

Abstract
Background
Right ventricular (RV) failure because of chronic pressure load is an important determinant of outcome in pulmonary hypertension. Progression towards RV failure is characterized by diastolic dysfunction, fibrosis and metabolic dysregulation. Metabolic modulation has been suggested as therapeutic option, yet, metabolic dysregulation may have various faces in different experimental models and disease severity. In this systematic review and meta‐analysis, we aimed to identify metabolic changes in the pressure loaded RV and formulate recommendations required to optimize translation between animal models and human disease.
Methods and Results
Medline and EMBASE were searched to identify original studies describing cardiac metabolic variables in the pressure loaded RV. We identified mostly rat‐models, inducing pressure load by hypoxia, Sugen‐hypoxia, monocrotaline (MCT), pulmonary artery banding (PAB) or strain (fawn hooded rats, FHR), and human studies. Meta‐analysis revealed increased Hedges’ g (effect size) of the gene expression of GLUT1 and HK1 and glycolytic flux. The expression of MCAD was uniformly decreased. Mitochondrial respiratory capacity and fatty acid uptake varied considerably between studies, yet there was a model effect in carbohydrate respiratory capacity in MCT‐rats.
Conclusions
This systematic review and meta‐analysis on metabolic remodeling in the pressure‐loaded RV showed a consistent increase in glucose uptake and glycolysis, strongly suggest a downregulation of beta‐oxidation, and showed divergent and model‐specific changes regarding fatty acid uptake and oxidative metabolism. To translate metabolic results from animal models to human disease, more extensive characterization, including function, and uniformity in methodology and studied variables, will be required.
Keywords: heart failure, metabolism, myocardial biology, pulmonary hypertension, remodeling
Subject Categories: Metabolism, Heart Failure, Remodeling, Myocardial Biology
Clinical Perspective
What Is New?
This is the first systematic review and meta‐analysis studying metabolic adaptation of the right ventricle in response to pressure overload and includes studies in both animal models and humans.
In the pressure‐loaded right ventricle, glucose uptake and glycolysis were shown to be increased, mediated by insulin‐independent mechanisms irrespective of the model used.
In contrast, changes in mitochondrial respiratory capacity were variable and depended on the animal model used.
What Are the Clinical Implications?
This study implies that in developing and testing future therapeutic options targeting metabolism of the pressure‐loaded right ventricle, one should account for causative factors.
To establish actual translation from experimental models to human disease, experimental methods and outcome parameters should be standardized and uniform.
Introduction
Right ventricular (RV) function is an important predictor for clinical outcome in a variety of cardiac diseases.1, 2, 3, 4 In patients with pulmonary hypertension (PH), RV failure is the main cause of death.2 Development of RV failure because of sustained pressure load is characterized by progressive diastolic dysfunction, changes in fibrotic content, and metabolic remodeling.5, 6, 7, 8, 9 The healthy adult myocardium primarily uses long‐chain fatty acids as substrates, in contrast to the fetal heart, which uses primarily glucose and lactate.10, 11, 12, 13 Under stress, the heart switches to a so‐called “fetal phenotype”, which includes a change in substrate utilization from oxidative metabolism towards glycolysis.12 While these changes may have advantages (ie, better ratio of ATP production versus oxygen use), they may also have disadvantages (eg, increase of stimulation of inflammatory cascades via intermediaries). The RV under pressure may be especially susceptible to changes in substrate utilization because of its unique physiological properties.14 The RV is a thin‐walled crescent‐shaped structure that under physiological conditions is coupled to low‐resistance pulmonary circulation. Increased pressure load in the RV, prevalent in PH, congenital heart disease, and also in left ventricle (LV) failure, causes a relatively high load for the RV. In addition, the RV may be more susceptible compared with the LV because of the relatively higher disadvantageous changes in coronary perfusion with increased afterload. Several studies have attempted to improve RV adaptation by metabolic modulation. Metabolic intervention tested whether direct or indirect stimulation of glucose oxidation by compounds such as dichloroacetate, ranolazine, trimetazidine, and 6‐diazo‐5‐oxo‐l‐norleucine, could be supportive in the pressure‐loaded RV.15, 16, 17, 18, 19, 20, 21 Indeed, these modulations seem to affect cardiac performance positively, but because of the limited number of studies, different models, different compounds, and different study parameters, consensus has not been reached, complicating translation to clinical practice.22, 23 To support the validated setup of clinical trials and to identify challenges and opportunities in evaluating metabolic findings in animal models for human disease, a comprehensive appreciation of all evidence collected in previous studies addressing metabolic adaptation of the RV to pressure load is necessary. The aim of this systematic review and meta‐analysis is to provide an overview of the current knowledge about metabolic remodeling, focusing on carbohydrate and fatty acid metabolism in the pressure‐loaded RV. Both experimental and clinical studies were included, taking into account the different models or type of disease, and the degree and duration of RV pressure load, and RV‐ and clinical function. In addition, we present an overview of the studies performed regarding interventions affecting metabolism in the RV under pressure.
Materials and Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Literature Search
We performed a systematic literature search in Medline and EMBASE on November 29, 2017. The search strategy and global methodological approach using Systematic Review Protocol for Animal Studies, version 2.0 formatted by SYRCLE24, 25 was published on the online platform of the working group Collaborative Approach to Meta‐Analysis and Review of Animal Data for Experimental Studies (CAMARADES) on December 13, 2016. The search strategy was composed to capture overlapping parts of the following domains: (1) RV; (2) pressure load; and (3) metabolism (Data S1).
Study Selection
Two researchers (A.M.C.K. and G.P.L.B.) independently screened the identified abstracts according to the following inclusion criteria: (1) English; (2) original article; (3) RV pressure load; (4) no reversible pressure load; (5) no mixed loading; and (6) RV metabolism. Full texts were screened for control group and sufficiency of the model by confirming increased pressure load by at least (1) increased RV pressure load (ie, RV systolic pressure or mean pulmonary artery pressure), or (2) hypertrophy (ie, RV weight, Fulton index (RV divided by LV+interventricular septum) or RV to body weight ratio). For inclusion of human studies, a control group for pressure load measurements was not required, since inclusion of individuals at study level did meet the criteria of international guidelines for pulmonary hypertension.26
Data Extraction
For the meta‐analysis inclusion, the study had to report on metabolic variables, which were investigated in at least 2 or more other studies. Variable of metabolism was defined as (1) mRNA expression of genes involved in substrate uptake of metabolism; (2) protein expression and/or activity of genes involved in substrate uptake of metabolism; or (3) metabolism measured in vivo or in vitro using either oxygraphy in isolated mitochondria (eg, Oroboros, Clark‐type electrode), oxygraphy in whole cells (eg, Seahorse) or in isolated hearts (eg, Langendorf). General upstream regulators also involved in metabolism (eg, mitogen‐activated protein kinase and AKT [protein kinase B]) were not included. In addition, study characteristics such as species, model/type of pressure load, and degree and duration of pressure load of selected studies were extracted. We extracted the mean, SD (if not presented, SE), and number of subjects (n) of the selected variables from all eligible studies. Universal Desktop Ruler (Avpsoft) was used to derive data from graphs. In case of missing information, authors were contacted. If response was lacking, we approached the data as follows: when the SD was unknown, the SD was calculated when mean difference, (corrected) P value, and number of used subjects were available; in case of unknown SD of the control groups, we used the SD of the experimental group; if the exact n was unknown, the greatest number given was used for the calculation of the SD.
Data Synthesis
Effect sizes, defined as Hedges’ g, with associated CI of 95% were calculated, after which multiple separate random effects meta‐analyses were performed using STATA 11. When the actual number of animals (n) used for a certain variable was unknown (ie, not reported in the manuscript and not acquired after contacting the author), the smallest n mentioned by the authors was used to calculate the Hedges’ g. Combined effect sizes of a particular variable were calculated for (1) the different models (shown by the gray squares) and (2) all studies describing the variable (shown by the black squares). Heterogeneity was assessed using Cochran's Q‐test and the i2 quantity. In order to explore the sources of heterogeneity, meta‐regression analyses were performed for duration and degree of pressure load if information was available for more than 2 groups. To perform meta‐regression analysis of a variable with duration, actual duration of pressure load had to be given (ie, variables were excluded from meta‐regression analysis if corresponding duration was defined as a time‐interval [eg, 2–6 weeks]). To be included for meta‐regression analyses concerning the degree of pressure load, RV loading had to be measured as actual pressure rather than increase in hypertrophy. Unfortunately, meta‐regression of cardiac or RV function was impossible because of lack of available data. In addition, differences between models were tested with unpaired t test or 1‐way analysis of variance with post‐hoc Tukey's correction.
Since they have different functions in biological processes, gene expression (at mRNA level) and protein expression of studied variables were separately included in the meta‐analysis. In some studies, mitochondrial content was tested by different measurement techniques within the same animals. To avoid overrepresentation of included subjects, the results of only 1 (the superior) technique/definition was included for meta‐analysis. We ranked the different definitions of mitochondrial content (which were used in the same animals) as follows: (1) ratio mitochondria to myofibrils, (2) mitochondrial yield, (3) citrate synthase activity, (4) citrate synthase at mRNA level, and (5) whole tissue citrate synthase activity. However, all results (from all different techniques) are visually shown in the figures.
If the study concerned did not provide the exact number of animals used for the test of a particular variable, the mean of the range of the number of animals reported in the concerning study was presented in our figures.
The number of included animals per model provided in the current figures may give a slight overestimation in case of multiple groups using the same control group.
Results
Identified Studies
In total, 1393 unique citations were identified, as shown in Figure 1. Based on title abstract screening, 1282 citations were excluded. Of the 111 articles selected for full text review, 86 articles concerned animal studies and 28 articles concerned human studies, and 3 articles described both (Table S1). After full text review, 35 studies were excluded because no control group for the metabolic variables was included (n=22), no increase in RV pressure was measured (n=11), or full text was not available (n=2). The former involved mostly the human studies. We included 28 studies for meta‐analysis (Table S1); 2 of the studies described both human and animal data (Piao, 201316 and Gomez‐Arroyo, 201327).
Figure 1.
