

ABSTRACT
Nervous system development is instructed by genetic programs and refined by distinct mechanisms that couple neural activity to gene expression. How these processes are integrated remains poorly understood. Here, we report that the regulated release of insulin-like peptides (ILPs) during development of the Caenorhabditis elegans nervous system accomplishes such an integration. We find that the p38 MAP kinase PMK-3, which is required for the differentiation of chemosensory BAG neurons, limits an ILP signal that represses expression of a BAG neuron fate. ILPs are released from BAGs themselves in an activity-dependent manner during development, indicating that ILPs constitute an autocrine signal that regulates the differentiation of BAG neurons. Expression of a specialized neuronal fate is, therefore, coordinately regulated by a genetic program that sets levels of ILP expression during development, and by neural activity, which regulates ILP release. Autocrine signals of this kind might have general and conserved functions as integrators of deterministic genetic programs with activity-dependent mechanisms during neurodevelopment.
KEY WORDS: C. elegans, Sensory neuron, Nervous system development, Insulin signaling
Summary: Insulin signaling is involved in C. elegans nervous system development as part of a mechanism that integrates intrinsic and activity-dependent gene expression programs.
INTRODUCTION
The nervous system comprises a multitude of neuron types, each endowed with unique physiology, connectivity, and molecular characteristics. This diversity in neuronal form and function is required for neural circuits to support complex brain functions and behaviors (Bargmann and Marder, 2013). Accordingly, understanding how neuronal diversity is generated during development remains a major question in neuroscience. A remarkable feature of nervous system development is that it is coordinately regulated both by rigidly specified genetic programs and by neural activity. Genetic programs pattern the developing nervous system and instruct the differentiation of thousands of different neuronal subtypes with unique molecular signatures and circuitries (Philippidou and Dasen, 2013). By contrast, neural activity regulates the differentiation of developing neurons in response to cues from the environment and experience (Wamsley and Fishell, 2017). In the developing brain, and notably in sensory areas, neural activity induces transcriptional programs that promote proper neuronal connectivity and tune the excitability of component neurons (Yap and Greenberg, 2018). How these two different mechanisms – one specified by a developmental program and the other activity dependent – are integrated during nervous system development remains poorly understood.
The nematode Caenorhabditis elegans is a powerful model for the study of nervous system development. Although its nervous system comprises only 302 neurons, these neurons are highly diversified and form at least 118 distinct classes (White et al., 1986; Hobert et al., 2016). Invariant cell lineages generate a large majority of its neuron types (Sulston et al., 1983; Sulston and Horvitz, 1977), suggesting that genetic programs intrinsic to cell lineages are major drivers of neuronal differentiation in C. elegans. In support of this, many studies have identified transcription factors that act in specific lineages to promote specific neuronal fates (Hobert, 2016). However, there are also important roles for neuronal activity during development of the C. elegans nervous system. For example, there is a striking role for activity of embryonic AWC chemosensory neurons in determining their differentiation into functionally distinct subtypes (Troemel et al., 1999; Sagasti et al., 2001). Also, there is a critical period during which neural activity instructs circuit assembly in the C. elegans motor system (Barbagallo et al., 2017). Post-developmentally, neural and sensory activity are required for maintaining the proper morphology of chemosensory neurons (Peckol et al., 1999; Mukhopadhyay et al., 2008) and for expression of chemosensory receptors and neuropeptides that define specific chemosensory neuron fates (Peckol et al., 2001; Rojo Romanos et al., 2017; Gruner et al., 2014). Therefore, like the vertebrate nervous system, development of the C. elegans nervous system requires both stereotyped programs of gene expression and activity-dependent gene regulation.
We have studied mechanisms required for the development of a pair of C. elegans sensory neurons – the BAGs – which sense microbe-derived carbon dioxide (CO2) to control foraging behaviors (Brandt and Ringstad, 2015). Properly specified BAG neurons are equipped with a chemotransduction apparatus used to sense CO2, which includes the receptor-type guanylyl cyclase GCY-9 (Smith et al., 2013), the neurotransmitter glutamate (Serrano-Saiz et al., 2013) and a specific set of neuropeptides (Kim and Li, 2004). Previous studies identified transcription factors that promote the BAG neuron fate (Brandt et al., 2012; Guillermin et al., 2011; Gramstrup Petersen and Pocock, 2013; Rojo Romanos et al., 2015). However, mutants for these transcription factors still generate BAG neurons that differentiate to some extent. Therefore, other mechanisms that promote a BAG fate must exist. Through a screen for additional regulators of the BAG fate we identified the p38 MAP kinase (MAPK) PMK-3 (Brandt and Ringstad, 2015). Interestingly, although pmk-3 mutants are strongly defective in BAG neuron function, their gene expression defects are restricted to a subset of BAG-specific genes. PMK-3 is also required during development, and post-developmental expression of PMK-3 does not restore gene expression or function to pmk-3 mutant BAG cells (Brandt and Ringstad, 2015). Although it is clear that PMK-3 functions in BAG development, how PMK-3 promotes the differentiation of BAG neurons is unknown. p38 MAPKs have many functions in the nervous system (Thomas and Huganir, 2004), but these functions are often part of injury or stress responses, and roles for p38 MAPKs in neuronal differentiation remain poorly understood.
To determine how PMK-3 functions in BAG neuron development, we isolated and characterized pmk-3 suppressor mutations. We discovered that a major complementation group of suppressor mutations comprises alleles of unc-31, which encodes a factor required for the regulated secretion of neuropeptides and hormones. Loss of unc-31 restores gene expression to pmk-3 mutant BAG cells by interfering with the regulated release of insulin-like peptides (ILPs), which are overexpressed in pmk-3 mutants and repress expression of a BAG cell fate. ILPs are released from BAG neurons themselves and therefore function as an inhibitory autocrine signal during BAG neuron development. This mechanism combines a gene regulatory program, which sets the levels of ILP expression, and neural activity, which controls ILP release, to regulate the differentiation of a specific neuron type. We propose that similar mechanisms might function widely during nervous system development to integrate neural activity with genetically specified developmental programs.
RESULTS
pmk-3 mutant sensory neurons are defective in expression of a functionally important neuropeptide
pmk-3 mutants generate BAG neurons with the appropriate nuclear position and cellular morphology, but fail to express the BAG neuron-specific neuropeptide gene flp-17 (Fig. 1A). FLP-17 peptides activate the Gi/o-coupled receptor EGL-6 to inhibit motor neurons in the C. elegans egg-laying system (Ringstad and Horvitz, 2008). FLP-17 neuropeptides are also required for BAG neuron-dependent CO2 avoidance behavior (Guillermin et al., 2017; Lee et al., 2017). To determine whether the behavioral defect of pmk-3 mutants can be explained by their failure to express flp-17 neuropeptides, we compared the CO2-avoidance defects of pmk-3 and flp-17 mutants. Both pmk-3 and flp-17 mutants were severely defective for CO2 avoidance (Fig. 1B). However pmk-3 mutants had, on average, a lower CO2 avoidance index than flp-17 mutants, although this difference was not statistically significant (pmk-3 avoidance index of 0.06288 versus flp-17 avoidance index of 0.2811; P=0.1380, ordinary one-way ANOVA followed by Tukey's multiple comparisons test). Interestingly, we found that egl-6 mutants, which lack the known receptor for FLP-17 peptides, were wild type for CO2 avoidance (Fig. 1B), indicating that FLP-17 neuropeptides act through another receptor to mediate CO2 avoidance.
Fig. 1.
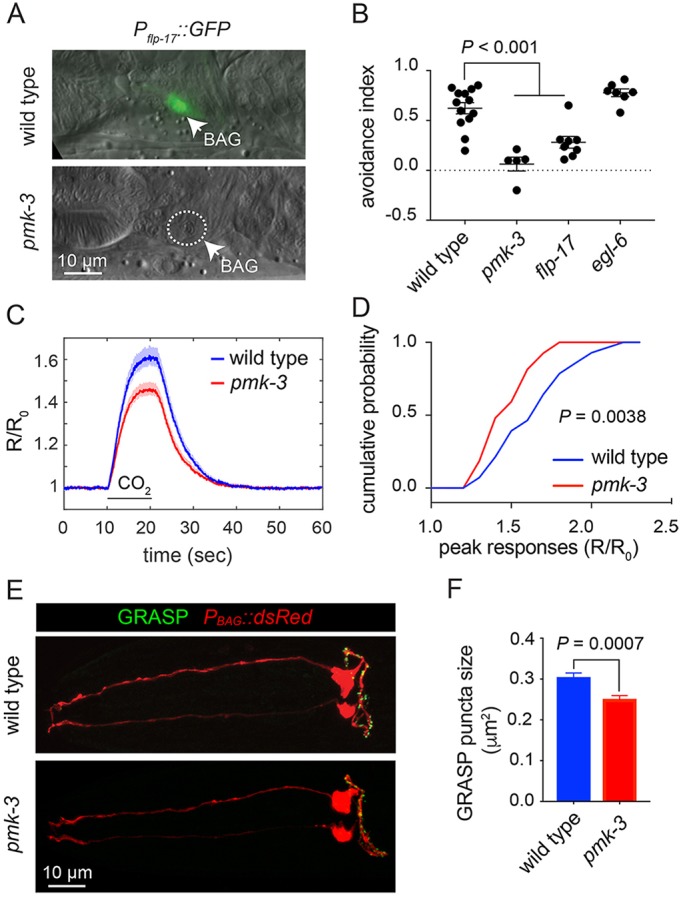
PMK-3 mutation affects neuropeptide expression, chemotransduction and synapse morphology of chemosensory BAG neurons. (A) Overlaid DIC and fluorescence micrographs of wild-type and pmk-3(wz31) mutant animals expressing Pflp-17::GFP, a marker of differentiated BAG cells. (B) CO2-avoidance indices of the wild type, and pmk-3(ok169), flp-17(n4894) and egl-6(n4536) mutants. n≥5 independent trials. P<0.0001 for wild type versus pmk-3, and P=0.0007 for wild type versus flp-17 (ordinary one-way ANOVA followed by Tukey's multiple comparisons test). Error bars represent s.e.m. (C) Average calcium responses of wild-type and pmk-3(ok169) mutant BAG cells to 10% CO2 stimuli. Shaded area represents the mean±s.e.m. n>26 animals/genotype. R/R0 is the ratio of YFP/CFP emissions normalized to the pre-stimulus ratio. (D) Cumulative probability plots of the peak calcium responses (R/R0) in wild-type and pmk-3(ok169) mutant BAG cells to CO2 stimuli. P=0.0038 (unpaired t-test). (E) Fluorescence micrographs of BAG GRASP puncta, Pgcy-9::nlg-1::GFP1-10 Podr-2b::nlg-1::GFP11, in wild-type and pmk-3(ok169) mutant BAG cells overlaid with Pgcy-9::dsRed to label BAG cells. (F) Quantification of the size of GRASP puncta in wild-type and pmk-3(ok169) mutant BAG cells. n>28 animals/genotype. P=0.0007 (unpaired t-test). Error bars represent s.e.m.
