

Abstract
The dialogue between cancer cells and the surrounding fibroblasts, tumor‐associated macrophages (TAM), and immune cells can create a tumor microenvironment (TME) able to promote tumor progression and metastasis and induce resistance to anticancer therapies. Cancer cells, by producing growth factors and cytokines, can recruit and activate fibroblasts in the TME inducing their transdifferention in cancer‐associated fibroblasts (CAFs). Then, CAFs, in a reciprocal cross‐talk with cancer cells, sustain cancer growth and survival and support malignancy and tumor resistance to therapies. Therefore, the identification of the molecular mechanisms regulating the interplay between cancer cells and fibroblasts can offer an intriguing opportunity for novel diagnostic and therapeutic anticancer purpose. HIPK2 is a multifunctional tumor suppressor protein that modulates cancer cell growth and apoptosis in response to anticancer drugs and negatively regulates pathways involved in tumor progression and chemoresistance. HIPK2 protein downregulation is induced by hypoxia and hyperglycemia and HIPK2 knockdown favors tumor progression and resistance to therapy other than a pseudohypoxic, inflammatory, and angiogenic cancer phenotype. Therefore, we hypothesized that HIPK2 modulation in cancer cells could contribute to modify the tumor–host interaction. In support of our hypothesis, here we provide evidence that culturing human fibroblasts (hFB) with conditioned media derived from cancer cells undergoing HIPK2 knockdown (CMsiHIPK2) triggered their transdifferentiation CAF‐like, compared to hFB cultured with CM‐derived from HIPK2‐carrying control cancer cells. CAF transdifferentiation was identified by expression of several markers including α‐smooth muscle actin (α‐SMA) and collagen I and correlated with autophagy–mediated caveolin‐1 degradation. Although the molecular mechanisms dictating CAF‐transdifferentiation need to be elucidated, these results open the way to further study the role of HIPK2 in TME remodeling for prognostic and therapeutic purpose.
Keywords: cancer‐associated fibroblast (CAF), cancer progression, caveolin‐1, fibroblast transdifferentiation, HIPK2, reactive‐oxygen species (ROS)
Abbreviations
- CAFs
cancer‐associated fibroblasts
- CM
conditioned media
- COX‐2
cyclooxygenase‐2
- CQ
cloroquine
- ECM
extracellular matrix
- EMT
epithelial mesenchymal transition
- FAP
fibroblast activation protein
- hFB
human fibroblasts
- HIF‐1α
hypoxia inducible factor 1α
- HIPK2
homeodomain‐interacting protein kinase 2
- IL‐6
interleukin‐6
- NAC
N‐acetyl‐L‐cysteine
- Nox1
NADPH oxidase 1
- PGE2
prostaglandin E2
- PP2A
protein phosphatase 2
- ROS
reactive oxygen species
- RT‐PCR
reverse transcription (RT)‐PCR
- TAMs
Tumor‐Associated Macrophages
- TME
tumor microenvironment
- TNF‐α
tumor necrosis factor‐α
- α‐SMA
α‐smooth‐muscle actin
1. INTRODUCTION
The interplay between cancer cells and the surrounding tumor microenvironment (TME), including fibroblasts, immune system, vasculature, and extracellular matrix (ECM) components may contribute to cancer progression and resistance to anticancer therapies.1 The TME of most solid tumors, including colon, is characterized by “fibroblast‐like cells” that undergo transdifferentiation into cancer‐associated fibroblasts (CAFs), myofibroblasts‐like cells that express α‐smooth‐muscle actin (α‐SMA).2 CAF are characterized by a more rapid proliferative rate, enhanced collagen production, secretion of growth factors, and extracellular matrix (ECM) modulators3 and, when activated by tumor cells, secrete various growth factors, cytokines, and chemokines that promote tumor progression in an interplay with cancer cells.3