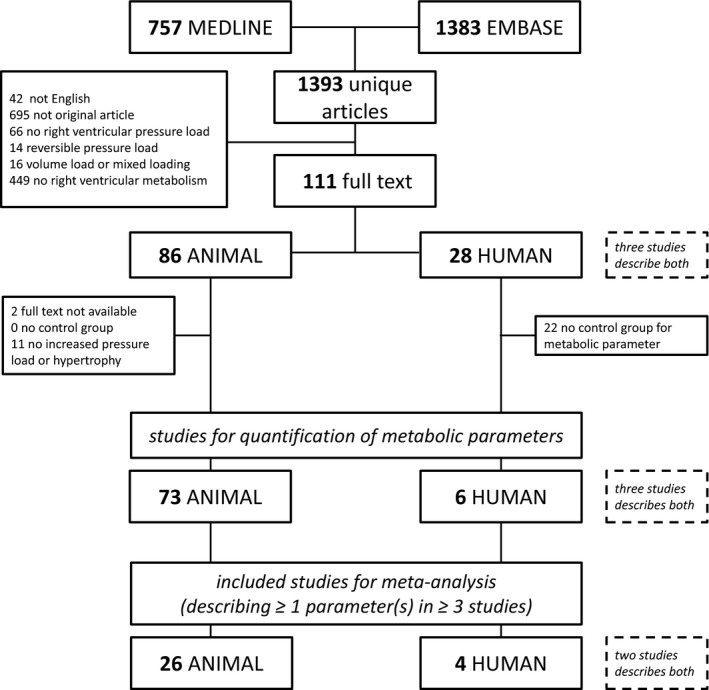
Flow chart of systematic study selection and inclusion meta‐analysis.
From 3 selected publications, 3 study groups were excluded (Balestra 2015, MCT3028; Rumsey 1999, 1 day29; and Zhang 2014, 2 weeks30), since pressure load and hypertrophy did not increase significantly or was not reported. All other groups had at least increased RV systolic pressure (Figure S1A), RV weight, Fulton index (Figure S1B), or RV/body weight ratio.
Glucose Transport and Glycolysis
We identified 3 variables of glucose transport that were described in 3 or more studies: fluorodeoxyglucose (FDG) uptake and expression of transporters GLUT1 and 4 (Figure 2). The uptake of the glucose‐analogue FDG was uniformly increased in animal models19, 31, 32 as well as in patients with PH33 (Figure 2A). Numerous studies investigated the expression of the major glucose transporters, GLUT1 and GLUT4, and correlated this with FDG uptake. Our meta‐analysis revealed that GLUT1 mRNA as well as protein level were significantly increased in the pressure‐loaded RV (Figure 2B). The increase in GLUT1 mRNA expression was universal in all models,15, 18, 21, 27, 34, 35, 36, 37 but protein levels were higher in the monocrotaline (MCT) model15 as compared with the hypoxia, pulmonary artery banding (PAB), and fawn hooded rat (FHR) models15, 16, 21 (P<0.05 for all groups). In contrast to GLUT1, the gene expression of GLUT434, 35, 36 and the GLUT4 proteins levels19, 36, 38, 39, 40 were not altered (Figure 2C). Meta‐regression analyses for FDG‐uptake, GLUT1 and GLUT4, revealed no statistical significant correlations with duration or degree of RV pressure load (Table S2). Meta‐regression of GLUT1 at protein level and GLUT4 at gene level with degree of RV pressure load is not performed because of missing pressure measurements in the studies concerned.
Figure 2.

Right ventricular uptake of carbohydrates. Forrest plots of FDG‐uptake (A), GLUT1 expression at mRNA (B‐1) and protein (B‐2) level, and GLUT4 expression at mRNA (C‐1) and protein (C‐2) level. Data are presented as Hedges’ g. Combined Hedges’ g are presented as squares: gray representing Hedges’ g of a specific model, black representing Hedges’ g of all included studies. Bars represent 95% CI. = indicates not statistically significant affected; ↓, decreased; CI, cardiac index; CO, cardiac output; FDG uptake, fluorodeoxyglucose uptake; GLUT, glucose transporter; i2, level of heterogeneity; MCT, monocrotaline; n, number of included animals; RVEF, right ventricular ejection fraction; TAPSE, tricuspid annular plane systolic movement; X, not included in meta‐analysis. *Significantly (P<0.05) increased compared with hypoxia, pulmonary artery banding‐ and fawn hooded rats‐models.
Glucose transport is coupled with glucose–phosphorylation by hexokinases, driving glucose into glycolysis. The mRNA expression of HK1 (Figure 3A) was significantly increased in all models.18, 21, 27, 29, 30 In addition, meta‐regression analysis showed a negative trend with the duration of RV pressure load (P=0.08) (Figure 3B). HK2 expression was not altered15, 16, 21, 27, 29, 30, 37 (Figure 3C) and meta‐regression analysis revealed no correlations with duration of degree of pressure load (Tables S2 and S3). Unfortunately, protein levels of HK1 were only determined in 1 study18 and HK2 protein levels were not determined at all, and therefore it is unclear how HK protein levels are affected by pressure overload. Glycolysis was studied on isolated hearts in a Langendorf perfusion system of 3 RV pressure overload models: MCT,15 PAB,21 and FHR.16 In addition, glycolysis was determined by Seahorse in RV preparations of the FHR model.16 Meta‐analysis of the data revealed that glycolysis was significantly increased in cardiac tissue of these RV pressure‐loaded hearts (Figure 3D).
Figure 3.
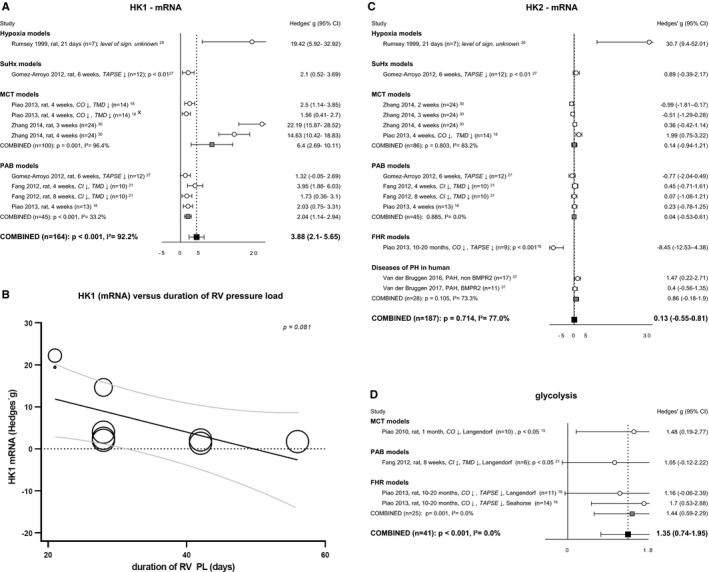
Glycolysis. Forrest plot of HK1 (A) and bubble plot showing meta‐regression analysis of HK1 expression at mRNA level with the duration of RV pressure load (B). Forrest plots of HK2 (C) expression at mRNA level and glycolytic flux measured with Seahorse or Langendorf (D). Data are presented as Hedges’ g. Combined Hedges’ g are presented as squares: gray representing Hedges’ g of a specific model, black representing Hedges’ g of all included studies. Bars represent 95% CI. Bubble size represents relative study precision, calculation based on SD. Black line represents regression line, gray lines represents 95% CI. = indicates not statistically significantly affected; ↓, decreased; 95% CI, cardiac index; CO, cardiac output; FHR, fawn hooded rats; HK, hexokinase; i2, level of heterogeneity; MCT, ; n, number of included animals; PAB, pulmonary artery banding; PH, pulmonary hypertension; RVEF, right ventricular ejection fraction; TAPSE, tricuspid annular plane systolic movement; X, not included in meta‐analysis.
Transport of Fatty Acids
Transporter cluster differentiation 36 (CD36), the main transporter of fatty acids across the plasma membrane, was only investigated in 3 studies (either RNA or protein)27, 37, 41 and hence did not meet the criteria for meta‐analysis. Transport of fatty acids over the mitochondrial membrane is highly regulated by carnitine palmitoyltransferases (CPT1 and CPT2) (outer and inner membrane, respectively). Only meta‐analysis of subunit CPT1B was possible, but revealed ambiguous and nonsignificant results16, 27, 34, 37 (Figure 4A). However, CPT1B mRNA negatively correlated with duration of pressure overload (Figure 4B).
Figure 4.
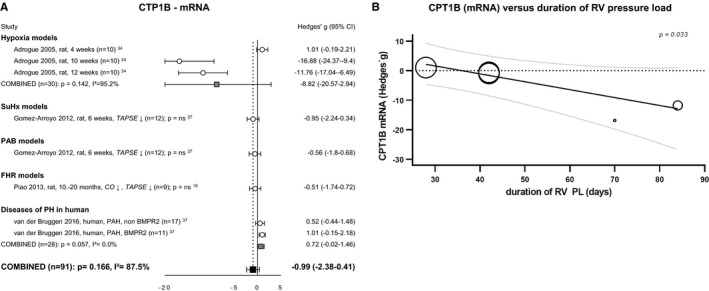
Right ventricular uptake of fatty acids. Forrest plot of CPT1B expression at mRNA level (A). Bubble plot showing the relation between CPT1B expression at mRNA level with duration of pressure load (B). Data are presented as Hedges’ g. Combined Hedges’ g are presented as squares: gray representing Hedges’ g of a specific model, black representing Hedges’ g of all included studies. Bars represent 95% CI. Bubble size represents relative study precision, calculation based on SD. Black line represents regression line, gray lines represents 95% CI. = indicates not statistically significant affected; ↓, decreased; CI, cardiac index; CO, cardiac output; CPT1B, carnitine palmitoyltransferase; FHR, fawn hooded rats; i2, level of heterogeneity; n, number of included animals; PAB, pulmonary artery banding; PH, pulmonary hypertension; PL, pressure load; RV, right ventricular; TAPSE, tricuspid annular plane systolic movement.