We asked whether PMK-3 regulates other aspects of BAG neuron physiology or structure. First, we tested whether pmk-3 mutant BAG cells exhibit sensory transduction defects using in vivo calcium imaging to measure their calcium responses to CO2 stimuli (Brandt et al., 2012). pmk-3 mutant BAG cells responded to CO2 stimuli, although their responses were smaller than those of the wild type (Fig. 1C,D, Fig. S1A). When wild-type and pmk-3 mutant cell responses were scaled to unity, we observed no difference between the dynamics of the calcium responses, indicating that the kinetics of cell activation were not altered by pmk-3 mutation (Fig. S1B). To determine whether PMK-3 is required for synapse formation between BAG and its downstream targets, we used GFP reconstitution across synaptic partners (GRASP) (Feinberg et al., 2008) to label BAG synapses in the wild type and in pmk-3 mutants. One component of the GRASP system was expressed in BAGs, using the BAG cell-specific and PMK-3-independent gcy-9 promoter (Brandt and Ringstad, 2015; Smith et al., 2013). We expressed the other GRASP component using regulatory sequences from odr-2, which is expressed by several interneurons post-synaptic to BAGs, including the AIB and RIG interneurons (Chou et al., 2001; White et al., 1986). A Pgcy-9::dsRed transgene was also used to mark the BAG cells in these experiments (Fig. 1E). GRASP puncta were observed throughout the axons of both wild-type and pmk-3 mutant BAG cells (Fig. 1E). We observed no significant difference in the number of puncta in pmk-3 mutants and in the wild type (Fig. S2). The average size of puncta in pmk-3 mutants, however, was 18% smaller than that of the wild type (Fig. 1F). From these data, we concluded that a major function for PMK-3 in BAG sensory neurons is to regulate the expression of neuropeptides, but our data also show that loss of PMK-3 affects chemotransduction and synapse morphology.
Genes required for Ca2+-dependent neural secretion suppress a neuropeptide gene expression defect of pmk-3 mutants
Our previous study indicated that PMK-3 is a p38 MAPK that acts downstream of a Toll-like receptor to promote BAG neuron development (Brandt and Ringstad, 2015), but how PMK-3 regulates expression of a BAG neuron fate was not known. To address this question, we performed a genetic screen for suppressors of the flp-17 expression defects of pmk-3 mutants. From two screens that covered approximately 25,000 mutagenized haploid genomes, we recovered 18 mutants in which expression of flp-17 was restored to pmk-3 mutants (Fig. 2A,B). Five of these mutants defined a major complementation group. These suppressor mutations increased in pmk-3 mutants the frequency of flp-17 reporter expression, i.e. the probability that BAG neurons expressed detectable reporter (Fig. 2C), and also significantly restored the levels of reporter expression (Fig. S3). We tested whether suppressor mutations affected another PMK-3-regulated gene, gcy-33, which encodes a guanylyl cyclase required for sensing hypoxia (Zimmer et al., 2009; Brandt and Ringstad, 2015). Unlike flp-17, gcy-33 expression was not restored in suppressed pmk-3 mutants (Fig. S4) indicating that these suppressor mutations do not fully restore gene expression to pmk-3 mutant BAG cells.
Fig. 2.
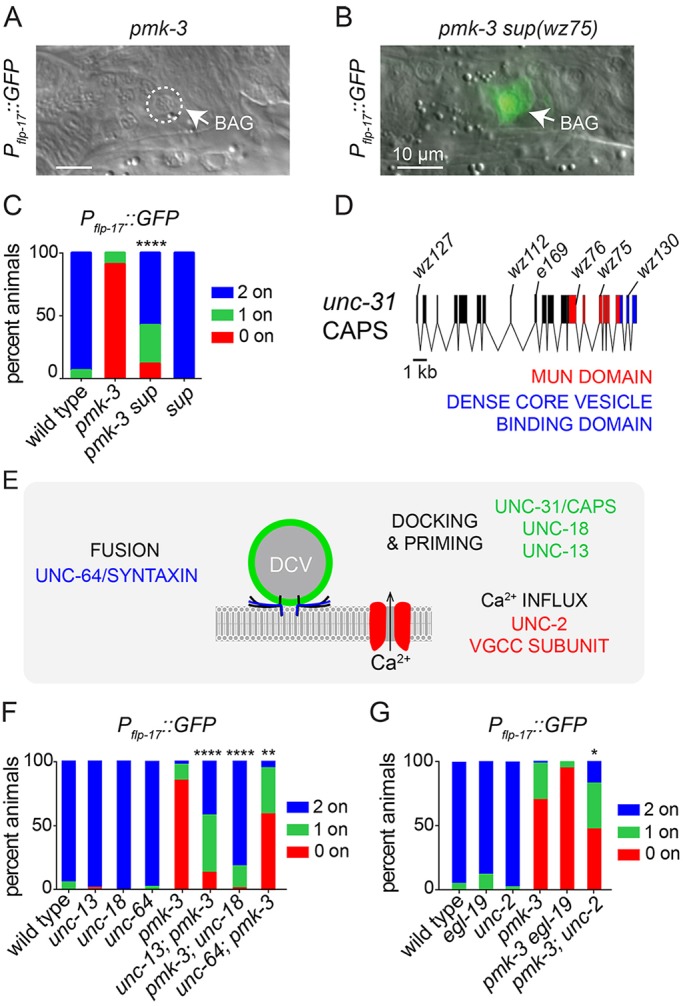
Mutation of genes required for calcium-regulated neuronal secretion suppress a gene-expression defect caused by loss of PMK-3. (A,B) Overlays of DIC and fluorescence micrographs of a pmk-3(wz31) mutant (A) and a pmk-3(wz31) mutant animal carrying the suppressor mutation wz75 [pmk-3 sup(wz75)] (B) expressing Pflp-17::GFP. Scale bars: 10 µm. (C) Penetrance of Pflp-17::GFP expression in the wild type, in pmk-3(wz31) and pmk-3(wz31) sup(wz75) mutant animals, and in animals carrying a mutation in the suppressor gene on its own, sup(e169). (D) Structure of the unc-31 genetic locus. wz75, wz76, wz112, wz127 and wz130 are five non-complementing alleles isolated by our pmk-3 suppressor screen, and e169 is the unc-31 reference allele. (E) Genes that function with UNC-31/CAPS to regulate secretion of dense core vesicles (DCVs). (F) Penetrance of Pflp-17::GFP expression in animals containing loss-of-function mutations in genes required for docking and priming or fusion of DCVs, with or without pmk-3 mutation. Data shown for pmk-3 represent the average penetrance of Pflp-17::GFP expression collected from pmk-3(wz31) and pmk-3(ok169) mutant animals. (G) Penetrance of Pflp-17::GFP expression in animals harboring loss-of-function mutations in VGCC subunits with and without pmk-3(ok169) mutation. n>25 animals/genotype. *P<0.05, **P<0.01, ****P<0.0001 (chi-square test).
We next tested whether suppressor mutations restored gene expression to mutants for transcription factors that promote a BAG fate. We tested three known regulators of the BAG fate: the ETS-family transcription factor ETS-5 (Guillermin et al., 2011; Brandt et al., 2012), the SOX transcription factor EGL-13 (Gramstrup Petersen et al., 2013) and the zinc finger-containing transcription factor EGL-46 (Rojo Romanos et al., 2015). ets-5 and egl-13 mutants were defective for flp-17 expression and their gene expression defects were not modified by the suppressor mutation (Fig. S5). egl-46 mutants were wild type for flp-17 expression, and the pmk-3 suppressor did not modify flp-17 expression in egl-46 mutants (Fig. S5). These findings suggest that the gene defined by these suppressor mutations specifically antagonizes a PMK-3-dependent mechanism, and does not generally de-repress expression of a BAG fate.
Using high-resolution single nucleotide polymorphism (SNP) mapping and whole-genome sequencing we identified the suppressor gene as unc-31 (Fig. 2D), which encodes the C. elegans homolog of calcium-dependent activator protein for secretion (CAPS; CADPS). UNC-31/CAPS is a phosphoinositide-binding protein required for docking and priming of dense core vesicles (DCVs), which mediate the regulated release of peptide hormones and growth factors from neurons (Speese et al., 2007; Zhou et al., 2007; Sieburth et al., 2007). Because of this well-known function of unc-31, we hypothesized that Ca2+-dependent secretion plays a role in BAG neuron development. We tested whether mutations that affect other crucial factors for neural secretion (Fig. 2E) also suppress pmk-3. Loss of UNC-13 or UNC-18, which like UNC-31 are required for docking and priming of synaptic and dense core vesicles (Weimer et al., 2003; Richmond et al., 1999), strongly restored the frequency of flp-17 expression to pmk-3 mutants (Fig. 2F). We also tested a partial loss-of-function allele of the syntaxin homolog UNC-64 (Saifee et al., 1998), and observed partial restoration of flp-17 expression to pmk-3 mutant BAG neurons (Fig. 2F).
DCV exocytosis is triggered by Ca2+ that enters cells through voltage-gated calcium channels (VGCCs). We therefore tested whether mutation of VGCC subunits suppresses the gene expression defect of pmk-3 mutants. Mutation of the L-type VGCC alpha-1 subunit gene egl-19 (Lee et al., 1997) did not restore flp-17 expression to pmk-3 mutants (Fig. 2G). By contrast, loss of the NPQ-type VGCC alpha-1 subunit gene unc-2 (Schafer and Kenyon, 1995), partly suppressed pmk-3 and significantly restored flp-17 expression (Fig. 2G, Fig. S6B). The VGCC alpha-2-delta subunit UNC-36 is a part of both L- and NPQ-type channels (Lee et al., 1997). Loss of UNC-36 did not affect the frequency with which pmk-3 mutants expressed flp-17, but did cause an increase in the expression levels of flp-17 when it was expressed in pmk-3 mutants (Fig. S6). Together, these data indicate that reductions in Ca2+-evoked secretion restore gene expression to pmk-3 mutant BAG neurons and suggest that some, but not all, of the Ca2+ trigger for secretion is provided by NPQ-type VGCCs.
Neural activity during development regulates neuropeptide gene expression in BAG neurons
PMK-3 functions cell-autonomously during a critical period of embryonic development to promote expression of flp-17 in BAG neurons (Brandt and Ringstad, 2015). We tested whether neural activity and regulated secretion also act cell-autonomously in BAG neurons during the same critical period. To dampen BAG activity, we overexpressed the inward rectifying potassium (K+) channel IRK-1 (Emtage et al., 2012) using the BAG-neuron-specific and PMK-3-independent gcy-9 promoter (Brandt and Ringstad, 2015; Smith et al., 2013). IRK-1 expression in BAGs strongly restored flp-17 expression to pmk-3 mutants (Fig. 3A). In parallel, we performed BAG-targeted knockdown of unc-31 using RNA interference (RNAi). Knockdown of unc-31 in BAGs also restored flp-17 expression to pmk-3 mutant BAG neurons (Fig. 3B). We compared the effects of BAG-targeted IRK-1 expression, or unc-31 knockdown, with the effects of disrupting neural activity and regulated secretion in all neurons using a pan-neuronal promoter. BAG-targeted inhibition of neural activity or regulated secretion suppressed pmk-3 as strongly as the corresponding pan-neuronal manipulations (Fig. 3A,B). Together, these data indicate that neural activity and the secretion factor UNC-31 function cell-autonomously to regulate flp-17 expression.
Fig. 3.
