

Abstract
Background/Aim: Human chronic periodontitis is a major health problem. Although some oral bacteria have been reported to be putative pathogens, Epstein–Barr virus (EBV) is reported to be associated with the progression of periodontitis. However, the role of EBV in the aetiology of periodontitis is unknown. Therefore, we investigated periodontal pathogenesis of EBV to confirm whether EBV-encoded latent membrane protein 1 (LMP1) induces Interleukin-8 (IL8) production in human gingival cells. Materials and Methods: Real-time polymerase chain reaction, luciferase assay, enzyme-linked immunosorbent assay (ELISA), and western blotting were performed for determining IL8 mRNA expression, nuclear factor kappa B (NF-ĸB) transcription, IL8 production, and the phosphorylation of NF-ĸB p65 and Inhibitor of kappa B alpha (IĸBα), respectively, in Ca9-22 human gingival epithelial cells. Two LMP1 mutants lacking C-terminal activating region (CATR) domains responsible for activating NF-ĸB were used. Results: Extremely high IL8 production was induced by LMP1 in time- and dose-dependent manner, where simultaneous phosphorylation of NF-κB p65 and IĸBα and transcription of NF-ĸB were observed. On the contrary, IL8 production and NF-ĸB transcription were drastically inhibited by dominant negative mutant of IĸBα. Moreover, the LMP1 mutants failed to induce IL8 production. Conclusion: Our findings suggest that due to CATR domains, LMP1 contributes to the progression of periodontitis via IL8 production attributable to NF-ĸB activation.
Keywords: EBV, LMP1, periodontitis, IL8, NF-ĸB, gingival epithelial cells
Chronic periodontitis, a chronic inflammatory and infectious disease causing the destruction of the periodontium including the alveolar bone, is prevalent worldwide (1,2). Mounting evidence has indicated that chronic periodontitis is a risk factor for pre-term birth, heart disease, diabetes, and atherosclerosis (1,2). Over the past decade, neutrophil infiltration in the periodontium has been revealed to be the major aetiology for periodontitis (2). Some oral endogenous bacteria are believed to trigger periodontitis via host–parasite interactions (2,3). However, periodontopathic bacteria, such as Porphyromonas gingivalis, are not always detected in periodontal lesions (4-6); therefore, the conventional theory based on bacterial aetiology alone cannot fully explain the aetiology of periodontitis.
A positive association has been reported between chronic periodontitis and Epstein–Barr virus (EBV) infection (7-11). EBV, a member of the herpesvirus family, infects many adults. During primary EBV infection, the virus undergoes lytic replication in B-cells and epithelial cells of the upper aerodigestive tract, where it later establishes latency (12,13). EBV can be reactivated and is commonly found in the saliva of infected people (9,10,14). Many reports have demonstrated that the amount of EBV DNA detected in periodontal pockets of patients with chronic periodontitis correlates with disease severity (7-11). We observed that EBV DNA was more frequently detected in patients with deeper periodontal pockets than in those with shallow ones or healthy controls (15,16). We also observed that P. gingivalis was able to induce EBV reactivation through epigenetic regulation (17). Additionally, a large number of EBV-encoded small RNA (EBER)-positive B-cells were found in the gingival tissues of patients with chronic periodontitis (15). Thus, EBV is epidemiologically involved in the aetiology of chronic periodontitis. However, no causal relationship between EBV and chronic periodontitis has been delineated.
The level of gingival epithelial EBV infection is correlated with the severity of chronic periodontitis (18). EBV-infected cells reportedly express EBERs and EBV-encoded latent membrane protein (LMP1) (18). LMP1 is composed of 386 amino acids; it comprises a short N-terminal cytoplasmic domain, six transmembrane-spanning domains, and a C-terminal cytoplasmic tail (19-21), and mediates cell growth as an oncoprotein (19,21). LMP1 expression is essential for EBV-mediated primary B-cell transformation in vitro and associated with a number of human malignancies (12,13,19,21). In epithelial cells, LMP1 induces the expression of anti-apoptotic proteins and cell surface antigens (22-25). Additionally, it induces the production of matrix metallopeptidase 9, cyclo-oxygenase-2, and pro-inflammatory cytokines in immune cells (26-29). Taking these findings together, this viral protein activates inflammatory and immune-regulatory responses in EBV-infected cells.
Interleukin-8 (IL8), a potent neutrophil chemoattractant and activator, is a mediator of human chronic periodontitis via the accumulation and degranulation of neutrophils, which causes the subsequent destruction of periodontium (30,31). Moreover, it strikingly upregulates the release of elastase by neutrophils, which significantly amplifies periodontal inflammation (32). IL8 thus plays a crucial role in the progression of chronic periodontitis.
In this study, to the best of our knowledge, we show for the first time that LMP1 attributable to its C-terminal activator regions (CTAR) domains in a human gingival epithelial cell line induces IL8 production via nuclear factor kappa B (NF-ĸB) activation. Moreover, we discuss whether LMP1 in gingival epithelial cells possibly contributes to the progression of human chronic periodontitis.
Materials and Methods
Reagents and plasmids. Antibodies against phospho-IĸBα (Ser32) and phospho-p65 (Ser32) (Cell Signaling Technology, Danvers, MA, USA) and IĸBα, p65, β-actin, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) were purchased. A reporter plasmid expressing firefly luciferase under the control of NF-ĸB (pGL3-5xĸB-luc) and dominant negative mutant of IĸBα (IĸBαΔN) were used as previously described (33). The LMP1 expression vector (pSG-LMP1), its mutants, and control vector (pSG) (20) were generous gifts from Dr Martin Rowe (School of Cancer Sciences, University of Birmingham, UK).
