

Abstract
The γ-secretase complex is composed of four membrane protein subunits, including presenilin as the catalytic component with aspartyl protease activity. The enzyme cleaves within the transmembrane domain of >70 different type I integral membrane proteins and has been dubbed “the proteasome of the membrane”. The most studied substrates include the Notch family of receptors, involved in cell differentiation, and the amyloid precursor protein (APP), involved in the pathogenesis of Alzheimer’s disease. A central mechanistic question is how γ-secretase recognizes helical transmembrane substrates and carries out processive proteolysis. Recent findings addressing substrate recognition and processing will be discussed, including the role of protease subunit nicastrin as a gatekeeper, the effects of Alzheimer-causing mutations in presenilin on processive proteolysis of APP, and evidence that three pockets in the active site (S1’, S2’, and S3’) determine carboxypeptidase cleavage of substrate in intervals of three residues.
Keywords: amyloid, Notch, protease, genetics, biochemistry, Alzheimer’s disease
Introduction
A variety of membrane-embedded proteases carry out hydrolysis within the confines of the lipid bilayer. These intramembrane-cleaving proteases (I-CliPs) (Wolfe, 2009b) include the S2P family of metalloproteases, the rhomboid family of serine proteases, and the presenilin and presenilin-like family of aspartyl proteases, together providing remarkable examples of convergent evolution in which nature solved the problem of proteolysis in the lipid bilayer using means similar to those employed for proteolysis in the aqueous environment. The most widely studied I-CliP is the γ-secretase complex, containing presenilin as the catalytic component (Wolfe, 2009a). Two conserved transmembrane aspartates in presenilin catalyze proteolysis within the transmembrane domain (TMD) of substrate. The three other components of the complex (nicastrin, Aph-1, and Pen-2) assemble together with presenilin, whereupon presenilin undergoes autoproteolysis into N-terminal fragment (NTF) and C-terminal fragment (CTF) subunits. Each presenilin subunit contributes one essential aspartate to what is now the mature, active γ-secretase complex.
The γ-secretase complex has over 70 known substrates, type I integral membrane protein that undergo prior ectodomain shedding by membrane-tethered proteases (Hemming et al., 2008). No clear sequence specificity has yet been noted. The many substrates and broad specificity has led to the dubbing of γ-secretase as “the proteasome of the membrane” (Kopan and Ilagan, 2004). In most cases, it appears that the role of this aspartyl protease complex is to clear out membrane-bound remnants left behind after ectodomain shedding. Although this would seem to be an essential housekeeping function, knockout or inhibition of γ-secretase is not toxic to cultured cells, suggesting other means of clearing membrane protein remnants.
Knockout or inhibition of γ-secretase does, however, have toxic consequences in vivo, due to the key role that the protease complex plays in signaling from the Notch family of receptors (Selkoe and Kopan, 2003). Proteolysis within the Notch TMD by γ-secretase (Fig. 1) is an essential step in the signaling mechanism (De Strooper et al., 1999), releasing the Notch intracellular domain for translocation to the nucleus and activation of transcription factors that express genes critical to cell-fate determinations. Knockout of γ-secretase components is embryonic lethal in metazoans, interfering with cell differentiation and proper development of the organism (Shen et al., 1997; Wong et al., 1997). Notch signaling in cell differentiation is also critical in adult organisms, as γ-secretase inhibitors developed for Alzheimer’s disease resulted in effects consistent with Notch deficiencies: immunosuppression, gastrointestinal toxicity, and skin lesions (Coric et al., 2015; Doody et al., 2013).
Figure 1. Intramembrane proteolysis of Notch and APP by γ-secretase.
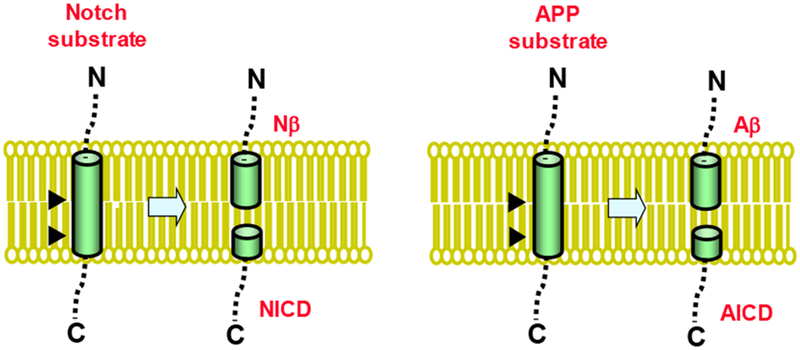
The γ-secretase complex cuts the single TMD of Notch substrate to release the Notch intracellular domain (NICD), which translocates to the nucleus and regulates gene expression. A short Notch-β (Nβ) peptide is also released. The protease cuts the single TMD of APP substrate to produce the amyloid-β peptide (Aβ) and APP intracellular domain (AICD). The identification of C-termini of Nβ and Aβ peptides and N-termini of the respective intracellular domains reveals that these substrates are cleaved at least twice by γ-secretase.