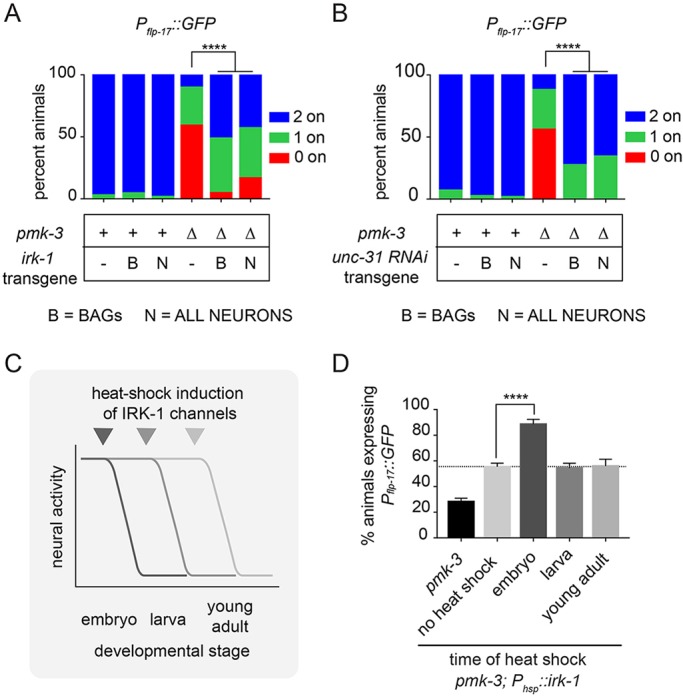
Electrical activity in BAG neurons during development regulates expression of a PMK-3-regulated gene. (A) Silencing BAG neural activity using Pgcy-9::irk-1 strongly restores the penetrance of Pflp–17::GFP expression to pmk-3(ok169) mutant BAG cells. Similar effects are observed when pan-neuronal activity is silenced using Prab-3::irk-1. (B) BAG-targeted knockdown of unc-31 using RNAi (Pgcy-9::unc-31 RNAi) is sufficient to strongly restore the penetrance of Pflp-17::GFP expression to pmk-3(ok169) mutant BAG cells, similar to pan-neuronal RNAi of unc-31 (Prab-3::unc-31 RNAi). n≥25 animals/genotype. ****P<0.0001 (chi-square test) (A,B). (C) Animals carrying a transgene for heat shock-inducible overexpression of irk-1, Phsp::irk-1, were heat shocked at different developmental stages (embryonic, larval or adult) to induce irk-1 expression and reduce neural activity. (D) Percentage of animals expressing Pflp-17::GFP in pmk-3(ok169) and pmk-3(ok169); Phsp::irk-1 mutant animals that had been heat shocked at the indicated developmental stages. Dashed line represents the shifted baseline of flp-17 expression in pmk-3; Phsp::irk-1 mutant animals in the absence of heat shock. n>95 animals/condition. ****P<0.0001 (ordinary one-way ANOVA followed by Dunnet's multiple comparison test). Bar graph data are plotted as mean ±s.e.m.
We also tested whether eliminating the sensory function of BAGs would suppress the effects of pmk-3 mutation. Loss of the receptor-type guanylyl cyclase GCY-9, which is required for BAG sensory function (Smith et al., 2013; Hallem et al., 2011), did not restore flp-17 expression to pmk-3 mutants (Fig. S7). These data suggest that the neural activity that regulates flp-17 expression in BAGs does not require BAG sensory function.
To determine when neural activity exerts its effect on flp-17 expression, we expressed irk-1 under the control of a heat shock-inducible promoter in pmk-3 mutants. irk-1 expression was induced in embryos, larvae or adults in order to reduce neural activity at specific developmental stages (Fig. 3C). flp-17 expression was subsequently measured when heat-shocked embryos or larvae became adults. The transgene used for inducible expression of IRK-1 caused some suppression of the pmk-3 phenotype even in the absence of heat shock, likely because of leaky expression of IRK-1 (Fig. 3D). However, heat-shock treatment of embryos restored flp-17 expression to pmk-3 mutants well above this baseline (Fig. 3D). By contrast, induction of IRK-1 expression during larval or adult stages did not significantly modify the pmk-3 gene expression defect (Fig. 3D). Heat-shock induction of IRK-1 expression broadly affects embryonic neurons and we cannot, therefore, completely rule out a non-cell-autonomous role for neural activity in regulating gene expression in BAGs. Taken together, however, our data suggest that, like PMK-3, regulated secretion by BAG neurons is required during development to regulate expression of BAG-specific neuropeptides.
pmk-3 mutation dysregulates expression of insulin-like genes in BAG neurons
Why do mutations in unc-31 and other genes required for regulated secretion suppress the effects of pmk-3 mutation? We hypothesized that pmk-3 mutant BAG neurons might overexpress a secreted factor that inhibits their own development. We sought evidence of such a secreted factor by transcriptionally profiling embryonic BAG neurons from the wild type and pmk-3 mutants. We purified GFP-marked BAG neurons and non-fluorescent control cells from the wild type and from pmk-3 mutants. After sequencing the BAG neuron transcriptome, we used the differential gene expression analysis program DESeq2 (Love et al., 2014) to (1) compare sequences from wild-type BAG neurons and non-BAG cells to identify BAG-enriched transcripts, and (2) to compare sequences from wild-type and pmk-3 mutant BAG neurons to determine how loss of pmk-3 affects gene expression. Genes known to have enriched expression in BAG neurons, such as those involved in sensory function (gcy-9, gcy–33, gcy-31 and flp-17) and BAG development (ets-5 and egl-13), were among the top BAG-enriched genes in our data set (Fig. S8A), which confirmed that we reliably isolated and sequenced BAG cell-derived transcripts from both the wild type and from pmk-3 mutants.
We identified 692 transcripts for which expression significantly differed between wild-type and pmk-3 mutant BAG cells (P<0.05) (Fig. 4A). Principal component analysis (PCA) showed that wild-type and pmk-3 mutant BAG transcriptomes are highly separable (Fig. 4B), indicating that pmk-3 is required for proper expression of many genes in developing BAG neurons. Notably, pmk-3 mutation did not affect expression of transcription factors known to generate and maintain the BAG cell identity, such as ets-5 and egl-13 (Fig. S8B,C).
Fig. 4.
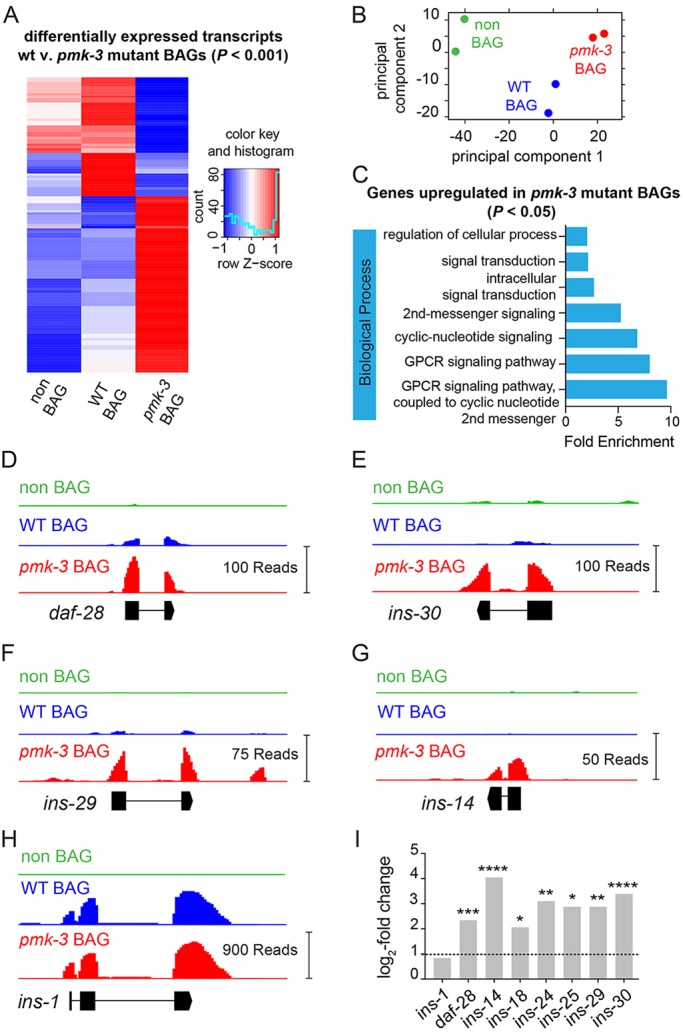
PMK-3 represses expression of genes encoding ILPs in BAG chemosensory neurons. (A) Heat map showing the relative expression of the most differentially expressed genes (P<0.001, 129 transcripts) between wild-type (WT) and pmk-3(wz31) mutant BAG cells. Expression of these transcripts in non-BAG cells is also shown. Colors represent standardized z-scores calculated from the average normalized DESeq2 read counts, with blue representing low expression and red representing high expression. (B) Principal component analysis of the transcriptomes from wild-type BAG cells, pmk–3(wz31) mutant BAG cells, and non-BAG cells. Each dot represents one biological replicate. (C) Biological processes significantly over-represented (P<0.05) amongst the subset of genes upregulated in pmk-3 mutant BAG cells. (D-H) Read coverage histograms for a subset of indicated ILPs that are significantly overexpressed in wild-type or pmk-3(wz31) mutant BAG cells (see Materials and Methods). (I) Fold changes of ILP gene expression in pmk-3(wz31) mutant BAG cells versus wild-type BAG cells. n=2 biological replicates. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. P-values were adjusted for FDR using DESeq2 (Love et al., 2014).
To identify gene classes affected in pmk-3 mutant BAG neurons, we analyzed genes upregulated (365 genes) and downregulated (327 genes) in pmk-3 mutant BAGs (P<0.05) for enriched gene ontology (GO) terms using the ‘protein analysis through evolutionary relationships’ (PANTHER) classification system (Mi et al., 2019). Amongst the genes upregulated in pmk-3 mutant BAG cells, we observed significant enrichment of the G protein-coupled receptor (GPCR) and cyclic nucleotide signaling pathways biological process GO terms (Fig. 4C). We also found strong enrichment of the terminal bouton, pre-synapse and dendrite cellular localization GO terms (Fig. S9A). Genes downregulated in pmk-3 mutant BAG neurons did not yield any significantly enriched biological process GO terms, but were enriched for nucleic acid binding molecular function GO terms and nuclear chromosome and chromosome telomeric region cellular component GO terms (Fig. S9B).
Strikingly, GO term analysis indicated that pmk-3 mutant BAG neurons overexpress genes linked to signaling pathways associated with peptide hormones and neural secretion. This observation supported our hypothesis that pmk-3 mutant BAG cells overexpress a secreted factor that inhibits their development. Inspection of the genes overexpressed in pmk-3 mutants revealed that pmk-3-mutant BAG cells overexpress a number of ILPs (Fig. 4I). The ILP gene daf-28, which is enriched in wild-type BAG neurons compared with non-BAG cells (Fig. S10), is upregulated in pmk-3 mutant BAGs (Fig. 4D). Also, ins-14, ins-29 and ins-30 transcripts, which we did not detect in wild-type BAGs, are clearly present in pmk-3 mutant BAG neurons (Fig. 4E-G). The ILPs affected by pmk-3 mutation are all agonists of the sole C. elegans insulin/insulin growth factor (IGF)-like receptor (InR) DAF-2 (Murphy and Hu, 2013; Kenyon et al., 1993; Kimura et al., 1997). ins-1, which encodes an ILP antagonist (Cornils et al., 2011), is enriched in wild-type BAGs (Fig. S10) but its expression is not affected by pmk-3 mutation (Fig. 4H,I). We next sought to determine whether excess ILP production by BAGs is a proximal cause of the BAG neuron defects associated with loss of PMK-3.
An autocrine insulin signaling pathway antagonizes PMK-3-dependent gene expression in BAG cells
To disrupt ILP production we used a dominant-negative ILP variant encoded by daf-28(sa191), which generates a non-functional ILP that further disrupts production of other ILPs (Li et al., 2003) (Fig. 5A). We found that daf-28(sa191) phenocopied mutations that affect regulated secretion and restored flp-17 expression to pmk-3 mutants (Fig. 5B). Notably, a deletion allele of daf-28 (Fig. 5A) also partially restored flp-17 expression to pmk-3 mutants (Fig. 5C), indicating that although BAGs produce several ILPs, DAF-28 has a non-redundant function in regulating BAG gene expression. We next overexpressed DAF-28(sa191) specifically in pmk-3 mutant BAG neurons to test whether ILP production in BAG neurons themselves is required for their development as our model predicts. Overexpressing DAF-28(sa191) in BAGs strongly restored flp-17 expression to pmk-3 mutants (Fig. 5B), suggesting that the pmk-3 phenotype is at least partly caused by dysregulated expression and release of ILPs.
Fig. 5.
