

Abstract
Historically microbiomes have been studied on the scale of the individual host, giving little consideration for the role of extra-host microbial populations in microbiome assembly. However, work in recent years has brought to light the importance of inter-host transmission and its influence on microbiome composition and dynamics. We now appreciate that microbiomes do not exist in isolation, but exchange constituents with the microbial communities of other hosts and the environment. Moving forward, fully understanding the role of transmission in microbiome assembly and dynamics will require a high-resolution view of the colonization and persistence patterns of particular microbial lineages (i.e. strains) across individuals and the environment. Yet, accomplishing this level of resolution will be an immense challenge, requiring improved sampling and bioinformatics approaches as well as employment of tractable experimental models. Insight gained from these investigations will contribute to our understanding of microbiome composition and variation, and lead to improved strategies for modulating microbiomes to improve human health.
Graphical Abstract
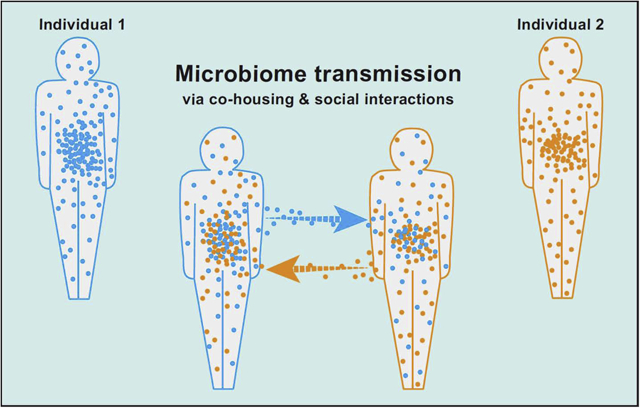
Introduction
Animals are colonized by communities of microorganisms (“microbiomes”) that serve essential roles in host health and development. In particular, the microbiome of the vertebrate gut is incredibly diverse, comprising hundreds of bacterial species representing thousands of different strains [1]. In recent decades, advances in DNA sequencing technologies have enabled extensive profiling of microbiomes and insight into how they vary both across individuals and within individuals over multiple scales of space and time [2]. From these studies, we have come to appreciate the incredibly complex and dynamic nature of these biologically important microbial consortia. In order to harness the microbiome to positively influence human health and treat or prevent microbiome-related diseases, we must be able to reliably predict or modulate microbiome communities. Thus, we must understand the ecological and evolutionary processes that govern microbiome membership and dynamics.
There is growing evidence that the process of transmission can be very important to microbiome composition and dynamics. By “transmission”, we mean the inter-host movement of microbes. Transmission can occur either directly (passed from one host to another via physical contact) or indirectly through various mechanisms. Indirect transmission may include an intermediate period of environmental persistence, and be facilitated by vectors (living) or fomites (non-living). Furthermore, mechanisms of transmission can be defined at a broader, ecological scale (e.g. horizontal vs. vertical transmission, deterministic vs. stochastic), or at a finer scale where genetic or phenotypic features of an organism that are relevant for transmission are identified (e.g. bacterial motility genes, or surface attachment molecules). An in-depth discussion of transmission mechanisms will not be presented here; rather we review recent research that demonstrates the contribution of transmission (by various mechanisms) to microbiome assembly and composition. Overall, this rapidly growing literature demonstrates the need to expand how we study microbiomes to a broader ecological scale, beyond the individual host, providing a new perspective for thinking about membership of the microbiome and microbial persistence within a host population.
Mounting evidence for the role of transmission in microbiome assembly
Understanding microbiome assembly (i.e. formation of the microbial community at a particular place and time) requires identification of the factors that influence variation across individuals and over time. Surprisingly, studies focused on teasing apart the relative contribution of host genetics and environmental factors on microbiome composition have found only a minor role for genetics in shaping the microbiome [3]. Furthermore, studies attempting to statistically correlate microbiome composition with numerous host factors such as diet, genetics, lifestyle, and clinical information, have not been able to explain more than a minor proportion (<20%) of the total variation across individuals [3–6]. One such study, conducted by Falony et al., included an impressive 503 potential host factors across nearly 4000 individuals, yet could explain only 16.4% of the total variation [6]. These findings highlight that factors or ecological processes not accounted for in these studies influence inter-host variation, and thus microbiome composition and function.
