

Abstract
Extracellular vesicles (exosomes, EVs) are cell membrane particles (30-200 nm) secreted by virtually all cells. During intercellular communication in the body, secreted EVs play crucial roles by carrying functional biomolecules (e.g., microRNAs and enzymes) into other cells to affect cellular function, including disease progression. We previously reported that the macropinocytosis pathway contributes greatly to the efficient cellular uptake of EVs. The activation of growth factor receptors, such as epidermal growth factor receptor (EGFR), induces macropinocytosis. In this study, we demonstrated the effects of gefitinib, a tyrosine kinase inhibitor of EGFR, on the cellular uptake of EVs. In EGFR-mutant HCC827 non-small cell lung cancer (NSCLC) cells, which are sensitive to gefitinib, macropinocytosis was suppressed by gefitinib treatment. However, the cellular uptake of EVs was increased by gefitinib treatment, whereas that of liposomes was reduced. In accordance with the results of the cellular uptake studies, the anti-cancer activity of doxorubicin (DOX)-loaded EVs in HCC827 cells was significantly increased in the presence of gefitinib, whereas the activity of DOX-loaded liposomes was reduced. The digestion of EV proteins by trypsin did not affect uptake, suggesting that the cellular uptake of EVs might not be mediated by EV proteins. These results suggest that gefitinib can enhance cell-to-cell communication via EVs within the tumor microenvironment. In addition, EVs show potential as drug delivery vehicles in combination with gefitinib for the treatment of patients harboring EGFR-mutant NSCLC tumors.
Keywords: Gefitinib, extracellular vesicles, exosomes, EGFR, non-small cell lung cancer
Graphical Abstract
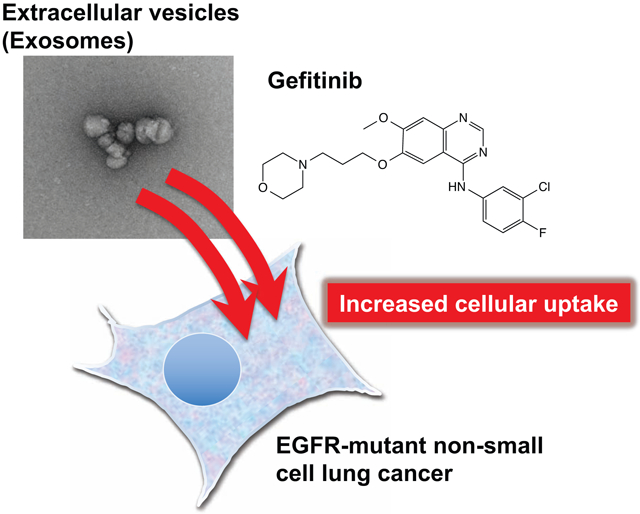
1. Introduction
Extracellular vesicles (exosomes, EVs) are cellular membrane-derived nanosized particles (30-200 nm) that are secreted by virtually all cells. In cell-to-cell communication, EVs, such as exosomes, play crucial roles by delivering functional biomolecules that are encapsulated in EVs (e.g., microRNAs and proteins) to other cells to effect cellular biological functions (Tan et al., 2013). Particularly in the research field of cancer, EV-mediated cellular communication has been actively investigated over the last decade, and EVs have been shown to influence cancer-related signaling pathways, including hypoxia-driven epithelial–mesenchymal transition (EMT), cancer stemness, angiogenesis, and metastasis (Azmi et al., 2013; Maia et al., 2018). Based on experimental findings, novel strategies to inhibit cancer metastasis via the blockade of cancer-derived EVs have been developed (Nishida-Aoki et al., 2017; Kosaka et al., 2013). However, detailed assessment and mechanism elucidation in terms of the influence of clinical cancer therapeutics on EV-based cell-to-cell communications is urgently needed.
Various mechanisms of cellular EV uptake have been proposed, including clathrin-mediated endocytosis, phagocytosis, and membrane fusion (Mulcahy et al., 2014). In addition, our research group recently reported that macropinocytosis contributes greatly to the efficient cellular uptake of EVs (Nakase et al., 2015). Macropinocytosis is a unique mode of endocytosis in which extracellular fluid is internalized in a clathrin- and caveolin-independent manner and relies on actin-dependent ruffle (lamellipodia) formation (> 1 μm in diameter) (Zwartkruis et al., 2013). Macropinocytosis can be induced by the expression of oncogenic K-Ras and the activation of growth factor receptors such as epidermal growth factor receptor (EGFR) (Swanson 2008). Macropinocytosis is mediated by appropriate signals when reorganizing the actin cytoskeleton that include the activation of Rho proteins, such as the small G-protein Rac1, and upstream effectors such as Ras (Zwartkruis et al., 2013; Maltese et al., 2015).