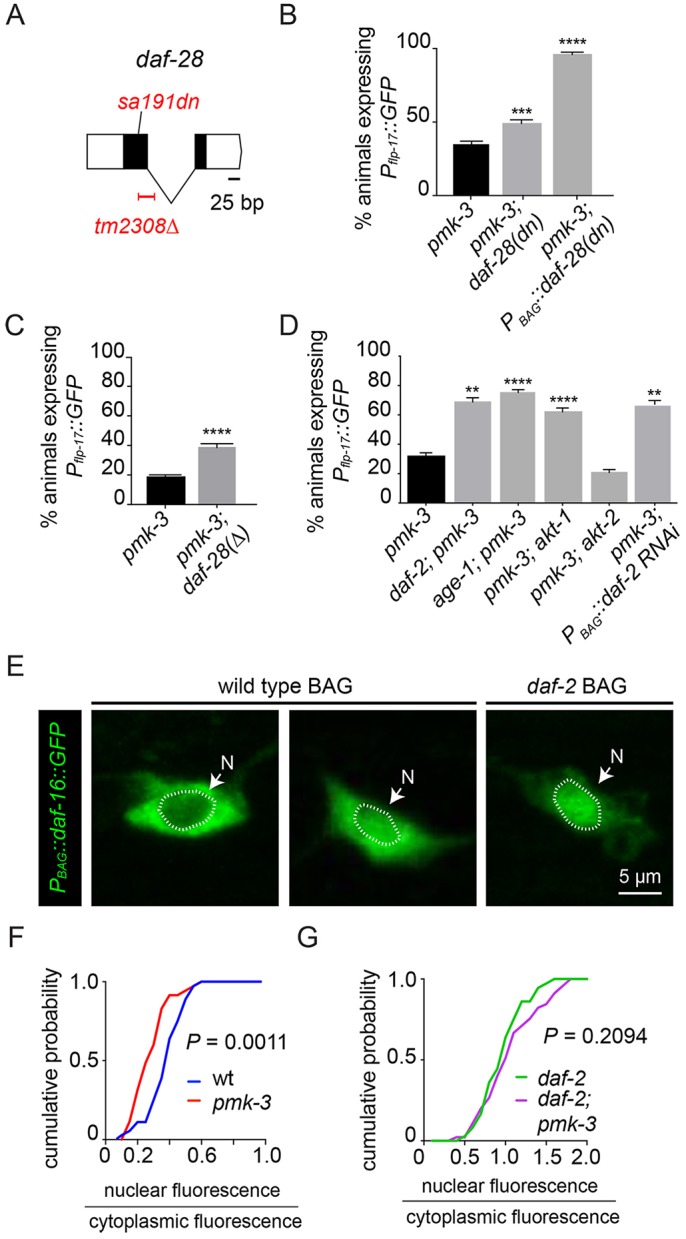
An autocrine insulin signal inhibits neuropeptide-gene expression in pmk-3 mutants. (A) Structure of the daf-28 locus showing the null allele, tm2308Δ, and the dominant-negative sa191 allele. (B) Disrupting insulin production in pmk-3(ok169) mutant BAG cells by overexpressing daf–28(sa191), PBAG::daf-28(sa191), strongly restores Pflp-17::GFP expression to pmk-3(ok169) mutants. The chromosomal daf-28(sa191) mutation also partially restores flp-17 expression to pmk-3(ok169) mutants. ***P=0.0002, ****P<0.0001 (unpaired t-test). (C) Deletion of daf-28 partially restores the percentage of pmk-3(ok169) mutant animals expressing Pflp-17::GFP. ****P<0.0001 (unpaired t-test). (D) Percentage of animals expressing Pflp-17::GFP in pmk-3(ok169), daf-2(e1370); pmk-3(ok169), age-1(hx546); pmk-3(ok169), pmk-3(ok169); akt-1(ok525) and pmk-3(ok169); akt-2(ok393) mutant animals, and in pmk-3(ok169) mutant animals harboring transgenes for BAG cell-specific RNAi of daf-2, PBAG::daf-2 RNAi. For the pmk-3 versus daf-2; pmk-3, pmk-3 versus age-1; pmk-3 and pmk-3 versus pmk-3; PBAG::daf-2 RNAi comparisons, P=0.0016, P<0.0001 and P=0.0047, respectively (unpaired t-test). For the pmk-3 versus pmk-3; akt-1 and pmk-3 versus pmk-3; akt-2 comparisons, P<0.0001 and P=0.9524, respectively (ordinary one-way ANOVA followed by Dunnet's multiple comparisons test). (E) Fluorescence micrographs of wild-type and daf-2(e1370) mutant animals expressing GFP-tagged DAF-16 in the BAG cell, PBAG::daf-16::GFP. Dotted line marks the boundary of the nucleus (N). (F) Cumulative probability plots of the nuclear-to-cytoplasmic DAF-16::GFP fluorescence ratios in wild-type and pmk-3(ok169) mutant BAG cells. n=36 cells from 27 wild-type animals and n=35 cells from 23 pmk-3 mutant animals. P=0.0011 (unpaired t-test). (G) Loss of PMK-3 does not alter the nuclear-to-cytoplasmic DAF-16::GFP fluorescence ratio in daf-2 mutants. n=46 cells from 30 daf-2(e1370) mutant animals and n=45 cells from 28 daf-2(e1370); pmk-3(ok169) mutant animals. P=0.2094 (unpaired t-test). Unless otherwise noted, n≥45 animals/genotype. Bar graph data are plotted as mean±s.e.m.
What cells respond to ILPs released from BAGs? ILPs might act on other cells that in turn signal back to BAGs. Alternatively, ILPs released from BAGs might function as autocrine signals to activate insulin-receptor signaling pathways in BAGs themselves. To distinguish these possibilities, we characterized the InR signaling pathway that regulates flp-17 expression in BAG neurons and determined its site of action. First, we interrogated genes with known functions in InR signaling for a role in regulation of flp-17 expression. The InR DAF-2 signals via the phosphoinositide-3 (PI3) kinase AGE-1 (Morris et al., 1996), and two serine/threonine kinases, AKT-1 and AKT-2 (Paradis and Ruvkun, 1998). Many genes in this canonical InR signaling pathway mutate to suppress pmk-3; loss-of-function mutations in daf-2, age-1 and akt-1 each strongly restored flp-17 expression to pmk-3 mutants (Fig. 5D). We observed no effect, however, of akt-2 mutation on the pmk-3 phenotype (Fig. 5D). In a wild-type background, loss of daf-2, age-1 or akt-1 did not affect flp-17 expression (Fig. S11). We note that our data also show that insulin pathway mutations are not completely epistatic to loss of pmk-3, indicating that other pathways are also affected by loss of PMK-3.
We next determined where the DAF-2 InR was required for its role in BAG-neuron development. We reduced daf-2 function in BAG neurons using cell-targeted RNAi. Like daf-2 chromosomal mutation, BAG-targeted daf-2 RNAi restored flp-17 expression to pmk-3 mutants (Fig. 5D). Together, these data indicate that ILPs released from BAGs constitute an autocrine signal that represses flp-17 expression.
pmk-3 mutant BAG cells experience increased insulin signaling
To independently test whether pmk-3-mutant BAG neurons experience excess InR signaling, we measured the InR-dependent accumulation of the Forkhead (FOXO) transcription factor DAF-16 in the BAG cell nucleus (Lee et al., 2001; Lin et al., 2001). We generated transgenic animals that express a DAF-16::GFP fusion specifically in BAG neurons, PBAG::daf-16::GFP, and we measured the ratio of nuclear to cytoplasmic DAF-16 as an index of InR signaling in BAGs. This ratio varied among wild-type BAGs, some of which had little nuclear DAF-16::GFP fluorescence whereas others had equal nuclear and cytoplasmic fluorescence (Fig. 5E, left, middle). Although pmk-3 mutant BAG neurons also displayed a range of nuclear-to-cytoplasmic DAF-16::GFP ratios, the distribution of ratios was significantly shifted (Fig. 5F), and on average pmk-3 mutant BAGs had more cytoplasmic DAF-16::GFP. As expected, mutation of the InR DAF-2 caused a dramatic accumulation of DAF-16::GFP in the nuclei of BAG neurons (Fig. 5E, right). In daf-2 mutants, there was no significant effect of pmk-3 mutation on DAF-16::GFP localization (Fig. 5G), suggesting that the effect of pmk-3 mutation requires the canonical InR DAF-2. These data provide additional evidence that pmk-3 mutant BAGs experience elevated insulin signaling.
A non-canonical InR signaling pathway regulates neuropeptide expression in BAG chemosensory neurons
Having used DAF-16 localization as a measure of InR activation, we next tested whether this transcription factor, which is a canonical effector of InR signaling, functions downstream of ILPs to regulate flp-17 expression. The daf-16 locus is tightly linked to the flp-17 reporter transgene that we use to assay the BAG fate. We therefore generated a new daf-16 deletion allele in a strain carrying the flp-17 reporter using CRISPR/Cas9 mutagenesis (Fig. S12A). When we measured the effect of daf-16 mutation in an otherwise wild-type strain we did not observe an effect on the frequency of animals that express flp-17, but did notice a significant decrease in flp-17 expression levels (Fig. S12B,C). DAF-16 is negatively regulated by DAF-2, and if DAF-16 were the relevant DAF-2 effector in regulating flp-17 expression one might expect to observe enhancement of the pmk-3 phenotype as a result of loss of DAF-16. Surprisingly, we observed that daf-16 mutation partially suppressed the pmk-3 phenotype (Fig. 6A). Importantly, however, the suppression of pmk-3 by knockdown of daf-2 still occurred in daf-16 mutants (Fig. 6A), indicating that DAF-2/InR regulates gene expression in BAGs in a DAF-16-independent manner.
Fig. 6.
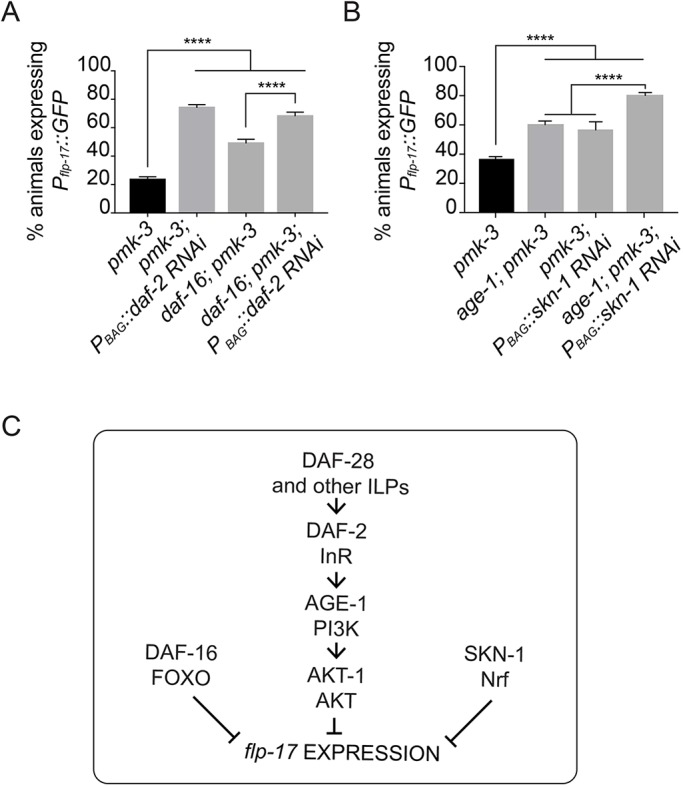
InR signaling regulates neuropeptide expression in BAG neurons independently of DAF-16 and SKN-1. (A) BAG cell-targeted knockdown of daf-2 using RNAi is sufficient to strongly restore Pflp-17::GFP expression to pmk-3(ok169) mutant BAG cells, even in the absence of daf-16(wz151). n≥45 animals/genotype. ****P<0.0001. For pmk-3; PBAG::daf-2 RNAi versus daf-16; pmk-3; PBAG::daf-2 RNAi, P=0.2612. P-values were calculated with an ordinary one-way ANOVA followed by Tukey's multiple comparisons test. (B) Loss of InR signaling, through mutation of age-1(hx546), and BAG cell-targeted knockdown of skn-1 (Pgcy-9::skn-1 RNAi) each partially restores flp-17 expression to pmk-3(ok169) mutant BAG cells. Reduced InR signaling and skn-1 expression has an additive effect on Pflp-17::GFP expression in pmk-3(ok169) mutants. n>250 animals/genotype. ****P<0.0001 (ordinary one-way ANOVA followed by Tukey's multiple comparisons test). (C) A model for insulin signaling, DAF-16 and SKN-1 in regulating expression of the BAG cell fate. Bar graph data are plotted as mean±s.e.m.