Factors not captured in the design of many of these studies are ones that measure the potential for transmission across individuals, and between individuals and the external environment. For example, they do not measure the extent of an individual’s inter-personal network and the type, frequency, and duration of physical interactions. However, several recent studies in both humans [7*–12] and non-human primates [13–16*] provide strong evidence for the contribution of transmission to microbiome assembly, and the need to measure these factors in future studies (Fig. 1). In particular, as discussed by Amato and colleagues in this issue (ref), the field of non-human primate studies is well suited for these endeavors because data on social interaction, shared environment, and other important factors such as diet are directly collected through observation. Tung et al. showed that social networks in wild baboons, and extent of social interaction, could predict microbiome structure, even after controlling for shared environment, diet, and relatedness [16*] (Figure 1a). Consistent with those findings, another study surveyed nine different non-human primate species and found that microbiomes varied with host species, but importantly also by social groups within species [14]. However, this was not seen within social groups of chimpanzees, suggesting that different ecological forces impact their microbiomes and that we should be cautious about generalizing conclusions across species [14].
Figure 1. Social groups and co-housing conditions facilitate microbiome transmission across individuals.

In all panels, individuals with similarly-dashed outlines represent close genetic relationships; small filled circles of varying colors represent phylogenetically diverse microbes. (a) Individuals belonging to the same social groups tend to share more microbial species with each other and their environment than individual from other groups. (b) Co-housed individuals have more similar microbiomes than individuals not co-housed, even when they are genetically closely related. (c) Zebrafish of different genotypes housed individually assemble microbiomes that are genotype-specific. However, microbiomes of co-housed fish are different from individually housed fish, irrespective of genotype.
In the human microbiome field, early studies observed that co-habitation correlated more strongly with microbiome similarity than genetic relatedness [17,18]. This opened the possibility for the role of transmission via a shared environment (Figure 1b); however, this was difficult to decouple from the influence of shared diet and lifestyle. Motivated by this question, more recent studies have designed experiments to better tease this apart. One such study, by Song et al., profiled the microbiomes of 159 people from 60 different families and found that cohabitation significantly influences microbiome composition by decreasing inter-individual variation among household members [7*]. The strongest signal was seen for cohabitating couples. Interestingly, the presence of a dog in the home amplified this homogenizing impact of co-habitation, especially for microbial communities of the skin. Looking even further into the effects of cohabitation, another study compared spouse and sibling pairs within the same household and found that spouses, especially those who reported having close relationships, shared more microbes than siblings or even spouses with less close relationships [8]. Moreover, they found that close relationships increased microbiome diversity and richness within those individuals. This is consistent with ecological theory demonstrating that dispersal among hosts bolsters microbial diversity [19*].
To get an even finer-resolution picture of the flow of microbes between hosts, improved bioinformatics tools have enabled strain-level resolution from microbiome sequencing data (reviewed in [20]). Achieving this level of phylogenetic accuracy from DNA sequencing data requires especially deep sequencing and specialized alignment and analysis tools, and so is not yet a standard in the field. When applied, however, this can be a powerful approach for tracking patterns of transmission within host populations. This was demonstrated beautifully in a recent study by Brito et al. where the researchers analyzed deeply-sequenced microbiome data from 287 people from 5 villages across the Fiji islands [21*]. Their results not only validate that transmission of strains occurs more frequently within households and between spouses, but also provided several new insights. For example, women’s microbiomes were more strongly influenced by inter-personal interactions than men’s. They also found no correlation of transmission patterns with specific bacterial phyla. This is suggestive of indirect or stochastic mechanisms of transmission, although these data cannot definitively differentiate between direct and indirect transmission events, or the directionality of transmission.
Harnessing the power of strain tracking from sequencing data, several other studies have made significant contributions to understanding mechanisms of transmission, particularly for investigating horizontal and vertical transmission of strains between mother and infant pairs in the first few months of life [22–25]. Here, vertical transmission is defined as being passed from parent to offspring, and includes those microbes passed via the egg or in the womb (the strictest definition of vertical transmission), but also those transmitted during or shortly after birth (e.g. from the birth canal, during breastfeeding). These studies incorporate longitudinal sampling, offering to date some of the most comprehensive studies for tracking commensal strain transmission and development of the infant microbiome. They highlight that vaginally-born infants acquire more maternal strains than cesarean-born infants [22], that infants are more likely to acquire dominant maternal strains [25] and that early maternally-acquired strains are often later replaced by strains from other sources [22–24]. Future studies like these will continue to illuminate how microbiomes are shaped over time and the mechanisms of transmission and persistence.
