

Abstract
Background
Mycobacterium smegmatis is a saprophytic bacterium frequently used as a genetic surrogate to study pathogenic Mycobacterium tuberculosis. The PrrAB two-component genetic regulatory system is essential in M. tuberculosis and represents an attractive therapeutic target. In this study, transcriptomic analysis (RNA-seq) of an M. smegmatis ΔprrAB mutant was used to define the PrrAB regulon and provide insights into the essential nature of PrrAB in M. tuberculosis.
Results
RNA-seq differential expression analysis of M. smegmatis wild-type (WT), ΔprrAB mutant, and complementation strains revealed that during in vitro exponential growth, PrrAB regulates 167 genes (q < 0.05), 57% of which are induced in the WT background. Gene ontology and cluster of orthologous groups analyses showed that PrrAB regulates genes participating in ion homeostasis, redox balance, metabolism, and energy production. PrrAB induced transcription of dosR (devR), a response regulator gene that promotes latent infection in M. tuberculosis and 21 of the 25 M. smegmatis DosRS regulon homologues. Compared to the WT and complementation strains, the ΔprrAB mutant exhibited an exaggerated delayed growth phenotype upon exposure to potassium cyanide and respiratory inhibition. Gene expression profiling correlated with these growth deficiency results, revealing that PrrAB induces transcription of the high-affinity cytochrome bd oxidase genes under both aerobic and hypoxic conditions. ATP synthesis was ~ 64% lower in the ΔprrAB mutant relative to the WT strain, further demonstrating that PrrAB regulates energy production.
Conclusions
The M. smegmatis PrrAB two-component system regulates respiratory and oxidative phosphorylation pathways, potentially to provide tolerance against the dynamic environmental conditions experienced in its natural ecological niche. PrrAB positively regulates ATP levels during exponential growth, presumably through transcriptional activation of both terminal respiratory branches (cytochrome c bc1-aa3 and cytochrome bd oxidases), despite transcriptional repression of ATP synthase genes. Additionally, PrrAB positively regulates expression of the dormancy-associated dosR response regulator genes in an oxygen-independent manner, which may serve to fine-tune sensory perception of environmental stimuli associated with metabolic repression.
Keywords: Mycobacterium smegmatis, Mycobacterium tuberculosis, prrAB, Two-component system, RNA-seq, Transcriptomics, Hypoxia, Respiration, Oxidative phosphorylation, ATP
Background
Two-component systems (TCSs) participate in signal transduction pathways and are ubiquitously found in bacteria, archaea, some lower eukaryotes and plants [1–4]. TCSs recognize specific environmental stimuli [5] and integrate an adaptive response, frequently by modulating transcription [6]. A prototypical TCS consists of a membrane-bound histidine kinase sensor and a cytoplasmic DNA-binding response regulator. In pathogenic bacteria, TCSs act as virulence factors that regulate diverse survival mechanisms, such as antibiotic resistance [7], phosphate limitation [8], low oxygen tension [9], and evasion of immune responses [10]. Though mammalian proteins bearing histidine kinase sequence motifs and activity [11] have been identified, response regulators appear absent in humans, opening the possibility for development of inhibitors targeting virulence-related or essential bacterial TCSs as novel therapeutic approaches.
Mycobacterium tuberculosis, the causative agent of tuberculosis, is an ancient disease of mankind and the leading cause of death from an infectious agent [12]. The M. tuberculosis genome harbors 11 paired TCSs, two orphaned histidine kinases, and six orphaned response regulators [13]. Of these TCSs, only MtrAB [14] and PrrAB [15] are essential for M. tuberculosis viability. The prrA response regulator and prrB histidine kinase genes are conserved across all fully-sequenced mycobacterial genomes, suggesting an evolutionary selective pressure to retain these TCS genes. M. tuberculosis prrAB is upregulated during the early stages of human macrophage infection [13] and under in vitro nitrogen limitation [15]. During infection in murine macrophages, prrAB is required for early replication and adaptation to the intracellular environment [16]. Capitalizing on findings that diarylthiazole compounds inhibit M. tuberculosis growth via the PrrAB TCS, Bellale et al. [17] exposed M. tuberculosis cultures to diarylthiazole and found that PrrAB modulates transcription of genes enabling metabolic adaptation to a lipid-rich environment, responsiveness to reduced oxygen tension, and production of essential ribosomal proteins and amino acid tRNA synthases.
Mycobacterium smegmatis strain mc2155 [18] is a non-pathogenic, rapid-growing, saprophytic mycobacterium that is used as a surrogate model to study M. tuberculosis genetics and mycobacterial TCSs. We recently demonstrated that prrAB is not essential in M. smegmatis and that PrrAB differentially regulates triacylglycerol biosynthetic genes during ammonium limitation [19]. The inability to generate an M. tuberculosis prrAB knockout mutant [15], the high degree of PrrA sequence identity (95%) between M. tuberculosis and M. smegmatis, and the presence of over 2000 homologous genes (51% of total genes in M. tuberculosis H37Rv) shared between these species prompted use of the M. smegmatis ΔprrAB mutant to better understand PrrAB transcriptional regulatory properties. A comprehensive profiling of the genes and pathways regulated by PrrAB in M. smegmatis would provide insights into the genetic adaptations that occur during M. tuberculosis infection and open new avenues for discovering novel therapeutic targets to treat tuberculosis.
In this study, we used RNA-seq-based transcriptomics analysis to obtain a global profile of the genes regulated by PrrAB in M. smegmatis. We compared the transcriptomic profiles of M. smegmatis WT, ΔprrAB mutant, and prrAB complementation strains during mid-logarithmic growth under standard laboratory conditions. Genes repressed by PrrAB were associated with broad aspects of metabolism and components of the F1F0 ATPase, while PrrAB induced genes involved in oxidoreductase activity, respiration, hypoxic response, and ion homeostasis. These data provide seminal information into the transcriptional regulatory properties of the mycobacterial PrrAB TCS and how PrrAB may be controlling molecular processes important in M. tuberculosis and other mycobacteria.
Results
Phylogenetic analyses of PrrA and PrrB in mycobacteria
Since prrAB orthologues are present in all mycobacterial species and prrAB is essential for viability in M. tuberculosis [15], it is reasonable to believe that PrrAB fulfills important regulatory properties in mycobacteria. We therefore questioned the evolutionary relatedness or distance between PrrA and PrrB proteins in mycobacteria. The M. tuberculosis H37Rv and M. smegmatis mc2155 PrrA and PrrB amino acid sequences share 93 and 81% identity, respectively. Maximum-likelihood phylogenetic trees, based on PrrA (Fig. 1a) and PrrB (Fig. 1b) multiple sequence alignments, were generated. Using the Gupta et al. [20] recent reclassification of mycobacterial species, the results suggested that, with a few exceptions, PrrA and PrrB evolved with specific mycobacterial clades (Fig. 1). While subtle differences in the PrrA or PrrB sequences may represent evolutionary changes as mycobacterial species of the same clade adapted to similar environmental niches, additional experiments are needed to determine if prrAB is essential in other pathogenic mycobacteria.
Fig. 1.

Maximum-likelihood phylogenetic analyses of mycobacterial (a) PrrA and (b) PrrB sequences based on the recent reclassification of mycobacterial species by Gupta et al. [20]. Blue squares, Fortuitum-Vaccae clade. Red triangles, Trivale clade. Green diamonds, Tuberculosis-Simiae clade. Yellow circles, Abscessus-Chelonae clade. Purple triangles, Terrae clade. M. smegmatis mc2155 and M. tuberculosis H37Rv are indicated by blue and green arrows, respectively. PrrA and PrrAB sequences were aligned using default MUSCLE algorithms [21] and phylogenetic tree was generated in MEGA 7 [22]
We next questioned if the distinct phylogenetic separations between clades could be mapped to specific PrrA or PrrB amino acid residues. We separately aligned mycobacterial PrrA and PrrB sequences in JalView using the default MUSCLE algorithm [21]. Within species of the Abscessus-Chelonae clade, two unique PrrA signatures were found: asparagine and cysteine substitutions relative to serine 38 (S38) and serine 49 (S49), respectively, of the M. smegmatis PrrA sequence (See Additional file 1: Figure S1). These Abscessus-Chelonae clade PrrA residues were not found at similar aligned sites in other mycobacteria (See Additional file 1: Figure S1). Similarly, members of the Abscessus-Chelonae clade (except Mycobacteriodes abscessus) harbored unique amino acid substitutions in PrrB, including glutamate, valine, lysine, aspartate, lysine, and valine corresponding to threonine 42 (T42), glycine 67 (G67), valine 90 (V90), methionine 318 (M318), alanine 352 (A352), and arginine (R371), respectively, of the M. smegmatis PrrB sequence (See Additional file 1: Figure S2).