Mitochondrial Function
Mitochondrial content
Mitochondrial content was studied using different assays and was subsequently expressed as the following: the ratio of mitochondrial DNA – nuclear (18S) DNA, the ratio of the number of mitochondria to myofibrils, mitochondrial yield (mg mitochondrial protein per gram RV), and citrate synthase activity or citrate synthase mRNA expression. Combining all the data from different models27, 28, 42, 43, 44, 45 and including all analyses, a significant decrease of mitochondrial content in the pressure‐loaded RV could be demonstrated (g=−0.60, P=0.016). However, several studies used data from the same experiment. After exclusion of the possible duplicate measurements (choosing most optimal determination, ranked according to order above), mitochondrial content tended to decrease, but lost its statistical significance (g=−0.68, P=0.054) (Figure 5A). Plotting duration against mitochondrial content suggests a curvilinear association, with a significant negative correlation in the first 6 weeks (Figure 5B). In addition, mitochondrial content is negatively correlated with the degree of RV pressure load (Figure S4).
Figure 5.

Mitochondrial function. Plots of mitochondrial content measured by mentioned methods (A). Bubble plot showing relation between mitochondrial content and duration of RV PL (B). Forrest plot of PDH activity as reflection of mitochondrial breakdown of pyruvate to acetyl‐CoA (C). Forrest plots of mitochondrial respiratory capacity for carbohydrate metabolites measured in isolated mitochondria (ADP‐driven) (D‐1) or intact cardiomyocytes (D‐2). Forrest plots of MCAD expression at mRNA level (E), as representative of the β‐oxidation. Forrest plots of mitochondrial respiratory capacity for fatty acids measured in isolated mitochondria (F‐1) and intact cardiomyocytes (F‐2). Data are presented as Hedges’ g. Combined Hedges’ g are presented as squares: gray representing Hedges’ g of a specific model, black representing Hedges’ g of all included studies. Bars represent 95% CI. Bubble size represents relative study precision, calculation based on SD. Gray bubbles are not included in meta‐analysis. Black line represents regression line, gray lines represent 95% CI. = indicates not statistically significant affected. ↓, decreased; ↓↓, decreased compared with decompensated group; CI, cardiac index; CO, cardiac output; FHR, fawn hooded rats; I2, level of heterogeneity; MCT, monocrotaline; n, number of included animals; PAB, pulmonary artery banding; PDH, pyruvate dehydrogenase; PL, pressure load; RVEF, RV ejection fraction; TAPSE, tricuspid annular plane systolic movement; X, not included in meta‐analysis. *Significantly (P<0.05) increased compared with PAB.
Glucose oxidation
Activity of pyruvate dehydrogenase (PDH), the enzyme converting pyruvate into acetyl‐CoA in the mitochondria, tended to be decreased in RV pressure load but did not reach statistical significance (g=−1.982, P=0.123)15, 16, 18, 21 (Figure 5C). A similar result was observed for PDK4, a negative regulator of PDH, (resp. g=−1.91, P=0.110), where meta‐analysis of expression at both mRNA16, 34, 35 and protein level16, 17, 32 was unchanged (Figure S2A, S2B). The same was true for PDK1 and PDK2 at protein level16, 17, 32 (Figure S2C, S2D). Heterogeneity was not explained by the duration or degree of pressure load (Tables S2 and S3), or the different models.
Respiratory capacity of glucose or pyruvate was reported in 7 articles. Analysis was divided in ADP‐driven respiratory state measured in isolated mitochondria with oxygraphy (Oroboros or Clark‐type) (n=2)20, 29 (Figure 5D‐1), and respiratory capacity measured in intact cardiomyocytes with Seahorse (n=2)16, 21 or isolated heart model (Langendorf) (n=3)15, 16, 18 (Figure 5D‐2). Subsequently, measurements in isolated mitochondria did not meet the inclusion criteria for meta‐analysis. Respiratory capacity measured by all methods showed a negative trend, albeit meta‐analysis of respiratory capacity for carbohydrates in intact cardiomyocytes did not reveal a significant decrease (g=−1.21 P=0.082). Respiratory capacity did increase in the MCT model compared with PAB (P<0.05) (Figure 5D). Meta‐regression analyses did not reveal correlations between respiratory capacity and duration or degree of RV pressure load.
Oxidative fatty acid metabolism
β‐Oxidation involved genes including ACADVL (1), EHHADH (2), HADHA (1), ACAA2 (3), ACAT1 (1), medium chain acyl CoA dehydrogenase (MCAD) (synonym ACADM) (6), ACADS (3), and ACOT2 (1) were all described, but only MCAD met the criteria for inclusion in meta‐analysis. MCAD at the mRNA level decreased in all models of RV pressure load (hypoxia P<0.001, SuHx P<0.01, and PAB P<0.05)5, 27, 34, 35, 46 (Figure 5E). No correlations with duration or degree of pressure load were observed (Tables S2 and S3). At the protein level, 3 studies27, 46, 47 were included in the meta‐analysis, which tended to decrease, but did not reach statistical significance (g=−2.02, P=0.141) (Figure S3).
Mitochondrial respiration regarding fatty acid oxidation measured in the ADP‐driven state (n=4) decreased, when tested in models of hypoxia29, 42 and SuHx20 (Figure 5F‐1). Respiratory capacity in intact cardiomyocytes was extracted from 2 publications showing contrary results in PAB21 compared with the FHR model16 (Figure 5F‐2).
Transcriptional Regulators of Metabolism
This systematic search identified several regulators of transcriptional regulators of metabolism (ie, PGC1α (5), PPARα (4), PPARγ (1), FOXO1 (1), Mef2c (1), HIF1α (4), and cMyc (1)) (numbers include both gene expression at mRNA level and protein expression). Meta‐analysis was performed for PGC1α and PPARα. PGC1α is best known as the master regulator of mitochondrial biogenesis and interacts with PPARα, which predominantly acts on lipid metabolism. Combined Hedges’ g of PGC1α mRNA expression27, 43 decreased (Figure S4B) and meta‐regression revealed a negative correlation with duration of pressure load (Figure S4C). Meta‐analysis for PGC1α protein expression did not reveal significant change (Figure S4D), but did show a model effect for MCT43 versus SuHx20, 27 (P<0.05) (Figure S4B). Combined Hedges’ g of PPARα mRNA expression27, 34, 35 during pressure load did not change significantly (Figure S4E) and no correlations with duration, degree, or model of RV pressure were observed. PPARα protein expression was studied once in SuHx rats, demonstrating a decrease (P<0.001).27
Results are summarized in Figure 6 and Table S4.
Figure 6.
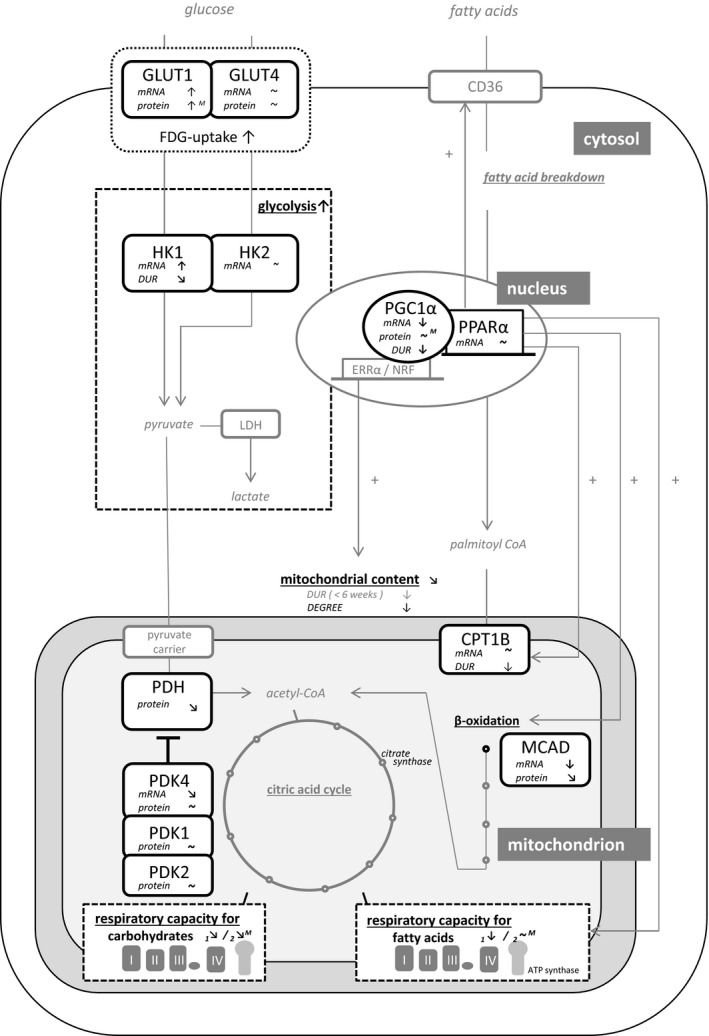
Metabolic changes in the pressure‐loaded right ventricle: summarizing results of multiple meta‐analyses. Black components are included in meta‐analysis. ~ indicates unchanged; ↑, significant increase or positive relation; ↗, positive trend (P<0.15); ↘, negative trend (P<0.15); ↓, significant decrease or negative relation; CD36, cluster differentiation 36 (cellular fat transporter); CPT1B, carnitine? palmitoyltransferase 1B; DUR, duration; ERRα, estrogen‐related receptor alpha; FDG‐uptake, fluorodeoxyglucose uptake; GLUT, glucose transporter; HK, hexokinase; LDH, lactate dehydrogenase; M, model effect; MCAD, medium chain acyl CoA dehydrogenase; NRF, nuclear respiratory factor; PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; PGC1α, PPAR gamma complex 1 alpha; PPARα, peroxisome proliferator‐activated receptor alpha.
