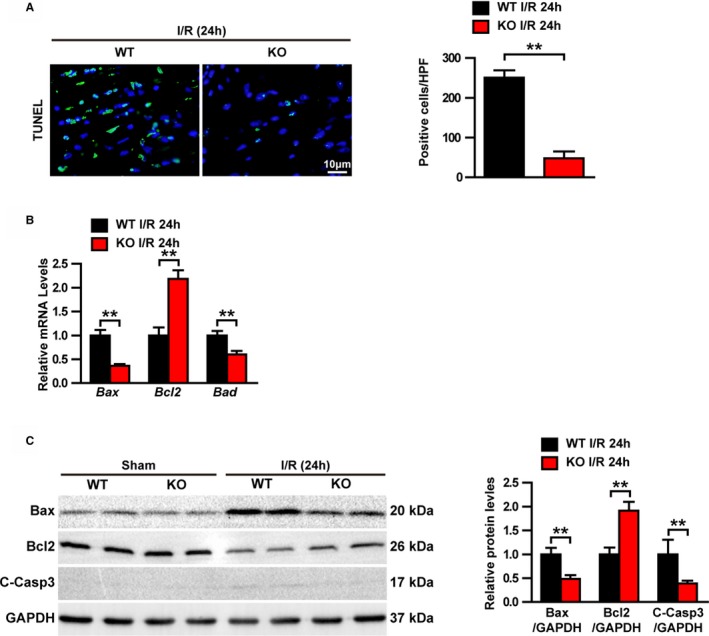
TRAF1 deficiency decreased I/R‐induced apoptosis. A, Immunofluorescence staining and positive cell‐count results of TUNEL in heart tissues (n=3 per group). Blue indicates nuclei; green indicates TUNEL positive staining. B, Relative mRNA levels of apoptosis‐related molecules in heart tissues (n=4 per group). C, Western blot and quantitative results of apoptosis‐related protein expression levels in sham and I/R groups. Results shown are representative of 3 blots, GAPDH served as loading control. For statistical analysis, a 2‐tailed Student t test was used for panels B and C. **P<0.01. Bax indicates BCL2 associated X, apoptosis regulator; Bcl2, BCL2 apoptosis regulator; C‐Casp3, cleaved caspase 3; HPF, high‐power filed; I/R, ischemia/reperfusion; KO, knockout; TRAF1, tumor necrosis factor receptor–associated factor 1; TUNEL, terminal deoxynucleotidyl transferase dUTP nick‐end labeling; WT, wild type.
TRAF1 Overexpression Exacerbates Inflammation and Apoptosis in Cardiomyocytes After H/R Stimulation In Vitro
Our results revealed that TRAF1 deletion inhibited the progress of myocardial I/R injury in systemic TRAF1‐KO mice, which TRAF1 was absent in all cell types. To further clarify whether TRAF1 achieves its function by regulating cardiomyocytes, we used adenovirus to overexpress TRAF1 in cultured primary NRCMs exposed to H/R stimulation. The NRCMs have multiple differences from adult cells, but their responses to inflammation, apoptosis, and oxidative stress are similar to those of I/R‐induced adult cells and are widely used in the study of contraction, ischemia, hypoxia, and toxicity of various compounds in hearts.25 Successful overexpression of TRAF1 was confirmed by Western blot (Figure 5A). Overexpression of TRAF1 significantly promoted inflammation induced by H/R stimulation, as shown by upregulation of expression of inflammatory factors and activation of the NF‐κB pathway in the AdTRAF1 H/R group compared with the AdGFP H/R group (Figure 5B and 5C). Furthermore, overexpression of TRAF1 also led to an increase in the mRNA level of proapoptotic‐related molecules and a decrease in the mRNA level of antiapoptotic molecules (Figure 5D). Accordingly, the elevated levels of the proapoptotic protein Bax and cleaved Casp3 and the decreased level of the antiapoptotic protein Bcl2 were observed in AdTRAF1 H/R group compared with controls (Figure 5E). These results indicate that TRAF1 exacerbates inflammation and apoptosis in cardiomyocytes after H/R stimulation in vitro.
Figure 5.