Gefitinib (Fig. 1a) is a tyrosine kinase inhibitor (TKI) of EGFR and has been approved for the treatment of non-small cell lung cancers (NSCLCs) with EGFR mutations (Berardi et al., 2013; Watanabe et al., 2015). Patients with a sensitizing exon 19 deletion or an exon 21 substitution mutation in EGFR are highly responsive to gefitinib (Wang et al., 2016). EGFR phosphorylation triggers the activation of the anti-apoptotic Ras signaling cascade, eventually leading to uncontrolled cell proliferation in EGFR-mutant cancer cells (Seshacharyulu et al., 2012; Oda et al., 2005). Gefitinib selectively binds to the kinase domain and inhibits the phosphorylation of EGFR, resulting in the suppression of downstream signals and the induction of apoptotic cell death (Wang et al., 2016; Seshacharyulu et al., 2012; Kobayashi, Ji et al., 2005; Kobayashi, Boggon et al., 2005; Campiglio et al., 2004).
Figure 1.

Effects of gefitinib treatment on cell death and micropinocytosis. (a) Chemical structure of gefitinib. (b, c) Viability of HCC827 cells treated with gefitinib (0 ~ 10 μM) for 24 h (b) and 72 h (c) at 37 °C. Data are presented as the mean (± SD) of four experiments. (d, e) Relative cellular uptake of FITC-dextran (70 kDa), a macropinocytosis marker, in HCC827 (d) and A549 (e) cells treated with gefitinib (0 ~ 100 nM) for 24 h at 37 °C. The uptake was determined using a flow cytometer. Data are presented as the mean (± SD) of three experiments. *p<0.05, ***p<0.001.
In this study, we assessed the effects of gefitinib treatment on EV-mediated intercellular communication. We hypothesized that blockade of the Ras signaling cascade would lead to the decreased cellular uptake of EVs via the suppression of macropinocytosis. As expected, gefitinib suppressed macropinocytosis in EGFR-mutant (exon 19 deletion) HCC827 cells that were sensitive to gefitinib but did not have any influence on EGFR wild-type A549 cells that were resistant to gefitinib. Unexpectedly, the cellular uptake of EVs increased following gefitinib treatment in HCC827 cells, whereas the cellular uptake of liposomes decreased following gefitinib treatment, in agreement with the results of the inhibition of micropinocytosis by gefitinib. Subsequently, we evaluated the influence of gefitinib pretreatment on the in vitro anti-cancer efficacy of doxorubicin (DOX)-loaded EVs and DOX-loaded liposomes. Moreover, we investigated the effect of EV membrane proteins on cellular uptake in the presence of gefitinib.
2. Materials and Methods
2.1. Materials
The following reagents were used in this study: gefitinib (Cell Signaling Technology, Inc., Danvers, MA, USA), human epidermal growth factor (EGF), penicillin-streptomycin, FITC-dextran (70 kDa) (Sigma-Aldrich Co., Inc., St. Louis, MO, USA), FITC-transferrin (Rockland Immunochemicals Inc., Limerick, PA, USA), RPMI-1640, minimum essential medium-α (α-MEM), fetal bovine serum (FBS) (Gibco, Life Technologies Corporation, Grand Island, NY, USA), exosome-free FBS (EXO-FBS, ATLAS Biological, Fort Collins, CO, USA), Dulbecco’s phosphate-buffered saline (PBS) and trypsin (0.5 g/L)/ethylenediaminetetraacetic acid (EDTA) (0.53 mmol/L) solution with phenol red (Nacalai Tesquen Inc., Kyoto, Japan), Hoechst 33342 (Invitrogen, Austin, TX, USA), dimethyl sulfoxide (DMSO), doxorubicin (Wako Pure Chemical Co., Inc., Osaka, Japan), the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific Inc., Rockford, IL, USA), the Premix WST-1(4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate) Cell Proliferation Assay System (Takara Bio Inc., Shiga, Japan), FITC-labeled liposomes (DOPC:CHOL:FITC-DHPE at a molar ratio of 54:45:1), doxorubicin-loaded liposomes (HSPC:CHOL:mPEG2000-DSPE at a molar ratio of 56.2:38.5:5.3) (FormuMax Scientific Inc., Sunnyvale, CA, USA), SDS-PAGE gel plates (Bio Craft Co., Tokyo, Japan), and silver stain reagent (Cosmo Bio Co., Tokyo, Japan).
2.2. Cell culture
HCC827 and A549 cell lines (American Type Culture Collection, Manassas, VA, USA), which are human NSCLC cell lines, were cultured in RPMI-1640 media containing 10% FBS and penicillin-streptomycin (100 units/mL and 100 μg/mL) in 100-mm cell culture dishes (Iwaki, Tokyo, Japan) and incubated at 37 °C in 5% CO2. Human cervical cancer-derived HeLa cells (Riken BRC Cell Bank, Ibaraki, Japan) were cultured in α-MEM containing 10% FBS in 100-mm dishes and incubated at 37 °C in 5% CO2.
2.3. Isolation of EVs
HeLa cells (2 × 106 cells) were seeded into 100-mm dishes in α-MEM (10 mL) containing 10% FBS and incubated for 1 day at 37 °C in 5% CO2. The cells were washed with serum-free α-MEM (five times, 5 mL) and incubated in α-MEM (10 mL) containing 10% exosome-free FBS for 2 days. The cell culture medium was collected, and the secreted EVs were isolated by the ultracentrifugation method. The collected cell culture medium was centrifuged (300 × g) for 10 min at 4 °C to remove the cell debris. The supernatant was centrifuged (2,000 × g) for 10 min at 4 °C and centrifuged again (10,000 × g) for 30 min at 4 °C to remove the microvesicles. The supernatant was then centrifuged twice (150,000 × g) for 70 min at 4 °C using an ultracentrifuge (Himac CP65β, Hitachi Koki, Tokyo, Japan), and the pellet was collected in PBS. The concentrations of the isolated EVs are described in terms of their protein concentrations, which were determined by BCA protein assays. The particle size distribution of the EVs was measured by a NanoSight LM10 with NTA2.3 Analytical Software (Malvern Panalytical, Malvern, UK). The encapsulation of FITC-labeled dextran into EVs was conducted as previously described (Nakase et al., 2015).