The serine/threonine homeodomain‐interacting protein kinase 2 (HIPK2) is a “caretaker” gene as its activation, usually in response to DNA damage, inhibits tumor growth while its inactivation, by hypoxia or hyperglycemia, increases tumor progression and chemoresistance.4 HIPK2 activation plays a crucial role in apoptotic function of p53 oncosuppressor, in order to induce cancer cell death5 and in p53 misfolding.6, 7 HIPK2 downregulates Wnt/β‐catenin signaling pathway5 that is often hyperactivated in cancer and strongly regulates cancer invasion by promoting the epithelial mesenchymal transition (EMT).8 HIPK2 corepresses vimentin, a driver for EMT and tumor invasion that induces chemotherapeutic resistance and poor prognosis.9 HIPK2 corepresses hypoxia inducible factor 1α (HIF‐1α),10 a major player in cancer progression through transactivation of HIF‐1 target genes involved in angiogenesis, glucose metabolism, chemotherapeutic resistance and invasion.11 On the other hand, HIPK2 depletion activates β4 integrin transcription and the Akt pathway, promoting anchorage‐independent growth and invasion12 and induces HIF‐1‐mediated cyclooxygenase‐2 (COX‐2) upregulation and prostaglandin E2 (PGE2) generation, promoting colon cancer cell invasion and immunosuppression.13 HIPK2 gene is rarely mutated while HIPK2 protein undergoes degradation by hypoxia10 or hyperglycemia,14 two conditions often associated with increased cancer progression and resistance to therapies, suggesting the need of cancer cells to inactivate HIPK2 for their survival.
The above background demonstrates the fundamental role of HIPK2 in controlling pathways involved in cancer progression and chemoresistance, however, the HIPK2 role in cancer cells crosstalk with the TME, and with fibroblasts in particular, has never been addressed. In this article, we provide evidence that culturing fibroblasts with conditioned media (CM) derived from HIPK2‐depleted colon cancer cells (CMsiHIPK2) increased fibroblasts transdifferentiation CAF‐like, compared to fibroblast cultured with CM derived from siRNA cancer cells (CMsiRNA). We hypothesize that loss of HIPK2 function may deregulate pathways in cancer cells that contribute to TME remodeling and in particular to fibroblasts transdifferentiation CAF‐like, in order to sustain tumor progression and chemotherapeutic resistance.
2. MATERIALS AND METHODS
2.1. Cell culture and reagents
The human RKO colon cancer cells, stably interfered for HIPK2 function by siRNA (siHIPK2) and control siRNA cells (siRNA)6 (Figure 1a) were routinely cultured in RPMI 1640 (Life Technologies‐Invitrogen) and human primary foreskin fibroblasts (FB1329, herein referred as hFB)15 were cultured in DMEM (Life Technologies‐Invitrogen), supplemented with 10% FBS (GIBCO‐BRL, Grand Island, NY), plus 100 units/ml penicillin/streptomycin, in 5% CO2 humidified incubator at 37°C. Fibroblasts were kept at low passages. The ROS inhibitor N‐acetyl‐L‐cysteine (NAC) (Sigma‐Aldrich, St Louis, MO) was used at 10 μM; inhibitor of autophagic protein degradation, cloroquine (CQ) (Sigma‐Aldrich) was used at 25 mM.
Figure 1.