γ-Secretase was first defined as a proteolytic activity: cleavage within the TMD of the amyloid precursor protein (APP) to produce the amyloid β-peptide (Aβ) that notoriously deposits in the brain in Alzheimer’s disease (Fig. 1) (Selkoe, 1994). Dominant missense mutations in the APP gene were found to cause early-onset, familial Alzheimer’s disease (FAD)(Chartier-Harlin et al., 1991; Goateetal., 1991; Murrell et al., 1991; Tanzi and Bertram, 2005). This hereditary form of the disease displays the same pathology, presentation, and progression seen in the much more common late-onset, sporadic form. The two dozen or so FAD mutations in APP are all found in and around the small Aβ region (4 kDa) of the large APP (~80 kDa) and alter the production or properties of Aβ. Thus, the discovery of these mutations bolstered the amyloid hypothesis of Alzheimer pathogenesis, which posits that the 42-residue form of Aβ (Aβ42) is the initiator of a cascade of events results in neurodegeneration (Selkoe and Hardy, 2016).
Presenilin-1 (PS1) and presenilin-2 (PS2) were subsequently discovered as sites of dominant FAD missense mutations (Levy-Lahad et al., 1995; Sherrington et al., 1995; Tanzi and Bertram, 2005). The function and biological roles of these multi-pass membrane proteins were completely unclear, and their sequences were unrelated to any proteins known at the time. The FAD mutations, however, were soon found to alter APP processing, increasing the proportion of the aggregation-prone Aβ42 (Citron et al., 1997). Knockout of PS1 in mice resulted in embryonic lethality with a Notch-deficient phenotype (Shen et al., 1997; Wong et al., 1997). However, embryonic fibroblasts could be cultured and used to show dramatic reduction in Aβ production from APP (De Strooper et al., 1998). Meanwhile, transition-state analogue inhibitors of γ-secretase based on the cleavage site in APP suggested that γ-secretase is an aspartyl protease (Wolfe et al., 1999a). These clues led to the discovery of the two conserved TMD aspartates as essential for γ-secretase activity and the recognition that presenilin is an unusual protease with a membrane-embedded active site (Wolfe et al., 1999b).
Presenilin on its own, however, did not have proteolytic activity, leading to the search for other components of γ-secretase and the discovery of nicastrin, Aph-1 and Pen-2 (Edbauer et al., 2003; Francis et al., 2002; Goutte et al., 2002; Kimberly et al., 2003; Takasugi et al., 2003; Yu et al., 2000). Aph-1 is thought to be a scaffold for the assembly of the complex, and Pen-2 provides the trigger for presenilin autoproteolysis. Nicastrin, in contrast, appears to play a role in substrate recognition, as described in the next section.
Role of nicastrin
Several years after the discovery of nicastrin as a component of the γ-secretase complex, Yu and colleagues reported the apparent homology of the nicastrin ectodomain with aminopeptidases (Shah et al., 2005). The absence of key residues needed for catalysis, however, indicated that nicastrin lacked protease activity. Nevertheless, nicastrin could be immunoprecipitated with Notch γ-secretase substrate, leading to the suggestion that this subunit is involved in recognition of substrate N-termini. That is, nicastrin had the binding properties of an aminopeptidase but without proteolytic activity. Recognition of a substrate N-terminus by nicastrin was thought to guide the substrate and its TMD into the active site on presenilin. The first reported high-resolution structure of the γ-secretase complex, determined by cryo-electron microscopy (cryo-EM), appeared to be consistent with this model: the TMDs of the complex were arranged in a horseshoe shape, with the active site on the concave side, while the aminopeptidase-like domain of nicastrin hovered over the this concave side (Lu et al., 2014).
This substrate receptor model for nicastrin function, however, was inconsistent with several recent observations (Bolduc et al., 2016a). First, mutations of Notch substrate N-terminus had no effect on the enzyme kinetics, affecting neither binding affinity or the rate of catalysis. Second, acetylation of Notch substrate, synthesized through native chemical ligation, also did not alter the enzyme kinetics, demonstrating that a free N-terminus is not required for substrate recognition and processing. Third, peptides based on the N-terminus of Notch substrate did not inhibit γ-secretase substrate cleavage; the nicastrin-as-substrate-receptor model would predict competition between these peptides and substrates for binding to nicastrin.
What did matter with respect to the substrate N-terminus was the length of the luminal/extracellular juxtamembrane domain. Near removal of this juxtamembrane domain provided the best substrate; lengthening of this domain resulted in progressively lower substrate binding and processing. Reduction of the disulfide bonds in the nicastrin ectodomain resulted in increased cleavage of the longer substrates. Taken together, these findings suggested that nicastrin serves as a gatekeeper for γ-secretase substrates, sterically blocking longer substrates from gaining access to the active site on presenilin (Bolduc et al., 2016a). This new model for the role of nicastrin is consistent with a revised structure of the γ-secretase complex (Bai et al., 2015b). In the original reported structure, the TMDs had been misassigned; with the correct assignment, the active site was now found on the convex side of the horseshoe-shaped arrangement of TMDs, with the ectodomain of nicastrin hovering 20 Å above the membrane and over the entry to the active site on presenilin (Fig. 2).
Figure 2. Nicastrin serves as a gatekeeper for substrate entry into the γ-secretase complex.