2.4. Western blotting analysis
To detect EV (exosomal) marker proteins, the isolated EVs were added to SDS sample buffer and boiled. The exosome samples were separated via 10% SDS-PAGE, transferred onto polyvinylidene fluoride (PVDF) membranes (GE Healthcare, Pittsburgh, PA, USA), and treated with anti-CD9 (EPR2949, Abcam, Cambridge, UK) or anti-CD63 antibody (TS63, Abcam, Cambridge, UK). Donkey anti-rabbit HRP-linked IgG (GE Healthcare), for CD9, or anti-mouse IgG HRP NA931V secondary antibody (GE Healthcare), for anti-CD63, was used. The immunoreactivity was detected using an Enhanced Chemiluminescence (ECL) Plus western blotting detection system (GE Healthcare) with an Amersham Imager 600 (GE Healthcare).
2.5. Electron microscopy
The exosomes were resuspended in PBS (30 μg/mL) and subsequently applied onto a carbon-coated grid (400 mesh) and washed with distilled water. Uranyl acetate was applied to the grid, which was incubated for 10 s at room temperature. Next, the reagent was removed with filter paper, and the grid was dried prior to imaging with a transmission electron microscope (TEM) (JEM1200EX, JEOL, Tokyo, Japan).
2.6. Encapsulation of DOX into EVs
To load DOX into EVs, isolated EVs (25 μg) were mixed with DOX (33 μg) in PBS (100 μL). The electroporation was performed as follows: two initial poring pulses (100 V, 5 msec) followed by five transfer pulses (20 V, 50 msec) were performed in a 1-cm electroporation cuvette at room temperature using a super electroporator NEPA21 TypeII (NEPA Genes, Tokyo, Japan). The removal of unencapsulated DOX was accomplished by washing and filtration using Amicon Ultra centrifugal filters (100 K device, Merck Millipore; triple washing with 500 μL PBS, 18,000 × g, 4 °C, 10 min). The concentration of DOX in the DOX-loaded EVs was confirmed using a spectrofluorometer (RF-5300PC, Shimadzu, Kyoto, Japan).
2.7. Cell viability
For the cytotoxicity studies using gefitinib, HCC827 or A549 cells (7.2 × 104 cells/well (600 μL/well) each) were incubated in 24-well microplates for 24 h at 37 °C. After the removal of the medium, the cells were treated with experimental sample (200 μL/well) containing EGF (100 nM) and penicillin-streptomycin (100 units/mL and 100 μg/mL) for 24 h or 72 h at 37 °C. After sample treatment, the cells were washed with PBS (triple washing, 200 μL) and treated with trypsin (0.1 g/L)-EDTA (0.11 mmol/L) (200 μL/well) at 37 °C for 10 min. Following the addition of RPMI-1640 medium (200 μL), the cell viability was determined using a OneCell Counter (Bio Medical Science Inc. Tokyo, Japan).
For the cytotoxicity studies using DOX-loaded EVs or liposomes, HCC827 or A549 cells (1.2 × 104 cells/well, 100 μL/well) were incubated in 96-well microplates for 24 h at 37 °C. After the removal of the medium, the cells were then treated with experimental sample (50 μL/well) containing EGF (100 nM) and penicillin-streptomycin (100 units/mL and 100 μg/mL) for 24 h at 37 °C. After the medium was replaced with fresh medium containing 10% FBS and penicillin-streptomycin (100 units/mL and 100 μg/mL), the cells were incubated for 48 h at 37 °C. The cells were then washed with PBS (three washes, 50 μL/wash) and treated with trypsin (0.1 g/L)-EDTA (0.11 mmol/L) at 37 °C for 10 min. Following the addition of RPMI-1640 medium (50 μL), the living cell number was counted using a OneCell Counter. The cell viability was calculated based on the ratio of the number of living cells in each well relative to that of the control. The experiments were repeated four times, and the average 50% growth inhibitory concentrations (IC50) were calculated.
2.8. Confocal microscopy
HCC827 or A549 cells (2.8 × 104 cells/well each (200 μL)) were plated on a μSlide 8 Well (Ibidi) and incubated for 24 h at 37 °C. After complete adhesion, the cells were treated with FITC-dextran-loaded EVs (10 μg/mL protein concentration) or FITC-labeled liposomes (5 μM lipid concentration, 200 μL/well) containing EGF (100 nM) and penicillin-streptomycin (100 units/mL and 100 μg/mL) for 24 h at 37 °C in the presence or absence of gefitinib (10 nM). The cells were stained with Hoechst 33342 (5 μg/mL) for 15 min at 37 °C prior to cell washing. The cells were then washed with fresh cell culture medium (three times, 200 μL) and analyzed using a FV1200 confocal laser scanning microscope (Olympus, Tokyo, Japan).