The observation of a DAF-16-independent function of DAF-2 prompted us to investigate whether an alternative effector of DAF-2, the Nrf-like transcription factor SKN-1 (Tullet et al., 2008), regulates flp-17 expression. In an otherwise wild-type background, mutation of skn-1 did not affect the frequency or levels of flp-17 expression (Fig. S12D,E). The skn-1 genetic locus is tightly linked to pmk-3, therefore to determine whether skn-1 regulates flp-17 expression downstream of insulin signaling in a pmk-3 mutant background, we performed BAG cell-targeted RNAi of skn-1 in animals mutant for pmk-3 and the downstream effector of the insulin receptor, the PI3 kinase age-1. We used age-1 mutants to assay for reversion because the suppressive effect of age-1 mutation on the pmk-3 phenotype was as strong as daf-2 mutation (Fig. 5D) and age-1; pmk-3 mutant animals did not arrest as dauer larvae and were, therefore, easier to grow than daf-2; pmk-3 mutant animals. Like daf-16 mutation, knockdown of skn-1 expression in pmk-3 mutant BAGs unexpectedly caused some restoration of flp-17 expression to pmk-3 mutant BAGs (Fig. 6B). Furthermore, expression of SKN-1 in BAGs was not required for suppression of the pmk-3 phenotype by mutation of age-1 (Fig. 6B). In fact, loss of age-1 and skn-1 had an additive effect on flp-17 expression in pmk-3 mutants (Fig. 6B). Together, these data show that daf-16 and skn-1 also suppress the pmk-3 mutant phenotype, and therefore most likely function in parallel to insulin signaling to regulate flp-17 expression (Fig. 6C). Strikingly, these data also provide evidence that, in BAG, DAF-2 functions through a non-canonical signaling pathway to repress expression of flp-17 (Fig. 6C).
Attenuating autocrine insulin signaling restores function to pmk-3 mutant BAG neurons
Mutation of PMK-3 dysregulates ILP expression in BAG neurons, but also affects expression of many other genes (Fig. 4A). To what extent is BAG neuron function affected by excess production of ILPs? To address this question, we tested whether disrupting autocrine insulin signaling in BAGs restores CO2 avoidance behavior to pmk-3 mutants. We again used the dominant-negative DAF-28(sa191) ILP variant to disrupt insulin production in BAG neurons of pmk-3 mutants, this time testing for effects of knocking down ILP production on behavior. Chromosomal daf-28(sa191) mutation significantly restored CO2 avoidance behavior to pmk-3 mutants (Fig. 7A). Overexpressing DAF-28(sa191) in pmk-3 mutant BAG cells also suppressed the CO2-avoidance defect of pmk-3 mutants (Fig. 7B), indicating that ILP production by BAGs is responsible for effects of pmk-3 mutation on BAG function and behavior. We further tested whether knockdown of daf-2 in BAGs could also restore behavior to pmk-3 mutants, as predicted by our model of autocrine ILP signaling regulating BAG differentiation. BAG neuron-specific daf-2 RNAi did not affect CO2 avoidance by wild-type worms (Fig. 7C), but restored CO2 avoidance to pmk-3 mutants (Fig. 7C). Together, these data show that the functional defects caused by loss of PMK-3, which are associated with widespread changes in gene regulation, can be rescued by targeting the ILP signaling pathway in BAG neurons.
Fig. 7.
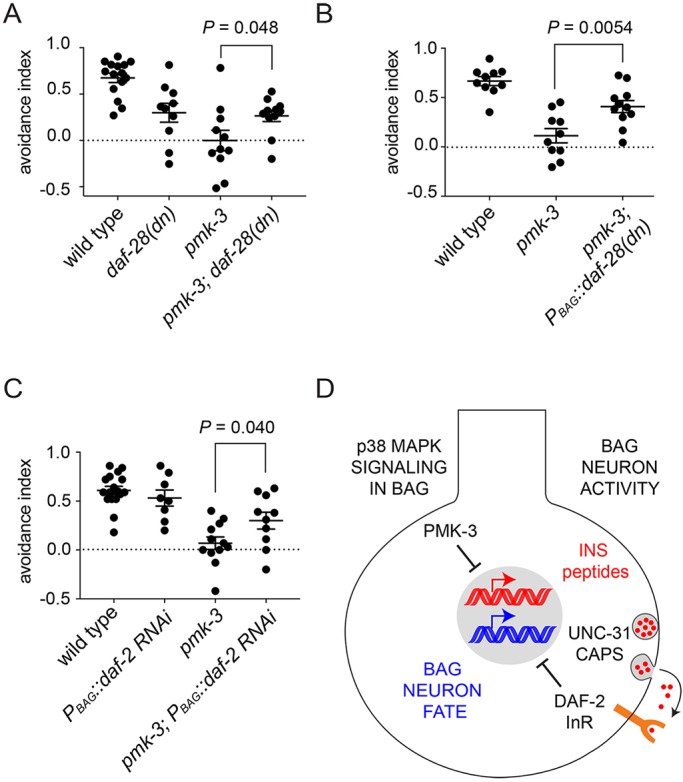
Disruption of autocrine insulin signaling restores function to pmk-3 mutant BAG neurons. (A) A dominant-negative ILP encoded by daf-28(sa191) restored CO2-avoidance behavior to pmk-3(ok169) mutant animals. (B) Reducing insulin secretion specifically in pmk-3(ok169) mutant BAG cells by overexpressing DAF-28(sa191) restored CO2 avoidance behavior. (C) BAG cell-targeted RNAi of the insulin receptor gene daf-2 restored CO2 avoidance behavior to pmk-3(ok169) mutant animals. (D) Model for p38 MAPK regulation of BAG neuron development via repression of an activity-dependent autocrine insulin signal. Bar graphs represent mean±s.e.m. n≥10 trials. P-values were calculated using an unpaired t-test.
DISCUSSION
We have found that the p38 MAP kinase PMK-3 controls development of C. elegans chemosensory BAG neurons through an unexpected mechanism: the regulation of an activity-dependent autocrine insulin signal (Fig. 7D). This insulin signal is regulated at the level of gene transcription. pmk-3 mutant sensory neurons overexpress transcripts encoding ILPs. As a consequence, their BAG cells experience elevated DAF-2/insulin receptor (InR) signaling, which antagonizes expression of a neuropeptide gene essential for their function. We further show that the neuronal defects caused by loss of PMK-3, and concomitant increases in ILP production, are abolished in mutants with defects in activity-dependent neuronal secretion. The autocrine insulin signal that controls expression of a chemosensory neuronal fate is, therefore, controlled both by a PMK-3-dependent gene regulatory mechanism and by neural activity, which controls secretion of ILPs. This mechanism neatly integrates the output of a program of gene expression with neural activity to regulate the differentiation of a specific neuronal fate.
Although ILP genes represent a fraction of the genes that are dysregulated by loss of PMK-3, we found that it is possible to restore BAG neuron function to pmk-3 mutants by manipulating insulin signaling only in BAG cells. Because of the central role for ILP signaling in the etiology of the pmk-3 mutant phenotype and because this mechanism converges on regulation of genes with defined roles in neuron function, it will be of great interest to determine the molecular mechanisms by which PMK-3 regulates ILP expression and how DAF-2/InR signaling subsequently regulates expression of FLP-17 neuropeptides. Notably, our study indicates that additional factors other than the canonical transcriptional regulators associated with ILP signaling, DAF-16 and SKN-1, are required for BAG development downstream of DAF-2. Unexpectedly, we find that DAF-16 and SKN-1 also regulate FLP-17 expression, but likely function in parallel to InR signaling. It will therefore also be of great interest to determine what regulates DAF-16 and SKN-1 in BAG cells and how they interact with the insulin signaling pathway to regulate BAG neuron differentiation.
Activity-dependent autocrine growth-factor signaling in the nervous system
A key feature of ILP signaling in BAG neuron development is that BAG neurons themselves supply the ILPs that regulate their differentiation, i.e. an autocrine signal regulates BAG development. Recent studies have revealed the importance of activity-dependent autocrine signals in nervous system development (reviewed by Herrmann and Broihier, 2018). In mammalian hippocampus, an activity-dependent autocrine insulin-like growth factor 2 (IGF2) signal stabilizes synapses made by dentate granule cells onto their postsynaptic partners (Terauchi et al., 2016). Likewise, in visual cortex, a subset of interneurons generates an activity-dependent autocrine IGF1 signal that regulates the strength of their inhibitory synaptic inputs (Mardinly et al., 2016). Other growth factors not related to insulin also function as autocrine signals during nervous system development. Brain-derived neurotrophic factor (BDNF) and bone morphogenetic protein (BMP)-like factor homolog have been identified as autocrine signals that regulate synapse development and function in mammalian hippocampus and at the insect neuromuscular junction, respectively (Harward et al., 2016; James et al., 2014).
A common feature of many of these autocrine signals is that they are activity dependent. As such, they translate neuronal activity into intracellular signals that regulate gene expression. There are well-studied mechanisms that couple neural activity to gene expression via intracellular calcium signaling, e.g. CREB signaling (West and Greenberg, 2011). Gene regulation by autocrine activity-regulated growth factor signals might, however, differ in functionally important ways. An autocrine signal might have a different threshold for activation by neural activity than CREB-dependent mechanisms, allowing for different patterns of neural activity to engage different gene regulatory mechanisms. Once engaged, autocrine signals might act on different timescales, and they likely regulate gene targets distinct from those regulated by CREB. Interestingly, some aspects of BAG neuron development are regulated by CREB (Rojo Romanos et al., 2017), indicating that neural activity controls the development of BAG neurons through multiple mechanisms.
Why would BAG neuron development be regulated in such a complex manner, invoking a host of transcription factors and an activity-dependent autocrine insulin signal? BAG neurons detect a product of microbial respiration – CO2 – and as such they are involved both in food-seeking and pathogen-avoidance behaviors (Brandt and Ringstad, 2015). Recent studies have demonstrated that BAG neuron function, and its connections to neural circuits that control foraging, are plastic and can be altered by physiological cues and experience (Rengarajan et al., 2019; Bhattacharya et al., 2019). Changes in BAG function are likely adaptive. For example, fasting switched BAGs from neurons that mediate avoidance to ones that mediate attraction (Rengarajan et al., 2019). Fasted animals, therefore, are attracted to the CO2 evolved by microbes that might serve as food. Development as a dauer larva, which is triggered by crowding, also changes BAG function by integrating BAGs into a circuit that mediates attraction to CO2 (Bhattacharya et al., 2019). This change also increases the attractiveness of bacteria and promotes food-seeking behaviors, this time in response to a life history in which food resources were limiting. Our data are, therefore, consistent with a model in which physiological signals during early development impact BAG development and function, perhaps to modulate food-seeking behaviors of larvae and adults.