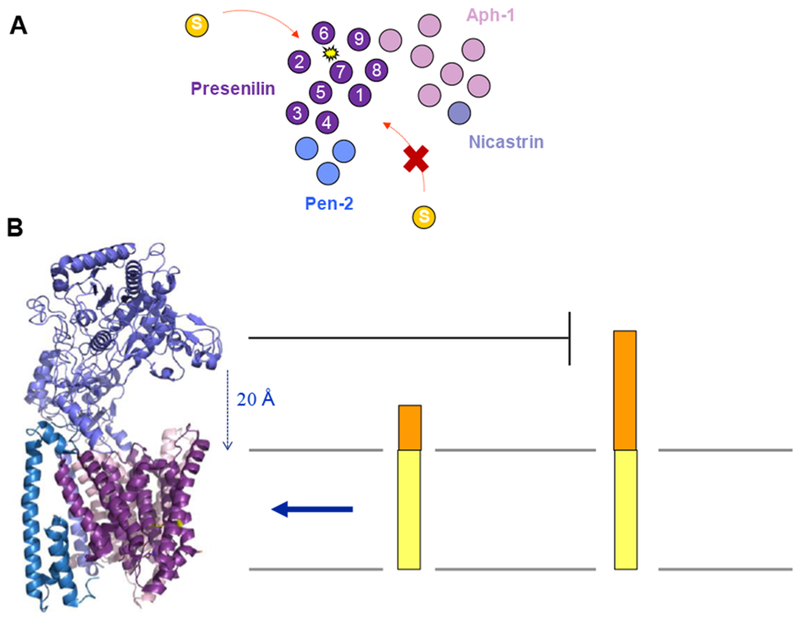
A. Revised cryo-EM structure of γ-secretase confirmed a horseshoe-shaped arrangement of TMDs but corrected their assignment. Approach of substrate into the active site cannot be from the concave side of the horseshoe, but rather from the convex side. B. The nicastrin ectodomain (lavender) projects out over the location of the active site in presenilin (purple). The 20 Å distance between this domain of nicastrin and the membrane sterically blocks approach of substrates with long luminal/extracellular domains. Structure of γ-secretase rendered from PDB ID: 5A63.
Effects of Alzheimer presenilin mutations
Well over 100 dominant missense mutations in the presenilins—the large majority in PS1—have now been identified as associated with FAD (Tanzi, 2012). For many years, the effects of presenilin mutations were described almost exclusively on Aβ42 and Aβ40, with Aβ40 being the major secreted form of Aβ and Aβ42 being the predominant protein component of amyloid plaques in the Alzheimer brain. However, there was a disconnect between the C-terminus of these Aβ peptides and the N-terminus of the APP intracellular domain (AICD) that is concomitantly produced by β-secretase, with some 7-10 APP TMD residues unaccounted for (Weidemann et al., 2002). The laboratory of Yasua lhara led the way in unraveling this conundrum with the discovery of longer, membrane-associated Aβ peptides, up to 49 residues in length. Ultimately, the Ihara team discovered that γ-secretase initially cuts the APP TMD to give a 48- or 49-residue Aβ peptide and then trims the C-terminus of these long Aβ peptides mostly in intervals of three amino acids (one step trims four amino acids)(Qi-Takahara et al., 2005). They ultimately demonstrated this phenomenon by detecting the tripeptides (and single tetrapeptide) by mass spectrometry (Takami et al., 2009). It is now clear that secreted Aβ peptides are primarily produced along two pathways: Aβ49→46→43→40 and Aβ48→45→42→38 (Fig. 3).
Figure 3. Processive proteolysis of APP by γ-secretase.
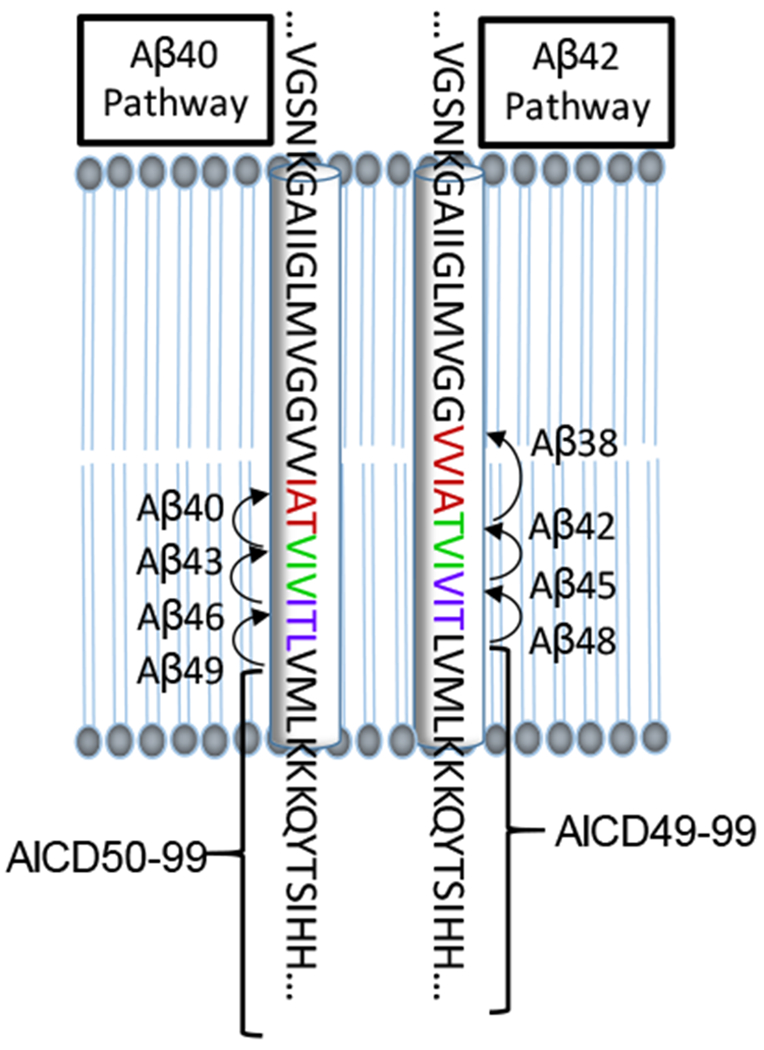
The 99-residue APP substrate is cleaved multiple times by the γ-secretase complex along two pathways. The Aβ40 pathway begins with cleavage to produce Aβ49, which is successively trimmed to Aβ46, Aβ43 and Aβ40. This pathway also produces AICD50-99. The Aβ42 pathway begins with cleavage to produce Aβ48, which is successively trimmed to Aβ45, Aβ42 and Aβ38. This pathway also produces AICD49-99.