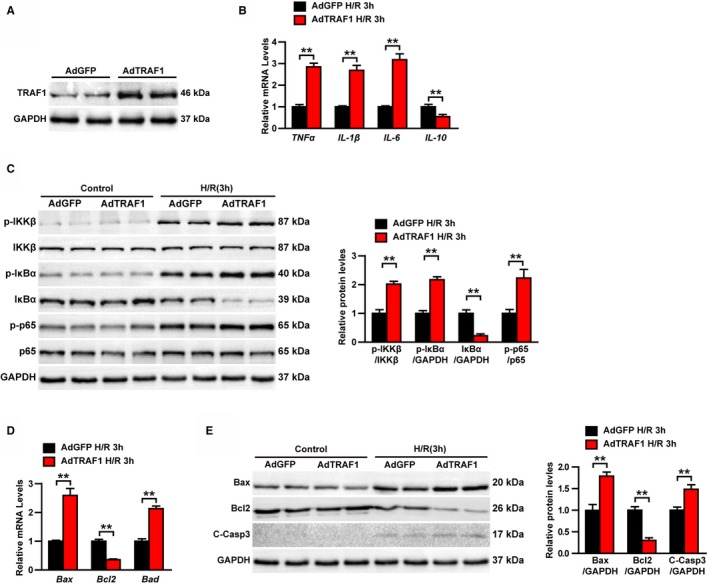
TRAF1 overexpression exacerbates inflammation and apoptosis in cardiomyocytes after H/R stimulation in vitro. A, Western blot results of TRAF1 in AdTRAF1 and AdGFP control group. B, Relative mRNA levels of cytokines, including TNF‐α, IL‐1β, IL‐6, and IL‐10, in AdTRAF1‐ and AdGFP‐group cardiomyocytes exposed to H/R for 3 hours. C, Western blot and quantitative results of NF‐κB signaling in AdTRAF1 and AdGFP control group cardiomyocytes exposed to H/R for 3 hours or not. D, Relative mRNA levels of apoptosis‐related molecules in AdTRAF1‐ and AdGFP‐group cardiomyocytes exposed to H/R for 3 hours. E, Western blot and quantitative results of apoptosis‐related protein expression levels in AdTRAF1‐ and AdGFP‐group cardiomyocytes exposed to H/R for 3 hours or not. For panels B and D, results shown are representative of 3 independent experiments. For panels (A, C, and E), results shown are representative of 3 blots. GAPDH served as loading control. For statistical analysis, a 2‐tailed Student t test was used for panels (B through E). **P<0.01. Ad indicates adenoviral vector encoded; Bax, BCL2 associated X, apoptosis regulator; Bcl2, BCL2 apoptosis regulator; C‐Casp3, cleaved caspase 3; GFP, green fluorescent protein; H/R, hypoxia reoxygenation; IkBα, nuclear factor κB inhibitor α; Ikkβ, inhibitor of nuclear factor κB kinase subunit β; IL, interleukin; NF‐κB, nuclear factor κB; p‐, phosphorylated; TNF‐α, tumor necrosis factor α; TRAF1, tumor necrosis factor receptor–associated factor 1.
TRAF1 Promotes Myocardial I/R–Induced Activation of ASK1–JNK/p38 Pathways
These results proved that TRAF1 can promote I/R‐induced cardiac injury, but the underlying mechanism remains to be studied further. Growing evidence demonstrates that the MAPK (mitogen‐activated protein kinase) signaling pathway plays important roles in inflammatory response and cardiomyocyte apoptosis during myocardial I/R.26, 27 Therefore, Western blot was performed to investigate the possible involvement of MAPK pathways in TRAF1‐associated molecular events during I/R progression. Although the phosphorylation levels of ASK1, ERK, JNK, and p38 were significantly increased 24 hours after I/R, only the activation of ASK1, JNK, and p38 was inhibited in the TRAF1‐KO group after I/R injury (Figure 6A). Conversely, overexpression of TRAF1 potentiated the H/R‐induced activation of ASK1, JNK, and p38 but had no effect on ERK activation (Figure 6B). Our results prove that TRAF1 promotes myocardial I/R–induced activation of ASK1–JNK/p38 pathways.
Figure 6.
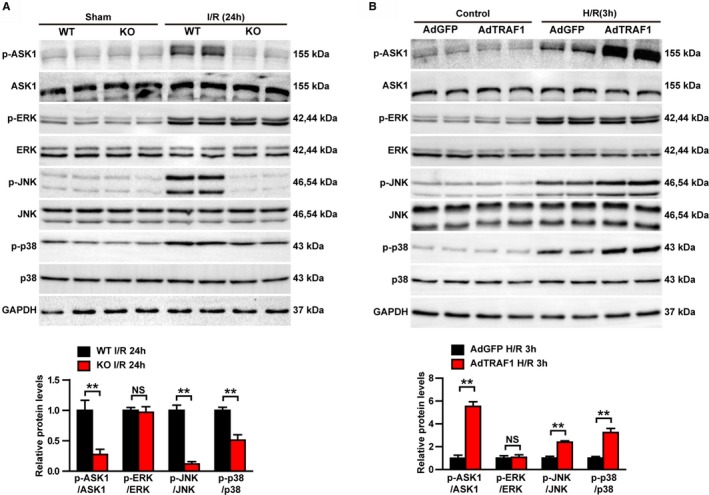
TRAF1 promotes activation of the ASK1–JNK/p38 signaling pathway after myocardial I/R. A, Western blot and quantitative results of phosphorylated and total ASK1, ERK, JNK, and p38 protein from heart tissue of mice in the indicated groups. B, Western blot and quantitative results of phosphorylated and total ASK1, ERK, JNK, and p38 protein in neonatal rat cardiomyocytes infected with AdGFP and AdTRAF1 exposed to H/R for 3 hours or not. Results shown are representative of 3 blots, and GAPDH served as loading control. For statistical analysis, a 2‐tailed Student t test was used for panels (B through E). **P<0.01. Ad indicates adenoviral vector encoded; ASK1, apoptosis signal‐regulating kinase 1; GFP, green fluorescent protein; H/R indicates hypoxia/reoxygenation; I/R, ischemia/reperfusion; JNK/p38, c‐Jun N‐terminal kinase/p38; KO, knockout; NS, not significant; p‐, phosphorylated; TRAF1, tumor necrosis factor receptor–associated factor 1; WT, wild type.