Effect of Interventions on Metabolism in the Pressure‐Loaded RV
Twenty studies described the effect of an intervention on metabolic parameter(s). Overall, these intervention studies aimed to decrease glycolysis by the increase of glucose oxidation. This could be established by recoupling of glycolysis with glucose oxidation, by, for example, dichloroacetate or 6‐diazo‐5‐oxo‐l‐norleucine, or indirectly by inhibition of fatty acid metabolism by, for example, trimetazidine or ranolazine. Seven studies included metabolic variables that were included in meta‐analyses above.15, 16, 17, 18, 19, 20, 21 Of these metabolic variables, effect sizes derived from certain metabolic variables of the intervention group treated with metabolic therapy compared with those of the intervention group without treatment are shown in Table S5. The effect of dichloroacetate on PDH activity was studied in 3 studies showing a significant increase in a FHR model,16 with contrary results regarding 2 MCT models.15, 17 The effects of therapeutic interventions on all other 21 reported variables were studied incidentally, precluding data synthesis and conclusions.
Discussion
In this systematic review on metabolism in the pressure‐loaded RV, we identified 26 animal and 4 human studies eligible for meta‐analysis. The systematic review combined with multiple separate meta‐analyses yielded a uniform increase in glucose uptake and glycolysis, whereas fatty acid uptake and changes in oxidative metabolism were less consistent. The effect of therapeutic interventions could not be analyzed because of the large variety of outcome variables used and compounds used.
In the current study, there are strong indications that glycolysis is increased in the pressure‐overloaded RV. Both gene expression of HK1, an important enzyme controlling the first step of glycolysis, and the capacity for glycolysis measured by Seahorse and Langendorf were significantly increased. In contrast, HK2 was unchanged. Previous studies in the LV have identified HK2 as a modulator of reactive oxygen species and described attenuating effects on cardiac hypertrophy.48, 49 HK2, involved in anabolic pathways by providing glucose‐6‐phosphate for glycogen synthesis, also fulfills a role in providing glucose‐6‐phosphate to the pentose phosphate pathway. Contrary to the many roles of HK2, HK1 primarily facilitates glycolysis.50, 51 HK1 is primarily expressed in neonatal cardiomyocytes and is associated with the fetal gene program,50, 52, 53 characterized by better resistance against an oxygen‐poor environment such as in the RV pressure load.5, 39, 54, 55, 56 The activation of the fetal gene program is also reflected in an increased expression of GLUT1, supporting increased glucose uptake, which increases the ability of increased glycolysis.16, 27, 32 Remarkably, HK1 and GLUT1 both concern insulin‐independent isoforms whereas HK2 and GLUT4 concern insulin‐dependent isoforms.57 The current meta‐analysis reveals a clear pattern in the pressure‐overloaded RV differentiating between the insulin‐independent versus insulin‐dependent profiles, directing to glycolysis by activation of insulin‐insensitive mechanisms.
The increase of glycolysis in the pressure‐loaded RV is also supported by the increased glucose uptake measured by FDG by positron emission tomography (PET)‐computed tomography. PET‐computed tomography has the ability to assess the actual uptake in vivo, whereas gene or protein expression of involved genes and respiratory capacity of isolated mitochondria are an approximation of the actual situation in vivo. However, FDG uptake represents glucose uptake rather than metabolic capacity itself. Studies describing FDG uptake that were excluded from meta‐analysis endorse our findings.58, 59, 60, 61, 62 In addition, increased RV FDG uptake has been associated with increased pressure load58, 60, 63, 64 and altered dimensions,60, 62, 64, 65 and inverse correlations with RV function,62, 63, 65 cardiac function,60 and clinical outcome.66, 67
Meta‐analysis of substrate‐specific oxidative metabolism in the pressure‐loaded RV reflects an ambiguous character. Glucose oxidation is regulated via pyruvate dehydrogenase kinase, which inhibits breakdown of pyruvate. The expression of pyruvate dehydrogenase kinase in response to pressure load in the RV varied widely with different models used (Figure S4A through S4D). In addition, the respiratory capacity for carbohydrates was also affected by the model used. Although cardiac performance was decreased in both MCT and PAB models to the same extent, respiratory capacity increased in MCT models, but decreased in pressure load only via PAB. Similarly, with respect to respiratory capacity for fatty acids, PAB models behaved differently from FHR, while there are no data from MCT models. Taken together, these data suggest that the RV oxidative capacity changes in response to pressure load are dependent upon methodological differences, and may be subsequently dependent on model or disease, cardiac function, and possibly on clinical severity. More cooperation between research groups and comparative studies between fixed RV‐PA uncoupling (in PAB) versus dynamic RV‐PA uncoupling (eg, in MCT) are needed to identify the systemic changes that may interfere with the cardiac response. Intriguingly, whereas there was variation in the respiratory capacity for fatty acids, the changes in 1 of the genes oxidizing fatty acids (MCAD) were uniform. Downregulation of the β‐oxidation was supported in the literature by decrease of other genes from the acyl‐coenzyme A (CoA) dehydrogenases family at both mRNA16, 27 and protein level.27, 46, 68 Downregulation of the oxidation phase has been suggested based on decreased expression of genes as HADH,5, 69 HADHA, HADHB, and EHHADH.5, 68, 70 In addition, malonyl‐CoA decarboxylase is described to be decreased in a model of hypoxia.34 Oxidative metabolism in general in the pressure‐loaded RV was studied in 2 studies and therefore is not included in the meta‐analysis. The clearance of11C‐acetate was used as representative of the tricarboxylic cycle. RV clearance rates correlated with the rate pressure product and oxygen consumption in idiopathic pulmonary arterial hypertension (iPAH),71 and appeared to be higher PH (chronic thromboembolic PH [CTEPH], pulmonary arterial hypertension (PAH), and PH with unclear multifactorial mechanisms) compared with controls.72 The current study stresses the need for further research in order to clarify changes caused by pressure load itself and changes as a result of the specific inducement of RV pressure load or a potential systemic disease.
The systematic literature search showed that processes involved in the transport of long‐chain fatty acids varied in different models and different cohorts of patients with PH. Gene expression of CD36, the transporter of long‐chain fatty acids across the cellular membrane, was decreased in SuHx rats, unaffected in PAB rats, and increased at protein level in patients with a BMPR2 mutation.27, 41 Studies measuring gene expression of fatty acid binding proteins (FABP1‐7) and fatty acids transporters (SLC7A1‐6) in the pressure‐loaded RV are scarce and were ambiguous.16, 31 We excluded studies describing actual fatty acid uptake measured with positron emission tomography tracers in the patient cohort without a control group. These studies also yielded various changes. Different cohorts representing different types of diseases, including precapillary PH and chronic obstructive lung disease, showed both pressure load–dependent73, 74 and –independent59, 60, 75 cellular uptake. Support of load‐dependent uptake was given by the reversibility of increased uptake after abolishing increased pressure load in patients with chronic thromboembolic PH.74 In addition, positive correlations between fatty acid uptake and markers of RV hypertrophy were observed60, 75 and, as shown for glucose uptake measured by positron emission tomography–computed tomography, uptake of free fatty acids has been inversely correlated with RV ejection fraction59, 75 as well. Although no correlation was found with cardiac index,74 fatty acid uptake has been positively correlated with clinical outcome, expressed by 6‐minute walking distance, New York Heart Association class, and mortality.74, 75 Mitochondrial uptake of long‐chain fatty acids in the healthy heart is predominately facilitated by CPT1B. CPT1B at the mRNA level negatively correlated with the duration of pressure load (Figure 4B). However, CPT1B expression in human forms of PAH tended to increase.37 Few studies described CPT1A, describing inconsistent results.14, 16, 27, 76 Although CPT1A was originally considered an insignificant player in muscle (including heart) tissue, recent publications identified increased CPT1A as a key step in early metabolic remodeling, which is linked to reduced fatty acid oxidation.77 Besides the contradictory results regarding fatty acid uptake between the different animal models and between different patient cohorts, no structural consistency was found between a specific animal model with a specific human disease. Nevertheless, a disease‐specific pattern seems to apply for intramyocardial lipid deposition. Published results indicate lipid accumulation based on decreased fatty acid oxidation and increased fatty acid uptake by increased translation of CD36 to plasma membrane in heritable PAH specifically,78, 79 whereas RV ceramide content in chronic hypoxia decreased.80 Unfortunately, only 3 studies reported intracardiac lipid deposition of various lipids, which made meta‐analysis impossible. Further research should aim for better understanding of the translational possibilities from experimental studies to human disease.
PGC1α acts on transcriptions factors such as the PPARs and is an important transcription factor of mitochondrial content. Coactivation of PGC1α with PPAR isoforms is known to induce activation of downstream genes regarding fatty acid handling including uptake and β‐oxidation, especially fat transporter genes CD36 and CPT1B, and β‐oxidation gene MCAD.81, 82, 83, 84 PPARα is the most studied PPAR in the heart and this also applies for the pressure‐loaded RV specifically.27, 34, 85 Nevertheless, data of PPARα expression in the pressure‐loaded RV is still limited and mostly showing statistically insignificant results (Figure S4D). This is in contrast to PGC1α, which is significantly negative affected in the pressure‐loaded RV and seems to be related to mitochondrial content in models of RV pressure load. It must be mentioned that the different studies identified mitochondrial content using different methods, since standardized methods are lacking. Future studies should clarify whether decreased mitochondrial content indeed is predominately established in models of SuHx and to what extent this mechanism is relevant for human PH disease. Remarkably, both PGC1α and PPARα are not identified in studies with unbiased approach by performing microarray5, 55, 86, 87, 88 or proteomics.87 This could imply that changes of PGC1α or PPARα are not causal for altered processes caused by RV pressure load.