Transcriptomics analysis of the M. smegmatis WT, ΔprrAB mutant, and complementation strains
We previously generated an M. smegmatis mc2155 prrAB deletion mutant (mc2155::ΔprrAB; FDL10) and its complementation strain (mc2155::ΔprrAB::prrAB; FDL15) [19]. Since the prrAB regulon and the environmental cue which stimulates PrrAB activity are unknown, a global transcriptomics approach was used to analyze differential gene expression in standard laboratory growth conditions. RNA-seq was used to determine transcriptional differences between the ΔprrAB mutant, mc2155, and the complementation strains during mid exponential growth, corresponding to an OD600 of ~ 0.6 (See Additional file 1: Figure S3), in supplemented Middlebrook 7H9 (M7H9) broth. Total RNA was isolated from three independent, biological replicates of each M. smegmatis strain. Based on multidimensional scaling (MDS) plot, one mc2155 biological replicate was deemed an outlier and excluded from subsequent analyses (details in Methods, see Additional file 1: Figure S4). Principal component analysis of the global expression patterns of the samples demonstrated that samples from the mc2155 and FDL15 complementation strains clustered together, apart from those of the FDL10 ΔprrAB strain with the majority of variance occurring along PC1 (See Additional file 1: Figure S5), indicating complementation with ectopically-expressed prrAB in the ΔprrAB background.
Identifying the PrrAB regulon
To identify differentially-expressed genes (DEGs), pair-wise comparisons of normalized read counts between the ΔprrAB mutant and WT (FDL10 vs. mc2155) as well as the ΔprrAB mutant and prrAB complementation (FDL10 vs. FDL15) datasets were performed using EdgeR. Deletion of prrAB resulted in induction of 95 genes and repression of 72 genes (q < 0.05), representing 167 transcriptional targets (Fig. 2a) that are repressed and induced, respectively, by PrrAB in the WT background (Fig. 2c). Less conservative comparisons revealed 683 DEGs (p < 0.05) between the WT and ΔprrAB mutant strains (See Additional file 1: Figure S6a). Between the ΔprrAB complementation and prrAB mutant strains, 67 DEGs (q < 0.05) were identified (Fig. 2b), representing 35 repressed and 32 induced genetic targets by the complementation of PrrAB (Fig. 2c), while less conservative comparisons (p < 0.05) revealed 578 DEGs (See Additional file 1: Figure S6a). Overall, pair-wise DEG analyses revealed that during mid-logarithmic M. smegmatis growth, PrrAB regulates transcription through a relatively balanced combination of gene induction and repression. In addition, comparison between the two DEG sets (i.e., for mc2155 vs. FDL10 and FDL15 vs. FDL10) datasets revealed 40 (Fig. 2e) and 226 (See Additional file 1: Figure S6b) overlapping DEGs at the significance levels of q < 0.05 and p < 0.05, respectively. Hierarchical clustering with the overlapping DEGs further illustrated that gene expression changes induced by the prrAB deletion were partially recovered by prrAB complementation (Fig. 2d). We randomly selected six DEGs for qRT-PCR analyses and verified the RNA-seq results for five genes in both the FDL10 vs. mc2155 and FDL10 vs. FDL15 comparisons (See Additional file 1: Figure S7). [See Additional file 2 for a complete list of DEGs between all pair-wise comparisons.]
Fig. 2.

Global DEG profiles (q < 0.05) between the mc2155 vs. FDL10 and FDL15 vs. FDL10 RNA-seq comparisons. Volcano plots of (a) FDL10 vs. mc2155 and (b) FDL10 vs. FDL15 group comparisons with red and blue dots representing differentially-expressed genes with p < 0.05 and q < 0.05, respectively. The horizontal hatched line indicates p = 0.05 threshold, while the left and right vertical dotted lines indicate log2 fold change of − 1 and + 1, respectively. c Repressed (blue) and induced (yellow) DEGs (q < 0.05) in mc2155 (WT) and FDL15 (prrAB complementation strain) compared to the FDL10 ΔprrAB mutant. d Average hierarchical clustering (FPKM + 1) of individual RNA-seq sample replicates. e Venn diagrams indicating 40 overlapping DEGs (q < 0.05) between mc2155 vs. FDL10 (WT vs. ΔprrAB mutant) and FDL15 vs. FDL10 (prrAB complementation strain vs. ΔprrAB mutant) strain comparisons
Gene ontology and clustering analyses
To infer function of the genes regulated by PrrAB, enrichment of gene ontology (GO) terms (biological processes and molecular functions) in the DEGs of the mc2155 vs. FDL10 comparison was assessed by the DAVID functional annotation tool (See Additional file 3 for a complete list of functional annotations returned from the DAVID results). The two sets of DEGs from the mc2155 vs. FDL10 comparison (See Additional file 1: Figure S6) were examined. In general, genes repressed by PrrAB were associated with numerous metabolic processes (Fig. 3a) and nucleotide binding (Fig. 3b), while PrrAB-induced genes were associated with ion or chemical homeostasis (Fig. 3c) and oxidoreductase, catalase, and iron-sulfur cluster binding activities (Fig. 3d). Similar GO enrichment terms in the two group comparisons (mc2155 vs. FDL10 and FDL15 vs. FDL10) suggested evidence of genetic complementation (Fig. 3; Additional file 1: Figure S8). GO term enrichment was also found for metabolism, nucleotide binding, oxidoreductase, and catalase activity, based on conservative (q < 0.05) DEG comparisons (See Additional file 1: Figures S9 and S10). The GO enrichment analyses suggested that during M. smegmatis exponential growth in M7H9 medium, PrrAB negatively regulates genes associated with diverse components of metabolic and biosynthetic processes and positively regulates expression of genes participating in respiration (qcrA, cydA, and cydB), ion transport (via the F1F0 ATPase), redox mechanisms, and recognition of environmental signals (dosR2) (Fig. 3; Additional file 1: Figures S8, S9, and S10).
Fig. 3.

GO term enrichment associated with DEGs (p < 0.05) that are (a, b) repressed (c, d) or induced by PrrAB in the WT background. GO terms categorized by (a, c) biological processes (BP) or (b, d) molecular function (MF). a GO terms share a common set of genes: MSMEG 3564, MSMEG 6422, and MSMEG 6467
Classification of genes (q < 0.05) based on clusters of orthologous groups (COGs) analyses were then performed using the online eggNOG mapper program. Of all COG categories in each gene list, 32% (n = 22) and 24% (n = 20) of genes repressed or induced by PrrAB, respectively, participate in diverse aspects of metabolism (Fig. 4), thus corroborating the GO results. Of the COG categories induced by PrrAB, 17% (n = 14) were associated with energy production and conversion (COG Category C). The relatively even proportions of COG categories associated with PrrAB-induced and repressed genes (Fig. 4) suggest that this TCS, as both transcriptional activator and repressor, fine-tunes diverse cellular functions to maximize and/or optimize growth potential during exponential replication.
Fig. 4.

COG analysis of DEGs (q < 0.05) induced (yellow) or repressed (blue) by PrrAB in the WT background. COGs from each category were normalized to represent the percent abundance of each category to all COGs returned in the induced or repressed analyses, respectively
PrrAB regulates dosR expression in M. smegmatis
Differential expression analysis revealed significant repression of MSMEG 5244 and MSMEG 3944, two orthologues of the dosR (devR) response regulator gene, in the ΔprrAB mutant strain (Fig. 2a). In M. tuberculosis, the hypoxia-responsive DosRS (DevRS) TCS (along with the DosT histidine kinase) induces transcription of ~ 50 genes that promote dormancy and chronic infection [23]. Here, we designate MSMEG 5244 as dosR1 (due to its genomic proximity to dosS) and MSMEG 3944 as dosR2. Among the 25 M. smegmatis homologues of the M. tuberculosis DosRS regulon genes, 7 genes were differentially expressed (+ 2-fold changes, q < 0.05) in pair-wise comparisons among the three strains (Fig. 5 and Additional file 4). Importantly, each of these M. smegmatis DosRS regulon homologues were induced by PrrAB in the WT and complementation backgrounds, corroborating the activity of the DosR as a positive transcriptional regulator [23].
Fig. 5.