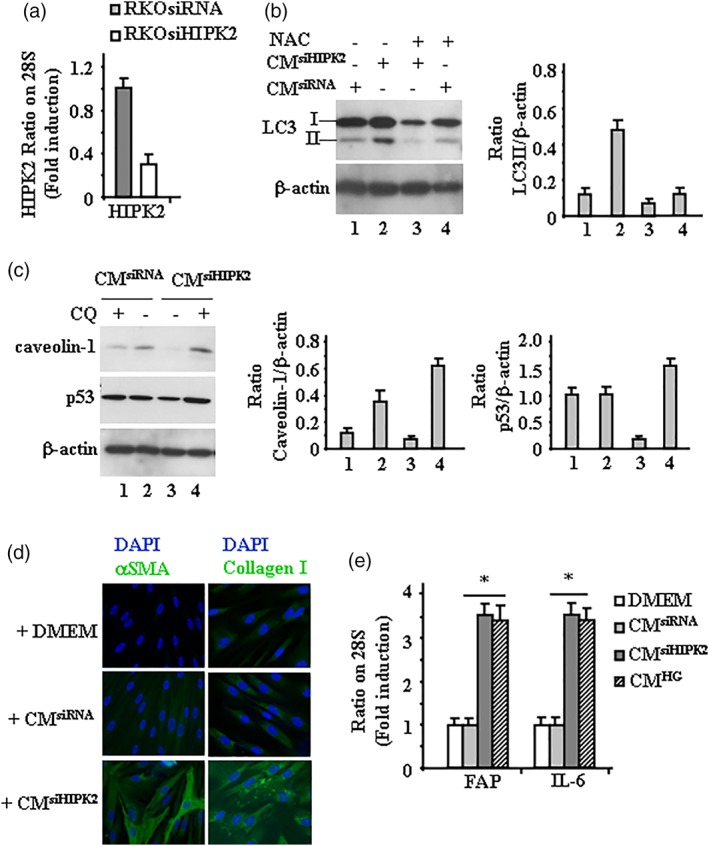
Human FB transdifferentiation by cancer cells‐derived CM. (a) RT‐PCR analysis of HIPK2 mRNA levels in RKOsiRNA and RKOsiHIPK2 cells. (b) Western blot showing LC3I/II levels in hFB cultured with CMsiHIPK2 or CMsiRNA with or without NAC. Anti‐β‐actin was used as protein loading control. Representative images are shown. Densitometric analysis was applied to quantify LC3II expression/β‐Actin ratio. (c) Western blot showing the indicated protein levels in FB cultured with CMsiHIPK2 or CMsiRNA with or without CQ. Anti‐β‐actin was used as protein loading control. Representative images are shown. Densitometric analysis was applied to quantify protein expression/β‐actin ratio. (d) Immunocytochemistry of αSMA and collagen type I in hFB cultured with CMsiHIPK2, CMsiRNA, or serum‐free DMEM. Representative images are shown. (e) RT‐PCR analysis of hFB cultured with CMsiHIPK2, CMsiRNA, or CMHG. The mRNA levels of FAP and IL‐6 genes were analyzed by densitometry and plotted as the mRNA/28S ratio. Data are the mean ± SD of three independent experiments. *p = .001
2.2. Treatment of cells with conditioned medium
For CM collection, RKO‐siRNA and RKO‐siHIPK2 cells were grown to over 70% confluence in 10 mm Petri dishes in DMEM with 10% FBS. The medium was refreshed with serum‐free DMEM and cells were cultured for 48 hr. For the experiments in high glucose (HG), RKO were cultured in DMEM medium containing 4.5 g/l D‐glucose (HG), as previously reported.16, 17 The media were collected and centrifuged at 1,000g for 10 min, and the CM from RKO‐siRNA (CMsiRNA), from RKO‐siHIPK2 (CMsiHIPK2) and from RKO high glucose (CMHG) cells were filtered with a 0.22‐μm membrane and stored at −80°C until use. At the same time, serum‐free DMEM was set as the control. When hFB reached subconfluence, the medium was replaced by CMsiRNA, CMsiHIPK2, CMHG, or serum‐free DMEM, then cultured for another 48 hr in the presence or absence of NAC or CQ.