Effect of TRAF1 on Cardiomyocyte Inflammation and Apoptosis Is Mediated by ASK1 Activity
To further elucidate whether ASK1 mediated the effect of TRAF1 on myocardial I/R injury, H/R‐stimulated AdTRAF1 and AdGFP NRCMs were administered GS‐4997, the inhibitor of ASK1. Western blot was used to detect the expression of TRAF1 and the phosphorylation level of ASK1 under normal conditions. The expression of TRAF1 in the AdTRAF1 and AdTRAF1 plus GS‐4997 groups was obviously increased, and the phosphorylation level of ASK1 in the GS4997‐administered group was dramatically decreased (Figure 7A). RT‐PCR and Western blotting revealed that TRAF1 overexpression induced upregulation of inflammatory factors and activation of the NF‐κB signaling pathway, and upregulation of apoptosis‐related molecules were blocked by inhibition of ASK1 (Figure 7B through 7E). These results demonstrate that the promotional effect of TRAF1 on inflammation and apoptosis in cardiomyocytes is mediated by ASK1 activity.
Figure 7.
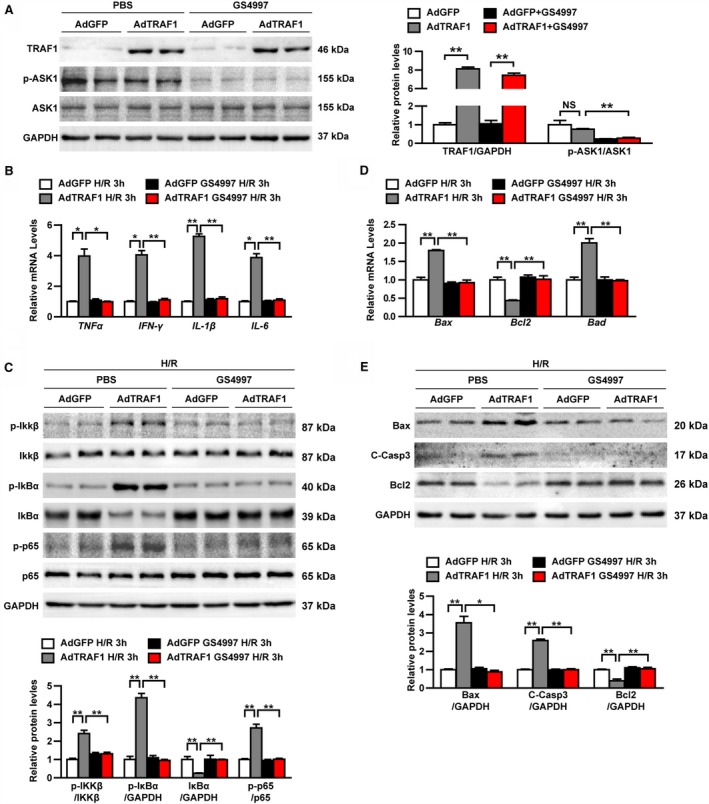
The effect of TRAF1 on cardiomyocyte inflammation and apoptosis is mediated by ASK1 activity. A, Western blot results and quantitative results of TRAF1, ASK1, and phosphorylated ASK1 in the indicated groups. B, Relative mRNA levels of cytokines, including TNF‐α, IFN‐γ, IL‐1β and IL‐6 in the indicated groups exposed to H/R for 3 hours. C, Western blot results and quantitative results of NF‐κB signaling in the indicated groups exposed to H/R for 3 hours. D, Relative mRNA levels of apoptosis‐related molecules in the indicated groups exposed to H/R for 3 hours. E, Western blot results and quantitative results of apoptosis‐related protein expression levels in the indicated groups exposed to H/R for 3 hours. For panels (A, C, and E), results shown are representative of 3 blots, and GAPDH served as loading control. For panels (B and D), results shown are representative of 3 independent experiments. For statistical analysis, 1‐way ANOVA was used for panels A through E. *0.01≤ P<0.05, **P<0.01. Ad indicates adenoviral vector encoded; ASK1, apoptosis signal‐regulating kinase 1; Bad, BCL2 associated agonist of cell death; Bax, BCL2 associated X, apoptosis regulator; Bcl2, BCL2 apoptosis regulator; C‐Casp3, cleaved caspase 3; GFP, green fluorescent protein; H/R, hypoxia/reoxygenation; IFN‐γ, interferon γ; IkBα, nuclear factor κB inhibitor α; Ikkβ, inhibitor of nuclear factor κB kinase subunit β; IL, interleukin; NF‐κB, nuclear factor κB; NS, not significant; p‐, phosphorylated; TNF‐α, tumor necrosis factor α; TRAF1, tumor necrosis factor receptor–associated factor 1.