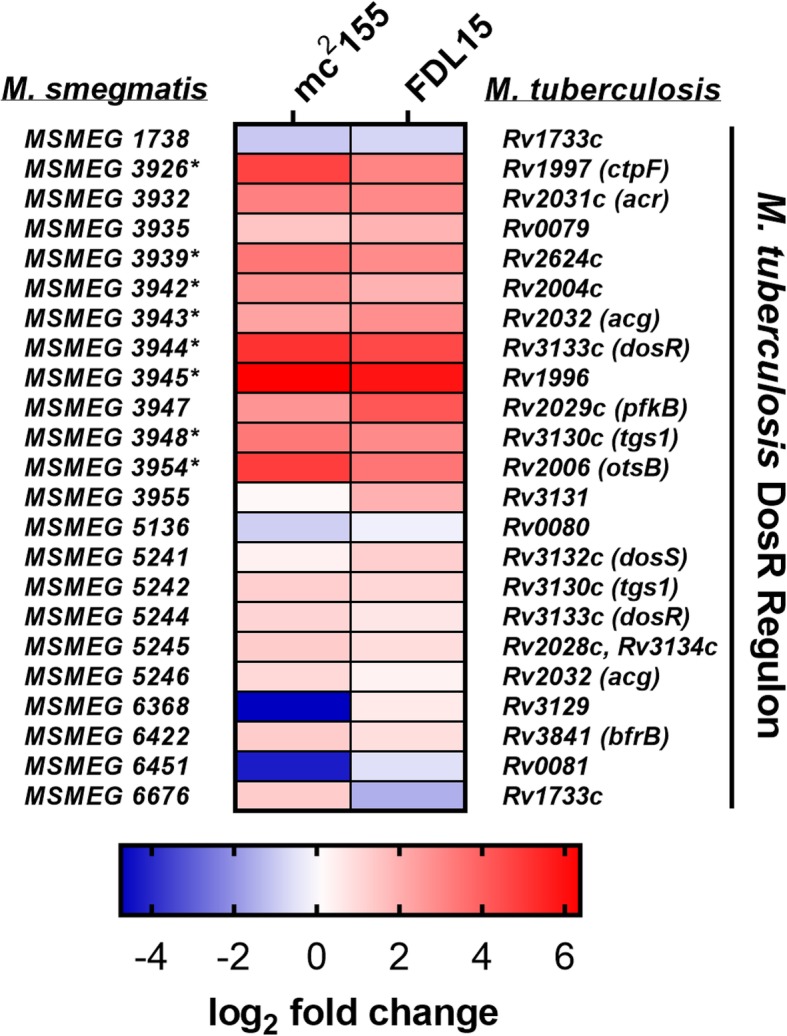
M. smegmatis PrrAB regulates dormancy-associated genes of the DosR regulon. Heatmap of M. smegmatis RNA-seq DEGs associated with M. tuberculosis dosR regulon homologues. Color bar indicates log2 fold change values corresponding to mc2155 vs. FDL10 (left tiles) and FDL15 vs. FDL10 (right tiles) DEGs. M. smegmatis genes differentially regulated (q < 0.05) are denoted by asterisks
PrrAB contributes to M. smegmatis adaptation to hypoxia
The cytochrome bd oxidase respiratory system is a high-affinity terminal oxidase that is important for M. smegmatis survival under microaerophilic conditions [24]. Because the cydA, cydB, and cydD genes were repressed in the ΔprrAB mutant during aerobic growth (Fig. 2a; Additional file 2), we questioned if the ΔprrAB mutant was more sensitive to hypoxia than the WT strain. Compared to WT and the prrAB complementation strains, the ΔprrAB mutant exhibited reduced viability (See Additional file 1: Figure S11a) and produced smaller colonies (See Additional file 1: Figure S11b) after 24 h hypoxia exposure. In contrast, cell viability and colony sizes were similar for all strains cultured under aerobic growth conditions (See Additional file 1: Figure S11).
Next, we questioned if differential expression of cydA, cydB, and cydD correlated with growth deficiencies in the ΔprrAB mutant during hypoxia. We compared transcriptional profiles of cydA, cydB, and cydD by qRT-PCR from each strain incubated in M7H9 broth under hypoxic and aerobic conditions for 24 h. After 24 h hypoxia, cydA and dosR2 expression was significantly decreased approximately 100-fold and 10-fold, respectively, in the ΔprrAB mutant relative to the WT strain (Fig. 6a, e). Expression levels of cydA and cydB were significantly reduced in the ΔprrAB mutant relative to the WT strain during aerobic growth (Fig. 6a, b). Furthermore, both dosR1 and dosR2 were significantly downregulated in the ΔprrAB mutant under aerobic conditions (Fig. 6d, e), further verifying the RNA-seq data (Additional file 2) and PrrAB-mediated regulation in both oxygen-rich and oxygen-poor environmental conditions.
Fig. 6.

PrrAB regulates cytochrome bd and dosR expression and is protective during hypoxia and cyanide-mediated respiratory inhibition. qRT-PCR of (a) cydA (MSMEG 3233), (b) cydB (MSMEG 3232), (c) cydD (MSMEG 3231), (d) dosR1 (MSMEG 5244), and (e) dosR2 (MSMEG 3944) RNA isolated from M. smegmatis strains cultured under aerobic (solid bars) or hypoxic (hatched bars) conditions for 24 h. Relative gene expression was calculated using the 2-ΔCt method and normalized to 16S rRNA for each strain and growth condition. qRT-PCR measurements for each gene and each condition (aerobic or hypoxic growth) was assessed in triplicate. *, p < 0.05; **, p < 0.01; ***, p < 0.001; one-way ANOVA, Dunnett’s multiple comparisons. (f) M. smegmatis growth in the presence (dashed lines) or absence (solid lines) of 1 mM cyanide (KCN). **, p < 0.01; ****, p < 0.0001; unpaired Student’s t tests. Values represent the mean ± SEM of data collected from three independent cultures
The ΔprrAB mutant is hypersensitive to cyanide exposure
Cyanide is a potent inhibitor of the aa3 cytochrome c oxidase in bacteria. Conversely, cytochrome bd oxidases in Escherichia coli [25], Pseudomonas aeruginosa [26], some staphylococci [27], and M. smegmatis [24] are relatively insensitive to cyanide inhibition. In the absence of alternative electron acceptors (e.g., nitrate and fumarate), aerobic respiratory capacity after cyanide-mediated inhibition of the M. smegmatis aa3 terminal oxidase would be provided by the cytochrome bd terminal oxidase (CydAB). Because cydA, cydB, and cydD were significantly repressed in the ΔprrAB mutant (Fig. 2a), as were most subunits of the cytochrome c bc1 – aa3 respiratory oxidase complex (See Additional file 2), we hypothesized that the ΔprrAB mutant would be hypersensitive to cyanide relative to the WT and complementation strains. Cyanide inhibited all three strains during the first 24 h (Fig. 6f). While the WT and complementation strains entered exponential growth after 24 h of cyanide exposure, the ΔprrAB mutant exhibited significantly delayed and slowed growth between 48 and 72 h (Fig. 6f). These data demonstrated that the ΔprrAB mutant strain had defects in alternative cytochrome bd terminal oxidase pathways, further supporting that genes controlling cytochrome c bc1 and aa3 respiratory oxidases are induced by PrrAB.
PrrAB positively regulates ATP levels
KEGG pathway analysis of DEGs (p < 0.05) induced by PrrAB revealed oxidative phosphorylation as a significantly enriched metabolic pathway (Additional file 3; enrichment = 3.78; p = 0.017). Further examination of the RNA-seq data generally revealed that genes of the terminal respiratory complexes (cytochrome c bc1-aa3 and cytochrome bd oxidases) were induced by PrrAB, whereas F1F0 ATP synthase genes were repressed by PrrAB (Fig. 7a). Therefore, we hypothesized that ATP levels would be greater in the ΔprrAB mutant relative to the WT and complementation strains despite the apparent downregulation of terminal respiratory complex genes (except ctaB) in the ΔprrAB mutant (Fig. 7a). While viability was similar between strains at the time of sampling (Fig. 7b), ATP levels ([ATP] pM/CFU) were 36 and 76% in the ΔprrAB mutant and complementation strains, respectively, relative to the WT strain (Fig. 7c). Ruling out experimental artifacts, we confirmed sufficient cell lysis with the BacTiter-Glo reagent (See Methods) and that normalized extracellular ATP in cell-free supernatants were similar to intracellular ATP levels (See Additional file 1: Figure S12). These data suggested that PrrAB positively regulates ATP levels during aerobic logarithmic growth, although prrAB complementation did not fully restore ATP to WT levels (Fig. 7c). Additionally, ATP levels correlated with PrrAB induction of respiratory complex genes rather than PrrAB-mediated repression than F1F0 ATP synthase genes (Fig. 7a). To verify the RNA-seq data which indicates PrrAB repression of nearly all F1F0 ATP synthase genes (Fig. 7a), we directly measured transcription of three genes in the atp operon: atpC (MSMEG 4935), atpH (MSMEG 4939), and atpI (MSMEG 4943).
Fig. 7.

PrrAB regulates oxidative phosphorylation genes and ATP levels in M. smegmatis. a Heatmap of genes participating in oxidative phosphorylation. Color bar indicates log2 fold change of gene expression between mc2155 vs. FDL10 (left column) and FDL15 vs. FDL10 (right column). M. smegmatis genes significantly regulated are denoted by asterisks (*, p < 0.05; **, q < 0.05) in at least one group comparison. b M. smegmatis viability (CFU/ml) at harvest and (c) corresponding ATP levels (pM/CFU) normalized to mc2155 were measured from exponentially-growing (OD600 ~ 0.6) aerobic cultures in M7H9 broth. ****, p < 0.0001; one-way ANOVA, Dunnett’s multiple comparisons
The qRT-PCR results revealed that PrrAB represses atpC, atpH, and atpI in the WT and prrAB complementation strains (See Additional file 1: Figure S13).