A study of five different PS1 FAD mutations, found in different regions of the protein and with different ages of disease onset, revealed that with four of these mutations, the initial APP TMD cleavage event by γ-secretase was reduced (Quintero-Monzon et al., 2011). However, one of these mutants showed initial cleavage unchanged from wild-type γ-secretase complex, providing a clear exception to the “loss-of-function” hypothesis for presenilin FAD pathogenesis (Shen and Kelleher, 2007). All five FAD-mutant protease complexes, in contrast, skewed the range of Aβ peptides produced toward longer Aβ peptides ≥45 residues (Quintero-Monzon et al., 2011). Moreover, these mutant complexes were dramatically deficient in their ability to trim synthetic Aβ49 to Aβ40 and synthetic Aβ48 to Aβ42 (Fernandez et al., 2014). Thus, all five FAD-mutant γ-secretase complexes possessed greatly reduced carboxypeptidase activity. The reduction of this specific function, not overall proteolytic activity, is apparently what correlates with disease-causing mutations. A recent study from Szaruga and colleagues revealed that presenilin FAD mutations increased the affinity of γ-secretase for longer Aβ peptides, providing a mechanistic explanation for the reduced trimming rate.
Active site pockets dictate trimming
The mechanism of this carboxypeptidase activity, trimming in intervals of three amino acids, remained unclear until recently (Bolduc et al., 2016b). The working hypothesis was that the active site on presenilin contains three substrate-binding pockets—S1’, S2’ and S3’—that measure the three residues fated for release—P1’, P2’ and P3’. To test this idea, the APP TMD was systematically mutated with phenylalanine. The side chain of Phe had been previously found to be untolerated when placed in the P2’ position of transition-state analogue inhibitors of γ-secretase (Esler et al., 2004). Phe was found to be unacceptable to the enzyme in every single P2’ position along the two Aβ peptide pathways (Bolduc et al., 2016b). Thus, Phe mutation at position 51 reduced Aβ production along the Aβ49→46→43→40 pathway, while mutation at position 50 reduced production along the Aβ48→45→42→389 pathway. Phe mutation at position 48 similarly reduced Aβ40 production by preventing trimming of Aβ49 to Aβ46, and Phe mutation at position 47 reduced Aβ42 production by blocking trimming of Aβ48 to Aβ45. Likewise, Phe mutation at position 45 reduced Aβ40 production by preventing trimming of Aβ46 to Aβ43, and Phe mutation at position 44 reduced Aβ42 by blocking its production from Aβ45. Interestingly, double Phe mutation at positions 50 and 51 dramatically slowed initial cleavage; however, this double mutant APP substrate still bound to the γ-secretase complex just as well as the wild-type substrate did.
These findings were consistent with previous evidence for the existence of an initial substrate docking site, distinct from the active site (Kornilova et al., 2005). Upon binding of the substrate TMD to this docking site, substrate can then move laterally, in whole or in part, into the active site for proteolysis. Double Phe mutation at positions 50 and 51 apparently leads to substrate trapped in the docking site and unable to move into the active site. Prior evidence using transition-state analogue inhibitors as probes strongly supported three S’ pockets (Esler et al., 2004). Together these findings offer a model for initial substrate cleavage and trimming by γ-secretase (Fig. 4): Substrate binds to the docking site, followed by movement into the active site to fill the three S’ pockets and set up the transition state for proteolysis. Initially formed long Aβ peptides then move in to fill these three S’ pockets to set up tripeptide trimming. This occurs iteratively until release of the secreted Aβ peptides.
Figure 4. Model for substrate processing by γ-secretase.
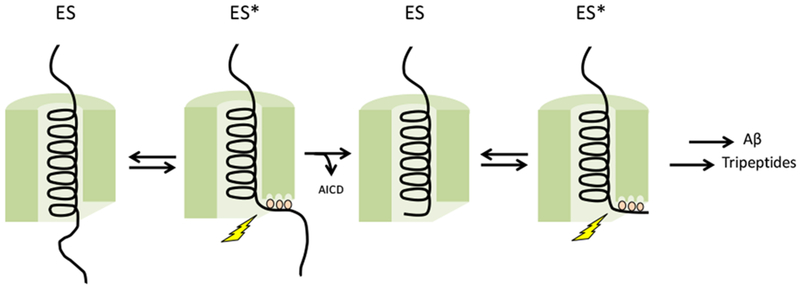
APP substrate initially docks on an exosite on presenilin. The active site contains three pockets—S1’, S2’, and S3’—that dictate initial endoproteolytic cleavage and carboxypeptidase trimming in intervals of three amino acids. Entry of substrate to fill these active-site pockets sets up the transition state for proteolysis.