Discussion
In this study, we characterized the adaptor protein TRAF1 as a novel mediator of the pathological process of myocardial I/R. In the absence of TRAF1, cardiac dysfunction and infarct size were significantly inhibited after I/R and cardiomyocyte apoptosis and inflammation were decreased. Conversely, TRAF1 overexpression significantly reduced cell activity after H/R stimulation. Mechanistically, these deleterious effects of TRAF1 were largely dependent on the activation of ASK1–JNK/p38 signaling. Therefore, TRAF1 might represent a promising therapeutic target for myocardial I/R.
Oxidative stress and ROS play important roles in myocardial I/R injury. Our investigation indicated that TRAF1 expression is induced during I/R injury through ROS, but the mechanism by which oxidative stress induces TRAF1 expression remains unclear. Studies have shown that H2O2 stimulation can reduce the expression level of microRNA miR‐214 in MC3T3‐E1 cells.28 In addition, miR‐214 expression was downregulated in liver tissues of I/R‐ and H/R‐induced hepatocytes, whereas TRAF1 expressions was upregulated; miR‐214 was proved to regulate the expression of TRAF1.29 Thus, the upregulation of TRAF1 expression induced by oxidative stress may be achieved by negative regulation of miR‐214. In addition, the expression of TRAF1 can also be regulated by other molecules, such as ATF3 (activating transcription factor 3) and miR‐1180.30, 31 Although our study demonstrated that oxidative stress induces the expression of TRAF1, other mechanisms may also be able to regulate the expression of TRAF1 and need further study.
Inflammatory cell infiltration and inflammatory response play important roles in myocardial I/R injury. Vascular endothelial cells and cardiomyocytes are activated during I/R, and the expression of adhesion molecules is increased, promoting lymphocytes infiltration.22 Inflammatory cells such as neutrophils and macrophages release a variety of proteolytic enzymes that can degrade the extracellular matrix of vascular endothelial cells and cause tissue damage.32 In addition, infiltrating inflammatory cells can also produce a series of inflammatory factors and chemokines such as TNF‐α, IL‐6, IL‐1β, CCL2, and Cxcl10 (C‐X‐C motif chemokine ligand 10).33 It was found that the levels of cytokines are directly related to the degree of damage to cardiac function and the amount of cell necrosis after ischemia.34 Therefore, inflammation can greatly exacerbate the development of cardiac I/R injury. In our investigation, TRAF1 deficiency inhibited the infiltration of Ly6G‐positive neutrophils and CD11b‐positive macrophages, decreased the expression of proinflammatory cytokines, inhibited the activation of NF‐κB signaling, and regulated the polarization of macrophages. In cardiomyocytes, TRAF1 can regulate the expression of inflammatory cytokines including TNF‐α, IL‐1β, IL‐6, and IL‐10. Inflammatory cytokines, such as IL‐10, have been shown to regulate the polarization of macrophages.35, 36 Consequently, the increased M1 polarization of macrophages observed in the present study may be due to the paracrine pathway induced by TRAF1 deficiency in cardiomyocytes. In addition, a previous study showed that Fusobacterium nucleatum AI‐2 (autoinducer 2) enhanced the mobility and M1 polarization of macrophages, possibly through TNFSF9 (TNF superfamily member 9)/TRAF1/p‐AKT/IL‐1β signaling, indicated that TRAF1 was involved in M1 polarization in macrophages.37 Because global knockout mice were used in this study, we do not exclude the possibility that TRAF1 may directly regulate macrophage polarization; this needs further investigation. All of these results demonstrate that TRAF1 has a powerful regulatory function on inflammatory responses during cardiac I/R. TRAF1 exacerbates myocardial injury by promoting the inflammatory response.
Apoptosis has been proposed as an important mechanism for a significant amount of cell death in reperfused ischemic myocardium.38 Investigations indicated that apoptosis increases significantly after myocardial I/R.10 Moreover, the numbers of apoptosis cells increased with the prolongation of ischemia and reperfusion time.39 Because of limited cardiac regeneration, apoptosis‐induced loss of cardiomyocytes leads directly to an increase in infarcted area, cardiac dysfunction, or even heart failure. The Bcl2 and caspase family proteases are the major regulatory genes of myocardial apoptosis.40, 41 Although TRAF1 was initially proposed to exhibit antiapoptotic properties,42 in this study, we showed that TRAF1 deletion can regulate cardiomyocyte apoptosis, as confirmed by the change of TUNEL‐positive cardiomyocytes, Casp3 activity, and the ratio of proapoptotic (Bax) and antiapoptotic (Bcl2) factors in vivo. Our in vitro study also confirmed that TRAF1 may trigger apoptosis directly in cardiomyocytes. This investigation provides a new therapeutic target for cardiac I/R injury and other cardiac diseases related to myocardial apoptosis.