2.9. Flow cytometry
HCC827 or A549 cells (7.2 × 104 cells, 1 mL) were plated into 24-well microplates (Iwaki) and incubated for 24 h at 37 °C. After the removal of the medium, the cells were treated with experimental sample (200 μL/well) containing EGF (100 nM) and penicillin-streptomycin (100 units/mL and 100 μg/mL) for 24 h at 37 °C. After sample treatment, the cells were washed with PBS (three washes, 200 μL/wash) and treated with trypsin (0.1 g/L)-EDTA (0.11 mmol/L, 200 μL/well) at 37 °C for 10 min. After the addition of PBS (200 μL), the cells were centrifuged at 400 × g for 5 min at 4 °C. After the removal of the supernatant, the cells were suspended in PBS (400 μL) and subjected to fluorescence analysis with a Guava EasyCyte flow cytometer (Merck Millipore, Billerica, MA, USA) using 488-nm laser excitation and a 525-nm emission filter. Living cells (10,000 cells/sample) were identified based on forward and side scatter analyses. The experiments were repeated three times, and the average cellular fluorescence intensity was calculated.
2.10. Protein digestion of the EV membrane by trypsin
FITC-dextran-loaded EVs (200 μg/mL) were incubated with trypsin (0.25 g/L)-EDTA (0.27 mmol/L) at 37 °C for 30 min. The digestion of proteins was confirmed by SDS-PAGE. The particle size distribution of the EVs was measured by a NanoSight LM10. The residual trypsin and EDTA were removed by washing and filtration using Amicon Ultra centrifugal filters (100 K device; washed twice with 500 μL PBS, 18,000 × g, 4 °C, 10 min).
2.11. Zeta potential
The zeta potential of each sample was detected in PBS according to the manufacturer’s instructions using an ELSZ-DN2 zeta potential analyzer (Otsuka Electronics, Osaka, Japan).
2.12. Statistical analyses
All statistical analyses were performed using GraphPad Prism software (v5, GraphPad, San Diego, CA, USA). For comparisons of two groups, an unpaired Student’s t-test was used for the verification of equal variances via an F-test. Welch’s correction was performed when the variances between groups were assumed to be unequal. For multiple comparison analyses, a one-way analysis of variance (ANOVA) followed by Dunnett’s or Tukey’s multiple comparison test was used. Differences were considered significant when the calculated p-value was < 0.05.
3. Results
3.1. Suppression of macropinocytosis by gefitinib treatment
As described above, it has been reported that macropinocytosis plays a crucial role in the efficient cellular uptake of EVs. Macropinocytosis is induced by activation of EGFR and, to the best of our knowledge, the effects of EGFR inhibitors such as gefitinib on macropinocytosis induction have not been studied. We evaluated the cellular uptake of FITC-dextran (70 kDa), a known marker of macropinocytosis (Falcone et al., 2006; Nakase et al., 2009). HCC827 is an NSCLC cell line with an activating EGFR mutation (del E746-A750) that renders the cells sensitive to gefitinib. A549 harbors wild-type EGFR and is resistant to gefitinib. In fact, cytotoxicity studies have revealed that the viability of HCC827 cells is affected by 100 nM gefitinib following 24 h and 72 h of exposure (Fig. 1b and c). On the other hand, the viability of A549 cells was not affected by gefitinib concentrations as high as 10 μM treatment following 72 h of exposure (Supplementary Fig. S1). Next, HCC827 and A549 cells were treated with FITC-dextran in the presence of gefitinib (0, 10, and 100 nM) for 24 h, and then the cellular uptake of FITC-dextran was evaluated by flow cytometry analysis (Fig. 1d and e). In the case of the HCC827 cells, the cellular uptake of FITC-dextran was reduced by gefltinib treatment (Fig. 1d), which indicates that gefitinib suppressed macropinocytosis. On the other hand, the cellular uptake of FITC-dextran was not affected by gefitinib treatment in A549 cells (Fig. 1e). These results suggest that gefitinib inhibits macropinocytosis in EGFR-mutant HCC827 cells.
3.2. Effects of gefitinib treatment on the cellular uptake of EVs and liposomes
Next, we compared the effects of gefitinib treatment on the cellular uptake of EVs to that of liposomes, which are artificial lipid nanoparticles. EVs were isolated from HeLa cells by differential ultracentrifugation. A NanoSight particle tracking system was used to evaluate the size distribution of the isolated EVs, which was determined to be 173 ± 9 nm (Supplementary Fig. S2a and b). Transmission electron microscopy (TEM) observations of isolated exosomes showed the vesicular structures (Supplementary Fig. S2c). The EV marker proteins CD63 and CD81 were detected in isolated EVs by western blot analysis (Supplementary Fig. S2d). FITC-dextran was encapsulated into the isolated EVs by electroporation, followed by ultrafiltration to remove the unencapsulated FITC-dextran. The zeta potential of the FITC-dextran-loaded EVs was −13.3 mV. In this study, we also used FITC-labeled liposomes, and their zeta potential was −10.2 mV.