Substrate TMD mimics as structural probes
In addition to transition-state analogue inhibitors directed to the protease active site, substrate-mimicking helical peptides were shown to be potent inhibitors that bind to an initial substrate docking site on presenilin that is distinct from the active site (Das et al., 2003; Kornilova et al., 2005). Affinity labeling reagents based on transition-state analogues (TSAs) and helical peptide inhibitors (HPIs) bound directly to presenilin NTF and CTF subunits; however TSAs could not block HPI affinity labeling probes, and HPIs could not compete with TSA affinity labeling probes. Thus, both the active site and docking site are apparently located at the interface between presenilin NTF and CTF subunits. Interestingly, extension of HPIs from 10 to 13 residues resulted in mutual competition of HPI and TSA for blocking affinity labeling of presenilin. Thus, although the active sites and docking sites on presenilin are distinct, they are proximal (apparently within the length of 3 amino acids).
We sought to develop full TMD substrate mimetics that combined HPI and TSA, connecting them through a variable linker. Specifically, we connected the C-terminus of a 10-residue HPI to the N-terminus of a 5-residue TSA containing residues P2 through P3’, with ω-aminoalkanoic acid linkers ranging from 0 to 10 atoms as spacers (Fig. 5). This design is consistent with γ-secretase carrying out the initial cleavage event three residues from the cytosolic end of TMD substrate. We envisioned that the HPI portion of the TMD substrate mimetics would bind to the initial substrate docking site and the TSA moiety would bind to the active site. In this way, the protease complex might be trapped in active form at its transition state bound to a full TMD mimetic. Several detailed structures of the γ-secretase complex solved by cryo-electron microscopy (cryo-EM) had recently been reported (Bai et al., 2015a; Bai et al., 2015b; Sun et al., 2015), the catalytic aspartates on presenilin were not quite close enough and coordinated for activation of a water molecule. Moreover, the structures did not provide insight into the nature of substrate recognition. The newly designed TMD mimetic inhibitors might therefore be important tools for elucidating the structural mechanism of γ-secretase.
Figure 5. Substrate TMD mimetic inhibitors of γ-secretase.
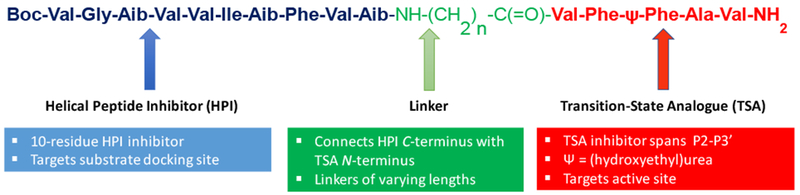
New design inhibitors mimic the full TMD of substrate and contain three parts: (1) a helical peptide inhibitor contain the helix-inducing residue α-aminoisobutyric acid (Aib), (2) a variable linker region composed of ω-aminoalkanoic acid, and (3) a pentapeptide transition-state analogue inhibitor containing a hydroxyethylurea moiety and three P’ residues to fill the three S’ pockets on γ-secretase.
We ultimately identified an HPI-TSA conjugate as a stoichiometric inhibitor of γ-secretase, inhibiting 1 nM of purified enzyme complex with an IC50 of 0.5 nM (S. Bhatterai, S. Devkota, K. Meneely, M. Xing, M. Wolfe, manuscript in preparation). Moreover, we found that this compound could compete with both TSA- and HPI-based affinity labeling reagents, consistent with the ability of the TMD mimetic to bind to both active site and docking site. Also consistent were inhibitor cross-competition kinetic experiments, which showed that the HPI-TSA conjugate competed with a TSA as well as an HPI inhibitor. In these experiments, the TSA and HPI inhibitors did not compete with each other.
Several key structural features were critical to the high potency of γ-secretase inhibition by the HPI-TSA conjugate. First, the helical conformation of the HPI is apparently required, as incorporation of two D-amino acids into the HPI portion resulted in eight-fold reduced potency. Second, the transition-state mimicking moiety is required, as its replacement with a peptide amide bond rendered the compound 22-fold less potent. Third, the linker length was critical, with the stoichiometric HPI-TSA inhibitor containing a 10-atom spacer, ω-aminononanoic acid. Interesting, this spacer is approximately the length of three amino acids, thought to be the distance between active site and docking site from previously reported affinity labeling experiments (Kornilova et al., 2005). Fourth, the identity of both amino- and carboxy-termini of the TMD mimetic affects potency: and N-terminal Boc group is preferred over acetyl, while a C-terminal amide is preferred over a methyl ester. These findings taken together show that the entire length of the HPI-TSA conjugate is involved in tight binding to the γ-secretase complex.