As shown in this review, metabolic modulation has been primarily focused on the reduction of glycolysis by activation of glucose oxidation. The most studied compound is dichloroacetate, which inhibits pyruvate dehydrogenase kinase and thereby indirectly stimulates activation of PDH. Interestingly, in the pressure‐loaded RV, the different isoforms of pyruvate dehydrogenase kinase and PDH encompass varied results (Figures S2 and 5). However, studies specifically focusing on interfering in the activity of these enzymes in the pressure‐loaded RV by dichloroacetate show positive effects on cell homeostasis, mitochondrial function, and cardiac function,15, 16, 17 with no effect on these functions in controls.15 In MCT and FHR, at respectively 6 weeks and >10 to 20 months of treatment, dichloroacetate leads to normalized levels of the upregulated PDK2 and PDK4, with restoration of PDH activity.16, 17 This was accompanied by normalization of FOXO1 levels, which were upregulated in disease in FHR animals and patients with PAH.16 This is consistent with the concept of activation of the fetal gene program and insulin‐independent mechanisms in the pressure‐loaded RV, since sustained FOXO1 activation in neonatal cardiomyocytes is known to diminish insulin signaling and impair glucose metabolism.89
Limitations
This study has some limitations that should be discussed. To guarantee actual pressure load on the RV, meta‐analysis includes both studies with proven increased pressure load by RV systolic pressure and mean pulmonary artery pressure, and by RVH. RVH was expressed as increased RV weight, Fulton index, or RV to body weight ratio. Although hypertrophy is a plausible effect of pressure load, the degree of hypertrophy within studies from the current literature search is independent of the actual degree of pressure load (data not shown). This might be explained by a predominant use of models of severe pressure load. This together with the fact that RVH based on weight is a widely supported confirmation of RV pressure overload resulted in RVH as an inclusion criterion in addition to increased pressure load.
In line with the statement of the Systematic Review Center for Laboratory animal Experimentation (SYRCLE),24 the aim of this meta‐analysis was to assess the general direction and magnitude of RV pressure load of the specific variable (rather than to obtain a precise point estimate explicitly) with additional exploration of the sources of heterogeneity by using meta‐regression analyses. We used effect size defined as Hedges' g. Hedges' g is the criterion standard in small samples (<10 samples per group), which includes a correction factor for small sample size bias,90, 91 and therefore is considered as a criterion standard in meta‐analysis of systematic reviews in animal data from experimental studies. However, we believe that the use of Hedges' g encompasses a specific point that should be addressed. Since the use of effect sizes implies standardized mean differences, calculations are based on a pooled SD, although unequal variances may be present. This may induce type I errors. However, the small and unequal sample sizes will likely cancel out this effect. An alternative statistic method would be statistics by using Z scores, but because we aimed to provide an overview of the results of the different studies, by the visualization by figures, this method was not preferred.
The interpretation of meta‐analysis results were challenged by substantial degrees of heterogeneity, which was partly explored by performing (1) meta‐regression analysis for duration and degree of pressure load, and (2) t tests or 1‐way analysis of variance of the results of the different models. This resulted in 3 significant correlations with duration and various differences between models. Only 1 correlation was found with the degree of RV pressure load, which could be because of the fact that included studies encompass significant loading conditions. Systematically testing for the effect of used species was impossible because only 1 study concerned animal species other than rat. This, however, contributed to large homogeneity at this particular point. Furthermore, we decided to use an almost similar approach for human as for animal studies in order to be able to apply the same methods regarding meta‐analysis. Subsequently, a number of clinical studies were excluded from meta‐analysis because of aspects regarding study design. Nevertheless, most of the excluded studies described FDG uptake and supported the presented results in the meta‐analyses. Other human studies that were excluded from the meta‐analyses described uptake of fatty acids, as has been described above.
Considerations Regarding Future Research
Because of the use of differing designs of the included studies, the power of the meta‐analysis is limited. In contrast to clinical trials, replication is still scarce in experimental research. The current study emphasizes the need for replication and the use of more standardization in models, methods, and outcome variables in studies that studied metabolic derangements in RV pressure load. This could be achieved in joint publications of different research groups. Available data describe to a certain extent the degree and duration of pressure load. In pursuing actual translation, absolute determination of pressure load will be necessary in both animals and humans, with the intention of differentiating between the actual component of pressure load and the cause of disease, including potential comorbidities. The cause of disease, or the character of the model, is important since models of PAH, such as hypoxia, SuHx, MCT, and FHR, may differ in their systemic effects and are known for differences in disease severity and cardiovascular interaction. These differences are driven by involvement of endothelial damage, level of inflammation, cytokine migration, and vasoconstriction. While isolated hypoxia with the absence of endothelial damage in the pulmonary vasculature induces mild PH only, FHR leads to more progressive PH, whereas SuHx and MCT will induce failure, with high rates of mortality in MCT. Exact mechanisms still need to be unraveled. The current meta‐analysis directs to further exploration of the role of diseases that expose the RV to altered insulin sensitivity or oxygen tension in remodeling during RV pressure load. The current overview shows that determination of protein expression is limited compared with gene expression, and often shows divergent results. Also, measurements of substrate activities are relatively scarce. We suggest that future studies in the pressure‐loaded RV should be more uniform and integral with respect to expression level (gene, protein, or activity level). The variables of metabolism to be studied should be uniform and those that are most optimal should be chosen based on research using unbiased approaches (ie, microarray, RNA sequences, proteomics, or metabolomics). Given the abovementioned observations, the translational applicability between, and within, animal models and human diseases of PH should most critically and carefully be considered.
Conclusions
This systematic review and meta‐analysis of metabolic variables in the pressure‐loaded RV showed a uniform increase in glucose uptake and glycolysis. Results regarding fatty acid uptake and changes in oxidative metabolism were divergent and model specific. To actually use metabolism as a therapeutic target in the RV exposed to increased pressure load in clinical practice, we need to learn more about model‐ and disease‐specific mechanisms of fatty acid uptake and mitochondrial impairment.
Disclosures
None.
Supporting information
Data S1. Supplemental Methods.
Table S1. List of Studies Studying Metabolic Parameters in the Pressure‐Loaded Right Ventricle Included for Full Text Review
Table S2. Meta‐Regression Analyses: Variables Vs Duration of RV Pressure Load
Table S3. Meta‐Regression Analyses: Variables Vs Degree of RV Pressure Load
Table S4. Level of Significance of Meta‐Analysis and Meta‐Regression
Table S5. Overview of Hedges’ g for Metabolic Parameters in Models of RV Pressure Load Subjected to Therapeutic Interventions
Figure S1. Models of Increased Pressure Load. Forest plots of right ventricular systolic pressure (A) and Fulton index (B). Data are presented as Hedges’ g (95% CI). Combined Hedges’ g are presented as squares: gray representing Hedges’ g of a specific model, black representing Hedges’ g of all included studies. Bars represent 95% CI. i2 indicates level of heterogeneity; n, number of included animals.
Figure S2. Forest plots of expression of PDK isoenzymes. PDK4 at both mRNA (A) and protein level (B), and PDK1 (C) and PDK2 (D) at protein level. Bars represent 95% CI. =, indicates not statistically significant affected; ↓, decreased; CI, cardiac index; CO, cardiac output; i2, level of heterogeneity; n, number of included animals; PDK, pyruvate dehydrogenase kinase; RVEF, RV ejection fraction; TAPSE, tricuspid annular plane systolic movement; X, not included in meta‐analysis.
Figure S3. Forest plot MCAD at protein level. Bars represent 95% CI. = indicates not statistically significant affected; ↓, decreased; CI, cardiac index; CO, cardiac output; i2, level of heterogeneity; MCAD, medium chain acyl CoA dehydrogenase; n, number of included animals; RVEF, RV ejection fraction; TAPSE, tricuspid annular plane systolic movement; X, not included in meta‐analysis.
Figure S4. Regulators of metabolism. Meta‐regression of mitochondrial content with degree of RV pressure load (A). PGC1α gene expression (B) and its relation with duration of pressure load shown by meta‐regression in a bubble plot (C). Forrest plot of PGC1α protein expression (D). Forrest plot of PPARα gene expression (E). Data are presented as Hedges’ g. Combined Hedges’ g are presented as squares: gray representing Hedges’ g of a specific model, black representing Hedges’ g of all included studies. Bars represent 95% CI. Bubble size represents relative study precision, calculation based on SD. Black line represents regression line, gray lines represents 95% CI. = indicates not statistically significant affected; ↓, decreased; CI, cardiac index; CO, cardiac output; i2, level of heterogeneity; n, number of included animals; RVEF, RV ejection fraction; TAPSE, tricuspid annular plane systolic movement. *Significantly (P<0.05) increased compared with SuHx‐model.
(J Am Heart Assoc. 2019;8:e012086 DOI: 10.1161/JAHA.119.012086.)
Beatrijs Bartelds is currently located at the Division of Pediatric Cardiology, Department of Pediatrics, Erasmus University Medical Center, Sophia Children's Hospital, Rotterdam, The Netherlands.