Discussion
TCSs provide transcriptional flexibility and adaptive responses to specific environmental stimuli in bacteria [28]. The mycobacterial PrrAB TCS is conserved across most, if not all, mycobacterial lineages and is essential for viability in M. tuberculosis [15], thus representing an attractive therapeutic target [17]. Here, we use an M. smegmatis ΔprrAB mutant [19] as a surrogate to provide insights into the essential nature and regulatory properties associated with the PrrAB TCS in M. tuberculosis. Our rationale for this approach is founded on the high degree of identity between the M. smegmatis and M. tuberculosis PrrA and PrrB sequences, including 100% identity in the predicted DNA-binding recognition helix of PrrA (See Additional file 1: Figure S14) [29].
Using BLAST queries of M. smegmatis PrrA and PrrB against 150 recently reclassified mycobacterial species, as proposed by Gupta et al. [20], all fully-sequenced mycobacterial genomes harbored prrA and prrB homologues, implying strong evolutionary conservation for the PrrAB TCS. Likely due to the incomplete genomic sequences [20], prrA was not found in Mycobacterium timonense and Mycobacterium bouchedurhonense genomes, while a prrB homolog was not identified in Mycobacterium avium subsp. silvaticum. Phylogenetic analyses showed that PrrA and PrrB sequences grouped closely, but not perfectly, within members of specific mycobacterial clades (Fig. 1), and members of the Abscessus-Chelonae clade harbored unique PrrA and PrrB amino acid substitutions (See Additional file 1: Figures S1 and S2). While it is unclear if these residues impact PrrA or PrrB functionality in the Abscessus-Chelonae clade, it may be possible to develop prrAB-based single nucleotide polymorphism genotyping or proteomic technologies for differentiating mycobacterial infections. Multiple sequence alignments of the M. smegmatis and M. tuberculosis PrrA DNA-binding recognition helices revealed 100% sequence conservation (See Additional file 1: Figure S14), suggesting a shared set of core genes regulated by PrrA in mycobacteria. Incorporation of a global approach, such as ChIP-seq, will be valuable for identifying and characterizing the essential genes directly regulated by PrrA in M. tuberculosis and other mycobacterial species.
We used RNA-seq-based transcriptomics analyses to define the M. smegmatis PrrAB regulon during exponential growth under standard laboratory conditions. We showed that in M. smegmatis, PrrAB deletion led to differential expression of 167 genes (q < 0.05), corresponding to ~ 2% of chromosomal genes, of which 95 genes are induced and 72 are repressed in the WT background (Fig. 2). Importantly, PrrAB differentially-regulated genes were involved in aerobic and microaerophilic respiration. The cytochrome c terminal oxidase bc1 (qcrCAB) and aa3 (ctaC) genes are essential in M. tuberculosis, but not in M. smegmatis, and mutants in the latter species are attenuated during exponential phase growth [30]. If M. tuberculosis PrrAB also regulates genes of the cytochrome c bc1 and/or aa3 respiratory complex, it could partially explain prrAB essentiality.
To corroborate the key findings from comparing the ΔprrAB mutant and WT strains, we included the prrAB complementation strain in our RNA-seq analyses. Of the 683 DEGs (p < 0.05) that were affected by the ΔprrAB mutation, expression changes of 10 genes were variably reversed in the ΔprrAB complementation strain. Induction of the three genes (MSMEG 5659, MSMEG 5660, and MSMEG 5661) adjacent to prrAB could be related to alteration of regulatory control sequences during generation of the knockout mutation. These results were unlikely due to poor RNA quality, as RNA integrity numbers (RIN) were consistently high (Additional file 5). We previously demonstrated similar prrA transcription and PrrA protein levels in the WT and complementation strains during aerobic mid-logarithmic growth in M7H9 broth [19], similar to the growth conditions employed in this study. The lack of full complementation seen in our RNA-seq results is likely affected by the low number of biological replicates analyzed. Baccerella et al. [31] demonstrated that sample number impacts RNA-seq performance to a greater degree relative to read depth. Although we found only 226 overlapping DEGs (p < 0.05) between the mc2155 vs. FDL10 and FDL15 vs. FDL10 group comparisons, global DEG regulation (i.e., relative ratios of up- or down-regulated genes), was similar. In both pairwise comparisons, 32 and 36% of all DEGs were induced by PrrAB in the WT and complementation backgrounds, respectively, while 68 and 64% of all DEGs were repressed by PrrAB in the WT and complementation backgrounds, respectively. These data indicate that complementation with prrAB in the deletion background restored global transcriptomic profiles to WT levels. Including additional biological replicates will improve the statistical reliability for better comparison of wild-type and complementation strains which, to the best of our knowledge, has not been previously reported in a transcriptomic study.
We found 40 DEGs (q < 0.05) that overlapped between the WT vs. ΔprrAB mutant and complementation vs. ΔprrAB mutant group comparisons (Fig. 2e). In this data set, enrichment analysis for the GO term “response to stimulus” contains genes of the DosR regulon (Additional file 6). A less-conservative approach using 226 overlapping DEGs (p < 0.05) revealed enrichment in GO terms related to respiratory pathways and ATP synthesis (Additional file 6), therefore corroborating our phenotypic and biochemical data (Figs. 6 and 7). It is interesting to postulate that these DEGs may accurately represent the PrrAB regulon in M. smegmatis under the conditions tested, as they are significantly represented in both WT and complementation strain group comparisons. Future studies are warranted to explore the utility of incorporating sequencing data from both WT and complementation strains to improve the reliability of transcriptomics experiments.
M. tuberculosis acclimates to an intramacrophage environment and the developing granuloma by counteracting the detrimental effects of hypoxia [32], nutrient starvation [33], acid stress [34], and defense against reactive oxygen and nitrogen species [35]. Adaptive measures to these environmental insults include activation of the dormancy regulon and upregulation of the high-affinity cytochrome bd respiratory oxidase [35], induction of the glyoxylate shunt and gluconeogenesis pathways [36], asparagine assimilation [37], and nitrate respiration [38]. As a saprophytic bacterium, M. smegmatis could encounter similar environmental stresses as M. tuberculosis, despite their drastically different natural environmental niches. Conserving the gene regulatory circuit of the PrrAB TCS for adaptive responses would thus be evolutionarily advantageous.
The hypoxia-responsive DosRS TCS controls the dormancy regulon in both M. tuberculosis [23] and M. smegmatis [39–41]. The M. smegmatis DosRS TCS regulates dormancy phenotypes similar to M. tuberculosis, including upregulation of the dosRS TCS [39], gradual adaptation to oxygen depletion [42], and upregulation of alanine dehydrogenase [43]. DosR is required for optimal viability in M. smegmatis after the onset of hypoxia [41]. Our RNA-seq and qRT-PCR data revealed that PrrAB induces both M. smegmatis dosR homologues (dosR1 and dosR2) during aerobic and hypoxic growth (Additional file 2, Fig. 2a, Fig. 6d, and Fig. 6e). Additionally, the RNA-seq data revealed that PrrAB induces genes associated with the M. tuberculosis DosR regulon [23, 44] (Fig. 5). Thus, it is possible that PrrAB also positively regulates dosR expression in M. tuberculosis, which would provide additional mechanisms of dosR control as previously demonstrated with PknB [45], PknH [46], NarL [47], and PhoP [48].