New structures of bound substrates
Two new cryo-EM structures of the γ-secretase complex have just been reported at the time of this writing, one with a bound Notch substrate and one with a bound APP substrate (Yang et al., 2019; Zhou et al., 2019). Both studies relied on two key modifications to capture substrate with protease: (1) the mutation of one of the two catalytic aspartates on presenilin to alanine, to prevent substrate cleavage, and (2) cysteine mutagenesis and disulfide crosslinking between substrate and presenilin. Both reported structures show the substrate surrounded by presenilin, with the mechanism of lateral entry of substrate unclear (Fig. 6). Both substrates were bound in very similar conformations, with the first two-thirds of the TMD in a helical conformation and the last third unwinding into a beta-strand and engaging the active site. Key parts of presenilin undergo conformational changes to interact with the substrate. This includes TMD 2, invisible in the apo-enzyme, becoming less mobile to the point of being observable. Also, a portion of TMD 6 becomes observable and interacts with the region of substrate TMD in the extended conformation. Relevant to the mechanism of FAD, many sites of presenilin mutations associated with the disease were found to interact with substrate, despite these sites being located in various regions in the linear sequence.
Figure 6. Structure of an APP substrate bound to the γ-secretase complex.
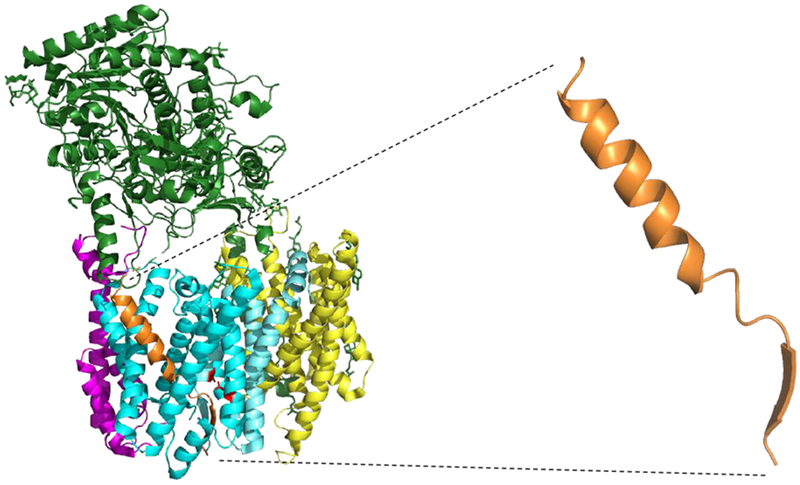
Newly reported cryo-EM structure of the γ-secretase complex bound to an APP substrate (Rendered from PDB ID: 6IYC). One of the active site aspartates is mutated to alanine, and both substrate and presenilin were mutated with cysteine to allow disulfide crosslinking. Blue: Presenilin; Yellow: Aph-1; Magenta: Pen-2; Green: Nicastrin; Orange: APP substrate. APP substrate alone is also shown to illustrate that the conformation of bound substrate resembles that of designed TMD substrates.
These two new structures offer a clear snapshot of substrates bound to the protease complex. However, as one of the catalytic aspartates is mutated, it remains unclear how the enzyme positions the scissile amide bond substrate for hydrolysis. Moreover, the covalent attachment of substrate to presenilin through disulfide linkage presents the possibility of artifacts, interactions that would not otherwise have been seen. Furthermore, as mentioned above, the new structures show substrate inside presenilin, without providing particular insight into the nature of lateral gating and substrate entry. Intriguingly, the conformation of bound substrate (Fig. 6) is similar to the newly designed TMD mimetic inhibitors: NMR studies reveal that the HPI portion is indeed helical like the first two-thirds of bound substrate (S. Bhatterai, S. Devkota, K. Meneely, M. Xing, M. Wolfe, manuscript in preparation). Thus, the new inhibitors are preorganized for interaction with γ-secretase. Moreover, the new TMD mimetic inhibitors, with both HPI and TSA moieties, should trap the enzyme in its active state and poised for catalysis and may reveal the HPI region bound to the external docking site, illuminating the lateral gating mechanism. Cryo-EM structure determination of the γ-secretase complex with these new chemical tools is in progress.
Perspective
Significant advances have been made toward understanding how the γ-secretase complex interacts with substrate and carries out intramembrane proteolysis. Evidence from substrate-based inhibitor probes and substrate mutagenesis supports a model in which the substrate TMD first binds to a docking exosite on presenilin. Subsequent lateral gating allows the substrate TMD to move in whole or in part to the inside of presenilin where the active site resides. The enzyme carries out successive proteolysis on its substrates, trimming initially formed intermediates containing most of the substrate TMD in intervals of three amino acids. Three active site pockets on the enzyme dictate this carboxypeptidase activity. New structures of the γ-secretase complex reveal key details of substrate binding. New TMD mimetic inhibitors have been developed as chemical tools for structural biology, with the potential to trap the enzyme in its transition state and reveal mechanisms for substrate gating.
Highlights.
γ-Secretase is a membrane protease complex that cleaves transmembrane substrates.
The enzyme processively proteolyzes substrates in intervals of 3 amino acids.
Subunit nicastrin serves as a gatekeeper for substrates based on length.
Alzheimer-causing mutations in the protease have reduced trimming function.
Three active-site pockets on the enzyme dictate trimming by 3 amino acids.