In the pathogenesis of myocardial I/R, MAPK signaling has been shown to participate in inflammatory and apoptotic responses through the activation and/or inactivation of the 3 critical subfamilies, ERK, JNK, and p38.43 In many studies, the ERK cascade specifically mediates cell growth and survival. In cultured neonatal cardiomyocytes, sustained activation of the ERK pathway mediates adaptive cytoprotection.44 Two other important branches of MAPK signaling, p38 and JNK cascades, function as specialized transducers of stress or injury responses, including responses to inflammatory cytokines, apoptosis, ultraviolet irradiation, heat shock, and I/R. Messadi DV et al reported that the activation of p38 and JNK1 signaling promoted cardiomyocytes apoptosis, inflammation, and fibrosis after myocardial infarction.45 In sympathetic nerve cells, inhibition of JNK phosphorylation using JNK inhibitors can significantly reduce apoptosis. Barancik et al found that inhibiting the activation of p38 using SB203580 protected against cell death in the myocardium.46 Activation of p38 also leads to apoptosis of brain granulosa cells.47 In our present study, we demonstrated that all 3 subunits of the MAPK family could be activated by myocardial I/R and H/R stimulation, but only JNK and p38 were inhibited by the TRAF1 deficiency and markedly enhanced by overexpression of TRAF1. Considerable evidence has indicated that ASK1 can regulate cellular remodeling and apoptosis through activation of MKK3/6 (MAPK kinase 3/6) and MKK4/7, which activate JNK1/2 and p38, respectively.48 In addition, ASK1 plays an important role in I/R injury in many tissues such as heart, kidney, and spinal cord.49, 50, 51 In cerebral and hepatic tissues, TRAF1 activated the ASK1/JNK pathway and promoted I/R injury.17, 18 Consistent with this finding, our results in this study revealed that I/R‐ or hypoxia‐elicited ASK1–JNK/p38 signaling activity was significantly enhanced by TRAF1 overexpression but almost completely suppressed by TRAF1 deficiency.
A limitation of our study was that TRAF1 global knockout mice were used to investigate the function of TRAF1 in myocardial I/R injury. Using TRAF1‐KO mice, we demonstrated that TRAF1 deficiency inhibited myocardial I/R–induced inflammatory response, apoptosis, and cardiac infarction. Subsequently, we revealed that overexpression of TRAF1 in primary neonatal cardiomyocytes promoted cardiomyocyte inflammation and apoptosis, indicating that TRAF1 played an important role in regulation of cardiomyocytes in response to the stimulation of H/R. However, TRAF1 is expressed in various cell types of the heart, including cardiomyocytes, fibroblasts, and endothelial cells.45, 52 Whether downregulation of TRAF1 in noncardiomyocytes contributes to the regulation of the cardiac injury process after I/R is still unclear. Moreover, there are some differences between neonatal and adult cardiomyocytes in function and structure. For example, neonatal cardiomyocytes are far less sensitive to calcium‐containing medium and are less able to culture for long periods of time in vitro compared with adult cardiomyocytes.53 The myofilament in neonatal cardiomyocytes is not fully developed, so adult cardiomyocytes are more suitable for the study of myofibrils.54 Although we demonstrated TRAF1 function in neonatal cardiomyocytes, our findings remain to be confirmed in adult cardiomyocytes.
In summary, our study demonstrates that TRAF1 deficiency has a protective effect on both inflammatory response and cell death after cardiac I/R. The regulating effect of TRAF1 is largely dependent on the regulation of ASK1–JNK/p38 signaling. Based on these findings, TRAF1 may be a novel therapeutic target for preventing I/R injuries that result from revascularization therapy after acute myocardial infarction.
Sources of Funding
This work was supported by Hubei Provincial Natural Science Foundation of China (no. 2017CFB211) and Health Commission of Hubei Province Scientific Research Project (no. WJ2019Q011).
Disclosures
None.
Supporting information
Figure S1. Sample images for hemodynamic measurements in the sham and ischemia/reperfusion groups.
Figure S2. Relative mRNA levels of macrophage marker molecules, including CD38, Gpr18 (G protein‐coupled receptor 18), Fpr2 (formyl peptide receptor 2), Egr2 (early growth response 2), and c‐Myc in the indicated groups (n=4 per group).
(J Am Heart Assoc. 2019;8:e012575 DOI: 10.1161/JAHA.119.012575.)