The M. tuberculosis respiration and oxidative phosphorylation pathways have increasingly gained attention as promising anti-tuberculosis therapeutic targets. Bedaquiline (TMC207), a recent FDA-approved mycobacterial F1F0 ATP synthase inhibitor, is active against drug-sensitive and drug-resistant M. tuberculosis strains [49, 50], as is Q203 (telacebec), a cytochrome c bc1 inhibitor, which has advanced to Phase 2 clinical trials [51]. Accumulating evidence suggests that the alternative terminal cytochrome bd oxidase system, encoded by the cydABDC genes in M. tuberculosis, is important during chronic infection and may represent a novel drug target. M. tuberculosis cydA mutants are hypersensitive to the bactericidal activity of bedaquiline [52], suggesting that combined therapeutic regimens simultaneously targeting the F1F0 ATP synthase and cytochrome bd oxidase represent promising anti-tuberculosis treatment strategies. Analysis of the DEGs (p < 0.05) induced by PrrAB (Additional file 3) revealed significant enrichment of the oxidative phosphorylation KEGG pathway, including genes encoding the cytochrome c bc1 (qcrA), cytochrome c aa3 (ctaC, ctaE), and cytochrome bd (cydB, cydD) terminal respiratory branches. We showed that the ΔprrAB mutant was more sensitive to hypoxic stress and cyanide inhibition relative to the WT and complementation strains (See Additional file 1: Figure S11 and Fig. 6), thus corroborating the transcriptomics results. Although 24 h hypoxia only caused a modest reduction in the ΔprrAB mutant after 24 h hypoxia exposure, relative to the WT and complementation strains, the ΔprrAB mutant small colony phenotype indicated a growth defect under these conditions (See Additional file 1: Figure S11). Additionally, qRT-PCR results demonstrated significantly lower expression of cydA and dosR2 in the ΔprrAB mutant relative to WT during hypoxic growth, further supporting the biological data. The combined results demonstrate that PrrAB contributes to optimal growth during and after hypoxic stress. We recently reported that the ΔprrAB mutant is hypersensitive to hypoxia during growth in low-ammonium medium [19]. Our current data suggest that the hypoxia growth defect exhibited by the ΔprrAB mutant is likely not medium-specific, but rather a global consequence of differential regulation of respiratory and/or the dosR regulon genes. Bacterial cytochrome bd oxidases are relatively insensitive to cyanide inhibition compared to the cytochrome c oxidase respiratory branch [53–55]. Growth of the ΔprrAB mutant in the presence of 1 mM potassium cyanide was similar to M. smegmatis cydA mutant growth under similar conditions [24]. Our data demonstrates that the M. smegmatis PrrAB TCS controls expression of aerobic and microaerophilic respiratory genes. Notably, to date, a master transcriptional regulator of respiratory systems in M. tuberculosis has not been discovered.
We found increased expression of the F1F0 ATP synthase genes, including atpA, atpD, atpF, atpG, and atpH, in the ΔprrAB mutant strain compared to WT (Fig. 7a; Additional file 1: Figure S13 and Additional file 2), leading us to hypothesize that ATP levels would be elevated in the ΔprrAB mutant. Conversely, ATP levels were lower in ΔprrAB mutant strain compared to the WT and complementation strains (Fig. 7c). Induction of atp genes in the ΔprrAB mutant may indicate a compensatory measure to maintain ATP homeostasis due to repression of the bc1-aa3 terminal respiratory complex (except ctaB) and hence, disruption of the transmembrane proton gradient.
Via comprehensive transcriptomics analyses, we demonstrated that PrrAB regulates expression of genes involved in respiration, environmental adaptation, ion homeostasis, oxidoreductase activity, and metabolism in M. smegmatis. The inability to induce transcription of the cydA, cydB, cydD, dosR1, and dosR2 genes likely led the ΔprrAB mutant to grow poorly after 24 h hypoxia exposure. An important goal of our RNA-seq study was to provide insight into the essential nature of PrrAB in M. tuberculosis using an M. smegmatis ΔprrAB mutant as a surrogate model while recognizing differences in their natural environmental niches, pathogenic potential, and genetic composition. From a therapeutic perspective, PrrAB could influence the sensitivity of M. tuberculosis to Q203 and/or bedaquiline by controlling expression of cytochrome bd oxidase, cytochrome c bc1 oxidase, and ATP synthase genes. Furthermore, it remains unknown whether diarylthiazoles directly target PrrB [17] or whether the prrB mutations associated with diarylthiazole resistance are compensatory in nature. Taken together, our study provides seminal information regarding the mycobacterial PrrAB TCS regulon as well as a powerful surrogate platform for in-depth investigations of this essential TCS in M. tuberculosis.
Conclusions
We used RNA-seq-based transcriptomics as an experimental platform to provide insights into the essential M. tuberculosis prrAB TCS using an M. smegmatis ΔprrAB mutant as a genetic surrogate. In M. smegmatis, PrrAB regulates high-affinity respiratory systems, intracellular redox and ATP balance, and the dosR TCS response regulator genes, all of which promote infectious processes in M. tuberculosis. Using these results, we may be able to exploit diarylthiazole compounds that putatively target the PrrB histidine kinase as synergistic therapies with bedaquiline. These results are informing the basis of prrAB essentiality in M. tuberculosis and advancing our understanding of regulatory systems that control metabolic, respiration, energy-generating, and dormancy pathways in mycobacteria. Exploitation of PrrAB as a drug target will advance the discovery and development of novel therapeutics to combat the global tuberculosis epidemic.
Methods
Bacterial strains and culture conditions
Genetic construction of the M. smegmatis FDL10 ΔprrAB deletion mutant and the FDL15 complementation strain was previously described [19]. All M. smegmatis strains (mc2155, FDL10, and FDL15) were routinely cultured in Middlebrook 7H9 broth (pH 6.8) supplemented with 10% albumin-dextrose-saline (ADS), 0.2% glycerol (v/v), and 0.05% Tween 80 (v/v), herein referred to as M7H9. M. smegmatis was incubated on Middlebrook 7H10 agar supplemented with 10% ADS and 0.5% glycerol, herein referred to as M7H10 agar, for CFU/ml enumeration.
Hypoxic growth conditions
M. smegmatis strains were initially cultured aerobically in M7H9 medium at 37 °C, 100 rpm to an OD600 ~ 0.6. Cells were diluted into fresh, pre-warmed M7H9 to an OD600 ~ 0.05, serially diluted in PBS (pH 7.4), and spot-plated onto M7H10 agar. The plates were transferred to a GasPak chamber containing two anaerobic GasPak sachets (Beckon Dickinson, Franklin Lakes, NJ, USA), sealed, and incubated at 37 °C for 24 h after the onset of hypoxia (~ 6 h), as indicated by decolorization of an oxygen indicator tablet included with the sachet. Plates were then incubated aerobically for an additional 48 h to allow colony outgrowth. Control plates were cultured under aerobic conditions for 48 h prior to counting and documenting colonies. Colonies were visualized using a dissecting microscope (Stereomaster, Fisher Scientific). All experiments were performed in triplicate.
Cyanide inhibition assays
M. smegmatis strains were grown in the presence of potassium cyanide (KCN) as described by [24] with modifications. Briefly, cultures were inoculated into prewarmed M7H9 broth to an OD600 ~ 0.05 and incubated at 37 °C, 100 rpm for 30 min. KCN, prepared in M7H9 broth, was then added to a final concentration of 1 mM and growth was allowed to resume. Negative control cultures using M7H9 broth without KCN addition were performed concurrently. Cultures were grown for 5 d with samples collected at 24 h intervals for OD600 measurements and CFU quantitation on M7H10 agar. All experiments were performed in triplicate.
ATP assays
M. smegmatis strains were cultured in M7H9 broth at 37 °C, 100 rpm. Cultures were sampled in 100 μl aliquots upon reaching an OD600 ~ 0.6, flash-frozen in a dry ice-ethanol bath, and stored at − 70 °C for 7 d. Cells were thawed at room temperature and ATP quantification was performed using the BacTiter-Glo kit (Promega, Madison, WI, USA). Fifty μl of cells were mixed with equal volumes of BacTiter-Glo reagent in opaque 96-well plates and incubated at room temperature for 5 min. ATP standard curves were included in the same plate. Relative luminescence was measured in a SpectraMax M5 plate reader (Molecular Devices, San Jose, CA, USA). To assess lysis efficiency, viability of all samples was confirmed after both freeze-thaw and processing in the BacTiter-Glo reagent by plating serial dilutions onto M7H10 agar followed by incubation at 37 °C for 48–72 h. Lysis efficiencies collected from three independent cultures of mc2155, FDL10, and FDL15 were 99.97% (± 0.03), 99.99% (± 0.04), and 99.99% (± 0.02), respectively. Cell viability was quantified for each sample at the time of harvest by plating serial dilutions onto M7H10 agar followed by incubation at 37 °C for 48 h before enumerating CFU/ml. Samples for extracellular ATP measurement were collected as described by Hirokana et al. [56]. Briefly, cells were harvested by centrifugation at 10,621 x g for 2 min at 4 °C. The supernatant was clarified via 0.22 μm filtration, and aliquots (100 μl) were flash-frozen in a dry ice-ethanol bath and stored at − 70 °C until further use. After thawing, ATP was measured using the BacTiter-Glo kit, as described above. Filtered supernatants were spot plated onto M7H10 agar and incubated at 37 °C for 3 d to verify lack of contaminating cells. All strains were analyzed in triplicate with two technical replicates each.