Acknowledgment
This work was supported by grant GM122894 from the National Institutes of Health to M.S.W.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bai XC, Rajendra E, Yang G, Shi Y, Scheres SH, 2015a. Sampling the conformational space of the catalytic subunit of human γ-secretase. Elife 4, pii: e11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai XC, Yan C, Yang G, Lu P, Ma D, Sun L, Zhou R, Scheres SH, Shi Y, 2015b. An atomic structure of human γ-secretase. Nature 525, 212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolduc DM, Montagna DR, Gu Y, Selkoe DJ, Wolfe MS, 2016a. Nicastrin functions to sterically hinder γ-secretase-substrate interactions driven by substrate transmembrane domain. Proc Natl Acad Sci U S A 113, E509–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolduc DM, Montagna DR, Seghers MC, Wolfe MS, Selkoe DJ, 2016b. The amyloid-β forming tripeptide cleavage mechanism of γ-secretase. Elife 5, pii: e17578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Crawford F, Houlden H, Warren A, Hughes D, Fidani L, Goate A, Rossor M, Roques P, Hardy J, et al. , 1991. Early-onset Alzheimer’s disease caused by mutations at codon 717 of the β-amyloid precursor protein gene. Nature 353, 844–846. [DOI] [PubMed] [Google Scholar]
- Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, Kholodenko D, Motter R, Sherrington R, Perry B, Yao H, Strome R, Lieberburg I, Rommens J, Kim S, Schenk D, Fraser P, St George Hyslop P, Selkoe DJ, 1997. Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid β-protein in both transfected cells and transgenic mice. Nat Med 3, 67–72. [DOI] [PubMed] [Google Scholar]
- Coric V, Salloway S, van Dyck CH, Dubois B, Andreasen N, Brody M, Curtis C, Soininen H, Thein S, Shiovitz T, Pilcher G, Ferris S, Colby S, Kerselaers W, Dockens R, Soares H, Kaplita S, Luo F, Pachai C, Bracoud L, Mintun M, Grill JD, Marek K, Seibyl J, Cedarbaum JM, Albright C, Feldman HH, Berman RM, 2015. Targeting Prodromal Alzheimer Disease With Avagacestat: A Randomized Clinical Trial. JAMA Neurol 72, 1324–1333. [DOI] [PubMed] [Google Scholar]
- Das C, Berezovska O, Diehl TS, Genet C, Buldyrev I, Tsai JY, Hyman BT, Wolfe MS, 2003. Designed helical peptides inhibit an intramembrane protease. J Am Chem Soc 125, 11794–11795. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R, 1999. A presenilin-1-dependent γ-secretase-like protease mediates release of Notch intracellular domain. Nature 398, 518–522. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F, 1998. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391, 387–390. [DOI] [PubMed] [Google Scholar]
- Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, He F, Sun X, Thomas RG, Aisen PS, Siemers E, Sethuraman G, Mohs R, 2013. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med 369, 341–350. [DOI] [PubMed] [Google Scholar]
- Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C, 2003. Reconstitution of γ-secretase activity. Nat Cell Biol 5, 486–488. [DOI] [PubMed] [Google Scholar]
- Esler WP, Das C, Wolfe MS, 2004. Probing pockets S2-S4’ of the γ-secretase active site with (hydroxyethyl)urea peptidomimetics. Bioorg Med Chem Lett 14, 1935–1938. [DOI] [PubMed] [Google Scholar]
- Fernandez MA, Klutkowski JA, Freret T, Wolfe MS, 2014. Alzheimer presenilin-1 mutations dramatically reduce trimming of long amyloid β-peptides (Aβ) by γ-secretase to increase 42-to-40-residue Aβ. J Biol Chem 289, 31043–31052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J, Nicoll M, Maxwell M, Hai B, Ellis MC, Parks AL, Xu W, Li J, Gurney M, Myers RL, Himes CS, Hiebsch R, Ruble C, Nye JS, Curtis D, 2002. aph-1 and pen-2 are required for Notch pathway signaling, γ-secretase cleavage of βAPP, and presenilin protein accumulation. Dev Cell 3, 85–97. [DOI] [PubMed] [Google Scholar]
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, et al. , 1991. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349, 704–706. [DOI] [PubMed] [Google Scholar]
- Goutte C, Tsunozaki M, Hale VA, Priess JR, 2002. APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc Natl Acad Sci U S A 99, 775–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemming ML, Elias JE, Gygi SP, Selkoe DJ, 2008. Proteomic profiling of γ-secretase substrates and mapping of substrate requirements. PLoS Biol 6, e257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimberly WT, LaVoie MJ, Ostaszewski BL, Ye W, Wolfe MS, Selkoe DJ, 2003. γ-Secretase is a membrane protein complex comprised of presenilin, nicastrin, aph-1, and pen-2. Proc Natl Acad Sci U S A 100, 6382–6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopan R, Ilagan MX, 2004. γ-Secretase: proteasome of the membrane? Nat Rev Mol Cell Biol 5, 499–504. [DOI] [PubMed] [Google Scholar]
- Kornilova AY, Bihel F, Das C, Wolfe MS, 2005. The initial substrate-binding site of γ-secretase is located on presenilin near the active site. Proc Natl Acad Sci U S A 102, 3230–3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, Crowley AC, Fu YH, Guenette SY, Galas D, Nemens E, Wijsman EM, Bird TD, Schellenberg GD, Tanzi RE, 1995. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269, 973–977. [DOI] [PubMed] [Google Scholar]