Insulin-like peptides and nervous system development
During development of C. elegans BAG neurons, ILPs inhibit expression of a fully differentiated and functional BAG neuron fate. This role for ILPs differs from their established function as regulators of cell proliferation during nervous system development (Fernandez and Torres-Alemán, 2012). During development of the Drosophila nervous system, ILPs are produced by glial cells in response to nutritive cues, and trigger adjacent neural stem cells to exit quiescence and begin dividing (Chell and Brand, 2010; Sousa-Nunes et al., 2011). Mammalian IGF1 functions as a mitogenic signal during corticogenesis, and recruits neural progenitors to the cell cycle (Mairet-Coello et al., 2009). A related factor – IGF2 – promotes the maintenance and expansion of neural stem cells in mammals (Ziegler et al., 2014). IGF2 also regulates the proliferation of cerebral cortical progenitors (Lehtinen et al., 2011). We observed a role for ILPs in nervous system development that is distinct from their known function as regulators of proliferation, and found that ILPs repress the differentiation of a specific neuron type. Both functions of ILPs in nervous system development have in common, however, that they delay the appearance of fully differentiated and functional neurons, either by promoting proliferation of neuronal stem cells at the expense of their differentiation, or by inhibiting expression of specific neuronal cell fates.
C. elegans express a large number of ILPs, many in the nervous system. Insulin-like receptor and its associated signaling factors are also expressed throughout the C. elegans nervous system, as is PMK-3, which we show regulates ILP gene expression. Therefore it seems likely that the ILP-dependent mechanism that regulates development of BAG neurons might also function to regulate the differentiation of other neuron types in C. elegans. Furthermore, the known molecular constituents of ILP signaling pathways are highly conserved between nematodes and vertebrates. It is therefore possible that ILPs regulate the expression of specific neuronal cell fates in the developing vertebrate nervous system as our study indicates they do in the nematode.
MATERIALS AND METHODS
Strains
All strains used in this study were cultivated under standard conditions (Brenner, 1974) at 20°C, and are listed in Table S1. The daf-16(wz151) deletion strain was generated by CRISPR/Cas9-mediated mutagenesis using Cas9 ribonucleoprotein (Paix et al., 2015), along with a co-CRISPR strategy to increase efficiency (Kim et al., 2014), as previously described (Zamanian et al., 2018). Transgenic animals were generated via microinjection as previously described (Mello et al., 1991).
Two alleles of pmk-3 were used in the paper: the deletion allele ok169, and the wz31 allele, which was isolated in a genetic screen for factors that regulate the BAG fate (Brandt and Ringstad, 2015) and contains a point mutation in the kinase domain. pmk-3(wz31) mutants were used for the initial genetic suppressor screen (see below), in early studies characterizing the nature of the suppressor mutation and related factors, and in the RNA-Seq experiment. pmk-3(ok169) mutants were used for all other experiments performed in this study. Similarly, two alleles of unc-31 were used: the reference allele, e169, and the wz75 allele, which was isolated in our pmk-3 suppressor screen (see below). wz75 is tightly linked to the pmk-3(wz31) chromosomal locus. Therefore, the e169 allele was used for all experiments testing the effects of loss of unc-31 alone.
Plasmids
Plasmids used in this study were made using Gibson assembly (Gibson et al., 2009) and are listed in Table S2. The daf-28(sa191) cDNA was purchased as a gene block from Integrated DNA Technologies and cloned into the pPD49.26 fire vector using Gibson cloning.
Microscopy
Animals were mounted on 2% agarose pads made in M9 medium and immobilized with 30 mM sodium azide. Fluorescence and differential interference contrast (DIC) micrographs were acquired with a Zeiss Axioimager M2 upright microscope equipped with an EM-CCD camera (Andor) using a 100×/1.4 NA objective. z-stacks were obtained with an LSM700 laser-scanning confocal microscope (Zeiss) using a 40×/1.4 NA objective. Maximum projections of image stacks were generated with Fiji (Schindelin et al., 2012).
CO2 avoidance assays
CO2 avoidance assays were performed as previously described (Brandt and Ringstad, 2015). Briefly, a total of 40-50 adult hermaphrodites were confined to a custom-made chamber on an unseeded 10-cm NGM plate fitted with inlets of air and 10% CO2. Gas mixes were pushed into the chamber at 1.5 ml/min using a syringe pump (New Era). After 35 min, an avoidance index (A.I.) was computed according to the following equation: 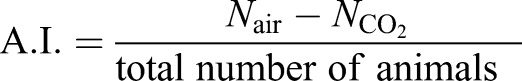 , where N is the number of animals in the indicated region.
, where N is the number of animals in the indicated region.
In vivo calcium imaging
In vivo calcium imaging was performed as previously described (Smith et al., 2013; Brandt and Ringstad, 2015) using the ratiometric calcium indicator YC3.60 (Nagai et al., 2004). Briefly, YC3.60 was excited with 435 nm light generated by a fast-switching monochromatic light source (Till Photonics) and CFP and YFP emissions were split and imaged side by side using a Dual View image splitter (Photometrics) and an Andor iXon EM-CCD camera. Imaging was performed on a Nikon Eclipse Ti microscope using a 20×/0.8 NA air objective. CO2 stimuli were delivered using a custom-built chamber that switched between control air (80% N2 and 20% O2) and high-CO2 air (70% N2, 20% O2 and 10% CO2). CFP/YFP emissions ratios were calculated after subtracting background fluorescence and after correcting for bleed-through of emissions from one channel into the other.
Quantification of GRASP puncta
The number and size of GRASP puncta were quantified using Fiji (Schindelin et al., 2012). z-stacks of BAG axons were thresholded and subjected to particle analysis, which automatically generated regions of interest (ROIs) around puncta for which the fluorescence intensity was above background. The number of puncta was defined as the number of particles found in axons, and the area of each particle was measured as a proxy for the size of a GRASP punctum. Each particle identified by the software was inspected to confirm that it contained only one GRASP punctum. If a particle comprised multiple puncta, ROIs were manually drawn around individual puncta.
Genetic suppressor screen for regulators of p38 MAPK-dependent BAG cell development
pmk-3 mutant animals carrying the BAG fate marker ynIs64[Pflp-17::GFP] were mutagenized with 47 mM ethyl methane sulfate (EMS) as described (Brenner, 1974), and screened in the F2 generation for restored GFP expression using a Leica M165FC fluorescence dissecting microscope. Approximately 10% of pmk-3 mutants expressed Pflp-17::GFP. Candidates were identified as suppressors of pmk-3 if more than 50% of their F3 progeny had restored GFP expression.
Mapping and cloning of suppressor alleles
An initial round of screening identified two non-complementing alleles, wz75 and wz76. High-resolution SNP mapping of wz75 was performed by crossing wz75 mutants to the polymorphic strain CB4856 and identifying crossovers using restriction fragment polymorphisms (snip-SNPs) (Davis et al., 2005), which placed wz75 in a 5 Mbp interval on LG IV. Whole-genome sequencing revealed that wz75 and wz76 mutants carry mutations in unc-31: wz75 contains a G→A mutation predicted to change Trp971 to an Amber stop, and wz76 contains a G→A mutation predicted to disrupt a splice-donor/acceptor site. Further screening and complementation tests identified three additional alleles of unc-31: wz112, wz127 and wz130. Whole-genome sequencing of wz112 and wz130 revealed that wz112 is a G→A missense mutation predicted to change Asp422 to Asn, and wz130 is a G→A nonsense mutation predicted to change Trp1114 to an Opal stop. Sanger sequencing of wz127 showed that it is a C→T mutation predicted to change Gln15 to an Amber stop.
Transgene expression analysis
Animals were immobilized as described above. Reporter transgene expression was quantified using a 20×/0.8 NA objective on a Zeiss Axioimager M2 upright microscope equipped with an EM-CCD camera (Andor). To determine the penetrance of transgene expression, we measured the number of BAG cells expressing the reporter in each animal. As an alternative method, we counted the percentage of animals expressing the reporter on a Leica M165FC fluorescence dissecting microscope. To determine the levels of reporter transgene expression, we measured the mean pixel values in a 30- or 50-pixel-radius circular ROI centered on the BAG soma using Fiji (Schindelin et al., 2012). For all experiments, data were collected over 3 days.
Heat-shock experiments
pmk-3(ok169) mutant animals carrying a Phsp16.41::irk-1 transgene were shifted twice to 37°C for 30 min, with 1 h at 20°C for recovery in between heat shocks. After heat shock, animals resumed growth at 20°C. To heat shock embryos, 50-75 gravid adults were placed on a 6 cm seeded NGM plate with Escherichia coli and allowed to lay eggs 1-2 h before and after being heat shocked. The adults were then removed from the plate 2 h after heat shock, and the remaining embryos resumed normal development at 20°C and were assayed for gene expression when they reached adulthood. To heat shock larvae, young larvae (L1/L2 stage) were manually picked onto assay plates, heat shocked as described, and then assayed for gene expression when they reached adulthood. To heat shock adults, young adults were picked onto assay plates, heat shocked, and then assayed 24 h after heat shock.
Cell culture and FACS sorting for RNA-Seq
Embryonic cell cultures were prepared as previously described (Christensen et al., 2002; Zhang et al., 2002) from wild-type and pmk-3(wz31) mutant animals expressing the BAG-specific and pmk-3-independent marker wzIs113[Pgcy-9::GFP]. In brief, embryos were isolated from synchronized populations of hypochlorite-treated hermaphrodites, and dissociated into single cells by chitinase treatment. Cells were re-suspended in L-15 medium supplemented with 10% fetal bovine serum (Sigma-Aldrich) and antibiotics, and passed through a 5 µm syringe filter (Millipore). Cells were plated onto poly-D-lysine-coated single-well chambered cover glasses (Lab-Tek II) and incubated overnight at 25°C.
GFP-labeled BAG neurons were isolated approximately 24 h after dissociation using fluorescence activated cell sorting (FACS); sorted cells were confirmed to be >90% GFP positive by direct inspection on a fluorescent microscope. Non-fluorescent, ‘non-BAG’, cells were also collected. Dead cells were marked with propidium iodide and excluded from sorted cells. Sorting was performed on a FACSAria Ilu SORP cell sorter using a 70 µm nozzle. Cells were sorted directly into RNA Extraction Buffer (10,000 cells/100 µl buffer), and RNA was purified using the Arcturus PicoPure RNA Isolation Kit (Thermo Fisher Scientific). RNA integrity and concentration were evaluated using an Agilent Bioanalyzer. RNA samples had an RNA Integrity score of at least 8, indicating that all samples were of high quality. Two biological replicates were prepared from each cell type.
RNA-Seq analysis of wild-type and pmk-3 mutant BAG neurons
cDNA libraries were prepared from RNA (1-2 ng) using the Ovation RNA-Seq System V2 (NuGEN), multiplexed, and sequenced as 100-bp paired end reads using the HiSeq 2500 (Illumina). Reads were aligned to the C. elegans genome and transcriptome (Wormbase WS243) using the STAR software package (Dobin et al., 2013). Gene expression quantification was performed using HTSeq (Anders et al., 2014). Differential expression analysis was done using the DESeq2 software package (Love et al., 2014) in R. Heat maps were generated using gplots (version 2.14.1, accessed 5 March, 2018) and RColorBrewer (ColorBrewer Palettes, version 1.1-2, accessed 5 March, 2018) R software packages. PCA analysis was performed using the DESeq2 software package (Love et al., 2014) in R. Read coverage histograms were generated from genomic alignments (BAM files) using Integrative Genomics Viewer (Thorvaldsdottir et al., 2013).