RNA isolation
For aerobic cultures, M. smegmatis strains mc2155, FDL10, and FDL15 were grown in 30 ml M7H9 at 37 °C, 100 rpm until mid-logarithmic phase (OD600 ~ 0.6). For hypoxic cultures, M. smegmatis strains were first grown (OD600 ~ 0.6) aerobically in M7H9. Each culture (15 ml) was then transferred a fresh tube, and methylene blue (1.5 μg/ml, final concentration) was added as an indicator of O2 depletion. Cultures were incubated in a sealed GasPak chamber containing two anaerobic sachets (Beckon Dickinson, Franklin Lakes, NJ, USA) for 24 h post-decolorization of the methylene blue in the media. Culture aliquots (15 ml) were harvested by centrifugation at 3441 x g for 10 min at 4 °C. The supernatant was discarded, and the cell pellet was resuspended in 1 ml TRIzol (Invitrogen), transferred to 2 ml screw cap tubes containing 500 mg of zirconia-silicate beads (0.1–0.15 mm), and placed on ice. Cells were mechanically disrupted 3X by bead beating (BioSpec Products) at the highest setting for 40 s and incubated on ice for at least 1 min between disruptions. The cell lysates were incubated at room temperature for 5 min, centrifuged at 13,000 x g for 1 min to separate cell debris, and the supernatant was transferred to a new microcentrifuge tube. Chloroform (200 μl) was added, and samples were vortexed for 15 s followed by 5 min incubation at 4 °C. The homogenate was centrifuged at 13,000 x g for 15 min at 4 °C and the upper, aqueous phase was transferred to a new microcentrifuge tube. RNA was precipitated with 500 μl isopropanol overnight at 4 °C. Total RNA was pelleted by centrifugation at 13,000 x g for 15 min at 4 °C, and the supernatant was discarded. RNA pellets were washed 2X with 70% ethanol and centrifuged at 13,000 x g for 5 min at 4 °C between washes. After evaporation of residual ethanol by air-drying, total RNA was resuspended in 100 μl nuclease-free H2O. Total RNA (10 μg) was treated with TURBO-DNase (Invitrogen, Carlsbad, CA) for 20 min at 37 °C to degrade residual genomic DNA. RNA samples were purified using the RNeasy Mini Kit (Qiagen, Germany) and eluted in 50 μl nuclease-free H2O. RNA yields were quantified by Nanodrop (Thermo Scientific, Waltham, MA), and quality was assessed by agarose gel electrophoresis and a 2100 Bioanalyzer (Agilent, Santa Clara, CA). RNA (250 ng) was subjected to PCR using primers directed at the 16S rRNA gene to confirm lack of residual genomic DNA.
RNA-seq library preparation
cDNA was generated from RNA using the Nugen Ovation RNA-seq System via single primer isothermal amplification and automated on the BRAVO NGS liquid handler (Agilent, Santa Clara, CA, USA). cDNA was quantified on the Nanodrop (Thermo Fisher Scientific) and was sheared to approximately 300 bp fragments using the Covaris M220 ultrasonicator. Libraries were generated using the Kapa Biosystem’s library preparation kit (Kapa Biosystems, Wilmington, MA, USA). Fragments were end-repaired and A-tailed and individual indexes and adapters (Bioo, catalogue #520999) were ligated on each separate sample. The adapter-ligated molecules were cleaned using AMPure beads (Agencourt Bioscience/Beckman Coulter, La Jolla, CA, USA), and amplified with Kapa’s HIFI enzyme (Kapa Biosystems, Wilmington, MA, USA). Each library was then analyzed for fragment size on an Agilent Tapestation and quantified by qPCR (KAPA Library Quantification Kit, Kapa Biosystems, Wilmington, MA, USA) using Quantstudio 5 (Thermo Fisher Scientific) prior to multiplex pooling.
Sequencing and data processing
Sequencing was performed on a 1 × 75 bp flow cell using the NextSeq500 platform (Illumina) at the ASU Genomics Core facility. The total number of 101,054,986 Illumina NextSeq500 paired-end reads were generated from nine RNA samples (i.e., triplicates for each strain). The total number of reads generated for each sample ranged from 7,729,602 to 14,771,490. RNA-seq reads for each sample were quality checked using FastQC v 0.10.1 and aligned to the Mycolicibacterium smegmatis MC2155 assembly obtained from NCBI (https://www.ncbi.nlm.nih.gov/assembly/GCF_000015005.1/) using STAR v2.5.1b. Cufflinks v2.2.1 was used to report FPKM (Fragments Per Kilobase of transcript per Million mapped reads) values and the read counts. As a quality check for the biological replicates, overall similarity of gene expression profiles were then assessed by MDS, in which distances correspond to leading log-fold changes between samples. The MDS analysis demarcated clearly one of the three mc2155 samples as an outlier that did not cluster with the other two mc2155 samples and the three FDL15 samples (See Additional file 1: Figure S3), and the sample was thus excluded from further analysis. Average genome-wide expression (FPKM) was 6.76 for the WT strain, 5.88 for the ΔprrAB mutant, and 6.38 for the complementation strain.
Bioinformatics analysis
Differential expression analysis was performed with EdgeR package from Bioconductor v3.2 in R 3.2.3. EdgeR applied an overdispersed Poisson model to account for variance among biological replicates. Empirical Bayes tagwise dispersions were also estimated to moderate the overdispersion across transcripts. Then, a negative binomial generalized log-linear model was fit to the read counts for each gene for all comparison pairs. For each pairwise comparison, genes with p values < 0.05 were considered significant and log2-fold changes of expression between conditions (logFC) were reported. False discovery rate was calculated following the Benjamini and Hochberg procedure [57], the expected proportion of false discoveries amongst the rejected hypotheses.
Principal component analysis (PCA) was done on the scaled data using the prcomp function in R. Clustering analysis was done using Cluster 3.0 software, in which normalized expression (FPKM + 1) values were log2 transformed and grouped using uncentered Pearson’s correlation distance and average linkage hierarchal clustering [58]. Data matrices and tree dendrograms were visualized in Java TreeView. Gene ontology (GO) term enrichment, KEGG pathways, and statistical analyses of differentially expressed genes were performed using the DAVID functional annotation tool (https://david.ncifcrf.gov/summary.jsp). Clusters of orthologous groups (COGs) were obtained by querying DEGs (q < 0.05) against the eggNOG Mapper database (http://eggnogdb.embl.de/#/app/emapper).
Quantitative RT-PCR (qRT-PCR)
cDNA libraries from each RNA sample (described above) were generated by reverse transcription of 1 μg total RNA using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA), according to the manufacturer’s instructions. Primer efficiency was validated against 10-fold dilution standard curves using a cutoff criterion for acceptable efficiency of 90–110% and coefficient of determination (R2) ≥ 0.997. Relative gene expression was calculated using the 2-ΔCt or 2-ΔΔCt method [59], as indicated, and using the 16S gene as an internal normalization reference. The primers used for qRT-PCR are described in Table S1 (See Additional file 1).
Phylogenetic analyses
The M. smegmatis mc2155 PrrA and PrrB sequences were separately queried in BLASTp (https://blast.ncbi.nlm.nih.gov/Blast.cgi) against all Mycobacteriacea (taxid: 1762). Sequences corresponding to the revised mycobacterial phylogenetic clade classification [20] were selected for further analysis. When multiple hits were returned from the same species, those corresponding to the lowest E-value were selected for alignment. Compiled PrrA and PrrB sequences were separately aligned in MEGA 7 (https://www.megasoftware.net/) using default MUSCLE algorithms. Maximum-likelihood phylogenetic trees were generated in MEGA 7 and visualized by iTOL [60].
Statistical analyses
We used one-way ANOVA to assess significant differences in cell viability, qRT-PCR gene expression, and ATP quantification assays. Statistical analyses were performed using GraphPad Prism 7 (GraphPad Software, San Diego, CA) and p-values of < 0.05 were considered statistically significant. For volcano plot data, the -log10 p-value of each DEG was plotted against the ratio of the mean log2-fold change of each differential expressed gene between FDL10 vs. mc2155 or FDL10 vs. FDL15.
Supplementary information
Additional file 1. Supplemental figures.
Additional file 2. M. smegmatis DEG data sets.
Additional file 3. DAVID gene ontology results from significant DEGs.
Additional file 4. DosR regulon DEG and comparison of genes betweeen M. smegmatis and M. tuberculosis.
Additional file 5. RNA Bioanalyzer results (RIN numbers and electrophoretic traces).
Additional file 6. DAVID gene ontology results from overlapping DEGs between mc2155 vs. FDL10 and FDL15 vs. FDL10 data sets.
Acknowledgements
We thank Jason Steel and the Genomics Core at the ASU Biodesign Institute for cDNA library preparations and for performing the Illumina sequencing. We thank Yannik Haller for assisting with the qRT-PCR experiments. We also appreciate the critical insight and suggestions from the anonymous reviewers, which ultimately led to an improved manuscript.
Abbreviations
- ADS
Albumin-dextrose-saline
- COG
Clusters of orthologous groups
- DEG
Differentially expressed gene
- FPKM
Fragments per kilobase of transcript per million mapped reads
- GO
Gene ontology
- KCN
Potassium cyanide
- logFC
log2 fold change
- M7H9
Middlebrook 7H9
- MDS
Multidimensional scaling
- PCA
Principal component analysis
- qRT-PCR
Quantitative reverse transcriptase PCR
- TCS
Two-component system
- WT
Wild-type
Authors’ contributions
SEH conceived and designed the study with JDM. JDM performed the experiments. SY and JGP processed and analyzed the RNA-seq data. JDM and SY performed the bioinformatics analyses. JDM and SEH analyzed the data. JDM, JGP, and SEH wrote the manuscript. All authors read and approved the final manuscript.