2.3. Immunoblot analysis
After treatment, hFB were harvested, washed in PBS, and resuspended in lysis buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA, pH 8.0, 150 mM KCl, 1 mM dithiothreitol, 1% Nonidet P‐40) plus protease and phosphatase inhibitors (Sigma‐Aldrich Chemical Company). After incubation on ice for 30 min, total cell lysates were centrifuges at 14,000g for 15 min at 4°C and the supernatant collected for western blot analyses. Samples were denatured in SDS sample buffer. Proteins were separated by loading 10–30 μg of total cell lysates on denaturing 8–20% SDS‐polyacrylamide gel (BioRad) and electro‐blotted onto polyvinylidene difluoride (PVDF) membranes (Millipore, Milan, Italy). Membranes were blocked with 5% nonfat dry milk in tris phosphate‐buffered saline tween (TBST) (50 mM Tris pH 7.5, 0.9% NaCl, 0.1% Tween 20) for 30 min at room temperature and probed with primary antibodies: mouse monoclonal LC3B (1:1000) (Sigma‐Aldrich, #L7543), mouse monoclonal p53 (1:1000) (DO‐1) (Santa Cruz Biotechnology, CA, #sc‐126), mouse monoclonal caveolin‐1 (7C8) (1:1000) (Santa Cruz Biotechnology, #sc‐53,564), and mouse monoclonal β‐actin (1:1000) (Calbiochem, San Diego, CA, #CP01). Secondary antimouse IgGs conjugated to horseradish peroxidase (Bio‐Rad Laboratories, Hercules, CA, #172‐1011) was used (1:10000 in TBST) to detect primary antibodies. Immunodetection was carried out using chemiluminescence substrates (ECL kit, Amersham Biosciences, Freiburg, Germany) and recorded using Hyperfilm ECL (Amersham Biosciences). Densitometry was performed on ECL results with ImageJ software (http://rsb.info.nih.gov/hihimage/) and relative band intensity normalized to β‐actin and plotted as protein expression/β‐actin ratio.
2.4. Indirect immunofluorescence assay (IF)
After treatment, fibroblasts were washed with PBS and fixed with 2% paraformaldehyde (Electron Microscopy Science) for 30 min at RT. Then fibroblasts were incubated with primary rabbit polyclonal anti alpha smooth muscle actin (αSMA) (Abcam, #ab5694), or rabbit polyclonal anti collagen I (Abcam, #ab34710) antibodies for 1 hr at RT, washed with PBS and incubated with the secondary antibody Goat Anti‐Rabbit IgG H&L (FITC) (Abcam, #ab6717), for 45 min. Cells were also stained with DAPI (4′,6′‐diamidino‐2‐phenylindole) (1 μg/ml) (Sigma‐Aldrich) and slides were observed by fluorescence microscope (Olympus BX53).
2.5. RNA extraction and semi‐quantitative reverse transcription (RT)‐PCR analysis
Cells were harvested in TRIzol Reagent (Life Technologies‐Invitrogen) and total RNA was isolated following the manufacturer's instructions. The first strand cDNA was synthesized from 2 μg of total RNA with MuLV reverse transcriptase kit (Applied Biosystems). Semi‐quantitative reverse‐transcribed (RT)‐PCR was carried out by using Hot‐Master Taq polymerase (Eppendorf) with 2 μl cDNA reaction and genes‐specific oligonucleotides under conditions of linear amplification. PCR products were run on a 2% agarose gel and visualized with ethidium bromide. The housekeeping 28S gene, used as internal standard, was amplified from the same cDNA reaction mixture. Densitometric analysis was applied to quantify mRNA levels compared to control gene expression.
2.6. Statistical analysis
Each experiment was performed at least three times. Results are reported as the mean ± SD. Statistical significance was determined using one‐way ANOVA analysis (post hoc Bonferroni) using GraphPad Prism software (San Diego, CA). A value of p < .05 was considered statistically significant.