Experimental animal models are tractable systems for studying transmission
Laboratory animal models are powerful systems for studying host-microbe interactions, particularly because they confer the ability to control and track features of host-microbe systems that are often not possible in natural settings [26]. A number of studies have used animal models (e.g. mouse, fly, zebrafish) to explore the role transmission plays in host-microbe interactions [27–31]. Different animal models provide different advantages for the study of transmission; for example, the mouse model is especially well-suited for studying direct (host-to-host) transmission, since mice are coprophagic [32], and the zebrafish has emerged as an especially powerful model for exploring environmentally-mediated transmission [33–35], because the environment external to the host can be sampled and manipulated with relative ease. Invertebrate models with short generation times (e.g. hydra, nematodes, etc.; [36]) are ideal for studying the role of transmission in host-microbe coevolution. The use of animal models in host-microbiome research, including transmission studies, has been reviewed elsewhere [26], Below we provide a few illustrative examples.
An open question in the microbiome field is the relative contribution of vertical (passed from parent to offspring) versus horizontal transmission to microbiome assembly. Moeller et al. investigated this by establishing 17 inbred mouse lines derived from two geographically distinct wild mouse populations and monitoring microbiome composition over many host generations [27]. Lines were maintained separately, but with opportunities for introduction of exogenous microbes through handling and housing conditions. Their data showed the strongest influence of vertical transmission on microbiome composition, however, a small subset of the taxa were correlated with horizontal transmission. Interestingly, vertically transmitted taxa tended to be obligate anaerobes, while horizontally transmitted taxa were more often aerobic, which could be explained by the importance of oxygen tolerance for mechanisms of horizontal transmission. These results contrast with the dominant influence of horizontal transmission for human infant microbiomes at later developmental stages ([23]), again demonstrating that conclusions from these studies may not translate across species.
Using zebrafish as a model, Burns et al. investigated how dispersal among hosts alters microbiome assembly [29*]. To do this, zebrafish of two different genotypes (wild-type and immune-deficient) were reared individually, in groups of the same genotype, or groups of mixed genotypes. Surprisingly, genotype-specific differences in microbiome composition were only apparent when fish were reared in isolation. These effects were mitigated when the fish were co-housed, either with the same genotype or mixed genotypes (Figure 1c). This result shows that the impact of inter-host dispersal on microbiome assembly is greater than the influence of an intact innate immune system. The importance of transmission in the zebrafish system was further demonstrated via the serial passage of a bacterial member of the zebrafish gut microbiome (Aeromonas sp.) through axenic zebrafish larvae [28*] (Figure 2a). Rather than adapting to the intra-host environment during serial passage, these bacteria first evolved an increased ability to migrate into the host from the aqueous environment and to transmit from host-to-host (Figure 2b).
Figure 2. Transmission impacts bacterial evolutionary strategies for optimizing host colonization.

(a) In an experimental evolution study, a bacterial symbiont was passaged through populations of zebrafish, each time selecting gut-associated bacterial populations to inoculate the water of a new host population. (b) The adaptation that evolved first in the experiment was the ability to migrate into the host more quickly than the ancestor.
Scales of persistence within the microbiome
The field of community ecology has long recognized that ecological dynamics within local communities of species are influenced by dispersal across communities at larger spatial scales, forming the basis of metacommunity theory [37]. This was originally developed in the context of communities of organisms living within abiotic environments (“patches”, e.g. rock pools). However, recently the concepts and theories born from metacommunity ecology have begun to be applied to the study of animal microbiomes, with the premise that individual hosts serve as patches connected via transmission [38–42]. These theories are now being refined to more appropriately capture the ecological features of host-associated microbiomes, such as feedback between the biotic host patches and the microbiome, and the impact of microbial persistence in the environment on microbiome composition [19*,43].
It is broadly recognized in ecology that one strategy for persistence is to specialize in dispersal between or across communities rather than competing to persist within a single community [44]. Although this has not been well documented in host-microbiome systems, there is evidence that this kind of strategy is employed in free-living microbial communities [45]. Even though the relevance of this for host-associated microbiomes has not been directly demonstrated, it suggests that the continual opportunity for reintroduction via transmission could enable dispersal specialists to persist long term within a network of local comminutes, such as a population of hosts (Figure 3a). For instance, if conditions are unfavorable for persistence within a particular host at a given time, it is possible to persist within the system (i.e. population of hosts and their environment) and have an opportunity to colonize a new host, or re-inoculate the same host when intra-host conditions change (Figure 3b). These dynamics make it difficult to differentiate between species that are capable of persisting long term and ones that are consistently present due to re-inoculation.
Figure 3. Transmission of dispersal-specialist species within a host population.

(a) Some microbial strains (blue) more readily transmit between hosts than other more long-term resident strains (pink). Through this mechanism, they can persist at a host population level even if they are poor competitors within a single host. (b) Transmission and colonization of a dispersal specialist (blue) may not be successful at one point in time (T1), however, this microbe can persist within the host population until conditions within the second host allow successful colonization and persistence (T2).