Cell culture and transfection. The gingival epithelial cell line Ca9-22 was purchased from RIKEN BioResource Center (Tsukuba, Japan) and maintained at 37˚C in Dulbecco’s modified Eagle’s medium (Sigma, St. Louis, MO, USA) with 10% heat-inactivated fetal bovine serum (Thermo Fisher Scientific, Rockford, IL, USA), penicillin, and streptomycin as previously described (34). Ca9-22 cells were transfected with pSG-LMP1 using Lipofectamine 2000 (Thermo Fisher Scientific), in accordance with the manufacturer’s instructions.
mRNA preparation and real-time polymerase chain reactions (PCR). Experimental procedures for RNA purification and real-time (RT)-PCR were performed as previously described (34). For cDNA synthesis, total RNA was reverse transcribed using an RNA PCR kit (PrimeScript; Takara Bio, Shiga, Japan). The resulting cDNA mixture was subjected to RT-PCR analysis using SYBR Premix Ex Taq solution (Takara Bio) containing sense and antisense primers. The following primer sequences were used: IL8, forward (5’-CTTGTCATTGCCAGCTGTGT-3’) and reverse (5’-TGACTGT GGAGTTTTGGCTG-3’); GAPDH, forward (5’-TGCACCACC AACTGCTAGC-3’) and reverse (5’-GGCATGGACTGTGGTCA TGAG-3’). PCR assays were performed using a TP-800 Thermal Cycler Dice Real-Time System (Takara Bio) and analysed using software provided by the device manufacturer. Thermal cycling conditions were 40 cycles at 95˚C for 5 s, 60˚C for 30 s, and 72˚C for 1 min. All RT-PCR experiments were performed in triplicate; the specificity of each product was verified by melting curve analysis. The calculated level of gene expression was normalized to that of GAPDH mRNA.
IL8 measurements. IL8 in Ca9-22 cell-culture supernatants were measured using a human enzyme-linked immunosorbent assay (ELISA) kit for IL8 (R&D systems, Minneapolis, MN, USA), according to the manufacturer’s instructions. All experiments were performed in triplicate, and data presented are representative of three independent experiments.
Preparation of cytoplasmic and nuclear extracts. Ca9-22 cells (2×105 cells/ml) were transfected with or without pSG-LMP1 for 24 h. Cells were washed with cold phosphate-buffered saline, resuspended in lysis buffer, and centrifuged. The supernatant was collected (whole-cell extract) and stored at −80˚C until use. Precipitated cells were resuspended in cytoplasmic lysis buffer (Chemicon International, Temecula, CA, USA) and incubated for 15 min on ice. The cells were vortexed and centrifuged (10 min, 20,000 × g), and the supernatant (cytoplasmic extract) was removed. Pelleted cells were washed twice with cytoplasmic buffer to remove any trace of proteins, resuspended in nuclear lysis buffer (Chemicon International), and incubated for 15 min on ice. Lysed nuclei were sonicated for 10 s and centrifuged (15 min, 20,000 × g); the supernatant (nuclear extract) was stored at −80˚C. The Pierce BCA Protein Assay–Reducing Agent Compatible kit (Thermo Fisher Scientific) was used for standardizing protein concentration.
Western blotting. Experimental procedures for western blotting were performed as previously described (34,35). Briefly, equal amounts of protein (15 μg) were separated by sodium dodecyl sulfate - poly acrylamide gel electrophoresis and transferred to a polyvinylidene fluoride membrane (EMD Millipore Corporation, Billerica, MA, USA). The membrane was probed and visualised using a SuperSignal West Pico enhanced chemiluminescence kit (Thermo Fisher Scientific).
Transient luciferase assay. Ca9-22 cells (4×105 cells/ml) were transfected with 200 ng of reporter plasmids (pGL3-5xĸB-luc) and 10 ng of the internal control plasmid (pRL-TK) expressing Renilla luciferase (Promega, Madison, WI, USA), with or without LMP1 or IĸBαΔN for 24 h, using Lipofectamine 2000. These cells were harvested using Passive Lysis Buffer (Promega); the extracts were assessed for luciferase activity using Dual-Luciferase Assay System (Promega) as previously described (17,35).
Statistical analysis. Mean values±standard deviation (SD) were calculated. Statistical analysis was performed using one-way analysis of variance with Tukey’s multiple comparisons test; p<0.05 was considered to be statistically significant.
Results
LMP1 expression induces IL8 production in a human gingival epithelial cell line. RT-PCR analysis was used to investigate the effects of LMP1 on IL8 expression in Ca9-22 cells. LMP1 transfection induced significant IL8 expression. The IL8 mRNA level was up-regulated time-dependently (Figure 1A) and dose-dependently (Figure 1B) in response to LMP1 transfection, but was not in cells transfected with the control vector. We next investigated the effects of LMP1 on IL8 protein production. As shown in Figure 1C and D, extremely high concentrations of IL8 were produced due to LMP1 in time- and dose-dependent manners. These results indicate that LMP1 in human gingival cells may be a potent inducer of IL8 production.