References
- 1. Norozi K, Wessel A, Alpers V, Arnhold JO, Geyer S, Zoege M, Buchhorn R. Incidence and risk distribution of heart failure in adolescents and adults with congenital heart disease after cardiac surgery. Am J Cardiol. 2006;97:1238–1243. [DOI] [PubMed] [Google Scholar]
- 2. Van Wolferen SA, Marcus JT, Boonstra A, Marques KMJ, Bronzwaer JGF, Spreeuwenberg MD, Postmus PE, Vonk‐Noordegraaf A. Prognostic value of right ventricular mass, volume, and function in idiopathic pulmonary arterial hypertension. Eur Heart J. 2007;28:1250–1257. [DOI] [PubMed] [Google Scholar]
- 3. Ghio S, Gavazzi A, Campana C, Inserra C, Klersy C, Sebastiani R, Arbustini E, Recusani F, Tavazzi L. Independent and additive prognostic value of right ventricular systolic function and pulmonary artery pressure in patients with chronic heart failure. J Am Coll Cardiol. 2001;37:183–188. [DOI] [PubMed] [Google Scholar]
- 4. Meyer P, Filippatos GS, Ahmed MI, Iskandrian AE, Bittner V, Perry GJ, White M, Aban IB, Mujib M, Italia LJD, Ahmed A. Effects of right ventricular ejection fraction on outcomes in chronic systolic heart failure. Circulation. 2010;121:252–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Borgdorff MAJ, Koop AMC, Bloks VW, Dickinson MG, Steendijk P, Sillje HHW, van Wiechen MPH, Berger RMF, Bartelds B. Clinical symptoms of right ventricular failure in experimental chronic pressure load are associated with progressive diastolic dysfunction. J Mol Cell Cardiol. 2015;79:244–253. [DOI] [PubMed] [Google Scholar]
- 6. Ryan JJ, Archer SL. The right ventricle in pulmonary arterial hypertension: disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ Res. 2014;115:176–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Borgdorff MAJ, Dickinson MG, Berger RMF, Bartelds B. Right ventricular failure due to chronic pressure load: What have we learned in animal models since the NIH working group statement? Heart Fail Rev. 2015;20:475–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Samson N, Paulin R. Epigenetics, inflammation and metabolism in right heart failure associated with pulmonary hypertension. Pulm Circ. 2017;7:572–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Koop AMC, Hagdorn QAJ, Bossers GPL, van T Leusden, Gerding A, van Weeghel M, Vaz FM, Koonen DPY, Silljé HHW, Berger RMF, Bartelds B. Right ventricular pressure overload alters cardiac lipid composition. Int J Cardiol. 2019;287:96–105. [DOI] [PubMed] [Google Scholar]
- 10. Bartelds B, Knoester H, Smid GB, Takens J, Visser GH, Penninga L, van der Leij FR, Beaufort‐Krol GC, Zijlstra WG, Heymans HS, Kuipers JR. Perinatal changes in myocardial metabolism in lambs. Circulation. 2000;102:926–931. [DOI] [PubMed] [Google Scholar]
- 11. Bartelds B, Knoester H, Beaufort‐Krol GC, Smid GB, Takens J, Zijlstra WG, Heymans HSA, Kuipers JRG. Myocardial lactate metabolism in fetal and newborn lambs. Circulation. 1999;99:1892–1897. [DOI] [PubMed] [Google Scholar]
- 12. Lopaschuk GD. Metabolic modulators in heart disease – Past Present and Future. Can J Cardiol. 2017;33:838–849. [DOI] [PubMed] [Google Scholar]
- 13. Neubauer S. The failing heart — an engine out of fuel. N Engl J Med. 2007;356:1140–1151. [DOI] [PubMed] [Google Scholar]
- 14. Sanz J, Sánchez‐Quintana D, Bossone E, Bogaard HJ, Naeije R. Anatomy, function, and dysfunction of the right ventricle: JACC state‐of‐the‐art review. J Am Coll Cardiol. 2019;73:1463–1482. [DOI] [PubMed] [Google Scholar]
- 15. Piao L, Fang Y‐H, Cadete VJJ, Wietholt C, Urboniene D, Toth PT, Marsboom G, Zhang HJ, Haber I, Rehman J, Lopaschuk GD, Archer SL. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: resuscitating the hibernating right ventricle. J Mol Med. 2010;88:47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Piao L, Sidhu VK, Fang Y‐H, Ryan JJ, Parikh KS, Hong Z, Toth PT, Morrow E, Kutty S, Lopaschuk GD, Archer SL. FOXO1‐mediated upregulation of pyruvate dehydrogenase kinase‐4 (PDK4) decreases glucose oxidation and impairs right ventricular function in pulmonary hypertension: therapeutic benefits of dichloroacetate. J Mol Med. 2013;91:333–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sun X‐Q, Zhang R, Zhang H‐D, Yuan P, Wang X‐J, Zhao Q‐H, Wang L, Jiang R, Jan Bogaard H, Jing Z‐C. Reversal of right ventricular remodeling by dichloroacetate is related to inhibition of mitochondria‐dependent apoptosis. Hypertens Res. 2016;39:302–311. [DOI] [PubMed] [Google Scholar]
- 18. Piao L, Fang Y‐H, Parikh K, Ryan JJ, Toth PT, Archer SL. Cardiac glutaminolysis: a maladaptive cancer metabolism pathway in the right ventricle in pulmonary hypertension. J Mol Med. 2013;91:1185–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Drozd K, Ahmadi A, Deng Y, Jiang B, Petryk J, Thorn S, Stewart D, Beanlands R, DeKemp RA, DaSilva JN, Mielniczuk LM. Effects of an endothelin receptor antagonist, Macitentan, on right ventricular substrate utilization and function in a Sugen 5416/hypoxia rat model of severe pulmonary arterial hypertension. J Nucl Cardiol. 2016;24:1–11. [DOI] [PubMed] [Google Scholar]
- 20. Liu A, Philip J, Vinnakota KC, Van den Bergh F, Tabima DM, Hacker T, Beard DA, Chesler NC. Estrogen maintains mitochondrial content and function in the right ventricle of rats with pulmonary hypertension. Physiol Rep. 2017;5:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fang Y‐H, Piao L, Hong Z, Toth PT, Marsboom G, Bache‐Wiig P, Rehman J, Archer SL. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: exploiting Randle's cycle. J Mol Med. 2012;90:31–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Khan SS, Cuttica MJ, Beussink‐Nelson L, Kozyleva A, Sanchez C, Mkrdichian H, Selvaraj S, Dematte JE, Lee DC, Shah SJ. Effects of ranolazine on exercise capacity, right ventricular indices, and hemodynamic characteristics in pulmonary arterial hypertension: a pilot study. Pulm Circ. 2015;5:547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Michelakis ED, Gurtu V, Webster L, Barnes G, Watson G, Howard L, Cupitt J, Paterson I, Thompson RB, Chow K, Regan DPO, Zhao L, Wharton J, Kiely DG, Kinnaird A, Boukouris AE, White C, Nagendran J, Freed DH, Wort SJ, Gibbs JSR, Wilkins MR. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci Transl Med. 2017;4583:1–13. [DOI] [PubMed] [Google Scholar]
- 24. de Vries RBM, Hooijmans CR, Langendam MW, van Luijk J, Leenaars M. A protocol format for the preparation, registration and publication of systematic reviews of animal intervention studies. Evidenc Based Preclin Med. 2015;1:e00007. [Google Scholar]
- 25. De Vries RBM, Wever KE, Avey MT, Stephens ML, Sena ES, Leenaars M. The usefulness of systematic reviews of animal experiments for the design of preclinical and clinical studies. ILAR J. 2014;55:427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Galiè N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, Beghetti M, Corris P, Gaine S, Gibbs JS, Gomez‐Sanchez MA, Jondeau G, Klepetko W, Opitz C, Peacock A, Rubin L, Zellweger M, Simonneau G. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2009;34:1219–1263. [DOI] [PubMed] [Google Scholar]
- 27. Gomez‐Arroyo J, Mizuno S, Szczepanek K, Van Tassell B, Natarajan R, Dos Remedios CG, Drake JI, Farkas L, Kraskauskas D, Wijesinghe DS, Chalfant CE, Bigbee J, Abbate A, Lesnefsky EJ, Bogaard HJ, Voelkel NF. Metabolic gene remodeling and mitochondrial dysfunction in failing right ventricular hypertrophy secondary to pulmonary arterial hypertension. Circ Heart Fail. 2013;6:136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Balestra GM, Mik EG, Eerbeek O, Specht PAC, van der Laarse WJ, Zuurbier CJ. Increased in vivo mitochondrial oxygenation with right ventricular failure induced by pulmonary arterial hypertension: mitochondrial inhibition as driver of cardiac failure? Respir Res. 2015;16:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rumsey WL, Abbott B, Bertelsen D, Mallamaci M, Hagan K, Nelson D, Erecinska M. Adaptation to hypoxia alters energy metabolism in rat heart. Am J Physiol. 1999;276:H71–H80. [DOI] [PubMed] [Google Scholar]
- 30. Zhang W‐H, Qiu M‐H, Wang X‐J, Sun K, Zheng Y, Jing Z‐C. Up‐regulation of hexokinase1 in the right ventricle of monocrotaline induced pulmonary hypertension. Respir Res. 2014;15:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Graham BB, Kumar R, Mickael C, Sanders L, Gebreab L, Huber KM, Perez M, Smith‐Jones P, Serkova NJ, Tuder RM. Severe pulmonary hypertension is associated with altered right ventricle metabolic substrate uptake. Am J Physiol. 2015;309:L435–L440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sutendra G, Dromparis P, Paulin R, Zervopoulos S, Haromy A, Nagendran J, Michelakis ED. A metabolic remodeling in right ventricular hypertrophy is associated with decreased angiogenesis and a transition from a compensated to a decompensated state in pulmonary hypertension. J Mol Med. 2013;91:1315–1327. [DOI] [PubMed] [Google Scholar]
- 33. Wang L, Li W, Yang Y, Wu W, Cai Q, Ma X, Xiong C, He J, Fang W. Quantitative assessment of right ventricular glucose metabolism in idiopathic pulmonary arterial hypertension patients: a longitudinal study. Eur Heart J Cardiovasc Imaging. 2016;17:1161–1168. [DOI] [PubMed] [Google Scholar]
- 34. Adrogue JV, Sharma S, Ngumbela K, Essop MF, Taegtmeyer H. Acclimatization to chronic hypobaric hypoxia is associated with a differential transcriptional profile between the right and left ventricle. Mol Cell Biochem. 2005;278:71–78. [DOI] [PubMed] [Google Scholar]