HCC827 or A549 cells were treated with FITC-dextran-loaded EVs (2 μg/mL) or FITC-labeled liposomes (5 μM) in the presence or absence of gefitinib (10 nM). After 24 h of incubation, the cellular uptake of FITC was evaluated by flow cytometric analysis (Fig. 2a and b, Supplementary Fig. S3 and S4). Surprisingly, the cellular uptake of EVs was increased following gefitinib treatment in HCC827 cells, despite the suppression of macropinocytosis (Fig. 2a). On the other hand, the cellular uptake of liposomes was reduced following gefitinib treatment (Fig. 2b). In A549 cells, there was no significant change in uptake regardless of the presence or absence of gefitinib treatment for both EVs and liposomes (Fig. 2a and b). Following flow cytometric analysis, confocal microscopic observation was conducted to visualize the cellular uptake of FITC-dextran-loaded EVs or FITC-labeled liposomes. When HCC827 cells were treated with FITC-dextran-loaded EVs, increased endosome-like dot signals from FITC in cells exposed to gefitinib could be observed (Fig. 2c). However, when HCC827 cells were heated with FITC-labeled liposomes, gefitinib treatment reduced the number of endosome-like dot signals from FITC (Fig. 2d). These results are in agreement with the results of the flow cytometry analysis. In A549 cells, FITC uptake was minimal under all experimental conditions (Supplementary Fig. S5).
Figure 2.

Effects of gefitinib treatment on the cellular uptake of EVs and liposomes. (a, b) Relative cellular uptake of FITC-dextran-loaded EVs (a) and FITC-labeled liposomes (b) in HCC827 or A549 cells in the presence or absence of gefitinib (10 nM) for 24 h at 37 °C using flow cytometry. Data are presented as the mean (± SD) of three experiments. (c, d) Confocal microscopic observations of HCC827 cells treated with FITC-dextran-loaded EVs (c) or FITC-labeled liposomes (d) in the same experimental conditions as those used in (a) and (b). ****p<0.0001.
3.3. Effects of gefitinib treatment on the in vitro cytotoxicity of anti-cancer reagent-encapsulated EVs or liposomes
Gefitinib demonstrated distinct and opposing effects on the cellular uptake of EVs and liposomes in HCC827 cells. To evaluate the biological activities of EVs and liposomes following gefitinib treatment, we analyzed the in vitro cytotoxicity of EVs and liposomes with the encapsulated chemotherapeutic doxorubicin (DOX). DOX was encapsulated into EVs by electroporation, followed by ultrafiltration to remove unencapsulated DOX. The efficiency of DOX encapsulation into EVs was calculated to be 0.5% based on the quantification of DOX using a spectrofluorometer. The recovery rate of free DOX (without electroporation) not encapsulated into EVs through the purification process was less than 0.1%. These results indicate that DOX was loaded in EVs efficiently.
HCC827 and A549 cells were treated with DOX-loaded EVs and liposomes in the presence or absence of gefitinib (10 nM). After 24 h of exposure and 48 h of incubation, the cell viability was evaluated by living cell counting analysis (Fig. 3). The cell viability was decreased in a DOX concentration-dependent manner in each cell line. In HCC827 cells treated with DOX-loaded EVs, the cell viability was significantly reduced in the presence of gefitinib, whereas treatment with gefitinib alone had no effect on cell viability (Fig. 1b and c). In contrast, the cell viability of HCC827 cells treated with DOX-loaded liposomes was increased following gefitinib treatment. The IC50 values of DOX-loaded EVs in the presence of gefitinib were approximately five times lower than the IC50 values in the absence of gefitinib (0.08 μM vs. 0.42 μM) (Supplementary Table S1). On the other hand, the IC50 value of DOX-loaded liposomes in the presence of gefitinib was more than three times greater than the IC50 value in the absence of gefitinib (0.95 μM vs. 0.28 μM). The cell viability of A549 cells was not affected by gefitinib treatment (Supplementary Table S1). These results were consistent with the results of the cellular uptake analysis by flow cytometry.
Figure 3.

Gefitinib treatment affects the in vitro cytotoxicity of DOX-loaded EVs and liposomes. (a, b) Viability of HCC827 (a) or A549 cells (b) treated with DOX-loaded EVs in the presence or absence of gefitinib (10 nM) for 24 h and 48 h post incubation at 37 °C. (c, d) Viability of HCC827 (c) or A549 cells (d) treated with DOX-loaded liposomes in the same experimental conditions as those used in (a) and (b). The data are the averages (± SD) of four experiments. ***p<0.001, ****p<0.0001.
3.4. Effect of EV protein digestion on cellular uptake
To elucidate the mechanism underlying the increased cellular uptake of EVs following gefitinib treatment, we evaluated the effect of the trypsin digestion of EV proteins on cellular uptake. FITC-dextran-loaded EVs were first incubated with trypsin to digest the proteins present in the EVs. SDS-PAGE analysis demonstrated an absence of higher molecular weight proteins following trypsin treatment, which indicates EV surface protein degradation (Fig. 4a). NanoSight measurements revealed that the average size and distribution of EVs was unaltered following trypsin digestion (Fig. 4b and c). In addition, trypsin digestion did not affect the zeta potential of the EVs (before trypsin digestion: −13.3 mV, after trypsin digestion: −13.4 mV). HCC827 cells were treated with trypsin-treated and trypsin-untreated FITC-dextran-loaded EVs (2 μg/mL) in the presence or absence of gefitinib. After 24 h of incubation, the cellular uptake of FITC-dextran-loaded EVs was increased after gefitinib treatment, regardless of protein digestion (Fig. 4d). The degree of increased cellular uptake was greater in cells treated with digested EVs relative to that in untreated cells.