While microbiome constituents are often defined in terms of being “resident” or “transient”, it is clear that microbes exist along a spectrum of colonization and persistence, with some able to maintain stable, long-lasting populations in a host, while others are much more short-lived [46–48]. However, current methods for studying the microbiome, particularly microbiome profiling, do not provide a high enough resolution understanding to define the colonization and persistence dynamics of most microbial lineages within and across individuals and their environment (see Box 1). In particular, the most widely used approach is fecal sampling, which does not capture any information about extra-host microbial populations, and furthermore is a poor representation of the community within the host. Nonetheless, in light of the work highlighted here demonstrating that microbiomes are connected via transmission and dispersal, it is clear that there is a need to shift our conceptualization of microbiome membership to one that encompasses the broader ecological context of host-microbe systems and considers microbial persistence at larger spatial scales.
Box 1. The challenges and limitations of microbiome profiling.
The majority of microbiome profiling studies use DNA sequencing approaches to analyze microbiome structure and composition. These studies were enabled by advancements in DNA sequencing technologies which allow high-throughput profiling of microbiome samples [49]. However, a sequencing dataset is only as valuable as the tools you have to analyze it, and the challenge of gleaning meaningful information from these complex data is more complicated than one would anticipate. Bioinformatics tools for analysis are continually being developed and refined, and can be tailored to answer specific research questions [50,51]. An enormous amount of genomic information can be acquired from a microbiome sample, yet the limitations of sampling and analysis must be kept in mind when interpreting the data. In essence, the level of resolution that can be achieved is limited in three major ways—spatially, temporally, and taxonomically. Each of these impacts how we interpret the data to understand the complexities and dynamics of the microbiome, especially for tracking individual strains.
Limits of spatial resolution
Spatial resolution in profiling studies is limited by the design and technical aspects of sampling. Fecal sampling is the most common approach for profiling the gut microbiome, primarily due to ease of collection and low cost. However, a fecal sample does not offer a complete representation of the microbial communities within the intestine. Numerous studies have demonstrated that luminal communities, which are primarily represented in fecal samples, are distinct from mucosa-associated communities [52–56]; reviewed in [57]. Furthermore, there are differences in the spatial distribution of taxa along the length of the gut (reviewed in [58]), thus a fecal sample primarily comprises microbes from the distal intestine. As a result, we know little about the microbial communities in the other regions of the intestine, including the proximal colon and the different compartments of the small intestine. In addition, sequencing results can be highly dependent on aspects of the fecal sample preparation, such as homogenization, storage, and DNA extraction [59]. Importantly, few studies sample microbial communities at larger spatial scales outside of hosts, such as from the environment or communities associated with food.
Limits of temporal resolution
Capturing a complete picture of the dynamics of individual strains within the microbiome requires temporal sampling. Temporal resolution can be considered at multiple scales, from hours to years, and even across generations of hosts [60–62]. The majority of microbiome studies sample only a single time point, although decreasing costs of sampling and analysis are leading to more studies that incorporate temporal dynamics into their experimental design.
Limits of taxonomic resolution
Taxonomic resolution is a major challenge in microbiome profiling studies. Until recently, the most common approach for profiling has been targeted 16S rRNA sequencing [63]. However, bacterial strains encoding near-identical 16S rRNA genes, can have significant differences in content across the rest of the genome [64]. Therefore, 16S rRNA sequencing is generally unable to resolve taxa beyond the species level, although new approaches are being developed to increase this resolution [65,66]. Recent advancements in DNA sequencing technologies have resulted in a marked decrease in cost and efficiency, promoting whole shotgun (i.e. metagenomic) sequencing. These data can provide strain-level resolution of the community, although this requires incredibly deep sequencing and complex analysis tools and so is not yet a standard in the field.
Recognizing the importance of transmission and changing the scale at which we view and study the microbiome has important implications for how we interpret microbiome data. To illustrate, it might be concluded that a low abundance but consistently detected bacterial strain is a fair competitor in the intestine with a small but stable population, when in fact it might be a very poor competitor that maintains its population by continued influx from external sources such as cohabiting individuals or an environmental reservoir. Just as importantly, this broader scale of persistence shifts how we delineate what constitutes an individual’s microbiome—it blurs the lines between hosts and their environment. It also creates an opportunity for feedback within the system such that hosts impact the microbial composition of the environment, which in turn changes the pool from which hosts recruit potential future colonizers. This intermediate environment could be conceptualized as a “cloud” of host influence within which hosts could enhance transmission across the population and to which microbes could adapt. From this new perspective, we can reimagine how microbes might adapt to being host-associated and the strategies they might use to maintain stable populations not within a particular host, but within a host population over long time periods, even across generations.