References
- 1. Hillis LD, Lange RA. Myocardial infarction and the open‐artery hypothesis. N Engl J Med. 2006;355:2475–2477. [DOI] [PubMed] [Google Scholar]
- 2. Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. [DOI] [PubMed] [Google Scholar]
- 3. Lefer DJ, Granger DN. Oxidative stress and cardiac disease. Am J Med. 2000;109:315–323. [DOI] [PubMed] [Google Scholar]
- 4. Kang SM, Lim S, Song H, Chang W, Lee S, Bae SM, Chung JH, Lee H, Kim HG, Yoon DH, Kim TW, Jang Y, Sung JM, Chung NS, Hwang KC. Allopurinol modulates reactive oxygen species generation and ca2+ overload in ischemia‐reperfused heart and hypoxia‐reoxygenated cardiomyocytes. Eur J Pharmacol. 2006;535:212–219. [DOI] [PubMed] [Google Scholar]
- 5. Chen Z, Chua CC, Ho YS, Hamdy RC, Chua BH. Overexpression of Bcl‐2 attenuates apoptosis and protects against myocardial I/R injury in transgenic mice. Am J Physiol Heart Circ Physiol. 2001;280:H2313–H2320. [DOI] [PubMed] [Google Scholar]
- 6. Sodha NR, Clements RT, Feng J, Liu Y, Bianchi C, Horvath EM, Szabo C, Stahl GL, Sellke FW. Hydrogen sulfide therapy attenuates the inflammatory response in a porcine model of myocardial ischemia/reperfusion injury. J Thorac Cardiovasc Surg. 2009;138:977–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Granger DN. Ischemia‐reperfusion: mechanisms of microvascular dysfunction and the influence of risk factors for cardiovascular disease. Microcirculation. 1999;6:167–178. [PubMed] [Google Scholar]
- 8. Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53:31–47. [DOI] [PubMed] [Google Scholar]
- 9. Liao YH, Xia N, Zhou SF, Tang TT, Yan XX, Lv BJ, Nie SF, Wang J, Iwakura Y, Xiao H, Yuan J, Jevallee H, Wei F, Shi GP, Cheng X. Interleukin‐17a contributes to myocardial ischemia/reperfusion injury by regulating cardiomyocyte apoptosis and neutrophil infiltration. J Am Coll Cardiol. 2012;59:420–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huttemann M, Helling S, Sanderson TH, Sinkler C, Samavati L, Mahapatra G, Varughese A, Lu G, Liu J, Ramzan R, Vogt S, Grossman LI, Doan JW, Marcus K, Lee I. Regulation of mitochondrial respiration and apoptosis through cell signaling: cytochrome C oxidase and cytochrome C in ischemia/reperfusion injury and inflammation. Biochem Biophys Acta. 2012;1817:598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee SY, Choi Y. TRAF1 and its biological functions. Adv Exp Med Biol. 2007;597:25–31. [DOI] [PubMed] [Google Scholar]
- 12. Inoue J, Ishida T, Tsukamoto N, Kobayashi N, Naito A, Azuma S, Yamamoto T. Tumor necrosis factor receptor‐associated factor (TRAF) family: adapter proteins that mediate cytokine signaling. Exp Cell Res. 2000;254:14–24. [DOI] [PubMed] [Google Scholar]
- 13. Wajant H, Henkler F, Scheurich P. The tnf‐receptor‐associated factor family—scaffold molecules for cytokine receptors, kinases and their regulators. Cell Signal. 2001;13:389–400. [DOI] [PubMed] [Google Scholar]
- 14. Sabbagh L, Srokowski CC, Pulle G, Snell LM, Sedgmen BJ, Liu YQ, Tsitsikov EN, Watts TH. A critical role for TNF receptor‐associated factor 1 and BIM down‐regulation in CD8 memory T cell survival. Proc Natl Acad Sci USA. 2006;103:18703–18708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chaudhary PM, Eby MT, Jasmin A, Kumar A, Liu L, Hood L. Activation of the NF‐kappab pathway by caspase 8 and its homologs. Oncogene. 2000;19:4451–4460. [DOI] [PubMed] [Google Scholar]
- 16. Rajandram R, Bennett NC, Wang ZQ, Perry‐Keene J, Vesey DA, Johnson DW, Gobe GC. Patient samples of renal cell carcinoma show reduced expression of TRAF1 compared with normal kidney and functional studies in vitro indicate TRAF1 promotes apoptosis: potential for targeted therapy. Pathology. 2012;44:453–459. [DOI] [PubMed] [Google Scholar]
- 17. Lu YY, Li ZZ, Jiang DS, Wang L, Zhang Y, Chen K, Zhang XF, Liu Y, Fan GC, Chen YJ, Yang QL, Zhou Y, Zhang XD, Liu DP, Li HL. traf1 is a critical regulator of cerebral ischaemia‐reperfusion injury and neuronal death. Nat Commun. 2013;4:2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang XF, Zhang R, Huang L, Wang PX, Zhang Y, Jiang DS, Zhu LH, Tian S, Zhang XD, Li H. TRAF1 is a key mediator for hepatic ischemia/reperfusion injury. Cell Death Dis. 2014;5:e1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu Y, Jiang XL, Liu Y, Jiang DS, Zhang Y, Zhang R, Chen YJ, Yang QL, Zhang XD, Fan GC, Li HL. Toll‐interacting protein (TOLLIP) negatively regulates pressure overload‐induced ventricular hypertrophy in mice. Cardiovasc Res. 2014;101:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Misra MK, Sarwat M, Bhakuni P, Tuteja R, Tuteja N. Oxidative stress and ischemic myocardial syndromes. Med Sci Monit. 2009;15:RA209–RA219. [PubMed] [Google Scholar]