Gene ontology (GO) term analysis
GO term enrichment analyses were performed using the PANTHER Overrepresentation Test (Mi et al., 2019). Genes upregulated in pmk-3 mutant BAG cells (log2-fold change of gene expression in pmk-3 versus wild-type BAGs>1, P<0.05) were analyzed separately from genes downregulated in pmk-3 mutant BAG cells (log2-fold change of gene expression in pmk-3 versus wild-type BAGs<1, P<0.05). Fold enrichment is defined as the ratio of the number of genes in the uploaded gene list that map to the GO term to the number of genes expected to map to that GO term based on the C. elegans reference gene list. False discovery rate (FDR)-corrected P-values were calculated using Fisher's exact test with the Benjamini–Hochberg FDR multiple test correction. The GO terms for which fold enrichment had an FDR-adjusted P-value <0.05 were shown.
DAF-16::GFP localization
z-stacks were collected from young larvae (L2/L3) expressing PBAG::daf-16::GFP, as described above. Image analysis was performed in Fiji (Schindelin et al., 2012). For each cell, the maximum sum projection of five stacks (∼1 µm/stack) was generated. ROIs were drawn around the nucleus and cell body in a summated projection image, and the total amount of DAF-16::GFP fluorescence was measured in each region. The amount of fluorescence in the cytoplasm was defined as the total amount of DAF-16::GFP fluorescence in the cell body minus the amount in the nucleus. We then computed the ratio of nuclear DAF-16::GFP fluorescence to cytoplasmic DAF-16::GFP fluorescence as a measure of insulin receptor signaling.
Statistical analysis
Standard error of the mean (s.e.m.) and P-values were calculated using GraphPad Prism Software. For categorical data, statistical significance was determined using a chi-square test. For all other statistical comparisons, we used an unpaired t-test (when two experimental groups were compared) or an ordinary one-way ANOVA corrected for multiple comparisons using the Dunnet's test (when all experimental groups were compared with a control population, e.g. pmk-3 mutants) or Tukey's test (when all possible comparisons were made).
Supplementary Material
Acknowledgements
We thank Heather Broihier for helpful comments and conversations, Oliver Hobert (Columbia University) and Jane E. Hubbard (NYU School of Medicine) for reagents, Farzana Khan and Eugene Rudensky for isolating unc-31 alleles from suppressor screens, Nechama Schneider for assisting with measurements of flp-17 expression levels, and Igor Dolgalev for assisting with bioinformatic analysis. Flow cytometry-assisted cell sorting was performed by the Cytometry and Cell Sorting Laboratory at NYU School of Medicine. Whole-genome sequencing was performed by the NYU School of Medicine Genome Technology Center. Some strains were provided by the CGC, which is funded by the NIH Office of Research Infrastructure Programs grant P40 OD010440, and the Mitani lab (Tokyo Women's Medical University, Japan).
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: L.B.H., J.P.B., N.R.; Methodology: L.B.H.; Formal analysis: L.B.H., J.P.B.; Investigation: L.B.H., J.P.B.; Data curation: L.B.H., N.R.; Writing - original draft: L.B.H., N.R.; Writing - review & editing: L.B.H., N.R.; Visualization: L.B.H., N.R.; Supervision: N.R.; Project administration: N.R.; Funding acquisition: L.B.H., N.R.
Funding
This work was supported by the National Institutes of Health (R35 GM122573 to N.R. and F31 NS100360 to L.B.H.). Deposited in PMC for release after 12 months.
Data availability
The datasets generated and analyzed for this study are available in the Gene Expression Omnibus repository under accession number GSE137267.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.182873.supplemental
References
- Anders S., Pyl P. T. and Huber W. (2014). HTSeq - a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166-169. 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbagallo B., Philbrook A., Touroutine D., Banerjee N., Oliver D., Lambert C. M. and Francis M. M. (2017). Excitatory neurons sculpt GABAergic neuronal connectivity in the C. elegans motor circuit. Development 144, 1807-1819. 10.1242/dev.141911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargmann C. I. and Marder E. (2013). From the connectome to brain function. Nat. Methods 10, 483-490. 10.1038/nmeth.2451 [DOI] [PubMed] [Google Scholar]
- Bhattacharya A., Aghayeva U., Berghoff E. G. and Hobert O. (2019). Plasticity of the electrical connectome of C. elegans. Cell 176, 1174-1189.e16. 10.1016/j.cell.2018.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt J. P. and Ringstad N. (2015). Toll-like receptor signaling promotes development and function of sensory neurons required for a C. elegans pathogen-avoidance behavior. Curr. Biol. 25, 2228-2237. 10.1016/j.cub.2015.07.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt J. P., Aziz-Zaman S., Juozaityte V., Martinez-Velazquez L. A., Petersen J. G., Pocock R. and Ringstad N. (2012). A single gene target of an ETS-family transcription factor determines neuronal CO2-chemosensitivity. PLoS ONE 7, e34014 10.1371/journal.pone.0034014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chell J. M. and Brand A. H. (2010). Nutrition-responsive glia control exit of neural stem cells from quiescence. Cell 143, 1161-1173. 10.1016/j.cell.2010.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou J. H., Bargmann C. I. and Sengupta P. (2001). The Caenorhabditis elegans odr-2 gene encodes a novel Ly-6-related protein required for olfaction. Genetics 157, 211-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen M., Estevez A., Yin X., Fox R., Morrison R., Mcdonnell M., Gleason C., Miller D. M. III and Strange K. (2002). A primary culture system for functional analysis of C. elegans neurons and muscle cells. Neuron 33, 503-514. 10.1016/S0896-6273(02)00591-3 [DOI] [PubMed] [Google Scholar]
- Cornils A., Gloeck M., Chen Z., Zhang Y. and Alcedo J. (2011). Specific insulin-like peptides encode sensory information to regulate distinct developmental processes. Development 138, 1183-1193. 10.1242/dev.060905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. W., Hammarlund M., Harrach T., Hullett P., Olsen S. and Jorgensen E. M. (2005). Rapid single nucleotide polymorphism mapping in C. elegans. BMC Genomics 6, 118 10.1186/1471-2164-6-118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M. and Gingeras T. R. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15-21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emtage L., Aziz-Zaman S., Padovan-Merhar O., Horvitz H. R., Fang-Yen C. and Ringstad N. (2012). IRK-1 potassium channels mediate peptidergic inhibition of Caenorhabditis elegans serotonin neurons via a G(o) signaling pathway. J. Neurosci. 32, 16285-16295. 10.1523/JNEUROSCI.2667-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg E. H., Vanhoven M. K., Bendesky A., Wang G., Fetter R. D., Shen K. and Bargmann C. I. (2008). GFP reconstitution across synaptic partners (GRASP) defines cell contacts and synapses in living nervous systems. Neuron 57, 353-363. 10.1016/j.neuron.2007.11.030 [DOI] [PubMed] [Google Scholar]
- Fernandez A. M. and Torres-Alemán I. (2012). The many faces of insulin-like peptide signalling in the brain. Nat. Rev. Neurosci. 13, 225-239. 10.1038/nrn3209 [DOI] [PubMed] [Google Scholar]
- Gibson D. G., Young L., Chuang R.-Y., Venter J. C., Hutchison C. A. III and Smith H. O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343-345. 10.1038/nmeth.1318 [DOI] [PubMed] [Google Scholar]
- Gramstrup Petersen J. and Pocock R. (2013). Neuronal cell fate decisions: O2 and CO2 sensing neurons require egl-13/Sox5. Worm 2, e27284 10.4161/worm.27284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramstrup Petersen J., Rojo Romanos T., Juozaityte V., Redo Riveiro A., Hums I., Traunmüller L., Zimmer M. and Pocock R. (2013). EGL-13/SoxD specifies distinct O2 and CO2 sensory neuron fates in Caenorhabditis elegans. PLoS Genet. 9, e1003511 10.1371/journal.pgen.1003511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruner M., Nelson D., Winbush A., Hintz R., Ryu L., Chung S. H., Kim K., Gabel C. V. and van der Linden A. M. (2014). Feeding state, insulin and NPR-1 modulate chemoreceptor gene expression via integration of sensory and circuit inputs. PLoS Genet. 10, e1004707 10.1371/journal.pgen.1004707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillermin M. L., Castelletto M. L. and Hallem E. A. (2011). Differentiation of carbon dioxide-sensing neurons in Caenorhabditis elegans requires the ETS-5 transcription factor. Genetics 189, 1327-1339. 10.1534/genetics.111.133835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillermin M. L., Carrillo M. A. and Hallem E. A. (2017). A single set of interneurons drives opposite behaviors in C. elegans. Curr. Biol. 27, 2630-2639.e6. 10.1016/j.cub.2017.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallem E. A., Spencer W. C., Mcwhirter R. D., Zeller G., Henz S. R., Rätsch G., Miller D. M., Horvitz H. R., Sternberg P. W. and Ringstad N. (2011). Receptor-type guanylate cyclase is required for carbon dioxide sensation by Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 108, 254-259. 10.1073/pnas.1017354108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harward S. C., Hedrick N. G., Hall C. E., Parra-Bueno P., Milner T. A., Pan E., Laviv T., Hempstead B. L., Yasuda R. and Mcnamara J. O. (2016). Autocrine BDNF-TrkB signalling within a single dendritic spine. Nature 538, 99-103. 10.1038/nature19766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann K. A. and Broihier H. T. (2018). What neurons tell themselves: autocrine signals play essential roles in neuronal development and function. Curr. Opin. Neurobiol. 51, 70-79. 10.1016/j.conb.2018.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobert O. (2016). Terminal selectors of neuronal identity. Curr. Top. Dev. Biol. 116, 455-475. 10.1016/bs.ctdb.2015.12.007 [DOI] [PubMed] [Google Scholar]
- Hobert O., Glenwinkel L. and White J. (2016). Revisiting neuronal cell type classification in Caenorhabditis elegans. Curr. Biol. 26, R1197-R1203. 10.1016/j.cub.2016.10.027 [DOI] [PubMed] [Google Scholar]
- James R. E., Hoover K. M., Bulgari D., Mclaughlin C. N., Wilson C. G., Wharton K. A., Levitan E. S. and Broihier H. T. (2014). Crimpy enables discrimination of presynaptic and postsynaptic pools of a BMP at the Drosophila neuromuscular junction. Dev. Cell 31, 586-598. 10.1016/j.devcel.2014.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon C., Chang J., Gensch E., Rudner A. and Tabtiang R. (1993). A C. elegans mutant that lives twice as long as wild type. Nature 366, 461-464. 10.1038/366461a0 [DOI] [PubMed] [Google Scholar]
- Kim K. and Li C. (2004). Expression and regulation of an FMRFamide-related neuropeptide gene family in Caenorhabditis elegans. J. Comp. Neurol. 475, 540-550. 10.1002/cne.20189 [DOI] [PubMed] [Google Scholar]
- Kim H., Ishidate T., Ghanta K. S., Seth M., Conte D. Jr., Shirayama M. and Mello C. C. (2014). A co-CRISPR strategy for efficient genome editing in Caenorhabditis elegans. Genetics 197, 1069-1080. 10.1534/genetics.114.166389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K. D., Tissenbaum H. A., Liu Y. and Ruvkun F. (1997). daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 277, 942-946. 10.1126/science.277.5328.942 [DOI] [PubMed] [Google Scholar]
- Lee R. Y. N., Lobel L., Hengartner M., Horvitz H. R. and Avery L. (1997). Mutations in the alpha1 subunit of an L-type voltage-activated Ca2+ channel cause myotonia in Caenorhabditis elegans. EMBO J. 16, 6066-6076. 10.1093/emboj/16.20.6066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee R. Y. N., Hench J. and Ruvkun G. (2001). Regulation of C. elegans DAF-16 and its human ortholog FKHRL1 by the daf-2 insulin-like signaling pathway. Curr. Biol. 11, 1950-1957. 10.1016/S0960-9822(01)00595-4 [DOI] [PubMed] [Google Scholar]