Conclusions
The transmission of pathogenic microbes has been studied for many decades but only in recent years are we beginning to understand its relevance for the microbiome. The first observations alluding to the influence of transmission on microbiome assembly came from microbiome profiling studies in humans and non-human primates showing correlations between microbiome composition and cohabitation or social networks. More recently, experiments using animal model systems to test hypotheses about microbiome transmission have substantiated these findings. We now have an appreciation for the relevance of transmission in the ecological and evolutionary dynamics of the microbiome. This has important implications for how we design experiments to study microbiome variation and dynamics and interpret sequencing data. Many questions remain to be answered (see “outstanding questions”). Future work should focus on designing better experimental approaches to elucidate mechanisms of transmission and identify the specific colonization and persistence patterns of microbiome constituents. Furthermore, the bourgeoning microbiome field will benefit from the application of theories and ideas from other fields, such as metacommunity theory from the field of community ecology. Importantly, these insights bring to light a new ecological perspective for studying the microbiome at a larger spatial scale, beyond the individual host.
Outstanding questions:
What are the mechanisms for transmission of microbiomes in different host species?
What are the time scales on which microbiomes transmit and what factors influence these rates?
How does transmission affect the colonization and persistence patterns of individual taxa?
What microbial traits contribute to transmission?
Are there ecological or evolutionary trade-offs between persistence and transmission?
Can we distinguish between long-term persistence in individual hosts and serial re-inoculation?
How do changes in host behavior or environment influence microbiome transmission?
Highlights.
Microbial transmission shapes microbiomes through social networks and co-housing.
The colonization and persistence dynamics of microbiome members is underexplored.
Microbiome profiling offers limited spatial, temporal, and taxonomic resolution.
Animal models provide tractable systems for studying microbiome transmission.
Microbiome studies should consider ecological contexts beyond individual hosts.
Acknowledgements:
We are grateful to Beth Miller and Karen Adair for thoughtful discussions and review of the manuscript.
Funding: This work was supported by the National Institutes of Health [grant number P01GM125576]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012. June 13;486(7402):207–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lloyd-Price J, Mahurkar A, Rahnavard G, Crabtree J, Orvis J, Hall AB, et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature. Nature Publishing Group; 2017. October 5;550(7674):61–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, et al. Environment dominates over host genetics in shaping human gut microbiota. Nature. Nature Publishing Group; 2018. February 28;:1–19. [DOI] [PubMed] [Google Scholar]
- 4.Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, et al. Human Genetics Shape the Gut Microbiome. Cell. Elsevier Inc; 2014. November 6;159(4):789–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen J, Bittinger K, Charlson ES, Hoffmann C, Lewis J, Wu GD, et al. Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics. 2012. June 17;28(16):2106–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Falony G, Joossens M, Vieira-Silva S, Wang J, Darzi Y, Faust K, et al. Population-level analysis of gut microbiome variation. Science. American Association for the Advancement of Science; 2016. April 29;352(6285):560–4. [DOI] [PubMed] [Google Scholar]
- 7.*.Song SJ, Lauber C, Costello EK, Lozupone CA, Humphrey G, Berg-Lyons D, et al. Cohabiting family members share microbiota with one another and with their dogs. eLife. 2013. April 16;2:e00458–22. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study was the first to demonstrate the influence of co-habitation on microbiome variation across body sites other than the intestine. They found that the skin microbiome was impacted the most strongly, especially in households with pets.