Funding
This work was partially supported by a Potts Memorial Foundation grant to SEH. The Potts Memorial Foundation was not involved in the design of the study, in the collection, analysis, and interpretation of data, or in writing the manuscript.
Availability of data and materials
The raw Illumina paired-end sequence data for the RNA-seq studies performed in this article are available at the NCBI Sequence Read Archive (SRA) under the BioProject number PRJNA532282 under accession numbers SAMN11393348, SAMN11393349, and SAMN11393350. The assembled genome sequence for Mycolicibacterium smegmatis MC2 155 can be found in the GenBank database under assembly accession GCA_000015005.1.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jason D. Maarsingh, Email: Jason.Maarsingh@asu.edu
Shanshan Yang, Email: Shanshan.Yang.1@asu.edu.
Jin G. Park, Email: Jin.Park.1@asu.edu
Shelley E. Haydel, Email: Shelley.Haydel@asu.edu
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12864-019-6105-3.
References
- 1.Zschiedrich CP, Keidel V, Szurmant H. Molecular mechanisms of two-component signal transduction. J Mol Biol. 2016;428(19):3752–3775. doi: 10.1016/j.jmb.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krell T. Exploring the (almost) unknown: Archaeal two-component systems. J Bacteriol. 2018;200(7):10.1128. doi: 10.1128/JB.00774-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schaap P, Barrantes I, Minx P, Sasaki N, Anderson RW, Benard M, et al. The Physarum polycephalum genome reveals extensive use of prokaryotic two-component and metazoan-type tyrosine kinase signaling. Genome Biol Evol. 2015;8(1):109–125. doi: 10.1093/gbe/evv237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu Z, Yuan L, Sundaresan V, Yu X. Arabidopsis CKI1 mediated two-component signaling in the specification of female gametophyte. Plant Signal Behav. 2018;13(10):e1469360. doi: 10.1080/15592324.2018.1469360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar A, Toledo JC, Patel RP, Lancaster JR, Jr, Steyn AJ. Mycobacterium tuberculosis DosS is a redox sensor and DosT is a hypoxia sensor. Proc Natl Acad Sci U S A. 2007;104(28):11568–11573. doi: 10.1073/pnas.0705054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Richmond GE, Evans LP, Anderson MJ, Wand ME, Bonney LC, Ivens A, et al. The Acinetobacter baumannii two-component system AdeRS regulates genes required for multidrug efflux, biofilm formation, and virulence in a strain-specific manner. mBio. 2016;7(2):e00430–e00416. doi: 10.1128/mBio.00430-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gebhardt MJ, Shuman HA. GigA and GigB are master regulators of antibiotic resistance, stress responses, and virulence in Acinetobacter baumannii. J Bacteriol. 2017;199(10):10.1128. doi: 10.1128/JB.00066-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelliher JL, Radin JN, Kehl-Fie TE. PhoPR contributes to Staphylococcus aureus growth during phosphate starvation and pathogenesis in an environment-specific manner. Infect Immun. 2018;86(10):10.1128. doi: 10.1128/IAI.00371-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mehra S, Foreman TW, Didier PJ, Ahsan MH, Hudock TA, Kissee R, et al. The DosR regulon modulates adaptive immunity and is essential for Mycobacterium tuberculosis persistence. Am J Respir Crit Care Med. 2015;191(10):1185–1196. doi: 10.1164/rccm.201408-1502OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herrera CM, Crofts AA, Henderson JC, Pingali SC, Davies BW, Trent MS. The Vibrio cholerae VprA-VprB two-component system controls virulence through endotoxin modification. mBio. 2014;5(6):10.1128. doi: 10.1128/mBio.02283-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Srivastava S, Li Z, Ko K, Choudhury P, Albaqumi M, Johnson AK, et al. Histidine phosphorylation of the potassium channel KCa3.1 by nucleoside diphosphate kinase B is required for activation of KCa3.1 and CD4 T cells. Mol Cell. 2006;24(5):665–675. doi: 10.1016/j.molcel.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 12.WHO . Global tuberculosis report 2017. Geneva: The World Health Organization; 2017. [Google Scholar]
- 13.Haydel SE, Clark-Curtiss JE. Global expression analysis of two-component system regulator genes during Mycobacterium tuberculosis growth in human macrophages. FEMS Microbiol Lett. 2004;236(2):341–347. doi: 10.1111/j.1574-6968.2004.tb09667.x. [DOI] [PubMed] [Google Scholar]
- 14.Zahrt TC, Deretic V. An essential two-component signal transduction system in Mycobacterium tuberculosis. J Bacteriol. 2000;182(13):3832–3838. doi: 10.1128/JB.182.13.3832-3838.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haydel SE, Malhotra V, Cornelison GL, Clark-Curtiss JE. The prrAB two-component system is essential for Mycobacterium tuberculosis viability and is induced under nitrogen-limiting conditions. J Bacteriol. 2012;194(2):354–361. doi: 10.1128/JB.06258-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ewann F, Jackson M, Pethe K, Cooper A, Mielcarek N, Ensergueix D, et al. Transient requirement of the PrrA-PrrB two-component system for early intracellular multiplication of Mycobacterium tuberculosis. Infect Immun. 2002;70(5):2256–2263. doi: 10.1128/IAI.70.5.2256-2263.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bellale E, Naik M, BV V, Ambady A, Narayan A, Ravishankar S, et al. Diarylthiazole: an antimycobacterial scaffold potentially targeting PrrB-PrrA two-component system. J Med Chem. 2014;57(15):6572–6582. doi: 10.1021/jm500833f. [DOI] [PubMed] [Google Scholar]
- 18.Snapper SB, Melton RE, Mustafa S, Kieser T, Jacobs WR., Jr Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol Microbiol. 1990;4(11):1911–1919. doi: 10.1111/j.1365-2958.1990.tb02040.x. [DOI] [PubMed] [Google Scholar]
- 19.Maarsingh JD, Haydel SE. Mycobacterium smegmatis PrrAB two-component system influences triacylglycerol accumulation during ammonium stress. Microbiology. 2018;164(10):1276–1288. doi: 10.1099/mic.0.000705. [DOI] [PubMed] [Google Scholar]
- 20.Gupta RS, Lo B, Son J. Phylogenomics and comparative genomic studies robustly support division of the genus Mycobacterium into an emended genus Mycobacterium and four novel genera. Front Microbiol. 2018;9:67. doi: 10.3389/fmicb.2018.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113–2105. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutignary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park HD, Guinn KM, Harrell MI, Liao R, Voskuil MI, Tompa M, et al. Rv3133c/dosR is a transcription factor that mediates the hypoxic response of Mycobacterium tuberculosis. Mol Microbiol. 2003;48(3):833–843. doi: 10.1046/j.1365-2958.2003.03474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kana BD, Weinstein EA, Avarbock D, Dawes SS, Rubin H, Mizrahi V. Characterization of the cydAB-encoded cytochrome bd oxidase from Mycobacterium smegmatis. J Bacteriol. 2001;183(24):7076–7086. doi: 10.1128/JB.183.24.7076-7086.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Korshunov S, Imlay KR, Imlay JA. The cytochrome bd oxidase of Escherichia coli prevents respiratory inhibition by endogenous and exogenous hydrogen sulfide. Mol Microbiol. 2016;101(1):62–77. doi: 10.1111/mmi.13372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cunningham L, Pitt M, Williams HD. The cioAB genes from Pseudomonas aeruginosa code for a novel cyanide-insensitive terminal oxidase related to the cytochrome bd quinol oxidases. Mol Microbiol. 1997;24(3):579–591. doi: 10.1046/j.1365-2958.1997.3561728.x. [DOI] [PubMed] [Google Scholar]
- 27.Voggu L, Schlag S, Biswas R, Rosenstein R, Rausch C, Gotz F. Microevolution of cytochrome bd oxidase in staphylococci and its implication in resistance to respiratory toxins released by Pseudomonas. J Bacteriol. 2006;188(23):8079–8086. doi: 10.1128/JB.00858-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Calva E, Oropeza R. Two-component signal transduction systems, environmental signals, and virulence. Microb Ecol. 2006;51(2):166–176. doi: 10.1007/s00248-005-0087-1. [DOI] [PubMed] [Google Scholar]
- 29.Nowak E, Panjikar S, Konarev P, Svergun DI, Tucker PA. The structural basis of signal transduction for the response regulator PrrA from Mycobacterium tuberculosis. J Biol Chem. 2006;281(14):9659–9666. doi: 10.1074/jbc.M512004200. [DOI] [PubMed] [Google Scholar]