- Lee J. S., Shih P.-Y., Schaedel O. N., Quintero-Cadena P., Rogers A. K. and Sternberg P. W. (2017). FMRFamide-like peptides expand the behavioral repertoire of a densely connected nervous system. Proc. Natl. Acad. Sci. USA 114, E10726-E10735. 10.1073/pnas.1710374114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen M. K., Zappaterra M. W., Chen X., Yang Y. J., Hill A. D., Lun M., Maynard T., Gonzalez D., Kim S., Ye P. et al. (2011). The cerebrospinal fluid provides a proliferative niche for neural progenitor cells. Neuron 69, 893-905. 10.1016/j.neuron.2011.01.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Kennedy S. G. and Ruvkun G. (2003). daf-28 encodes a C. elegans insulin superfamily member that is regulated by environmental cues and acts in the DAF-2 signaling pathway. Genes Dev. 17, 844-858. 10.1101/gad.1066503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K., Hsin H., Libina N. and Kenyon C. (2001). Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat. Genet. 28, 139-145. 10.1038/88850 [DOI] [PubMed] [Google Scholar]
- Love M. I., Huber W. and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mairet-Coello G., Tury A. and Dicicco-Bloom E. (2009). Insulin-like growth factor-1 promotes G(1)/S cell cycle progression through bidirectional regulation of cyclins and cyclin-dependent kinase inhibitors via the phosphatidylinositol 3-kinase/Akt pathway in developing rat cerebral cortex. J. Neurosci. 29, 775-788. 10.1523/JNEUROSCI.1700-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardinly A. R., Spiegel I., Patrizi A., Centofante E., Bazinet J. E., Tzeng C. P., Mandel-Brehm C., Harmin D. A., Adesnik H., Fagiolini M. et al. (2016). Sensory experience regulates cortical inhibition by inducing IGF1 in VIP neurons. Nature 531, 371-375. 10.1038/nature17187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello C. C., Kramer J. M., Stinchcomb D. and Ambros V. (1991). Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 10, 3959-3970. 10.1002/j.1460-2075.1991.tb04966.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H., Muruganujan A., Huang X., Ebert D., Mills C., Guo X. and Thomas P. D. (2019). Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 14, 703-721. 10.1038/s41596-019-0128-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris J. Z., Tissenbaum H. A. and Ruvkun G. (1996). A phosphatidylinositol-3-OH kinase family member regulating longevity and diapause in Caenorhabditis elegans. Nature 382, 536-539. 10.1038/382536a0 [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S., Lu Y., Shaham S. and Sengupta P. (2008). Sensory signaling-dependent remodeling of olfactory cilia architecture in C. elegans. Dev. Cell 14, 762-774. 10.1016/j.devcel.2008.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy C. T. and Hu P. J. (2013). Insulin/insulin-like growth factor signaling in C. elegans WormBook, 1-43. 10.1895/wormbook.1.164.1 [DOI] [PMC free article] [PubMed]
- Nagai T., Yamada S., Tominaga T., Ichikawa M. and Miyawaki A. (2004). Expanded dynamic range of fluorescent indicators for Ca(2+) by circularly permuted yellow fluorescent proteins. Proc. Natl. Acad. Sci. USA 101, 10554-10559. 10.1073/pnas.0400417101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paix A., Folkmann A., Rasoloson D. and Seydoux G. (2015). High efficiency, homology-directed genome editing in C. elegans using CRISPR/Cas9 ribonucleoprotein complexes. Genetics 201, 47-54. 10.1534/genetics.115.179382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis S. and Ruvkun G. (1998). Caenorhabditis elegans Akt/PKB transduces insulin receptor-like signals from AGE-1 KI3 kinase to the DAF-16 transcription factor. Genes Dev. 12, 2488-2498. 10.1101/gad.12.16.2488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peckol E. L., Zallen J. A., Yarrow J. C. and Barmann C. I. (1999). Sensory activity affects sensory axon development in C. elegans. Development 126, 1891-1902. [DOI] [PubMed] [Google Scholar]
- Peckol E. L., Troemel E. R. and Bargmann C. I. (2001). Sensory experience and sensory activity regulate chemosensory receptor gene expression in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 98, 11032-11038. 10.1073/pnas.191352498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippidou P. and Dasen J. S. (2013). Hox genes: choreographers in neural development, architects of circuit organization. Neuron 80, 12-34. 10.1016/j.neuron.2013.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengarajan S., Yankura K. A., Guillermin M. L., Fung W. and Hallem E. A. (2019). Feeding state sculpts a circuit for sensory valence in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 116, 1776-1781. 10.1073/pnas.1807454116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond J. E., Davis W. S. and Jorgensen E. M. (1999). UNC-13 is required for synaptic vesicle fusion in C. elegans. Nature 2, 959-964. 10.1038/14755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringstad N. and Horvitz H. R. (2008). FMRFamide neuropeptides and acetylcholine synergistically inhibit egg-laying by C. elegans. Nat. Neurosci. 11, 1168-1176. 10.1038/nn.2186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo Romanos T., Petersen J. G., Riveiro A. R. and Pocock R. (2015). A novel role for the zinc-finger transcription factor EGL-46 in the differentiation of gas-sensing neurons in Caenorhabditis elegans. Genetics 199, 157-163. 10.1534/genetics.114.172049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo Romanos T., Petersen J. G. and Pocock R. (2017). Control of neuropeptide expression by parallel activity-dependent pathways in Caenorhabditis elegans. Sci. Rep. 7, 38734 10.1038/srep38734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagasti A., Hisamoto N., Hyodo J., Tanaka-Hino M., Matsumoto K. and Bargmann C. I. (2001). The CaMKII UNC-43 activates the MAPKKK NSY-1 to execute a lateral signaling decision required for asymmetric olfactory neuron fates. Cell 105, 221-232. 10.1016/S0092-8674(01)00313-0 [DOI] [PubMed] [Google Scholar]
- Saifee O., Wei L. and Nonet M. L. (1998). The Caenorhabditis elegans unc-64 locus encodes a syntaxin that interacts genetically with synaptobrevin. Mol. Biol. Cell 9, 1235-1252. 10.1091/mbc.9.6.1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer W. R. and Kenyon C. J. (1995). A calcium-channel homologue required for adaptation to dopamine and serotonin in Caenorhabditis elegans. Nature 375, 73-78. 10.1038/375073a0 [DOI] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676-682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Saiz E., Poole R. J., Felton T., Zhang F., de La Cruz E. D. and Hobert O. (2013). Modular control of glutamatergic neuronal identity in C. elegans by distinct homeodomain proteins. Cell 155, 659-673. 10.1016/j.cell.2013.09.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieburth D., Madison J. M. and Kaplan J. M. (2007). PKC-1 regulates secretion of neuropeptides. Nat. Neurosci. 10, 49-57. 10.1038/nn1810 [DOI] [PubMed] [Google Scholar]
- Smith E. S. J., Martinez-Velazquez L. and Ringstad N. (2013). A chemoreceptor that detects molecular carbon dioxide. J. Biol. Chem. 288, 37071-37081. 10.1074/jbc.M113.517367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa-Nunes R., Yee L. L. and Gould A. P. (2011). Fat cells reactivate quiescent neuroblasts via TOR and glial insulin relays in Drosophila. Nature 471, 508-512. 10.1038/nature09867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speese S., Petrie M., Schuske K., Ailion M., Ann K., Iwasaki K., Jorgensen E. M. and Martin T. F. J. (2007). UNC-31 (CAPS) is required for dense-core vesicle but not synaptic vesicle exocytosis in Caenorhabditis elegans. J. Neurosci. 27, 6150-6162. 10.1523/JNEUROSCI.1466-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston J. E. and Horvitz H. R. (1977). Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev. Biol. 56, 110-156. 10.1016/0012-1606(77)90158-0 [DOI] [PubMed] [Google Scholar]
- Sulston J. E., Schierenberg E., White J. G. and Thomson J. N. (1983). The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol. 100, 64-119. 10.1016/0012-1606(83)90201-4 [DOI] [PubMed] [Google Scholar]
- Terauchi A., Johnson-Venkatesh E. M., Bullock B., Lehtinen M. K. and Umemori H. (2016). Retrograde fibroblast growth factor 22 (FGF22) signaling regulates insulin-like growth factor 2 (IGF2) expression for activity-dependent synapse stabilization in the mammalian brain. eLife 5, e12151 10.7554/eLife.12151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas G. M. and Huganir R. L. (2004). MAPK cascade signalling and synaptic plasticity. Nat. Rev. Neurosci. 5, 173-183. 10.1038/nrn1346 [DOI] [PubMed] [Google Scholar]
- Thorvaldsdottir H., Robinson J. T. and Mesirov J. P. (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178-192. 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troemel E. R., Sagasti A. and Bargmann C. I. (1999). Lateral signaling mediated by axon contact and calcium entry regulates asymmetric odorant receptor expression in C. elegans. Cell 99, 387-398. 10.1016/S0092-8674(00)81525-1 [DOI] [PubMed] [Google Scholar]
- Tullet J. M. A., Hertweck M., An J. H., Baker J., Hwang J. Y., Liu S., Oliveira R. P., Baumeister R. and Blackwell T. K. (2008). Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell 132, 1025-1038. 10.1016/j.cell.2008.01.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamsley B. and Fishell G. (2017). Genetic and activity-dependent mechanisms underlying interneuron diversity. Nat. Rev. Neurosci. 18, 299-309. 10.1038/nrn.2017.30 [DOI] [PubMed] [Google Scholar]
- Weimer R. M., Richmond J. E., Davis W. S., Hadwiger G., Nonet M. L. and Jorgensen E. M. (2003). Defects in synaptic vesicle docking in unc-18 mutants. Nat. Neurosci. 6, 1023-1030. 10.1038/nn1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West A. E. and Greenberg M. E. (2011). Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harb. Perspect. Biol. 3, a005744 10.1101/cshperspect.a005744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J. G., Southgate E., Thomson J. N. and Brenner S. (1986). The structure of the nervous system of the nematode Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 314, 1-340. 10.1098/rstb.1986.0056 [DOI] [PubMed] [Google Scholar]
- Yap E.-L. and Greenberg M. E. (2018). Activity-regulated transcription: bridging the gap between neural activity and behavior. Neuron 100, 330-348. 10.1016/j.neuron.2018.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian M., Cook D. E., Zdraljevic S., Brady S. C., Lee D., Lee J. and Andersen E. C. (2018). Discovery of genomic intervals that underlie nematode responses to benzimidazoles. PLoS Negl. Trop. Dis. 12, e0006368 10.1371/journal.pntd.0006368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Ma C., Delohery T., Nasipak B., Foat B. C., Bounoutas A., Bussemaker H. J., Kim S. K. and Chalfie M. (2002). Identification of genes expressed in C. elegans touch receptor neurons. Nature 418, 331-335. 10.1038/nature00891 [DOI] [PubMed] [Google Scholar]
- Zhou K.-M., Dong Y.-M., Ge Q., Zhu D., Zhou W., Lin X.-G., Liang T., Wu Z.-X. and Xu T. (2007). PKA activation bypasses the requirement for UNC-31 in the docking of dense core vesicles from C. elegans neurons. Neuron 56, 657-669. 10.1016/j.neuron.2007.09.015 [DOI] [PubMed] [Google Scholar]
- Ziegler A. N., Chidambaram S., Forbes B. E., Wood T. L. and Levison S. W. (2014). Insulin-like growth factor-II (IGF-II) and IGF-II analogs with enhanced insulin receptor-a binding affinity promote neural stem cell expansion. J. Biol. Chem. 289, 4626-4633. 10.1074/jbc.M113.537597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer M., Gray J. M., Pokala N., Chang A. J., Karow D. S., Marletta M. A., Hudson M. L., Morton D. B., Chronis N. and Bargmann C. I. (2009). Neurons detect increases and decreases in oxygen levels using distinct guanylate cyclases. Neuron 61, 865-879. 10.1016/j.neuron.2009.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.