2.7. HIPK2‐modulated pathways are involved in fibroblasts transdifferentiation
We have previously shown that loss of HIPK2 function in cancer cells increases metabolic profiles of phosphocholine, lactate, myo‐inositol and total creatine, involved in glycolysis, tricarboxylic acid cycle, and phosphatidylcholine metabolism, suggesting glycolytic activation.18 Metabolic reprogramming is characterized by increased cellular glucose uptake, hyperglycolysis, and lactate production and is considered a hallmark of cancer leading to tumor progression and adaptive/acquired resistance to antitumor therapy,19 another tumor promoting hallmark of cancer cells depleted of HIPK2 function. In addition, we previously found that HIPK2 knockdown upregulates NADPH oxidase 1 (Nox1), a homolog of the catalytic subunit of the superoxide‐generating NADPH oxidase, involved in tumor progression and generation of reactive oxygen species (ROS).20 The ROS production and the lactic acid release by cancer cells play an important role in fibroblast transdifferentiation into CAFs21 by inducing autophagy in stromal fibroblasts that reduces caveolin‐1 expression promoting the activation of fibroblasts into CAFs.22 Caveolin‐1 is highly expressed in fibroblasts, adipocytes, and endothelial cells and is involved in the regulation of several cellular processes, including cell signaling.23 Importantly, caveolin‐1 in fibroblasts can undergo autophagic degradation induced by tumor cells crosstalk and reduced caveolin‐1 expression is a hallmark of the aggressive CAF phenotype in cancer patients.24, 25 Autophagy is usually upregulated in cancer cells to cope with the shortage of nutrients and the hypoxic conditions in which these cells are forced to survive are among the main intracellular signal transducers sustaining autophagy.26 Here, for the first time we report that autophagy was induced in fibroblasts cultured with CMsiHIPK2, compared to fibroblasts cultured with CMsiRNA and that autophagy was counteracted by the ROS scavenger NAC (Figure 1b), suggesting that the oxidative stress of HIPK2 knockdown cancer cells could contribute to fibroblasts activation in the stumor stroma. Compared to control CMsiRNA, culturing fibroblasts with CMsiHIPK2 reduced caveolin‐1 levels that were rescued by blocking autophagy with CQ (Figure 1c); interestingly, we also found reduction of p53 levels in fibroblasts cultured with CMsiHIPK2 (Figure 1c, compare Lane 1 with Lane 3). Although the molecular mechanisms need to be clarified in our setting, loss of p53 in stromal fibroblasts has been shown to enhance tumor growth and metastasis and has been found in patients‐derived CAFs.27 The transdifferentiation CAF‐like was assessed by increased αSMA and collagen type I expression in fibroblasts cultured with CMsiHIPK2 compared to fibroblasts cultured with CMsiRNA (Figure 1d); moreover, we found upregulation of fibroblast activation protein (FAP) and cytokine IL‐6 expression in fibroblasts cultured with CMsiHIPK2 compared to fibroblasts cultured with CMsiRNA (Figure 1e); fibroblasts cultured with CM of RKO cells cultured in high glucose condition (CMHG) showed upregulation of FAP and cytokine IL‐6 expression (Figure 1e), similarly to fibroblasts cultured with CMsiHIPK2, in support of our findings that high glucose degrades HIPK2.14 The serine protease FAP is expressed in the tumor stroma but not in healthy tissues, which makes it an attractive candidate for targeting CAFs, thus studies involving targeting of FAP are under way and IL‐6 in the TME contributes to tumor cell growth, angiogenesis, invasion, and metastasis.28 Altogether, these findings make us to hypothesize that HIPK2 modulation in cancer cells could contribute to TME remodeling by promoting fibroblasts transdifferentiation CAF‐like although a direct link, the underlying molecular mechanisms and whether HIPK2 kinase activity plays a role, need to be clarified.