Figure 1. Latent membrane protein 1 (LMP1) promotes interleukin-8 (IL8) production in a human gingival epithelial cell line. Ca9-22 cells were transfected with pSG-LMP1 (0.2 μg) or pSG (0.2 μg) as control vector (ContV) for different times (A, C) and with pSG-LMP1 at different concentrations (0.05, 0.1, or 0.2 μg) for 24 h (B, D). A, B: The cells were harvested and the level of IL8 mRNA was determined using reverse transcription polymerase chain reaction analysis with specific primers. C, D: IL8 released into the culture supernatants was determined using enzyme-linked immunosorbent assay. The values are presented as mean±SD; n=3. **Significantly different at p<0.0001).

LMP1 activates NF-ĸB transcription in Ca9-22 cells. NF-ĸB is an inducible cellular transcription factor that regulates a variety of cellular genes involved in controlling inflammatory and immune responses (36). NF-ĸB normally binds to its inhibitor IĸB present in the cytoplasm. Upon stimulation, intracellular signalling activates the IĸB kinase complex, which sequentially phosphorylates two serine residues (Ser32/36) in IĸBα (36). This results in the degradation of IĸB by the 26S proteasome and consequent nuclear translocation of NF-ĸB.
As NF-ĸB activity is important in mediating IL8 production (36,37), we next examined whether LMP1 activated NF-ĸB in Ca9-22 cells. We analysed the phosphorylation of NF-ĸB (p65) and IĸBα using western blotting. As shown in Figure 2A, the level of phosphorylated IĸBα increased, and that of IĸBα conversely decreased due to LMP1. In parallel with this, phosphorylation increased at Ser536 of the NF-ĸB p65 subunit, which plays a key role in the transcriptional competence of NF-ĸB and the nuclear translocation of p65 (36). To further examine whether NF-ĸB activation by LMP1 in Ca9-22 cells occurs at the transcriptional level, we performed luciferase assays using a reporter plasmid whose expression is proportional to NF-ĸB activity. As shown in Figure 2B, LMP1 up-regulated NF-ĸB transcription in a dose-dependent manner. These results indicate that LMP1 activates NF-ĸB in human gingival epithelial cells and that it may induce IL8 production.
Figure 2. Latent membrane protein 1 (LMP1) induces nuclear factor kappa B (NF-ĸB) activation. Ca9-22 cells were transfected with pSG-LMP1 or pSG (0.05 or 0.2 μg) as control vector (ContV). A: Proteins in the cytoplasmic extract (left panel) and the nuclear extract (right panel) were separated by polyacrylamide gel electrophoresis and then immunoblotted. Purity of the cytoplasmic and nuclear extracts was confirmed using antibodies specific for histone H3 as a representative of nuclear proteins and β-actin as a representative of cytoplasmic proteins. B: Ca9-22 cells were transfected with pSG-LMP1 (0.05, 0.1, or 0.2 μg) and 20 ng of the pGL3-5xĸB-luc reporter plasmid. The cells were harvested and luciferase (Luc) activity was measured 24 hours after transfection. The values are presented as mean±SD; n=3. Significantly different at *p<0.05 and **p<0.0001.

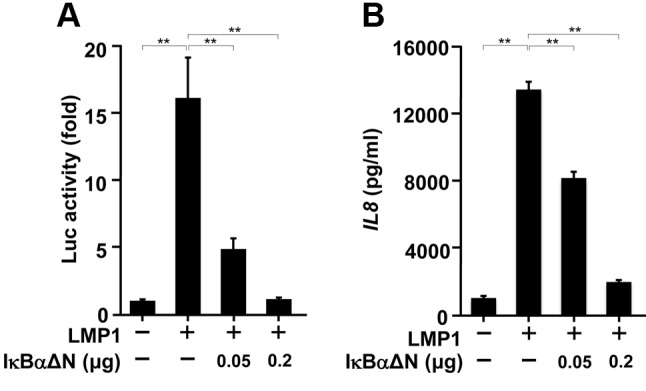
NF-ĸB is involved in LMP1-induced IL8 production from Ca9-22 cells. We next examined whether NF-ĸB was involved in LMP1-induced IL8 production in Ca9-22 cells. We used a dominant negative mutant of IĸBα (IĸBαΔN) to interfere with NF-ĸB activity (38). As shown in Figure 3A, LMP1-induced NF-ĸB activity was inhibited by IĸBαΔN in a dose-dependent manner. In parallel with this, LMP1-induced IL8 production was also inhibited (Figure 3B). These results suggest that NF-ĸB is a potent regulator of LMP1-induced IL8 production from human gingival epithelial cells.
Figure 3. The inhibitor of kappa B alpha (IĸBα) mutant inhibits EBVencoded latent membrane protein 1 latent membrane protein 1 (LMP1)- induced nuclear factor kappa B (NF-ĸB) activation and interleukin-8 (IL8) production. Ca9-22 cells were transfected with IĸBαΔNexpressing plasmid (0.05 or 0.2 μg), 0.2 μg of pSG-LMP1, and 20 ng of pGL3-5xĸB-luc reporter plasmid for 24 h. A: Luciferase (Luc) assay was then performed. B: Ca9-22 cells were transfected with IĸBαΔN (0.05 or 0.2 μg) and 0.2 μg of pSG-LMP1 for 24 h. Enzyme-linked immunosorbent assay test for IL8 was then performed. The values are presented as mean±SD; n=3. **Significantly different at p<0.0001.