- Lu P, Bai XC, Ma D, Xie T, Yan C, Sun L, Yang G, Zhao Y, Zhou R, Scheres SH, Shi Y, 2014. Three-dimensional structure of human γ-secretase. Nature 512, 166–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell J, Farlow M, Ghetti B, Benson MD, 1991. A mutation in the amyloid precursor protein associated with hereditary Alzheimer’s disease. Science 254, 97–99. [DOI] [PubMed] [Google Scholar]
- Qi-Takahara Y, Morishima-Kawashima M, Tanimura Y, Dolios G, Hirotani N, Horikoshi Y, Kametani F, Maeda M, Saido TC, Wang R, Ihara Y, 2005. Longer forms of amyloid beta protein: implications for the mechanism of intramembrane cleavage by γ-secretase. J Neurosci 25, 436–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintero-Monzon O, Martin MM, Fernandez MA, Cappello CA, Krzysiak AJ, Osenkowski P, Wolfe MS, 2011. Dissociation between the processivity and total activity of γ-secretase: implications for the mechanism of Alzheimer’s disease-causing presenilin mutations. Biochemistry 50, 9023–9035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D, Kopan R, 2003. Notch and Presenilin: regulated intramembrane proteolysis links development and degeneration. Annu Rev Neurosci 26, 565–597. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, 1994. Cell biology of the amyloid β-protein precursor and the mechanism of Alzheimer’s disease. Annu Rev Cell Biol 10, 373–403. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Hardy J, 2016. The amyloid hypothesis of Alzheimer’s disease at 25 years.EMBO Mol Med 8, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S, Lee SF, Tabuchi K, Hao YH, Yu C, LaPlant Q, Ball H, Dann CE 3rd, Sudhof T, Yu G, 2005. Nicastrin functions as a γ-secretase-substrate receptor. Cell 122, 435–447. [DOI] [PubMed] [Google Scholar]
- Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S, 1997. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell 89, 629–639. [DOI] [PubMed] [Google Scholar]
- Shen J, Kelleher RJ 3rd, 2007. The presenilin hypothesis of Alzheimer’s disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci U S A 104, 403–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin c., Li G, Holman K, Tsuda T, Mar L, Foncin JF, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Da Silva HA, Haines JL, Perkicak-Vance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, St George-Hyslop PH, 1995. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375, 754–760. [DOI] [PubMed] [Google Scholar]
- Sun L, Zhao L, Yang G, Yan C, Zhou R, Zhou X, Xie T, Zhao Y, Wu S, Li X, Shi Y, 2015. Structural basis of human γ-secretase assembly. Proc Natl Acad Sci U S A 112, 6003–6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takami M, Nagashima Y, Sano Y, Ishihara S, Morishima-Kawashima M, Funamoto S, Ihara Y, 2009. γ-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of β-carboxyl terminal fragment. J Neurosci 29, 13042–13052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, Thinakaran G, Iwatsubo T, 2003. The role of presenilin cofactors in the γ-secretase complex. Nature 422, 438–441. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, 2012. The genetics of Alzheimer disease. Cold Spring Harb Perspect Med 2, pii: a006296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanzi RE, Bertram L, 2005. Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell 120, 545–555. [DOI] [PubMed] [Google Scholar]
- Weidemann A, Eggert S, Reinhard FB, Vogel M, Paliga K, Baier G, Masters CL, Beyreuther K, Evin G, 2002. A novel var epsilon-cleavage within the transmembrane domain of the Alzheimer amyloid precursor protein demonstrates homology with Notch processing. Biochemistry 41,2825–2835. [DOI] [PubMed] [Google Scholar]
- Wolfe MS, 2009a. γ-Secretase in biology and medicine. Semin Cell Dev Biol 20, 219–224. [DOI] [PubMed] [Google Scholar]
- Wolfe MS, 2009b. Intramembrane-cleaving proteases. J Biol Chem 284, 13969–13973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe MS, Xia W, Moore CL, Leatherwood DD, Ostaszewski B, Donkor IO, Selkoe DJ, 1999a. Peptidomimetic probes and molecular modeling suggest Alzheimer’s γ-secretases are intramembrane-cleaving aspartyl proteases. Biochemistry 38, 4720–4727. [DOI] [PubMed] [Google Scholar]
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ, 1999b. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 398, 513–517. [DOI] [PubMed] [Google Scholar]
- Wong PC, Zheng H, Chen H, Becher MW, Sirinathsinghji DJ, Trumbauer ME, Chen HY, Price DL, Van der Ploeg LH, Sisodia SS, 1997. Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature 387, 288–292. [DOI] [PubMed] [Google Scholar]
- Yang G, Zhou R, Zhou Q, Guo X, Yan C, Ke M, Lei J, Shi Y, 2019. Structural basis of Notch recognition by human γ-secretase. Nature 565, 192–197. [DOI] [PubMed] [Google Scholar]
- Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, Song YQ, Rogaeva D, Chen F, Kawarai T, Supala A, Levesque L, Yu H, Yang DS, Holmes E, Milman P, Liang Y, Zhang DM, Xu DH, Sato C, Rogaev E, Smith M, Janus C, Zhang Y, Aebersold R, Farrer LS, Sorbi S, Bruni A, Fraser P, St George-Hyslop P, 2000. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and βAPP processing. Nature 407, 48–54. [DOI] [PubMed] [Google Scholar]
- Zhou R, Yang G, Guo X, Zhou Q, Lei J, Shi Y, 2019. Recognition of the amyloid precursor protein by human γ-secretase. Science 363, eaaw0930. [DOI] [PubMed] [Google Scholar]