- 21. Zhao ZQ. Oxidative stress‐elicited myocardial apoptosis during reperfusion. Curr Opin Pharmacol. 2004;4:159–165. [DOI] [PubMed] [Google Scholar]
- 22. Park JL, Lucchesi BR. Mechanisms of myocardial reperfusion injury. Ann Thorac Surg. 1999;68:1905–1912. [DOI] [PubMed] [Google Scholar]
- 23. Tak PP, Firestein GS. NF‐kappab: a key role in inflammatory diseases. J Clin Invest. 2001;107:7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gottlieb RA, Engler RL. Apoptosis in myocardial ischemia‐reperfusion. Ann N Y Acad Sci. 1999;874:412–426. [DOI] [PubMed] [Google Scholar]
- 25. Chlopcikova S, Psotova J, Miketova P. Neonatal rat cardiomyocytes—a model for the study of morphological, biochemical and electrophysiological characteristics of the heart. Biomed Pap Med Fac Univ Palacky Olomouc, Czech Repub. 2001;145:49–55. [PubMed] [Google Scholar]
- 26. Arthur JSC, Ley SC. Mitogen‐activated protein kinases in innate immunity. Nat Rev Immunol. 2013;13:679–692. [DOI] [PubMed] [Google Scholar]
- 27. Peter AT, Dhanasekaran N. Apoptosis of granulosa cells: a review on the role of MAPK‐signalling modules. Reprod Domest Anim. 2003;38:209–213. [DOI] [PubMed] [Google Scholar]
- 28. Lu XZ, Yang ZH, Zhang HJ, Zhu LL, Mao XL, Yuan Y. Mir‐214 protects mc3t3‐e1 osteoblasts against h2o2‐induced apoptosis by suppressing oxidative stress and targeting atf4. Eur Rev Med Pharmacol Sci. 2017;21:4762–4770. [PubMed] [Google Scholar]
- 29. Huang X, Gao Y, Qin J, Lu S. Mir‐214 down‐regulation promoted hypoxia/reoxygenation‐induced hepatocyte apoptosis through TRAF1/ASK1/JNK pathway. Dig Dis Sci. 2019;64:1217–1225. [DOI] [PubMed] [Google Scholar]
- 30. Li M, Zhai G, Gu X, Sun K. ATF3 and PRAP1 play important roles in cisplatin‐induced damages in microvascular endothelial cells. Gene. 2018;672:93–105. [DOI] [PubMed] [Google Scholar]
- 31. Zhu D, Gao W, Zhang Z. Microrna‐1180 is associated with growth and apoptosis in prostate cancer via TNF receptor associated factor 1 expression regulation and nuclear factor‐kappab signaling pathway activation. Oncol Lett. 2018;15:4775–4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bourboulia D, Stetler‐Stevenson WG. Matrix metalloproteinases (MMPS) and tissue inhibitors of metalloproteinases (TIMPS): positive and negative regulators in tumor cell adhesion. Semin Cancer Biol. 2010;20:161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nossuli TO, Lakshminarayanan V, Baumgarten G, Taffet GE, Ballantyne CM, Michael LH, Entman ML. A chronic mouse model of myocardial ischemia‐reperfusion: essential in cytokine studies. Am J Physiol Heart Circ Physiol. 2000;278:H1049–H1055. [DOI] [PubMed] [Google Scholar]
- 34. Yeh CH, Chen TP, Wu YC, Lin YM, Jing Lin P. Inhibition of NFkappaB activation with curcumin attenuates plasma inflammatory cytokines surge and cardiomyocytic apoptosis following cardiac ischemia/reperfusion. J Surg Res. 2005;125:109–116. [DOI] [PubMed] [Google Scholar]
- 35. Jung M, Ma Y, Iyer RP, DeLeon‐Pennell KY, Yabluchanskiy A, Garrett MR, Lindsey ML. Il‐10 improves cardiac remodeling after myocardial infarction by stimulating M2 macrophage polarization and fibroblast activation. Basic Res Cardiol. 2017;112:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lopes RL, Borges TJ, Zanin RF, Bonorino C. IL‐10 is required for polarization of macrophages to M2‐like phenotype by mycobacterial DnaK (heat shock protein 70). Cytokine. 2016;85:123–129. [DOI] [PubMed] [Google Scholar]
- 37. Wu J, Li K, Peng W, Li H, Li Q, Wang X, Peng Y, Tang X, Fu X. Autoinducer‐2 of fusobacterium nucleatum promotes macrophage m1 polarization via tnfsf9/il‐1beta signaling. Int Immunopharmacol. 2019;74:105724. [DOI] [PubMed] [Google Scholar]
- 38. Hamacher‐Brady A, Brady NR, Gottlieb RA. The interplay between pro‐death and pro‐survival signaling pathways in myocardial ischemia/reperfusion injury: apoptosis meets autophagy. Cardiovasc Drugs Ther. 2006;20:445–462. [DOI] [PubMed] [Google Scholar]