Figure 4.

Effects of trypsin digestion on the cellular uptake of EVs following gefitinib treatment. (a) SDS-PAGE analysis of FITC-dextran-loaded EVs before and after treatment with trypsin for 30 min at 37 °C. (b) Particle size distribution of FITC-dextran-loaded EVs before and after treatment with trypsin. (c) Table summarizing the average sizes of EVs. Data are presented as the mean (± SD) of three measurements. (d) Relative cellular uptake of FITC-dextran-loaded EVs with or without trypsin treatment in HCC827 cells in the presence or absence of gefitinib (25 and 50 nM) for 24 h at 37 °C using a flow cytometer. The data are the averages (± SD) of three experiments. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
4. Discussion and conclusion
In the current study, we determined whether gefitinib inhibits macropinocytosis in a gefitinib-sensitive, EGFR-mutant HCC827 NSCLC cell line. The EGFR wild-type A549 cells were shown to be insensitive to gefitinib-mediated macropinocytosis inhibition via the evaluation of the cellular uptake of FITC-dextran, a known macropinocytosis marker (Fig. 1d and e). This was thought to be due to the gefitinib-mediated inhibition of EGFR phosphorylation and the blockage of subsequent Ras signaling activation. The Ras signaling pathway is known to be involved in macropinocytosis induction (Zwartkruis et al., 2013; Swanson, 2008; Maltese et al., 2015). This blocking effect was observed even at concentrations of gefitinib (10 nM) that did not affect cell viability (Fig. 1b and c).
Next, we evaluated the effects of gefitinib treatment on the cellular uptake of EVs, since it has been reported that EVs communicate within the tumor microenvironment and can induce cancer malignancy via the acceleration of tumor growth, angiogenesis, and metastasis (Azmi et al., 2013). In the current study, we compared EVs to liposomes, which were considered a control lipid nanoparticle. Liposomes are also suitable as a drug delivery vehicle control. EVs have attracted attention as drug delivery carriers due to a number of advantageous properties, including reduced immunogenicity, low cytotoxicity, and their intrinsic targeting potential (Mulcahy et al., 2014; Ha et al., 2016; Hoshino et al., 2015). Unexpectedly, the cellular uptake of EVs was increased following gefitinib treatment in HCC827 cells (Fig. 2a and c), whereas that of liposomes was reduced (Fig. 2b and d). In addition, we compared the in vitro cytotoxicity of DOX-loaded EVs and liposomes. Here, we evaluated cell viability by counting the living cell number, since we reported in a previous paper that the cell proliferation reagent WST-1, a tetrazolium salt that detects activation of succinate-tetrazolium reductase, did not reflect the living cell number when HCC827 cells were treated with gefitinib at a high cell density because of the enhancement of mitochondrial biological activity, including the possible increased activation of succinate-tetrazolium reductase, by gefitinib treatment (Takenaka et al., 2017). In agreement with the results of the uptake study, the cell viability of HCC827 cells treated with DOX-loaded EVs decreased significantly in the presence of gefitinib (Fig. 3a). In the case of DOX-loaded liposomes, cell viability was increased in the presence of gefitinib, although the difference was not significant (Fig. 4c). Interestingly, the IC50 value of DOX-loaded EVs was more than ten times lower than that of DOX-loaded liposomes in HCC827 cells in the presence of gefitinib (0.08 μM vs. 0.95 μM) (Supplementary Table S1). These results indicate the following: first, cellular uptake pathways specific to EVs might be activated by gefitinib treatment despite the inhibition of macropinocytosis, which suggests the possibility that cell-to-cell communication via EVs might be enhanced within the tumor microenvironment. Second, gefitinib influences the cellular uptake of EVs and liposomes as well as the intracellular activity of bioactive agents loaded in the carriers. Finally, EVs may represent a superior delivery vehicle for doxorubicin compared to liposomes. For example, an EV-based drug delivery system might overcome the clinical inefficiencies of combinational gefitinib treatment with standard chemotherapies such as paclitaxel, carboplatin, and cisplatin (Giaccone et al., 2004; Herbst et al., 2004).
Next, we evaluated the effect of the protein digestion of EVs on cellular uptake. It has been reported that the cellular uptake mechanism of EVs may depend on the proteins and glycoproteins found on the surfaces of both vesicles and target cells (Mulcahy et al., 2014; Christianson et al., 2013; Morelli et al., 2004). We found that the cellular uptake of trypsin-digested EVs was enhanced by gefitinib treatment (Fig. 4d). This result indicates that EV-membrane proteins do not contribute to the increased cellular uptake of EVs in the presence of gefitinib. Toda et al. reported that the cellular uptake of glioblastoma-derived EVs into parent cells was not affected by the protein digestion of EVs, and lipid molecules were critical for the effective incorporation of EVs (Toda et al., 2015). Similarly, EV membrane components other than proteins, such as lipids, might have influenced our results. Moreover, Fig. 4d demonstrates that the cellular uptake of digested EVs is further enhanced by gefitinib treatment. A potential explanation for this phenomenon is that the trypsin-mediated digestion of EVs leads to the exposure of amino acid sequences that are favorable for cellular uptake or the degradation of proteins that contribute to the suppression of cellular uptake. However, further research is needed to understand the precise requirements for the gefitinib-mediated enhancement of EV cellular uptake.