2.8. HIPK2 is degraded by high glucose condition
Given its pivotal role in negatively regulating several pathways involved in tumor progression, a functional HIPK2 protein is required for an efficient anticancer therapy response and to restrain tumor progression. Interestingly, we previously demonstrated that HIPK2 can undergo protein degradation in high glucose condition, inhibiting p53 apoptotic activity in response to chemotherapeutic drugs.14, 16, 17 This is a novel identified intratumoral condition impairing HIPK2 activity. As a proof of principle, HIPK2 degradation was achieved in cancer cells cultured in the presence of hyperglycemic sera derived from patients with type‐2 diabetes compared to normo‐glycemic sera derived from healthy donors.14 The relevance of these findings comes from the fact that high blood glucose levels are associated with type‐2 diabetes, metabolic syndrome, and obesity and high glycemic load strongly correlates with higher recurrence of colon cancer and resistance to chemotherapy‐induced cell death.29 Long‐term hyperglycemia, indeed, may also activate pro‐inflammatory factors such as interleukin‐6 (IL‐6), tumor necrosis factor‐a (TNF‐α), and cyclooxygenase‐2 (COX‐2)30 known to promote development and progression of cancer; thus, chronic inflammation is considered a hallmark of cancer.31
3. CONCLUSIONS
Overall, these data allow us to hypothesize that the loss of HIPK2 in cancer cells might deregulate pathways involved in cross‐talk with the TME in order to support tumor growth and resistance to therapies, although the molecular mechanisms of such interplay need to be clarified. We can hypothesize a role for HIF‐1, upregulated by HIPK2 knockdown32 or by HIPK2 degradation in high glucose or hypoxic conditions;14, 33 for intracellular ROS production following HIPK2 knockdown and NADPH oxidase upregulation;20 or for glycolytic activation following HIPK2 knockdown.18 The physiological conditions that trigger HIPK2 degradation are hypoxia and hyperglycemia. Hypoxia is a common metabolic condition found in solid tumors and impairs tumor chemosensitivity, induces angiogenesis and metastasis and reduces p53 apoptotic function.33, 34 Similarly, hyperglycemia has been shown to reduce cancer cell response to therapies, conferring resistance to drug‐induced cell death.29, 30 Interestingly, we have shown that it is possible to target hypoxia and hyperglycemia to rescue both HIPK2 and p53 function, restoring tumor response to therapies, in vitro and in vivo10, 35, 36 (Figure 2a). Therefore, these data strongly support our hypothesis that a mutual interaction may exist between cancer cells that have lost HIPK2 function and fibroblasts activation in the TME. It would be interesting to validate whether this interplay may sustain cancer progression, chemoresistance, invasion, and angiogenesis, likely in a reciprocal interplay (Figure 2b), in order to develop novel therapeutic anticancer approaches.
Figure 2.
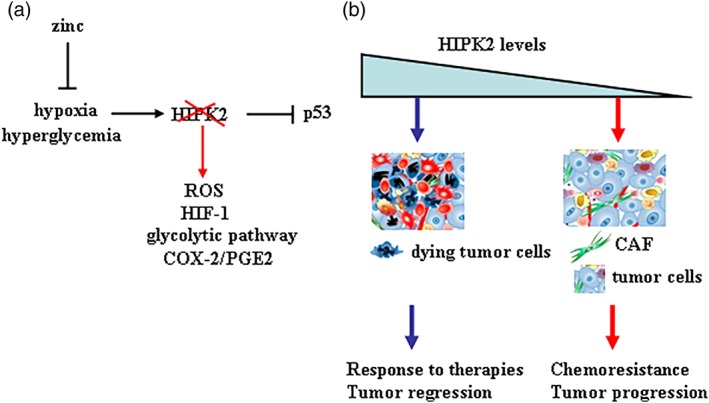
Schematic representation of HIPK2 function in TME. (a) Hypoxia and hyperglycemia induce HIPK2 degradation that consequently leads to impairment of p53 apoptotic function and induction of ROS, HIF‐1 activity, and so on. Zinc supplementation can counteract the effects of hypoxia and hyperglycemia on HIPK2/p53 axis. (b) Hypothetic model of interplay between cancer cells and the TME, in particular with the stromal fibroblasts, with respect of HIPK2 levels in cancer cells
The cross‐talk between tumor cells and CAFs has been also shown to produce immunosuppressive cytokines that promote polarization of M2‐like tumor‐associated macrophages (TAMs), with pro‐tumorigenic properties.28 Therefore, it would be interesting to evaluate whether the presence of CAF and TAM in patients‐derived tissues could correlate them with HIPK2 levels and with those of caveolin‐1 in CAF, in order to consider them markers of tumor progression. In addition, in vitro‐co‐culture systems comparing patients‐derived CAF with fibroblasts of normal tissues or culturing fibroblasts with primary cancer cells expressing different levels of HIPK2, would confirm the role of this interplay. The characterization of such dialogue may allow to define targetable pathways to be combined with biomarkers of chemoresistance and immunosuppression to be translated in clinic.
CONFLICT OF INTERESTS
The authors declare no potential conflict of interest.