- 39. Freude B, Masters TN, Robicsek F, Fokin A, Kostin S, Zimmermann R, Ullmann C, Lorenz‐Meyer S, Schaper J. Apoptosis is initiated by myocardial ischemia and executed during reperfusion. J Mol Cell Cardiol. 2000;32:197–208. [DOI] [PubMed] [Google Scholar]
- 40. Gustafsson AB, Gottlieb RA. BCL‐2 family members and apoptosis, taken to heart. Am J Physiol‐Cell Physio. 2007;292:C45–C51. [DOI] [PubMed] [Google Scholar]
- 41. Weiland U, Haendeler J, Ihling C, Albus U, Scholz W, Ruetten H, Zeiher AM, Dimmeler S. Inhibition of endogenous nitric oxide synthase potentiates ischemia‐reperfusion‐induced myocardial apoptosis via a caspase‐3 dependent pathway. Cardiovasc Res. 2000;45:671–678. [DOI] [PubMed] [Google Scholar]
- 42. Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS. Nf‐kappa b antiapoptosis: induction of TRAF1 and TRAF2 and c‐IAP1 and c‐IAP2 to suppress caspase‐8 activation. Science. 1998;281:1680–1683. [DOI] [PubMed] [Google Scholar]
- 43. Haq SEA, Clerk A, Sugden PH. Activation of mitogen‐activated protein kinases (p38‐MAPKs, SAPKs/JNKs and ERKs) by adenosine in the perfused rat heart. FEBS Lett. 1998;434:305–308. [DOI] [PubMed] [Google Scholar]
- 44. Yue TL, Wang CL, Gu JL, Ma XL, Kumar S, Lee JC, Feuerstein GZ, Thomas H, Maleeff B, Ohlstein EH. Inhibition of extracellular signal‐regulated kinase enhances ischemia/reoxygenation‐induced apoptosis in cultured cardiac myocytes and exaggerates reperfusion injury in isolated perfused heart. Circ Res. 2000;86:692–699. [DOI] [PubMed] [Google Scholar]
- 45. Messadi DV, Doung HS, Zhang Q, Kelly AP, Tuan TL, Reichenberger E, Le AD. Activation of NFkappaB signal pathways in keloid fibroblasts. Arch Dermatol Res. 2004;296:125–133. [DOI] [PubMed] [Google Scholar]
- 46. Barancik M, Htun P, Strohm C, Kilian K, Schaper W. Inhibition of the cardiac p38‐MAPK pathway by SB203580 delays ischemic cell death. J Cardiovasc Pharmacol. 2000;35:474–483. [DOI] [PubMed] [Google Scholar]
- 47. Harada J, Sugimoto M. An inhibitor of P38 and JNK map kinases prevents activation of caspase and apoptosis of cultured cerebellar granule neurons. Jpn J Pharmacol. 1999;79:369–378. [DOI] [PubMed] [Google Scholar]
- 48. Takeda K, Matsuzawa A, Nishitoh H, Ichijo H. Roles of MAPKKK ASK1 in stress‐induced cell death. Cell Struct Funct. 2003;28:23–29. [DOI] [PubMed] [Google Scholar]
- 49. Toldo S, Breckenridge DG, Mezzaroma E, Van Tassell BW, Shryock J, Kannan H, Phan D, Budas G, Farkas D, Lesnefsky E, Voelkel N, Abbate A. Inhibition of apoptosis signal‐regulating kinase 1 reduces myocardial ischemia‐reperfusion injury in the mouse. J Am Heart Assoc. 2012;1:e002360 DOI: 10.1161/JAHA.112.002360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang Y, Ji HX, Zheng JN, Pei DS, Hu SQ, Qiu SL. Protective effect of selenite on renal ischemia/reperfusion injury through inhibiting ASK1‐MKK3‐P38 signal pathway. Redox Rep. 2009;14:243–250. [DOI] [PubMed] [Google Scholar]
- 51. Wang P, Cao XJ, Nagel DJ, Yin GY. Activation of ASK1 during reperfusion of ischemic spinal cord. Neurosci Lett. 2007;415:248–252. [DOI] [PubMed] [Google Scholar]
- 52. Zirlik A, Bavendiek U, Libby P, MacFarlane L, Gerdes N, Jagielska J, Ernst S, Aikawa M, Nakano H, Tsitsikov E, Schonbeck U. TRAF‐1, ‐2, ‐3, ‐5, and ‐6 are induced in atherosclerotic plaques and differentially mediate proinflammatory functions of CD40 l in endothelial cells. Arterioscler Thromb Vasc Biol. 2007;27:1101–1107. [DOI] [PubMed] [Google Scholar]
- 53. Ehler E, Moore‐Morris T, Lange S. Isolation and culture of neonatal mouse cardiomyocytes. J Vis Exp. 2013; 6:e50154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Eppenberger ME, Hauser I, Baechi T, Schaub MC, Brunner UT, Dechesne CA, Eppenberger HM. Immunocytochemical analysis of the regeneration of myofibrils in long‐term cultures of adult cardiomyocytes of the rat. Dev Biol. 1988;130:1–15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Sample images for hemodynamic measurements in the sham and ischemia/reperfusion groups.
Figure S2. Relative mRNA levels of macrophage marker molecules, including CD38, Gpr18 (G protein‐coupled receptor 18), Fpr2 (formyl peptide receptor 2), Egr2 (early growth response 2), and c‐Myc in the indicated groups (n=4 per group).