- 8.Dill-McFarland KA, Tang Z-Z, Kemis JH, Kerby RL, Chen G, Palloni A, et al. Close social relationships correlate with human gut microbiota composition. Sci Rep. Springer US; 2019. January 3;:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ly M, Jones MB, Abeles SR, Santiago-Rodriguez TM, Gao J, Chan IC, et al. Transmission of viruses via our microbiomes. Microbiome. Microbiome; 2016. November 29;:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martínez I, Stegen JC, Maldonado-Gómez MX, Eren AM, Siba PM, Greenhill AR, et al. The gut microbiota of rural papua new guineans: composition, diversity patterns, and ecological processes. Cell Rep. 2015. April 28;11(4):527–38. [DOI] [PubMed] [Google Scholar]
- 11.Li SS, Zhu A, Benes V, Costea PI, Hercog R, Hildebrand F, et al. Durable coexistence of donor and recipient strains after fecal microbiota transplantation. Science. 2016. April 29;352(6285):586–9. [DOI] [PubMed] [Google Scholar]
- 12.Sharma A, Richardson M, Cralle L, Stamper CE, Maestre JP, Stearns-Yoder KA, et al. Longitudinal homogenization of the microbiome between both occupants and the built environment in a cohort of United States Air Force Cadets. Microbiome; 2019. May 1;:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moeller AH, Foerster S, Wilson ML, Pusey AE, Hahn BH, Ochman H. Social behavior shapes the chimpanzee pan-microbiome. Science Advances. American Association for the Advancement of Science; 2016. January 1;2(1):e1500997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gogarten JF, Davies TJ, Benjamino J, Gogarten JP, Graf J, Mielke A, et al. Factors influencing bacterial microbiome composition in a wild non- human primate community in Taï National Park, Côte d’Ivoire. ISME J. Springer US; 2018. September 18;:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grieneisen LE, Livermore J, Alberts S, Tung J, Archie EA. Group Living and Male Dispersal Predict the Core Gut Microbiome in Wild Baboons. Integrative and Comparative Biology. 2017. September 29;57(4):770–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.*.Tung J, Barreiro LB, Burns MB, Grenier J-C, Lynch J, Grieneisen LE, et al. Social networks predict gut microbiome composition in wild baboons. eLife. eLife Sciences Publications Limited; 2015. March 16;4:e05224. [DOI] [PMC free article] [PubMed] [Google Scholar]; Using data collected during long-term observation studies of two social groups of wild baboons, this is the first study to demonstrate that social groups and networks can be used to predict gut microbiome species.
- 17.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, et al. Human gut microbiome viewed across age and geography. Nature. 2012. May 9;486(7402):222–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature. 2009. January 22;457(7228):480–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.*.Miller ET, Svanbäck R, Bohannan BJM. Microbiomes as Metacommunities: Understanding Host-Associated Microbes through Metacommunity Ecology. Trends Ecol Evol (Amst). Elsevier Ltd; 2018. September 25;:1–10. [DOI] [PubMed] [Google Scholar]; The authors present a compelling argument for the application of metacommunity theory for understanding microbiome variation, offering ways to refine current modeling approaches to be more relevant to host-microbe systems.
- 20.Brito IL, Alm EJ. Tracking Strains in the Microbiome: Insights from Metagenomics and Models. Front Microbiol. Frontiers; 2016. May 19;7(109):170–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.*.Brito IL, Gurry T, Zhao S, Huang K, Young SK, Shea TP, et al. Transmission of human-associated microbiota along family and social networks. Nat Microbiol. 2019;4(6):964–71. [DOI] [PMC free article] [PubMed] [Google Scholar]; In this study, strain-level resolution of microbiome sequencing data across a unique cohort of families in the Fiji islands demonstrates that transmission patterns are influenced by co-habitation and can even predict inter-personal relationships. This is the first study to demonstrate a role of gender in strain transmission.
- 22.Korpela K, Costea P, Coelho LP, Kandels-Lewis S, Willemsen G, Boomsma DI, et al. Selective maternal seeding and environment shape the human gut microbiome. Genome Research. Cold Spring Harbor Lab; 2018. April;28(4):561–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nayfach S, Rodriguez-Mueller B, Garud N, Pollard KS. An integrated metagenomics pipeline for strain profiling reveals novel patterns of bacterial transmission and biogeography. Genome Research. Cold Spring Harbor Lab; 2016. November;26(11):1612–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferretti P, Pasolli E, Tett A, Asnicar F, Gorfer V, Fedi S, et al. Mother-to-Infant Microbial Transmission from Different Body Sites Shapes the Developing Infant Gut Microbiome. Cell Host & Microbe. Elsevier; 2018. July 11;24(1):133–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yassour M, Jason E, Hogstrom LJ, Arthur TD, Tripathi S, Siljander H, et al. Strain-Level Analysis of Mother-to-Child Bacterial Transmission during the First Few Months of Life. Cell Host & Microbe. 2018. July 11;24(1):146–154.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Douglas AE. Simple animal models for microbiome research. Nature Publishing Group. Springer US; 2019. August 14;:1–12. [DOI] [PubMed] [Google Scholar]
- 27.Moeller AH, Suzuki TA, Phifer-Rixey M, Nachman MW. Transmission modes of the mammalian gut microbiota. Science. American Association for the Advancement of Science; 2018. October 26;362(6413):453–7. [DOI] [PubMed] [Google Scholar]
- 28.*.Robinson CD, Klein HS, Murphy KD, Parthasarathy R, Guillemin K, Bohannan BJM. Experimental bacterial adaptation to the zebrafish gut reveals a primary role for immigration. Gore J, editor. PLoS Biol. Public Library of Science; 2018. December 10;16(12):e2006893–26. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates the utility of using experimental evolution to investigate selective pressures within host-microbe systems, specifically for bacteria colonizing the vertebrate gut.