In conclusion, our results demonstrate that gefitinib increases the cellular uptake of EVs in EGFR-mutant NSCLCs despite the suppression of macropinocytosis. Gefitinib may enhance EV-mediated intercellular communication within the tumor microenvironment, thereby influencing cancer malignancy. From a drug delivery perspective, we have provided evidence to support the use of EVs as a drug delivery vehicle in combination with gefitinib.
Supplementary Material
Acknowledgements
The study was supported in part by JSPS KAKENHI (JP16H02612, JP19H05553 for I.N.) and JST CREST (J19ZZ00066 for I.N.). This study was also supported by the Leading University as a Base for Human Resource Development in Nanoscience and Nanotechnology, Osaka Prefecture University. S.S.K. was supported by the National Institutes of Health (R21CA178301 and R01CA169259), the American Cancer Society (RSG-13-047), and the Harvard Stem Cell Institute Blood Program (DP-0110-12-00). The electron microscopy analyses were technically supported by Filgen (Aichi, Japan). The manuscript preparation was assisted by Kayo Hirano (Osaka Prefecture University).
Abbreviations:
- EVs
extracellular vesicles
- EGFR
epidermal growth factor receptor
- NSCLC
non-small cell lung cancer
- DOX
doxorubicin
- EMT
hypoxia-driven epithelial–mesenchymal transition
- TKI
tyrosine kinase inhibitor
- α-MEM
minimum essential medium α
- FBS
fetal bovine serum
- PBS
phosphate buffered saline
- EDTA
ethylenediaminetetraacetic acid
- DMSO
dimethyl sulfoxide
- PVDF
polyvinylidene fluoride
- HRP
horseradish peroxidase
- TEM
transmission electron microscope
- IC50
50% growth inhibitory concentrations
- ANOVA
one-way analysis of variance
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Appendix A. Supplementary data
Supplementary data to this article can be found online at ~.
Competing financial interests: The authors have no competing interests as defined by Elsevier, or other interests that might be perceived to influence the results and/or discussion reported in this paper.
References
- Azmi AS, Bao B and Sarkar FH, 2013. Exosomes in cancer development, metastasis and drug resistance: A comprehensive review. Cancer Metastasis Rev. 32, 623–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berardi R, Santoni M, Morgese F, Ballatore Z, Savini A, Onofri A, Mazzanti P, Pistelli M, Pierantoni C, De Lisa M, Caramanti M, Pagliaretta S, Pellei C and Cascinu S, 2013. Novel small molecule EGFR inhibitors as candidate drugs in non-small cell lung cancer. Onco. Targets Ther 6, 563–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campiglio M, Locatelli A, Olgiati C, Normanno N, Somenzi G, Viganò L, Fumagalli M, Ménard S and Gianni L, 2004. Inhibition of proliferation and induction of apoptosis in breast cancer cells by the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor ZD1839 (‘Iressa’) is independent of EGFR expression level. J. Cell Physiol 198, 259–268. [DOI] [PubMed] [Google Scholar]
- Christianson HC, Svensson KJ, van Kuppevelt TH, Li JP and Belting M, 2013. Cancer cell exosomes depend on cell-surface heparan sulfate proteoglycans for their internalization and functional activity. Proc. Natl. Acad. Sci. USA 110, 17380–17385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcone S, Cocucci E, Podini P, Kirchhausen T, Clementi E and Meldolesi J, 2006. Macropinocytosis: regulated coordination of endocytic and exocytic membrane traffic events. J. Cell Sci 119, 4758–4769. [DOI] [PubMed] [Google Scholar]
- Giaccone G, Herbst RS, Manegold C, Scagliotti G, Rosell R, Miller V, Natale RB, Schiller JH, Von Pawel J, Pluzanska A, Gatzemeier U, Grous J, Ochs JS, Averbuch SD, Wolf MK, Rennie P, Fandi A and Johnson DH, 2004. Gefitinib in combination with gemcitabine and cisplatin in advanced non-small-cell lung cancer: a phase III trial--INTACT 1. J. Clin. Oncol 22, 777–784. [DOI] [PubMed] [Google Scholar]
- Ha D, Yang N and Nadithe V, 2016. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: current perspectives and future challenges. Acta Pharm. Sin. B 4, 287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst RS, Giaccone G, Schiller JH, Natale RB, Miller V, Manegold C, Scagliotti G, Rosell R, Oliff I, Reeves JA, Wolf MK, Krebs AD, Averbuch SD, Ochs JS, Grous J, Fandi A and Johnson DH, 2004. Gefitinib in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: a phase III trial--INTACT 2. J. Clin. Oncol 22, 785–794. [DOI] [PubMed] [Google Scholar]
- Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, Molina H, Kohsaka S, Di Giannatale A, Ceder S, Singh S, Williams C, Soplop N, Uryu K, Pharmer L, King T, Bojmar L, Davies AE, Ararso Y, Zhang T, Zhang H, Hernandez J, Weiss JM, Dumont-Cole VD, Kramer K, Wexler LH, Narendran A, Schwartz GK, Healey JH, Sandstrom P, Labori KJ, Kure EH, Grandgenett PM, Hollingsworth MA, de Sousa M, Kaur S, Jain M, Mallya K, Batra SK, Jarnagin WR, Brady MS, Fodstad O, Muller V, Pantel K, Minn AJ, Bissell MJ, Garcia BA, Kang Y, Rajasekhar VK, Ghajar CM, Matei I, Peinado H, Bromberg J and Lyden D, 2015. Tumour exosome integrins determine organotropic metastasis. Nature 527, 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG and Halmos B, 2005. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med 352, 786–792. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Ji H, Yuza Y, Meyerson M, Wong KK, Tenen DG and Halmos B, 2005. An alternative inhibitor overcomes resistance caused by a mutation of the epidermal growth factor receptor. Cancer Res. 65, 7096–7101. [DOI] [PubMed] [Google Scholar]
- Kosaka N, Iguchi H, Hagiwara K, Yoshioka Y, Takeshita F, and Ochiya T, 2013. Neutral sphingomyelinase 2 (nSMase2)-dependent exosomal transfer of angiogenic microRNAs regulate cancer cell metastasis. J. Biol. Chem 288, 10849–10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maia J, Caja S, Strano Moraes MC, Couto N and Costa-Silva B, 2018. Exosome-Based Cell-Cell Communication in the Tumor Microenvironment. Front. Cell Dev. Biol 6, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltese WA and Overmeyer JH, 2015. Non-apoptotic cell death associated with perturbations of macropinocytosis. Front. Physiol 6, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli AE, Larregina AT, Shufesky WJ, Sullivan ML, Stolz DB, Papworth GD, Zahorchak AF, Logar AJ, Wang Z, Watkins SC, Falo LD Jr. and Thomson AW, 2004. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood 104, 3257–3266. [DOI] [PubMed] [Google Scholar]
- Mulcahy LA, Pink RC and Carter DR, 2014. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 3, 24641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakase I and Futaki S, 2015. Combined treatment with a pH-sensitive fusogenic peptide and cationic lipids achieves enhanced cytosolic delivery of exosomes. Sci. Rep 5, 10112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakase I, Hirose H, Tanaka G, Tadokoro A, Kobayashi S, Takeuchi T and Futaki S, 2009. Cell-surface accumulation of flock house virus-derived peptide leads to efficient internalization via macropinocytosis. Mol. Ther 17, 1868–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakase I, Kobayashi NB, Takatani-Nakase T and Yoshida T, 2015. Active macropinocytosis induction by stimulation of epidermal growth factor receptor and oncogenic Ras expression potentiates cellular uptake efficacy of exosomes. Sci. Rep 5, 10300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida-Aoki N, Tominaga N, Takeshita F, Sonoda H, Yoshioka Y and Ochiya T, 2017. Disruption of circulating extracellular vesicles as a novel therapeutic strategy against cancer metastasis. Mol. Ther 25, 181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda K, Matsuoka Y, Funahashi A and Kitano H, 2005. A comprehensive pathway map of epidermal growth factor receptor signaling. Mol. Syst. Biol 1, 0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshacharyulu P, Ponnusamy MP, Haridas D, Jain M, Ganti AK and Batra SK, 2012. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets 16, 15–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson JA, 2008. Shaping cups into phagosomes and macropinosomes. Nat. Rev. Mol. Cell Biol 9, 639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenaka T, Katayama M, Sugiyama A, Hagiwara M, Fujii I, Takatani-Nakase T, Kobayashi SS and Nakase I, 2017. Gefitinib enhances mitochondrial biological functions in nsclcs with egfr mutations at a high cell density. Anticancer Res. 37, 4779–4788. [DOI] [PubMed] [Google Scholar]
- Tan A, Rajadas J and Seifalian AM, 2013. Exosomes as nano-theranostic delivery platforms for gene therapy. Adv. Drug Deliv. Rev 65, 357–367. [DOI] [PubMed] [Google Scholar]
- Toda Y, Takata K, Nakagawa Y, Kawakami H, Fujioka S, Kobayashi K, Hattori Y, Kitamura Y, Akaji K and Ashihara E, 2015. Effective internalization of U251-MG-secreted exosomes into cancer cells and characterization of their lipid components. Biochem. Biophys. Res. Commun 456, 768–773. [DOI] [PubMed] [Google Scholar]
- Wang J, Wang B Chu H and Yao Y, 2016. Intrinsic resistance to EGFR tyrosine kinase inhibitors in advanced non-small-cell lung cancer with activating EGFR mutations. Onco. Targets Ther 9, 3711–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Inoue A, Nukiwa T and Kobayashi K, 2015. Comparison of Gefitinib versus chemotherapy in patients with non-small cell lung cancer with exon 19 deletion. Anticancer Res. 35, 6957–6961. [PubMed] [Google Scholar]
- Zwartkruis FJ and Burgering BM, 2013. Ras and macropinocytosis: trick and treat. Cell Res. 23, 982–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.