ACKNOWLEDGMENTS
This study was supported by grants from the Italian Association for Cancer Research (IG 2013 11377, IG 2015 Id.16742) and Fondi Ateneo ex 60%,University “G. d'Annunzio.”
Garufi A, Traversi G, Cirone M, D'Orazi G. HIPK2 role in the tumor–host interaction: Impact on fibroblasts transdifferentiation CAF‐like. IUBMB Life. 2019;71:2055–2061. 10.1002/iub.2144
Funding information Italian Association for Cancer Research (AIRC), Grant/Award Numbers: IG 2013 11377, IG 2015 Id.16742
REFERENCES
- 1. Wang M, Zhao J, Zhang L, et al. Role of tumor microenvironment in tumorigenesis. J Cancer. 2017;8:761–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cirri P, Chiarugi P. Cancer associated fibroblasts: The dark side of the coin. Am J Cancer Res. 2011;1:482–497. [PMC free article] [PubMed] [Google Scholar]
- 3. Tao L, Huang G, Song H, Chen Y, Chen L. Cancer associated fibroblasts: An essential role in the tumor microenvironment. Oncol Lett. 2017;14:2611–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Feng Y, Zhou L, Sun X, Li Q. Homeodomain‐interacting protein kinase 2 (HIPK2): A promising target for anti‐cancer therapies. Oncotarget. 2017;8:20452–20461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. D'Orazi G, Rinaldo C, Soddu S. Updates on HIPK2: A resourceful oncosuppressor for clearing cancer. J Exp Clin Cancer Res. 2012;31:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Puca R, Nardinocchi L, Gal H, et al. Reversible dysfunction of wild‐type p53 following homeodomain‐interacting protein kinase‐2 knockdown. Cancer Res. 2008;15:3707–3714. [DOI] [PubMed] [Google Scholar]
- 7. Stanga S, Lanni C, Govoni S, Uberti D, D'Orazi G, Racchi M. Unfolded p53 in the pathogenesis of Alzheimer's disease: Is HIPK2 the link? Aging (Albany NY). 2010;2:545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Krishnamurthy N, Kurzock R. Targeting the Wnt/beta‐catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat Rev. 2018;62:50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nodale C, Sheffer M, Jacob‐Hirsch J, et al. HIPK2 downregulates vimentin and inhibits breast cancer cell invasion. Cancer Biol Ther. 2012;13:198–205. [DOI] [PubMed] [Google Scholar]
- 10. Nardinocchi L, Puca R, Givol D, D'Orazi G. HIPK2‐a therapeutical target to be (re)activated for tumor suppression: Role in p53 activation and HIF‐1alpha inhibition. Cell Cycle. 2010;9:1–6. [DOI] [PubMed] [Google Scholar]
- 11. Soni S, Padwad YS. HIF‐1 in cancer therapy: Two decade long story of a transcription factor. Acta Oncol. 2017;56:503–515. [DOI] [PubMed] [Google Scholar]
- 12. Bon G, Di Carlo SE, Folgiero V, et al. Negative regulation of beta(b) integrin transcription by homeodomain‐interacting protein kinase e and p53 impairs tumor progression. Cancer Res. 2009;69:5978–5986. [DOI] [PubMed] [Google Scholar]
- 13. Garufi A, Pistritto G, Ceci C, et al. Targeting COX‐2/PGE(2) pathway in HIPK2 knockdown cancer cells: Impact on dendritic cell maturation. PLoS One. 2012;7:e48342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baldari S, Garufi A, Granato M, et al. Hyperglicemia triggers HIPK2 protein degradation. Oncotarget. 2017;8:1190–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baldari S, Ubertini V, Garufi A, D'Orazi G, Bossi G. Targeting MKK3 as a novel anticancer strategy: Molecular mechanisms and therapeutical implications. Cell Death Dis. 2015;6:e1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Garufi A, D'Orazi G. High glucose dephosphorylates serine 46 and inhibits p53 apoptotic activity. J Exp Clin Cancer Res. 2014;33:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Garufi A, Pistritto G, Baldari S, Toietta G, Cirone M, D'Orazi G. p53‐dependent PUMA to DRAM antagonistic interplay as a key molecular switch in cell‐fate decision in normal/high glucose conditions. J Exp Clin Cancer Res. 2017;36:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Garufi A, Ricci A, Trisciuoglio D, et al. Glucose restriction induces cell death in parental but not in HIPK2 depleted RKO colon cancer cells: Molecular mechanisms and implications for tumor therapy. Cell Death Dis. 2013;4:e639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yoshida GJ. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J Exp Clin Cancer Res. 2015;34:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Puca R, Nardinocchi L, Starace G, et al. Nox1 is involved in p53 deacetylation and suppression of its transcriptional activity and apoptosis. Free Rad Biol Med. 2010;48:1338–1346. [DOI] [PubMed] [Google Scholar]
- 21. Arcucci A, Ruocco MR, Granato G, Sacco AM, Montagnani S. Cancer: An oxidative crosstalk between solid tumor cells and cancer associated fibroblasts. BioMed Res Inter. 2016;4502846:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gonzalez CD, Alvarez S, Ropolo A, Rosenzvit C, Gonzalez Bagnes MF, Vaccaro MI. Autophagy, Warburg, and Warburg reverse effects in human cancer. BioMed Res Inter. 2014;2014:926729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen D, Che G. Value of caveolin‐1 in cancer progression and prognosis: Emphasis on cancer‐associated fibroblasts, human cancer cells and mechanism of caveolin‐1 expression. Oncol Lett. 2014;8:1409–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Martinez‐Outschoorn UE, Pavlides S, Whitaker‐Menezes D, et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin‐1 degradation. Cell Cycle. 2010;12:2423–2433. [DOI] [PubMed] [Google Scholar]
- 25. Shen XJ, Zhang H, Tang GS, et al. Caveolin‐1 is a modulator of fibroblast activation and a potential biomarker for gastric cancer. Int J Biol Sci. 2015;11:370–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Diff. 2015;22:377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bar J, Feniger‐Barish R, Lukashchuk N, et al. Cancer cells suppress p53 in adjacent fibroblasts. Oncogene. 2009;28:933–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Roma‐Rodrigues C, Mendes R, Baptista PV, Fernandes AR. Targeting tumor microenvironment for cancer therapy. Int J Mol Sci. 2019;20:840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ryu TY, Park J, Scherer PE. Hyperglycemia as a risk factor for cancer progression. Diabetes Metab J. 2014;38:330–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Duan W, Shen X, Lei J, et al. Hyperglycemia, a neglected factor during cancer progression. BioMed. Res. Inter. 2014;2014:461917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Korniluk A, Koper O, Kemona H, Dymicka‐Piekarska V. From inflammation to cancer. Ir J Med Sci. 2017;186:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nardinocchi L, Puca R, Guidolin D, et al. Transcriptional regulation of hypoxia‐inducible factor 1alpha by HIPK2 suggests a novel mechanism to restrain tumor growth. Biochim Biophys Acta. 2009;1793:368–377. [DOI] [PubMed] [Google Scholar]
- 33. Nardinocchi L, Puca R, D'Orazi G. HIF‐1α antagonizes p53‐mediated apoptosis by triggering HIPK2 degradation. Aging (Albany NY). 2011;3:33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Petrova V, Annicchiarico‐Petruzzelli M, Melino G, Amelio I. The hypoxic tumour microenvironment. Oncogene. 2018;7:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Margalit O, Simon AJ, Yakubov E, et al. Zinc supplementation augments in vivo antitumor effect of chemotherapy by restoring p53 function. Int J Cancer. 2012;131:E562–E568. [DOI] [PubMed] [Google Scholar]
- 36. Garufi A, Trisciuoglio D, Cirone M, D'Orazi G. ZnCl2 sustains the adriamycin‐induced cell death inibite by high glucose. Cell Death Dis. 2016;7:e2280. [DOI] [PMC free article] [PubMed] [Google Scholar]