- 29.*.Burns AR, Miller E, Agarwal M, Rolig AS, Milligan-Myhre K, Seredick S, et al. Interhost dispersal alters microbiome assembly and can overwhelm host innate immunity in an experimental zebrafish model. Proc Natl Acad Sci USA. 2017. October 2;8:201702511–6. [DOI] [PMC free article] [PubMed] [Google Scholar]; This work demonstrates that animal models are valuable systems for testing hypotheses about the influence of inter-host dispersal on microbiome composition. Here, it is shown that transmission among hosts has a greater impact on microbiome variation than that of the innate immune system.
- 30.Martino ME, Joncour P, Leenay R, Gervais H, Shah M, Hughes S, et al. Bacterial Adaptation to the Host’s Diet Is a Key Evolutionary Force Shaping Drosophila-Lactobacillus Symbiosis. Cell Host & Microbe. 2018. July 11;24(1):109–119.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stagaman K, Burns AR, Guillemin K, Bohannan BJ. The role of adaptive immunity as an ecological filter on the gut microbiota in zebrafish. ISME J. Nature Publishing Group; 2017. July;11(7):1630–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nguyen TLA, Vieira-Silva S, Liston A, Raes J. How informative is the mouse for human gut microbiota research? Dis Model Mech. 2015. January;8(1):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burns AR, Guillemin K. The scales of the zebrafish: host-microbiota interactions from proteins to populations. Current Opinion in Microbiology. 2017. August;38:137–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wiles TJ, Wall ES, Schlomann BH, Hay EA, Parthasarathy R, Guillemin K. Modernized Tools for Streamlined Genetic Manipulation and Comparative Study of Wild and Diverse Proteobacterial Lineages. Ruby EG, editor. MBio. American Society for Microbiology; 2018. October 9;9(5):e01877–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Milligan-Myhre K, Charette JR, Phennicie RT, Stephens WZ, Rawls JF, Guillemin K, et al. Study of Host–Microbe Interactions in Zebrafish Third Edition Vol. 105, Methods in Cell Biology. Elsevier Inc; 2011. 30 p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petersen JM, Osvatic J. Microbiomes In Natura: Importance of Invertebrates in Understanding the Natural Variety of Animal-Microbe Interactions. mSystems. American Society for Microbiology Journals; 2018. April 24;3(2):3229–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leibold MA, Holyoak M, Mouquet N, Amarasekare P, Chase JM, Hoopes MF, et al. The metacommunity concept: a framework for multi-scale community ecology. Ecology Letters. John Wiley & Sons, Ltd (10.1111); 2004. July 1;7(7):601–13. [Google Scholar]
- 38.Burns AR, Stephens WZ, Stagaman K, Wong S, Rawls JF, Guillemin K, et al. Contribution of neutral processes to the assembly of gut microbial communities in the zebrafish over host development. ISME J. 2016. March;10(3):655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adair KL, Douglas AE. ScienceDirect Making a microbiome: the many determinants of host-associated microbial community composition. Current Opinion in Microbiology. Elsevier Ltd; 2017. February 1;35:23–9. [DOI] [PubMed] [Google Scholar]
- 40.Griffin NW, Ahern PP, Cheng J, Heath AC, Ilkayeva O, Newgard CB, et al. Prior Dietary Practices and Connections to a Human Gut Microbial Metacommunity Alter Responses to Diet Interventions. Cell Host & Microbe. 2017. January 11;21(1):84–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Venkataraman A, Bassis CM, Beck JM, Young VB, Curtis JL, Huffnagle GB, et al. Application of a neutral community model to assess structuring of the human lung microbiome. MBio. 2015. January 20;6(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeng Q, Rodrigo A. Neutral models of short-term microbiome dynamics with host subpopulation structure and migration limitation. Microbiome; 2018. April 26;:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller ET, Bohannan BJM. Life Between Patches: Incorporating Microbiome Biology Alters the Predictions of Metacommunity Models. Front Ecol Evol. Frontiers; 2019. July 18;7:1–13. [Google Scholar]
- 44.Levins R, Culver D. Regional Coexistence of Species and Competition between Rare Species. Proc Natl Acad Sci USA. 1971. June;68(6):1246–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dini-Andreote F, van Elsas JD, Olff H, Salles JF. Dispersal-competition tradeoff in microbiomes in the quest for land colonization. Sci Rep. 2018. Jun 21;8(1):9451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martinson JNV, Pinkham NV, Peters GW, Cho H, Heng J, Rauch M, et al. Rethinking gut microbiome residency and the Enterobacteriaceae in healthy human adults. ISME J. Springer US; 2019. May 10;:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schloissnig S, Arumugam M, Sunagawa S, Mitreva M, Tap J, Zhu A, et al. Genomic variation landscape of the human gut microbiome. Nature. Nature Publishing Group; 2013. January 3;493(7430):45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, et al. The Long-Term Stability of the Human Gut Microbiota. Science. 2013. July 4;341(6141):1237439–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kulski JK. Next-Generation Sequencing — An Overview of the History, Tools, and “Omic” Applications. In: Next Generation Sequencing - Advances, Applications and Challenges. IntechOpen; 2016. [Google Scholar]