- 35. Sharma S, Taegtmeyer H, Adrogue J, Razeghi P, Sen S, Ngumbela K, Essop MF. Dynamic changes of gene expression in hypoxia‐induced right ventricular hypertrophy. Am J Physiol. 2004;286:H1185–H1192. [DOI] [PubMed] [Google Scholar]
- 36. Sivitz WI, Lund DD, Yorek B, Grover‐McKay M, Schmid PG. Pretranslational regulation of two cardiac glucose transporters in rats exposed to hypobaric hypoxia. Am J Physiol. 1992;263:E562–E569. [DOI] [PubMed] [Google Scholar]
- 37. van der Bruggen CE, Happé CM, Dorfmüller P, Trip P, Spruijt OA, Rol N, Hoevenaars FP, Houweling AC, Girerd B, Marcus JT, Mercier O, Humbert M, Handoko ML, van der Velden J, Vonk Noordegraaf A, Bogaard HJ, Goumans M‐J, de Man FS. Bone morphogenetic protein receptor type 2 mutation in pulmonary arterial hypertension: a view on the right ventricle. Circulation. 2016;133:1747–1760. [DOI] [PubMed] [Google Scholar]
- 38. Bruns DR, Dale Brown R, Stenmark KR, Buttrick PM, Walker LA. Mitochondrial integrity in a neonatal bovine model of right ventricular dysfunction. Am J Physiol. 2015;308:L158–L167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Paulin R, Sutendra G, Gurtu V, Dromparis P, Haromy A, Provencher S, Bonnet S, Michelakis ED. A miR‐208‐Mef2 axis drives the decompensation of right ventricular function in pulmonary hypertension. Circ Res. 2015;116:56–69. [DOI] [PubMed] [Google Scholar]
- 40. Broderick TL, King TM. Upregulation of GLUT‐4 in right ventricle of rats with monocrotaline—induced pulmonary hypertension. Med Sci Monit. 2008;14:BR261–BR264. [PubMed] [Google Scholar]
- 41. Talati MH, Brittain EL, Fessel JP, Penner N, Atkinson J, Funke M, Grueter C, Jerome WG, Freeman M, Newman JH, West J, Hemnes AR. Mechanisms of lipid accumulation in the bone morphogenetic protein receptor type 2 mutant right ventricle. Am J Respir Crit Care Med. 2016;194:719–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nouette‐Gaulain K, Malgat M, Rocher C, Savineau J‐P, Marthan R, Mazat J‐P, Sztark F. Time course of differential mitochondrial energy metabolism adaptation to chronic hypoxia in right and left ventricles. Cardiovasc Res. 2005;66:132–140. [DOI] [PubMed] [Google Scholar]
- 43. Enache I, Charles A‐L, Bouitbir J, Favret F, Zoll J, Metzger D, Oswald‐Mammosser M, Geny B, Charloux A. Skeletal muscle mitochondrial dysfunction precedes right ventricular impairment in experimental pulmonary hypertension. Mol Cell Biochem. 2013;373:161–170. [DOI] [PubMed] [Google Scholar]
- 44. Lauva IK, Brody E, Tiger E, Kent RL, Copper G IV, Marino TA. Control of myocardial tissue components and cardiocyte organelles in pressure‐overload hypertrophy of the cat right ventricle. Am J Anat. 1986;177:71–80. [DOI] [PubMed] [Google Scholar]
- 45. Olivetti G, Ricci R, Lagrasta C, Maniga E, Sonnenblick EH, Anversa P. Cellular basis of wall remodeling in long‐term pressure overload‐induced right ventricular hypertrophy in rats. Circ Res. 1988;63:648–657. [DOI] [PubMed] [Google Scholar]
- 46. Sack MN, Disch DL, Rockman HA, Kelly DP. A role for Sp and nuclear receptor transcription factors in a cardiac hypertrophic growth program. Proc Natl Acad Sci USA. 1997;94:6438–6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sheikh AM, Barrett C, Villamizar N, Alzate O, Valente AM, Herlong JR, Craig D, Lodge A, Lawson J, Milano C, Jaggers J. Right ventricular hypertrophy with early dysfunction: a proteomics study in a neonatal model. J Thorac Cardiovasc Surg. 2009;137:1146–1153. [DOI] [PubMed] [Google Scholar]
- 48. Mccommis KS, Douglas DL, Krenz M, Baines CP. Cardiac‐specific hexokinase 2 overexpression attenuates hypertrophy by increasing pentose phosphate pathway flux. J Am Heart Assoc. 2013;2:e000355 DOI: 10.1161/JAHA.113.000355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wu R, Wyatt E, Chawla K, Tran M, Ghanefar M, Laakso M, Epting CL, Ardehali H. Hexokinase II knockdown results in exaggerated cardiac hypertrophy via increased ROS production. EMBO Mol Med. 2012;4:633–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Calmettes G, John SA, Weiss JN, Ribalet B. Hexokinase—mitochondrial interactions regulate glucose metabolism differentially in adult and neonatal cardiac myocytes. J Gen Physiol. 2013;142:425–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001;15:1406–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fritz HL, Smoak IW, Branch S. Hexokinase I expression and activity in embryonic mouse heart during early and late organogenesis. Histochem Cell Biol. 1999;112:359–365. [DOI] [PubMed] [Google Scholar]
- 53. St John JC, Ramalho‐Santos J, Gray HL, Petrosko P, Rawe VY, Navara CS, Simerly CR, Schatten GP. The expression of mitochondrial DNA transcription factors during early cardiomyocyte in vitro differentiation from human embryonic stem cells. Cloning Stem Cells. 2005;7:141–153. [DOI] [PubMed] [Google Scholar]
- 54. Reddy S, Zhao M, Hu D‐Q, Fajardo G, Katznelson E, Punn R, Spin JM, Chan FP, Bernstein D. Physiologic and molecular characterization of a murine model of right ventricular volume overload. Am J Physiol Heart Circ Physiol. 2013;304:H1314–H1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Reddy S, Zhao M, Hu D‐Q, Fajardo G, Hu S, Ghosh Z, Rajagopalan V, Wu JC, Bernstein D. Dynamic microRNA expression during the transition from right ventricular hypertrophy to failure. Physiol Genomics. 2012;44:562–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Drake JI, Bogaard HJ, Mizuno S, Clifton B, Xie B, Gao Y, Dumur CI, Fawcett P, Voelkel NF, Natarajan R. Molecular signature of a right heart failure program in chronic severe pulmonary hypertension. Am J Respir Cell Mol Biol. 2011;45:1239–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Postic C, Leturque A, Printz RL, Maulard P, Loizeau M, Granner DK, Girard J. Development and regulation of glucose transporter and hexokinase expression in rat. Am J Physiol. 1994;266:E548–E559. [DOI] [PubMed] [Google Scholar]
- 58. Frille A, Steinhoff KG, Hesse S, Grachtrup S, Wald A, Wirtz H, Sabri O, Seyfarth H‐J. Thoracic [18F]fluorodeoxyglucose uptake measured by positron emission tomography/computed tomography in pulmonary hypertension. Med (Baltimore). 2016;95:e3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ohira H, deKemp R, Pena E, Davies RA, Stewart DJ, Chandy G, Contreras‐Dominguez V, Dennie C, Mc Ardle B, Mc Klein R, Renaud JM, DaSilva JN, Pugliese C, Dunne R, Beanlands R, Mielniczuk LM. Shifts in myocardial fatty acid and glucose metabolism in pulmonary arterial hypertension: a potential mechanism for a maladaptive right ventricular response. Eur Heart J Cardiovasc Imaging. 2016;17:1424–1431. [DOI] [PubMed] [Google Scholar]
- 60. Sakao S, Daimon M, Voelkel NF, Miyauchi H, Jujo T, Sugiura T, Ishida K, Tanabe N, Kobayashi Y, Tatsumi K. Right ventricular sugars and fats in chronic thromboembolic pulmonary hypertension. Int J Cardiol. 2016;219:143–149. [DOI] [PubMed] [Google Scholar]
- 61. Bokhari S, Raina A, Rosenweig EB, Schulze PC, Bokhari J, Einstein AJ, Barst RJ, Johnson LL. PET imaging may provide a novel biomarker and understanding of right ventricular dysfunction in patients with idiopathic pulmonary arterial hypertension. Circ Cardiovasc Imaging. 2011;4:641–647. [DOI] [PubMed] [Google Scholar]
- 62. Saygin D, Highland KB, Farha S, Park M, Sharp J, Roach EC, Tang WHW, Thomas JD, Erzurum SC, Neumann DR, DiFilippo FP. Metabolic and functional evaluation of the heart and lungs in pulmonary hypertension by gated 2‐[18F]‐Fluoro‐2‐deoxy‐D‐glucose positron emission tomography. Pulm Circ. 2017;7:428–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nakaya T, Ohira H, Tsujino I. Right heart morphology, function and metabolism in pulmonary hypertension. Respir Circ. 2016;64:543–547. [Google Scholar]
- 64. Oikawa M, Kagaya Y, Otani H, Sakuma M, Demachi J, Suzuki J, Takahashi T, Nawata J, Ido T, Watanabe J, Shirato K. Increased [18F]fluorodeoxyglucose accumulation in right ventricular free wall in patients with pulmonary hypertension and the effect of epoprostenol. J Am Coll Cardiol. 2005;45:1849–1855. [DOI] [PubMed] [Google Scholar]
- 65. Lundgrin EL, Park MM, Sharp J, Tang WHW, Thomas JD, Asosingh K, Comhair SA, DiFilippo FP, Neumann DR, Davis L, Graham BB, Tuder RM, Dostanic I, Erzurum SC. Fasting 2‐deoxy‐2‐[18F]fluoro‐D‐glucose positron emission tomography to detect metabolic changes in pulmonary arterial hypertension hearts over 1 year. Ann Am Thorac Soc. 2013;10:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Li W, Wang L, Xiong C‐M, Yang T, Zhang Y, Gu Q, Yang Y, Ni X‐H, Liu Z‐H, Fang W, He J‐G. The prognostic value of 18F‐FDG uptake ratio between the right and left ventricles in idiopathic pulmonary arterial hypertension. Clin Nucl Med. 2015;40:859–863. [DOI] [PubMed] [Google Scholar]
- 67. Tatebe S, Fukumoto Y, Oikawa‐Wakayama M, Sugimura K, Satoh K, Miura Y, Aoki T, Nochioka K, Miura M, Yamamoto S, Tashiro M, Kagaya Y, Shimokawa H. Enhanced [18F]fluorodeoxyglucose accumulation in the right ventricular free wall predicts long‐term prognosis of patients with pulmonary hypertension: a preliminary observational study. Eur Heart J Cardiovasc Imaging. 2014;15:666–672. [DOI] [PubMed] [Google Scholar]
- 68. Faber MJ, Dalinghaus M, Lankhuizen IM, Bezstarosti K, Verhoeven AJM, Duncker DJ, Helbing WA, Lamers JMJ. Time dependent changes in cytoplasmic proteins of the right ventricle during prolonged pressure overload. J Mol Cell Cardiol. 2007;43:197–209. [DOI] [PubMed] [Google Scholar]