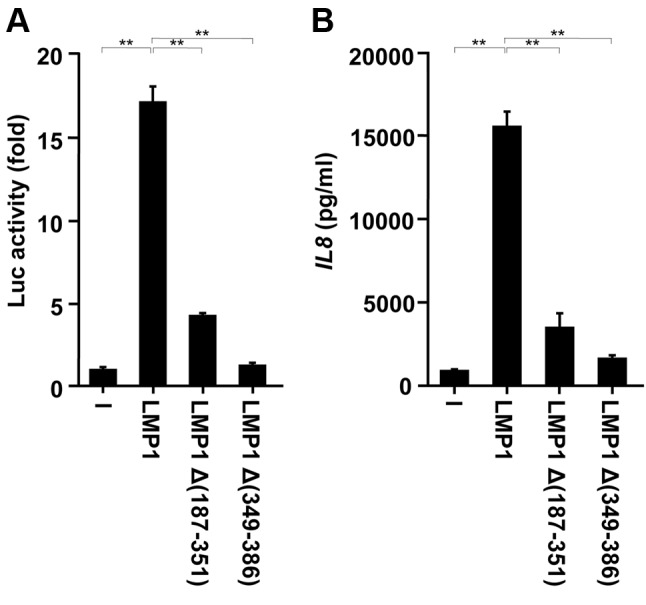
The CTAR1 and CTAR2 domains of LMP1 contribute to LMP1-induced IL8 production from Ca9-22 cells. Previous studies have identified two functional domains in the LMP1 C-terminal cytoplasmic tail, CTAR1 (amino acids 187-231) and 2 (amino acids 351-386), which are important for NF-ĸB activation (20,25,39). Proximal CTAR1 interacts with several tumor necrosis factor (TNF) receptor-associated factors (TRAFs) and induces NF-ĸB signalling (25,39). Distal CTAR2 binds to the TNF receptor-associated death domain (TRADD) protein and mediates NF-ĸB signalling (20). Thus, we employed two LMP1 mutants, LMP1Δ(187-351) lacking CTAR1 and LMP1Δ(349-386) lacking CTAR2, to analyse the effects of such mutations on NF-ĸB activation. The expression of LMP1Δ(187-351) or LMP1Δ(349-386) significantly reduced NF-ĸB activity (Figure 4A) and reduced IL8 production by approximately 80-90% compared with expression of wild-type LMP1 (Figure 4B). Therefore, both TRAF-interacting CTAR1 and TRADD-interacting CTAR2 should be necessary for LMP1-induced IL8 production from human gingival epithelial cells.
Figure 4. C-terminal activator region 1 (CTAR1) and CTAR2 domains of latent membrane protein 1 (LMP1) are involved in LMP1-induced nuclear factor kappa B (NF-ĸB) activation and interleukin-8 (IL8) production. Ca9-22 cells were transfected with 0.2 μg of the wild-type or the indicated mutant LMP1 plasmids together with 20 ng of the pGL3-5xĸB-luc reporter plasmid for 24 h. A: Luciferase (Luc) assay was then performed as shown. B: Ca9-22 cells were transfected with wild-type or mutant (Δ) LMP1 plasmids for 24 h. Cell culture supernatants were prepared and assayed for IL8 protein. The values are presented as mean±SD; n=3. **Significantly different at p<0.0001.
Discussion
Although emerging evidence suggests an association between EBV and human chronic periodontitis, whose major aetiology has recently been revealed to be neutrophil infiltration into the periodontium, how EBV relates to the progression of periodontitis is not understood. To clarify this issue, it would be important to identify and characterize any complex pro-inflammatory cytokine network involved in periodontal pathogenesis and its link to EBV. We, therefore, first examined whether EBV LMP1 in a human gingival cell line, Ca9-22, induced IL8 production. As expected, LMP1 induced IL8 production. In addition, for IL8, the activation of NF-ĸB was necessary. In parallel with this, mRNA expression of other NF-ĸB-drived pro-inflammatory cytokines, namely, IL1β, IL6, and TNFα, was also up-regulated by LMP1 (data not shown).
Like other infectious diseases, adhesion to and subsequent invasion into the epithelia by pathogens are critical steps in the initiation of periodontitis. Bacterial adhesion to gingival epithelium reportedly induces the production of various immune-response mediators (1,40). Among these, IL8, a potent neutrophil chemoattractant and activator, is such a mediator of human chronic periodontitis that acts via its involvement in the accumulation and degranulation of neutrophils, which causes subsequent destruction of the periodontium (30,31). Many investigators have demonstrated the presence of IL8 in gingival crevicular fluid and pro-inflammatory cytokine levels in gingival crevicular fluid being closely associated with the severity of inflammation and periodontal destruction (41-44). Moreover, primed by IL1β, IL8 strikingly up-regulates neutrophils for elastase release, giving rise to the significant increase of periodontal inflammation (32). IL8 is produced excessively or continuously in response to accumulated periodontopathic bacteria and their products in human gingival crevices or periodontal pockets and is an important determinant of the progression of chronic periodontitis (2). In the present study, extremely high levels of IL8 were induced due to LMP1 in Ca9-22 cells. This suggests that LMP1 contributes to the progression of chronic periodontitis by promoting the production of pro-inflammatory cytokines, IL8 in particular.
Genes of several pro-inflammatory cytokines, including chemokines, contain NF-ĸB-binding sites in their proximal promoters (36). Hence, NF-ĸB activation possibly also plays a key role in virus-dependent cytokine expression and pathogenesis with inflammation. In the present study, LMP1 stimulated simultaneous phosphorylation of IĸBα and NF-ĸB p65 and the transcription of NF-ĸB. Additionally, IĸBαΔN inhibited LMP1-induced NF-ĸB activation and subsequent IL8 production in a dose-dependent manner. This suggests that NF-ĸB is a potent regulator of LMP1-induced IL8 production from human gingival epithelial cells. In this connection, this NF-ĸB-dependent IL8 production has been reported to be associated with distinct human cells of the periodontium, such as a cervical squamous cell carcinoma cell line and umbilical vein endothelial cells (45,46).