- 50.Kim Y, Koh I, Rho M. Deciphering the human microbiome using next-generation sequencing data and bioinformatics approaches. Methods. Elsevier Inc; 2015. June 1;79–80(C):52–9. [DOI] [PubMed] [Google Scholar]
- 51.Hamady M, Knight R. Microbial community profiling for human microbiome projects: Tools, techniques, and challenges. Genome Research. 2009. July 1;19(7):1141–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zmora N, Zilberman-Schapira G, Suez J, Mor U, Dori-Bachash M, Bashiardes S, et al. Personalized Gut Mucosal Colonization Resistance to Empiric Probiotics Is Associated with Unique Host and Microbiome Features. Cell. Elsevier Inc; 2018. September 6;174(6):1388–1405.e21. [DOI] [PubMed] [Google Scholar]
- 53.Mark Welch JL, Hasegawa Y, McNulty NP, Gordon JI, Borisy GG. Spatial organization of a model 15-member human gut microbiota established in gnotobiotic mice. Proceedings of the National Academy of Sciences. National Acad Sciences; 2017. October 24;114(43):E9105–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang L, Wu W, Lee Y-K, Xie J, Zhang H. Spatial Heterogeneity and Co-occurrence of Mucosal and Luminal Microbiome across Swine Intestinal Tract. Front Microbiol. Frontiers; 2018. January 26 9 1055–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jones RB, Zhu X, Moan E, Murff HJ, Ness RM, Seidner DL, et al. Inter-niche and inter-individual variation in gut microbial community assessment using stool, rectal swab, and mucosal samples. Sci Rep. Springer US; 2018. February 28;:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yasuda K, Oh K, Ren B, Tickle TL, Franzosa EA, Wachtman LM, et al. Biogeography of the Intestinal Mucosal and Lumenal Microbiome in the Rhesus Macaque. Cell Host & Microbe. Elsevier Inc; 2015. March 11;17(3):385–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tropini C, Earle KA, Huang KC, Sonnenburg JL. The Gut Microbiome: Connecting Spatial Organization to Function. Cell Host & Microbe. Elsevier Inc; 2017. April 12;21(4):433–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hillman ET, Lu H, Yao T, Nakatsu CH. Microbial Ecology along the Gastrointestinal Tract. Microbes and environments. Japanese Society of Microbial Ecology · The Japanese Society of Soil Microbiology; 2017;32(4):300–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu W-K, Chen C-C, Panyod S, Chen R-A, Wu M-S, Sheen L-Y, et al. Optimization of fecal sample processing for microbiome study - The journey from bathroom to bench. Journal of the Formosan Medical Association. Elsevier Ltd; 2019. February 1;118(2):545–55. [DOI] [PubMed] [Google Scholar]
- 60.Uhr GT, Dohnalová L, Thaiss CA. The Dimension of Time in Host-Microbiome Interactions. mSystems. 2019. February 26;4(1):1079–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.David LA, Materna AC, Friedman J, Campos-Baptista MI, Blackburn MC, Perrotta A, et al. Host lifestyle affects human microbiota on daily timescales. Genome Biol. 2014;15(7):R89–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Groussin M, Mazel F, Sanders JG, Smillie CS, Lavergne SEB, Thuiller W, et al. Unraveling the processes shaping mammalian gut microbiomes over evolutionary time. Nature Communications. Nature Publishing Group; 2017. February 18;8:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pollock J, Glendinning L, Wisedchanwet T, Watson M. The Madness of Microbiome: Attempting To Find Consensus “Best Practice” for 16S Microbiome Studies. Liu S-J, editor. Applied and Environmental Microbiology. American Society for Microbiology; 2018. April 1;84(7):3225–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Janda JM, Abbott SL. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. Journal of Clinical Microbiology. American Society for Microbiology Journals; 2007. September;45(9):2761–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Segota I, bioRxiv TL, 2019. A high-resolution pipeline for 16S-sequencing identifies bacterial strains in human microbiome. biorxivorg. [Google Scholar]
- 66.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nature Methods. Nature Publishing Group; 2016. May 23;13(7):581–3. [DOI] [PMC free article] [PubMed] [Google Scholar]