- 30.Matsoso LG, Kana BD, Crellin PK, Lea-Smith DJ, Pelosi A, Powell D, et al. Function of the cytochrome bc1-aa3 branch of the respiratory network in mycobacteria and network adaptation occurring in response to its disruption. J Bacteriol. 2005;187(18):6300–6308. doi: 10.1128/JB.187.18.6300-6308.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baccarella A, Williams CR, Parrish JZ, Kim CC. Empirical assessment of the impact of sample number and read depth on RNA-Seq analysis workflow performance. BMC Bioinformatics. 2018;19(1):423. doi: 10.1186/s12859-018-2445-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aly S, Wagner K, Keller C, Malm S, Malzan A, Brandau S, et al. Oxygen status of lung granulomas in Mycobacterium tuberculosis-infected mice. J Pathol. 2006;210(3):298–305. doi: 10.1002/path.2055. [DOI] [PubMed] [Google Scholar]
- 33.Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, et al. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J Exp Med. 2003;198(5):693–704. doi: 10.1084/jem.20030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rohde KH, Veiga DF, Caldwell S, Balazsi G, Russell DG. Linking the transcriptional profiles and the physiological states of Mycobacterium tuberculosis during an extended intracellular infection. PLoS Path. 2012;8(6):e1002769. doi: 10.1371/journal.ppat.1002769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Voskuil MI, Visconti KC, Schoolnik GK. Mycobacterium tuberculosis gene expression during adaptation to stationary phase and low-oxygen dormancy. Tuberculosis. 2004;84(3–4):218–227. doi: 10.1016/j.tube.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 36.Timm J, Post FA, Bekker LG, Walther GB, Wainwright HC, Manganelli R, et al. Differential expression of iron-, carbon-, and oxygen-responsive mycobacterial genes in the lungs of chronically infected mice and tuberculosis patients. Proc Natl Acad Sci U S A. 2003;100(24):14321–14326. doi: 10.1073/pnas.2436197100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gouzy A, Larrouy-Maumus G, Bottai D, Levillain F, Dumas A, Wallach JB, et al. Mycobacterium tuberculosis exploits asparagine to assimilate nitrogen and resist acid stress during infection. PLoS Path. 2014;10(2):e1003928. doi: 10.1371/journal.ppat.1003928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tan MP, Sequeira P, Lin WW, Phong WY, Cliff P, Ng SH, et al. Nitrate respiration protects hypoxic Mycobacterium tuberculosis against acid- and reactive nitrogen species stresses. PLoS One. 2010;5(10):e13356. doi: 10.1371/journal.pone.0013356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mayuri B. G, das TK, Tyagi JS. Molecular analysis of the dormancy response in Mycobacterium smegmatis: expression analysis of genes encoding the DevR-DevS two-component system, Rv3134c and chaperone alpha-crystallin homologues. FEMS Microbiol Lett. 2002;211(2):231–237. doi: 10.1111/j.1574-6968.2002.tb11230.x. [DOI] [PubMed] [Google Scholar]
- 40.Bagchi G. Mayuri, Tyagi JS. Hypoxia-responsive expression of Mycobacterium tuberculosis Rv3134c and devR promoters in Mycobacterium smegmatis. Microbiology. 2003;149:2303–2305. doi: 10.1099/mic.0.C0120-0. [DOI] [PubMed] [Google Scholar]
- 41.O'Toole R, Smeulders MJ, Blokpoel MC, Kay EJ, Lougheed K, Williams HD. A two-component regulator of universal stress protein expression and adaptation to oxygen starvation in Mycobacterium smegmatis. J Bacteriol. 2003;185(5):1543–1554. doi: 10.1128/JB.185.5.1543-1554.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dick T, Lee BH, Murugasu-Oei B. Oxygen depletion induced dormancy in Mycobacterium smegmatis. FEMS Microbiol Lett. 1998;163(2):159–164. doi: 10.1111/j.1574-6968.1998.tb13040.x. [DOI] [PubMed] [Google Scholar]
- 43.Feng Z, Caceres NE, Sarath G, Barletta RG. Mycobacterium smegmatis L-alanine dehydrogenase (Ald) is required for proficient utilization of alanine as a sole nitrogen source and sustained anaerobic growth. J Bacteriol. 2002;184(18):5001–5010. doi: 10.1128/JB.184.18.5001-5010.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Voskuil MI, Schnappinger D, Visconti KC, Harrell MI, Dolganov GM, Sherman DR, et al. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J Exp Med. 2003;198(5):705–713. doi: 10.1084/jem.20030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bae HJ, Lee HN, Baek MN, Park EJ, Eom CY, Ko IJ, et al. Inhibition of the DevSR two-component system by overexpression of Mycobacterium tuberculosis PknB in Mycobacterium smegmatis. Mol Cells. 2017;40(9):632–642. doi: 10.14348/molcells.2017.0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chao JD, Papavinasasundaram KG, Zheng X, Chavez-Steenbock A, Wang X, Lee GQ, et al. Convergence of Ser/Thr and two-component signaling to coordinate expression of the dormancy regulon in Mycobacterium tuberculosis. J Biol Chem. 2010;285(38):29239–29246. doi: 10.1074/jbc.M110.132894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Malhotra V, Agrawal R, Duncan TR, Saini DK, Clark-Curtiss JE. Mycobacterium tuberculosis response regulators, DevR and NarL, interact in vivo and co-regulate gene expression during aerobic nitrate metabolism. J Biol Chem. 2015;290(13):8294–8309. doi: 10.1074/jbc.M114.591800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vashist A, Malhotra V, Sharma G, Tyagi JS, Clark-Curtiss JE. Interplay of PhoP and DevR response regulators defines expression of the dormancy regulon in virulent Mycobacterium tuberculosis. J Biol Chem. 2018;293(42):16413–16425. doi: 10.1074/jbc.RA118.004331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Diacon AH, Pym A, Grobusch M, Patientia R, Rustomjee R, Page-Shipp L, et al. The diarylquinoline TMC207 for multidrug-resistant tuberculosis. New Engl J Med. 2009;360(23):2397–2405. doi: 10.1056/NEJMoa0808427. [DOI] [PubMed] [Google Scholar]
- 50.Andries K, Verhasselt P, Guillemont J, Gohlmann HWH, Neefs JM, Winkler H, et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science. 2005;307(5707):223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 51.Butler MS, Blaskovich MA, Cooper MA. Antibiotics in the clinical pipeline at the end of 2015. J Antibiot. 2017;70(1):3–24. doi: 10.1038/ja.2016.72. [DOI] [PubMed] [Google Scholar]
- 52.Berney M, Hartman TE, Jacobs WR., Jr A Mycobacterium tuberculosis cytochrome bd oxidase mutant is hypersensitive to bedaquiline. mBio. 2014;5(4):e01275–e01214. doi: 10.1128/mBio.01275-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Megehee JA, Hosler JP, Lundrigan MD. Evidence for a cytochrome bcc-aa3 interaction in the respiratory chain of Mycobacterium smegmatis. Microbiology. 2006;152(3):823–829. doi: 10.1099/mic.0.28723-0. [DOI] [PubMed] [Google Scholar]
- 54.Hammer ND, Schurig-Briccio LA, Gerdes SY, Gennis RB, Skaar EP. CtaM is required for menaquinol oxidase aa3 function in Staphylococcus aureus. mBio. 2016;7(4). 10.1128/mBio.00823-00816. [DOI] [PMC free article] [PubMed]
- 55.Hirai T, Osamura T, Ishii M, Arai H. Expression of multiple cbb3 cytochrome c oxidase isoforms by combinations of multiple isosubunits in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2016;113(45):12815–12819. doi: 10.1073/pnas.1613308113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hironaka I, Iwase T, Sugimoto S, Okuda K, Tajima A, Yanaga K, et al. Glucose triggers ATP secretion from bacteria in a growth-phase-dependent manner. Appl Environ Microbiol. 2013;79(7):2328–2335. doi: 10.1128/AEM.03871-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc Ser B. 1995;57(1):289–300. [Google Scholar]
- 58.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95(25):14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCt method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 60.Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44(W1):W242–W245. doi: 10.1093/nar/gkw290. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Supplemental figures.
Additional file 2. M. smegmatis DEG data sets.
Additional file 3. DAVID gene ontology results from significant DEGs.
Additional file 4. DosR regulon DEG and comparison of genes betweeen M. smegmatis and M. tuberculosis.
Additional file 5. RNA Bioanalyzer results (RIN numbers and electrophoretic traces).
Additional file 6. DAVID gene ontology results from overlapping DEGs between mc2155 vs. FDL10 and FDL15 vs. FDL10 data sets.
Data Availability Statement
The raw Illumina paired-end sequence data for the RNA-seq studies performed in this article are available at the NCBI Sequence Read Archive (SRA) under the BioProject number PRJNA532282 under accession numbers SAMN11393348, SAMN11393349, and SAMN11393350. The assembled genome sequence for Mycolicibacterium smegmatis MC2 155 can be found in the GenBank database under assembly accession GCA_000015005.1.