- 69. Baandrup JD, Markvardsen LH, Peters CD, Schou UK, Jensen JL, Magnusson NE, Ørntoft TF, Kruhøffer M, Simonsen U. Pressure load: the main factor for altered gene expression in right ventricular hypertrophy in chronic hypoxic rats. PLoS One. 2011;6:e15859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Faber MJ, Dalinghaus M, Lankhuizen IM, Bezstarosti K, Dekkers DHW, Duncker DJ, Helbing WA, Lamers JMJ. Proteomic changes in the pressure overloaded right ventricle after 6 weeks in young rats: Correlations with the degree of hypertrophy. Proteomics. 2005;5:2519–2530. [DOI] [PubMed] [Google Scholar]
- 71. Wong YY, Raijmakers P, Van Campen J, Van Der Laarse WJ, Knaapen P, Lubberink M, Ruiter G, Noordegraaf AV, Lammertsma AA. 11C‐acetate clearance as an index of oxygen consumption of the right myocardium in idiopathic pulmonary arterial hypertension: a validation study using 15O‐labeled tracers and PET. J Nucl Med. 2013;54:1258–1262. [DOI] [PubMed] [Google Scholar]
- 72. Yoshinaga K, Ohira H, Tsujino I, Oyama‐Manabe N, Mielniczuk L, Beanlands RSB, Katoh C, Kasai K, Manabe O, Sato T, Fujii S, Ito YM, Tomiyama Y, Nishimura M, Tamaki N. Attenuated right ventricular energetics evaluated using 11C‐acetate PET in patients with pulmonary hypertension. Eur J Nucl Med Mol Imaging. 2014;41:1240–1250. [DOI] [PubMed] [Google Scholar]
- 73. Matsushita T, Ikeda S, Miyahara Y, Yakabe K, Yamaguchi K, Furukawa K, Iwasaki T, Shikuwa M, Fukui J, Kohno S. Use of [123I]‐BMIPP myocardial scintigraphy for the clinical evaluation of a fatty‐acid metabolism disorder of the right ventricle in chronic respiratory and pulmonary vascular disease. J Int Med Res. 2000;28:111–123. [DOI] [PubMed] [Google Scholar]
- 74. Sakao S, Miyauchi H, Voelkel NF, Sugiura T, Tanabe N, Kobayashi Y, Tatsumi K. Increased right ventricular fatty acid accumulation in chronic thromboembolic pulmonary hypertension. Ann Am Thorac Soc. 2015;12:1465–1472. [DOI] [PubMed] [Google Scholar]
- 75. Nagaya N, Goto Y, Satoh T, Uematsu M, Hamada S, Kuribayashi S, Okano Y, Kyotani S, Shimotsu Y, Fukuchi K, Nakanishi N, Takamiya M, Ishida Y. Impaired regional fatty acid uptake and systolic dysfunction in hypertrophied right ventricle. J Nucl Med. 1998;39:1676–1680. [PubMed] [Google Scholar]
- 76. Drake JI, Gomez‐Arroyo J, Dumur CI, Kraskauskas D, Natarajan R, Bogaard HJ, Fawcett P, Voelkel NF. Chronic carvedilol treatment partially reverses the right ventricular failure transcriptional profile in experimental pulmonary hypertension. Physiol Genomics. 2013;45:449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lewandowski ED, Fischer SK, Fasano M, Banke NH, Walker LA, Huqi A, Wang X, Lopaschuk GD, O'Donnell JM. Acute liver carnitine palmitoyltransferase I overexpression recapitulates reduced palmitate oxidation of cardiac hypertrophy. Circ Res. 2013;112:57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Talati M, Hemnes A. Fatty acid metabolism in pulmonary arterial hypertension: role in right ventricular dysfunction and hypertrophy. Pulm Circ. 2015;5:269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Brittain EL, Talati M, Fessel JP, Zhu H, Penner N, Calcutt MW, West JD, Funke M, Lewis GD, Gerszten RE, Hamid R, Pugh ME, Austin ED, Newman JH, Hemnes AR. Fatty acid metabolic defects and right ventricular lipotoxicity in human pulmonary arterial hypertension. Circulation. 2016;133:1936–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bitar FF, Bitar H, El Sabban M, Nasser M, Yunis KA, Tawil A, Dbaibo GS. Modulation of ceramide content and lack of apoptosis in the chronically hypoxic neonatal rat heart. Pediatr Res. 2002;51:144–149. [DOI] [PubMed] [Google Scholar]
- 81. Huss JM, Gigue V, Kelly DP. Estrogen‐related receptor alpha directs peroxisome proliferator‐activated receptor alpha signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Society. 2004;24:9079–9091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Burkart EM, Welch MJ, Kelly DP, Burkart EM, Sambandam N, Han X. Nuclear receptors PPAR b/d and PPAR a direct distinct metabolic regulatory programs in the mouse heart Find the latest version: nuclear receptors PPAR β/δ and PPAR α direct distinct metabolic regulatory programs in the mouse heart. J Clin Invest. 2007;117:3930–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Yang J, Sambandam N, Han X, Gross RW, Courtois M, Kovacs A, Febbraio M, Finck BN, Kelly DP. CD36 deficiency rescues lipotoxic cardiomyopathy. Circ Res. 2007;100:1208–1217. [DOI] [PubMed] [Google Scholar]
- 84. Duncan JG, Bharadwaj KG, Fong JL, Mitra R, Sambandam N, Courtois MR, Lavine KJ, Goldberg IJ, Kelly DP. Rescue of cardiomyopathy in peroxisome proliferator‐activated receptor‐alpha transgenic mice by deletion of lipoprotein lipase identifies sources of cardiac lipids and peroxisome proliferator‐activated receptor‐a activators. Circulation. 2010;121:426–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 2004;18:1692–1700. [DOI] [PubMed] [Google Scholar]
- 86. Urashima T, Zhao M, Wagner R, Fajardo G, Farahani S, Quertermous T, Bernstein D, Quertermous T, Molecular BD. Molecular and physiological characterization of RV remodeling in a murine model of pulmonary stenosis. Am J Physiol Heart Circ Physiol. 2008;295:H1351–H1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Friehs I, Cowan DB, Choi Y‐H, Black KM, Barnett R, Bhasin MK, Daly C, Dillon SJ, Libermann TA, McGowan FX, del Nido PJ, Levitsky S, McCully JD. Pressure‐overload hypertrophy of the developing heart reveals activation of divergent gene and protein pathways in the left and right ventricular myocardium. Am J Physiol Heart Circ Physiol. 2013;304:H697–H708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kreymborg Kg, Uchida S, Gellert P, Schneider A, Boettger T, Voswinckel R, Wietelmann A, Szibor M, Weissmann N, Ghofrani AH, Schermuly R, Schranz D, Seeger W, Braun T. Identification of right heart‐enriched genes in a murine model of chronic outflow tract obstruction. J Mol Cell Cardiol. 2010;49:598–605. [DOI] [PubMed] [Google Scholar]
- 89. Ni YG, Wang N, Cao DJ, Sachan N, Morris DJ, Gerard RD, Kuro‐o M, Rothermel BA, Hill JA. FoxO transcription factors activate Akt and attenuate insulin signaling in heart by inhibiting protein phosphatases. Proc Natl Acad Sci. 2007;104:20517–20522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hedges L, Olkin I. Statistical methods for meta‐analysis. Orlando: Academic Press; 1985. [Google Scholar]
- 91. Vesterinen HM, Sena ES, Egan KJ, Hirst TC, Churolov L, Currie GL, Antonic A, Howells DW, Macleod MR. Meta‐analysis of data from animal studies: a practical guide. J Neurosci Methods. 2014;221:92–102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental Methods.
Table S1. List of Studies Studying Metabolic Parameters in the Pressure‐Loaded Right Ventricle Included for Full Text Review
Table S2. Meta‐Regression Analyses: Variables Vs Duration of RV Pressure Load
Table S3. Meta‐Regression Analyses: Variables Vs Degree of RV Pressure Load
Table S4. Level of Significance of Meta‐Analysis and Meta‐Regression
Table S5. Overview of Hedges’ g for Metabolic Parameters in Models of RV Pressure Load Subjected to Therapeutic Interventions
Figure S1. Models of Increased Pressure Load. Forest plots of right ventricular systolic pressure (A) and Fulton index (B). Data are presented as Hedges’ g (95% CI). Combined Hedges’ g are presented as squares: gray representing Hedges’ g of a specific model, black representing Hedges’ g of all included studies. Bars represent 95% CI. i2 indicates level of heterogeneity; n, number of included animals.
Figure S2. Forest plots of expression of PDK isoenzymes. PDK4 at both mRNA (A) and protein level (B), and PDK1 (C) and PDK2 (D) at protein level. Bars represent 95% CI. =, indicates not statistically significant affected; ↓, decreased; CI, cardiac index; CO, cardiac output; i2, level of heterogeneity; n, number of included animals; PDK, pyruvate dehydrogenase kinase; RVEF, RV ejection fraction; TAPSE, tricuspid annular plane systolic movement; X, not included in meta‐analysis.
Figure S3. Forest plot MCAD at protein level. Bars represent 95% CI. = indicates not statistically significant affected; ↓, decreased; CI, cardiac index; CO, cardiac output; i2, level of heterogeneity; MCAD, medium chain acyl CoA dehydrogenase; n, number of included animals; RVEF, RV ejection fraction; TAPSE, tricuspid annular plane systolic movement; X, not included in meta‐analysis.
Figure S4. Regulators of metabolism. Meta‐regression of mitochondrial content with degree of RV pressure load (A). PGC1α gene expression (B) and its relation with duration of pressure load shown by meta‐regression in a bubble plot (C). Forrest plot of PGC1α protein expression (D). Forrest plot of PPARα gene expression (E). Data are presented as Hedges’ g. Combined Hedges’ g are presented as squares: gray representing Hedges’ g of a specific model, black representing Hedges’ g of all included studies. Bars represent 95% CI. Bubble size represents relative study precision, calculation based on SD. Black line represents regression line, gray lines represents 95% CI. = indicates not statistically significant affected; ↓, decreased; CI, cardiac index; CO, cardiac output; i2, level of heterogeneity; n, number of included animals; RVEF, RV ejection fraction; TAPSE, tricuspid annular plane systolic movement. *Significantly (P<0.05) increased compared with SuHx‐model.