Regarding LMP1-related NF-ĸB activation, the mutation analysis of LMP1 has identified CTAR1 and CTAR2 as independent effectors of NF-ĸB activation (20,25,39). CTAR1 binds to TRAF1, -2, -3, and -5 through a consensus TRAF-binding motif, while CTAR2 binds to the TRADD (20,25,39). In the present study, we found for the first time that LMP1Δ(187-351) and LMP1Δ(349-386), lacking CTAR1 and CTAR2, respectively, significantly reduced LMP1-induced IL8 production. Therefore, LMP1, particularly its CTAR domains, may be responsible for IL8 production from EBV-infected human gingival epithelial cells.
Taking our findings together, we confirmed that NF-ĸB activation was responsible for LMP1-induced IL8 production by the gingival epithelial cell line. However, this was only obtained from a single cell line. Although Ca9-22 has been employed in many periodontal studies (47-49), it is important to confirm what we found in this study in other gingival epithelial cells to clarify the physiological relevance of our findings.
Chronic periodontitis and EBV infection in the periodontium quite often coincide, where high numbers of EBER-positive B-cells and EBER-positive gingival epithelial cells are observed in the gingival tissues of patients with periodontal disease (15,18). Moreover, the involvement of EBV in the aetiology of human chronic periodontitis is supported by several observations: Anti-herpesvirus drug treatment reportedly resulted in an undetectable level of EBV with simultaneous periodontal improvement (10), and increased concentrations of pro-inflammatory cytokines in sera were consistently observed in EBV-infected patients (21,40). In addition, as described in our previous report, butyric acid produced by periodontopathic bacteria may cause EBV reactivation in the periodontium of EBV-infected individuals (17). Furthermore, the present study showed that LMP1 in the human gingival epithelial cell line Ca9-22 markedly induced IL8 production attributable to NF-ĸB activation.
Considering the observations mentioned above and the involvement of infiltrated neutrophils in the aetiology of human chronic periodontitis, it is suggested that inducible IL8 production by EBV LMP1 in the human gingival epithelium contributes to human chronic periodontitis. Therefore, EBV, as well as periodontopathic bacteria, should be considered as therapeutic targets when treating human chronic periodontitis. Further studies are needed to determine whether EBV is as an independent aetiological agent of human chronic periodontitis or whether it acts in combination with periodontopathic bacteria. The development of new treatments and superior preventative methods should be supported by a better understanding of the role of EBV in the aetiology of human chronic periodontitis.
Conflicts of Interest
The Authors declare that there are no conflicts of interest in regard to this study.
Authors’ Contributions
N.W., K.N., and R.K. performed the experiments, analysed the data, contributed reagents/materials/analysis tools, prepared figures, and reviewed drafts of the article. A.K., O.T., and T.K. contributed reagents/materials/analysis tools, analyzed the data, and reviewed drafts of the paper. H.S., M.T., Y.O., and S.S. contributed to discussion, analyzed the data, and reviewed drafts of the article. K.I. conceived and designed the experiments, performed the experiments, analysed the data, authored or reviewed drafts of the article.
Acknowledgements
The Authors thank Dr Martin Rowe for the LMP1 plasmids. This work was supported by JSPS KAKENHI (No.16K11526), Sato Fund, Dental Research Center, Nihon University School of Dentistry, and a Nihon University Multidisciplinary Research Grant for 2017.
References
- 1.Hajishengallis G. Periodontitis: From microbial immune subversion to systemic inflammation. Nat Rev Immunol. 2015;15(1):30–44. doi: 10.1038/nri3785. PMID: 25534621. DOI: 10.1038/nri3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Darveau RP. Periodontitis: A polymicrobial disruption of host homeostasis. Nat Rev Microbiol. 2010;8(7):481–490. doi: 10.1038/nrmicro2337. PMID: 20514045. DOI: 10.1038/nrmicro2337. [DOI] [PubMed] [Google Scholar]
- 3.Holt SC, Ebersole JL. Porphyromonas gingivalis, Treponema denticola and Tannerella forsythia: The “red complex”, a prototype polybacterial pathogenic consortium in periodontitis. Periodontol 2000. 2005;38(1):72–122. doi: 10.1111/j.1600-0757.2005.00113.x. DOI: 10.1111/j.1600-0757.2005.00113.x. [DOI] [PubMed] [Google Scholar]
- 4.Abusleme L, Dupuy AK, Dutzan N, Silva N, Burleson JA, Strausbaugh LD, Gamonal J, Diaz PI. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J. 2013;7(5):1016–1025. doi: 10.1038/ismej.2012.174. PMID: 23303375. DOI: 10.1038/ismej.2012.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ledder RG, Gilbert P, Huws SA, Aarons L, Ashley MP, Hull PS, McBain AJ. Molecular analysis of the subgingival microbiota in health and disease. J Appl Environ Microbiol. 2007;73(2):516–523. doi: 10.1128/AEM.01419-06. PMID: 17085691. DOI: 10.1128/AEM.01419-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riep B, Edesi-Neuß L, Claessen F, Skarabis H, Ehmke B, Flemmig TF, Bernimoulin JP, Göbel UB, Moter A. Are putative periodontal pathogens reliable diagnostic markers. J Clin Microbiol. 2009;47(6):1705–1711. doi: 10.1128/JCM.01387-08. PMID: 19386852. DOI: 10.1128/JCM.01387-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Contreras A, Nowzari H, Slots J. Herpesviruses in periodontal pocket and gingival tissue specimens. Oral Microbiol Immunol. 2000;15(1):15–18. doi: 10.1034/j.1399-302x.2000.150103.x. PMID: 11155159. DOI: 10.1034/j.1399-302x.2000.150103.x. [DOI] [PubMed] [Google Scholar]
- 8.Lu H, Zhu C, Li F, Xu W, Tao D, Feng X. Putative periodontopathic bacteria and herpesviruses in pregnant women: a case–control study. Sci Rep. 2016;6:27796–27796. doi: 10.1038/srep27796. PMID: 27301874. DOI: 10.1038/srep2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slots J, Saygun I, Sabeti M, Kubar A. Epstein–Barr virus in oral diseases. J Periodontal Res. 2006;41(4):235–244. doi: 10.1111/j.1600-0765.2006.00865.x. PMID: 16827715. DOI: 10.1111/j.1600-0765.2006.00865.x. [DOI] [PubMed] [Google Scholar]
- 10.Saygun I, Kubar A, Özdemir A, Slots J. Periodontitis lesions are a source of salivary cytomegalovirus and Epstein–Barr virus. J Periodontal Res. 2005;40(2):187–191. doi: 10.1111/j.1600-0765.2005.00790.x. PMID: 15733155. DOI: 10.1111/j.1600-0765.2005.00790.x. [DOI] [PubMed] [Google Scholar]
- 11.Konstantinidis A, Sakellari D, Papa A, Antoniadis A. Real-time polymerase chain reaction quantification of Epstein–Barr virus in chronic periodontitis patients. J Periodontal Res. 2005;40(4):294–298. doi: 10.1111/j.1600-0765.2005.00796.x. PMID: 15966906. DOI: 10.1111/j.1600-0765.2005.00796.x. [DOI] [PubMed] [Google Scholar]
- 12.Schmidt CW, Misko IS. The ecology and pathology of Epstein–Barr virus. Immunol Cell Biol. 1995;73:489–504. doi: 10.1038/icb.1995.79. PMID: 8713470. DOI: 10.1038/icb.1995.79. [DOI] [PubMed] [Google Scholar]
- 13.Thorley-Lawson DA, Gross A. Persistence of the Epstein–Barr virus and the origins of associated lymphomas. N Engl J Med. 2004;350(13):1328–1337. doi: 10.1056/NEJMra032015. PMID: 15044644. DOI: 10.1056/NEJMra032015. [DOI] [PubMed] [Google Scholar]
- 14.Ikuta K, Satoh Y, Hoshikawa Y, Sairenji T. Detection of Epstein–Barr virus in salivas and throat washings in healthy adults and children. Microbes Infect. 2000;2(2):115–120. doi: 10.1016/s1286-4579(00)00277-x. PMID: 10742683. DOI: 10.1016/S1286-4579(00)00277-X. [DOI] [PubMed] [Google Scholar]
- 15.Kato A, Imai K, Ochiai K, Ogata Y. Higher prevalence of Epstein–Barr virus DNA in deeper periodontal pockets of chronic periodontitis in Japanese patients. PLoS One. 2013;8(8):e71990. doi: 10.1371/journal.pone.0071990. PMID: 23991022. DOI: 10.1371/journal.pone.0071990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kato A, Imai K, Ochiai K, Ogata Y. Prevalence and quantitative analysis of Epstein–Barr virus DNA and Porphyromonas gingivalis associated with Japanese chronic periodontitis patients. Clin Oral Investig. 2015;19(7):1605–1610. doi: 10.1007/s00784-014-1387-y. PMID: 29233156. DOI: 10.1186/s12903-017-0438-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Imai K, Inoue H, Tamura M, Cueno ME, Inoue H, Takeichi O, Kusama K, Saito I, Ochiai K. The periodontal pathogen Porphyromonas gingivalis induces the Epstein–Barr virus lytic switch transactivator ZEBRA by histone modification. Biochimie. 2012;94(3):839–846. doi: 10.1016/j.biochi.2011.12.001. PMID: 22178321. DOI: 10.1016/j.biochi.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 18.Vincent-Bugnas S, Vitale S, Mouline CC, Khaali W, Charbit Y, Mahler P, Prêcheur I, Hofman P, Maryanski JL, Doglio A. EBV infection is common in gingival epithelial cells of the periodontium and worsens during chronic periodontitis. PLoS One. 2013;8(2):e80336. doi: 10.1371/journal.pone.0080336. PMID: 24367478. DOI: 10.1371/ journal.pone.0080336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McFarland ED, Izumi KM, Mosialos G. Epstein–Barr virus transformation: involvement of latent membrane protein 1-mediated activation of NF-ĸB. Oncogene. 1999;18(49):6959–6964. doi: 10.1038/sj.onc.1203217. PMID: 10602470. DOI: 10.1038/sj.onc.1203217. [DOI] [PubMed] [Google Scholar]
- 20.Floettmann JE, Rowe M. Epstein–Barr virus latent membrane protein-1 (LMP1) C-terminus activation region 2 (CTAR2) maps to the far C-terminus and requires oligomerisation for NF-ĸB activation. Oncogene. 1997;15(15):1851–1858. doi: 10.1038/sj.onc.1201359. PMID: 9362452. DOI: 10.1038/sj.onc.1201359. [DOI] [PubMed] [Google Scholar]
- 21.Klein G. Epstein–Barr virus strategy in normal and neoplastic B-cells. Cell. 1994;77(6):791–793. doi: 10.1016/0092-8674(94)90125-2. PMID: 8004668. DOI: 10.1016/0092-8674(94)90125-2. [DOI] [PubMed] [Google Scholar]
- 22.Henderson S, Rowe M, Gregory C, Croom-Carter D, Wang F, Longnecker R, Kieff E, Rickinson A. Induction of BCL-2 expression by Epstein–Barr virus latent membrane protein 1 protects infected B-cells from programmed cell death. Cell. 1991;65(7):1107–1115. doi: 10.1016/0092-8674(91)90007-l. PMID: 1648447. DOI: 10.1016/0092-8674(91)90007-l. [DOI] [PubMed] [Google Scholar]
- 23.Laherty CD, Hu HM, Opipari AW, Wang F, Dixit VM. The Epstein–Barr virus LMP1 gene product induces A20 zinc finger protein expression by activating nuclear factor ĸ B. J Biol Chem. 1997;267(34):24157–24160. PMID: 1332946. [PubMed] [Google Scholar]
- 24.Dawson CW, Rickinson AB, Young LS. Epstein–Barr virus latent membrane protein inhibits human epithelial cell differentiation. Nature. 1990;344(6268):777–780. doi: 10.1038/344777a0. PMID: 2158628. DOI: 10.1038/344777a0. [DOI] [PubMed] [Google Scholar]
- 25.Huen DS, Henderson SA, Croom-Carter D, Rowe M. The Epstein–Barr virus latent membrane protein-1 (LMP1) mediates activation of NF-ĸB and cell surface phenotype via two effector regions in its carboxy-terminal cytoplasmic domain. Oncogene. 1995;10(3):549–560. PMID: 7845680. [PubMed] [Google Scholar]
- 26.Murono S, Inoue H, Tanabe T, Joab I, Yoshizaki T, Furukawa M, Pagano JS. Induction of cyclooxygenase-2 by Epstein–Barr virus latent membrane protein 1 is involved in vascular endothelial growth factor production in nasopharyngeal carcinoma cells. Proc Natl Acad Sci USA. 2001;98:6905–6910. doi: 10.1073/pnas.121016998. PMID: 11381123. DOI: 10.1073/pnas.121016998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoshizaki T, Sato H, Furukawa M, Pagano JS. The expression of matrix metalloproteinase 9 is enhanced by Epstein–Barr virus latent membrane protein 1. Proc Natl Acad Sci USA. 1998;95(7):3621–3626. doi: 10.1073/pnas.95.7.3621. PMID: 9520415. DOI: 10.1073/pnas.95.7.3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hatzivassiliou E, Mosialos G. Cellular signaling pathways engaged by the Epstein–Barr virus transforming protein LMP1. Front Biosci. 2002;7:d319–d329. doi: 10.2741/hatziva. PMID: 11779697. [DOI] [PubMed] [Google Scholar]
- 29.Eliopoulos AG, Dawson CW, Mosialos G, Floettmann JE, Rowe M, Armitage RJ, Dawson J, Zapata JM, Kerr DJ, Wakelam MJ, Reed JC. CD40-induced growth inhibition in epithelial cells is mimicked by Epstein–Barr virus-encoded LMP1: involvement of TRAF3 as a common mediator. Oncogene. 1996;13(10):2243–2254. PMID: 8950992. [PubMed] [Google Scholar]
- 30.Bickel M. The role of interleukin-8 in inflammation and mechanisms of regulation. J Periodontol. 1993;64:456–460. PMID: 8315568. [PubMed] [Google Scholar]
- 31.Takashiba S, Takigawa M, Takahashi K, Myokai F, Nishimura F, Chihara T, Kurihara H, Nomura Y, Murayama Y. Interleukin-8 is a major neutrophil chemotactic factor derived from cultured human gingival fibroblasts stimulated with interleukin-1 β or tumor necrosis factor α. Infect Immun. 1992;60(12):5253–5258. doi: 10.1128/iai.60.12.5253-5258.1992. PMID: 1452358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brandolini L, Sergi R, Caselli G, Boraschi D, Locati M, Sozzani S, Bertini R. Interleukin-1 β primes interleukin-8-stimulated chemotaxis and elastase release in human neutrophils via its type I receptor. Eur Cytokine Netw. 1997;8(2):173–178. PMID: 9262966. [PubMed] [Google Scholar]
- 33.Asamitsu K, Yamaguchi T, Nakata K, Hibi Y, Victoriano AF, Imai K, Onozaki K, Kitade Y, Okamoto T. Inhibition of human immunodeficiency virus type 1 replication by blocking IĸB kinase with noraristeromycin. J Biochem. 2008;144(5):581–589. doi: 10.1093/jb/mvn104. PMID: 9519889. DOI: 10.1089/aid.1998.14.293. [DOI] [PubMed] [Google Scholar]
- 34.Imai K, Kamio N, Cueno ME, Saito Y, Inoue H, Saito I, Ochiai K. Role of the histone H3 lysine 9 methyltransferase Suv39 h1 in maintaining Epstein–Barr virus latency in B95-8 cells. FEBS J. 2014;281(9):2148. doi: 10.1111/febs.12768. PMID: 24588869. DOI: 10.1111/febs.12768. [DOI] [PubMed] [Google Scholar]
- 35.Imai K, Togami H, Okamoto T. Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J Biol Chem. 2010;285(22):16538–16545. doi: 10.1074/jbc.M110.103531. PMID: 20335163. DOI: 10.1074/ jbc.M110.103531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hayden MS, Ghosh S. Signaling to NF-kB. Genes Develop. 2004;18(18):2195–2224. doi: 10.1101/gad.1228704. DOI: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 37.Mukaida N. Interleukin-8: An expanding universe beyond neutrophil chemotaxis and activation. Int J Hematol. 2000;72(4):391–398. PMID: 11197203. [PubMed] [Google Scholar]
- 38.O’Brien DP, Oltz EM, Van Ness BG. Coordinate transcription and V(D)J recombination of the ĸ immunoglobulin light-chain locus: NF-ĸB-dependent and -independent pathways of activation. Mol Cell Biol. 1997;17(7):3477–3487. doi: 10.1128/mcb.17.7.3477. DOI: 10.1128/MCB.17.7.3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaye KM, Devergne O, Harada JN, Izumi KM, Yalamanchili R, Kieff E, Mosialos G. Tumor necrosis factor receptor associated factor 2 is a mediator of NF-ĸ B activation by latent infection membrane protein 1, the Epstein–Barr virus transforming protein. Proc Natl Acad Sci USA. 1996;93(20):11085–11090. doi: 10.1073/pnas.93.20.11085. PMID: 8855313. DOI: 10.1073/pnas.93.20.11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Slots J. Herpesviral-bacterial interactions in periodontal diseases. Periodontol 2000. 2010;52(1):117–140. doi: 10.1111/j.1600-0757.2009.00308.x. PMID: 20017799. DOI: 10.1111/j.1600-0757.2009.00308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Payne JB, Reinhardt RA, Masada MP, DuBois LM, Allison AC. Gingival crevicular fluid IL-8: Correlation with local IL-1 β levels and patient estrogen status. J Periodontal Res. 1993;28(6):451–453. PMID: 8254463. [PubMed] [Google Scholar]
- 42.Fitzgerald JE, Kreutzer DL. Localization of interleukin-8 in human gingival tissues. Oral Microbiol Immunol. 1995;10(5):297–303. doi: 10.1111/j.1399-302x.1995.tb00158.x. PMID: 8596673. [DOI] [PubMed] [Google Scholar]
- 43.Özmeriç N, Bal B, Baloş K, Berker E, Bulut Ş. Levels of interleukin-1 β and interleukin-8 in gingival crevicular fluids in adult periodontitis. J Periodontol. 1995;66(10):852–859. doi: 10.1902/jop.1995.66.10.852. PMID: 8537867. DOI: 10.1902/jop.1995.66.10.852. [DOI] [PubMed] [Google Scholar]
- 44.Jin L, Söder B, Corbet EF. Interleukin-8 and granulocyte elastase in gingival crevicular fluid in relation to periodonto-pathogens in untreated adult periodontitis. J Periodontol. 2000;71(6):929–939. doi: 10.1902/jop.2000.71.6.929. PMID: 10914796. DOI: 10.1902/jop.2000. 71.6.929. [DOI] [PubMed] [Google Scholar]
- 45.Yoshizaki T, Horikawa T, Qing-chun R, Wakisaka N, Takeshita H, Sheen TS, Lee SY, Sato H, Furukawa M. Induction of interleukin-8 by Epstein–Barr virus latent membrane protein-1 and its correlation to angiogenesis in nasopharyngeal carcinoma. Clin Cancer Res. 2001;7:1946–1951. PMID: 11448908. [PubMed] [Google Scholar]
- 46.Xiong A, Clarke-Katzenberg RH, Valenzuela G, Izumi KM, Millan MT. Epstein–Barr virus latent membrane protein 1 activates nuclear factor-ĸ B in human endothelial cells and inhibits apoptosis. Transplantation. 2004;78(1):41–49. doi: 10.1097/01.tp.0000129805.02631.ef. PMID: 15257037. DOI: 10.1097/01.tp.0000129805.02631.ef. [DOI] [PubMed] [Google Scholar]
- 47.Hieke C, Kriebel K, Engelmann R, Müller-Hilke B, Lang H, Kreikemeyer B. Human dental stem cells suppress PMN activity after infection with the periodontopathogens Prevotella intermedia and Tannerella forsythia. Sci Rep. 2016;6:39096–39096. doi: 10.1038/srep39096. DOI: 10.1038/srep39096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Suzuki K, Sakiyama Y, Usui M, Obama T, Kato R, Itabe H, Yamamoto M. Oxidized low-density lipoprotein increases interleukin-8 production in human gingival epithelial cell line Ca9-22. J Periodontal Res. 2010;45(4):488–495. doi: 10.1111/j.1600-0765.2009.01263.x. DOI: 10.1111/j.1600-0765.2009.01263.x. [DOI] [PubMed] [Google Scholar]
- 49.Takeuchi H, Setoguchi T, Machigashira M, Kanbara K, Izumi Y. Hydrogen sulfide inhibits cell proliferation and induces cell-cycle arrest via an elevated p21 Cip1 level in Ca9-22 cells. J Periodontal Res. 2008;43(1):90–95. doi: 10.1111/j.1600-0765.2007.00999.x. PMID: 20412422. DOI: 10.1111/j.1600-0765.2009.01263.x. [DOI] [PubMed] [Google Scholar]