

Abstract
In a systematic study of the Au‐catalyzed reaction of o‐alkynylphenols with aryldiazonium salts, we find that essentially the same reaction conditions lead to a change in mechanism when a light source is applied. If the reaction is carried out at room temperature using a AuI catalyst, the diazonium salt undergoes electrophilic deauration of a vinyl AuI intermediate and provides access to substituted azobenzofurans. If the reaction mixture is irradiated with blue LED light, C−C bond formation due to N2‐extrusion from the diazonium salt is realized selectively, using the same starting materials without the need for an additional photo(redox) catalyst under aerobic conditions. We report a series of experiments demonstrating that the same vinyl AuI intermediate is capable of producing the observed products under photolytic and thermal conditions. The finding that a vinyl AuI complex can directly, without the need for an additional photo(redox) catalyst, result in C−C bond formation under photolytic conditions is contrary to the proposed mechanistic pathways suggested in the literature till date and highlights that the role of oxidation state changes in photoredox catalysis involving Au is thus far only poorly understood and may hold surprises for the future. Computational results indicate that photochemical activation can occur directly from a donor–acceptor complex formed between the vinyl AuI intermediate and the diazonium salt.
Keywords: diazo compound, homogeneous gold catalysis, mechanistic divergence, photochemistry, vinyl gold intermediate
The Au‐catalyzed reaction of o‐alkynylphenols with aryldiazonium salts undergoes a change in mechanism when a light source is applied. If the reaction is carried out at room temperature using a AuI catalyst, the diazonium salt undergoes electrophilic deauration of a vinyl AuI intermediate generating substituted azobenzofurans. If the reaction mixture is irradiated with blue LED light, C−C bond formation due to N2‐extrusion from the diazonium salt is observed.
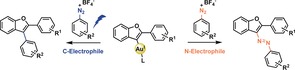
Photoredox catalysis has emerged over the past years to a highly competitive research area and many advances have been made.1 In particular, Au‐catalyzed transformations2 that make use of light in combination with diazonium salts have been identified.3 In these transformations diazonium salts commonly serve as aryl donors, leading to the extrusion of N2 (for representative examples see Scheme 1 A).3k, 3v, 3w In 2016 the group of Fensterbank reported the synthesis of arylated benzofurans from o‐alkynylphenols combining a photo(redox) catalyst (Ru(bpy)3(PF6)2) and a Au catalyst.3s This reaction interested us in two ways: 1) Some of us recently reported reactions involving diazonium salts that did not require a photo(redox) catalyst3h, 3v and wondered if this reaction could also be carried out in the absence of an additional photo(redox) catalyst. 2) We were curious if there was a way to probe the involvement of a vinyl Au intermediate.2f, 4, 5
Scheme 1.
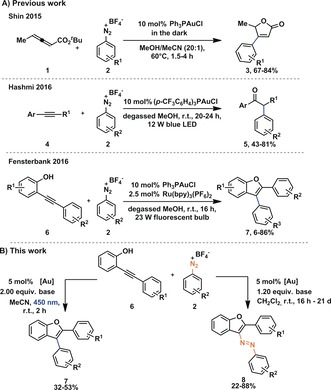
A) Selected examples for arylative Au photoredox catalysis.3s, 3v, 3w B) Divergence in Au‐catalyzed reactions leading to formation of arylated benzofurans 7 (N2‐extrusion) without additional photo(redox) catalyst and substituted azobenzofurans 8 (N2‐retention).
Our investigation led us to find that under very similar reaction conditions o‐alkynylphenols 6 can be either converted to arylated benzofurans 7 photolytically or in the absence of a light source to the formation of azobenzofurans 8. With this observation we demonstrate the first application of diazonium salts as electrophiles with vinyl AuI intermediates that do not lead to N2‐extrusion and form a C−N bond (Scheme 1 B).
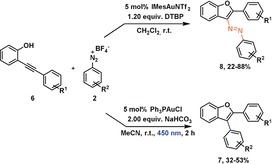
We began our studies by first exploring the reactivity of o‐alkynylphenol 6Me towards diazonium salt 2H by simply varying the base, the counter anion of the Au complex and the solvent using blue LED light (Table 1). In these experiments we omitted the previously used Ru(bpy)3(PF6)2 additive. We quickly realized that simple Ph3PAuCl in combination with NaHCO3 results in reasonable yields of the desired product 7Me. Unlike reported before,3s changing from the chloride anion to the weakly coordinating anion Tf2N− had very little effect on the outcome of the reaction. We note that these reactions are operationally extremely straightforward. All reagents were simply combined in a vial without precautions to exclude moisture or oxygen before being exposed to a light source.
Table 1.
Comparison of reactions conditions for the mechanistic divergence.
|
Entry |
Catalyst Base |
Yield 7Me [%] Irradiation with blue‐LED[a,c] |
Yield 8Me [%] No irradiation[a,b] |
|
|---|---|---|---|---|
|
|
|
MeCN |
CH2Cl2 |
MeCN |
|
1 |
Ph3PAuCl NaHCO3 |
46[d,e] |
5 |
7 |
|
|
|
|
|
|
|
2 |
Ph3PAuNTf2 NaHCO3 |
54[d,e] |
22[d] |
11 |
|
|
|
|
|
|
|
3 |
Ph3PAuCl DTBP |
4 |
not observed |
not observed |
|
|
|
|
|
|
|
4 |
Ph3PAuNTf2 DTBP |
11 |
19 |
23 |
[a] Averages of duplicate runs are given. [b] General conditions: 6Me (50 μmol), [Au] (5.00 mol %), 2H (100 μmol), base (100 μmol), solvent (500 μL), r.t., 24 h, determined via 1H NMR spectroscopy using benzyl acetate as internal standard. [c] General conditions: 6Me (100 μmol), [Au] (5.00 mol %), 2H (200 μmol), base (200 μmol), solvent (1 mL), r.t., 2 h, 450 nm light source, determined via GC‐MS using hexamethylbenzene as internal standard. [d] Full conversion of starting material. [e] With Au and a ruthenium photo(redox) catalyst present, similar yields were obtained with related substrates, see Ref. 3s.
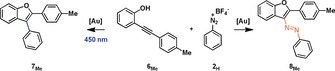
Interestingly, we observed small amounts of the azobenzene product 8Me in the absence of light. Notably, the yield of product 8Me increased when the less coordinating anion Tf2N− was used. An attempt to use a more soluble base 2,6‐di‐tert‐butyl‐pyridine (DTBP) had a detrimental effect on the reaction under photolytic conditions and changed little with regards to the formation of the azobenzene product 8Me. There are some conclusions that can be delineated from these experiments. Under photolytic conditions the use of simple Ph3PAuCl leads to arylated benzofuran 7Me, albeit in the absence of a photo(redox)catalyst and under aerobic conditions. The finding of azobenzofuran 8Me in the absence of light gives a strong indication that the reaction proceeds via a vinyl AuI intermediate and establishes, to the best of our knowledge, the first observation of a nitrogen‐based electrophile in C−N bond formation from a vinyl AuI complex. Control experiments confirm that the products 7Me and 8Me are not formed in the absence of a Au catalyst and that 7Me is not formed under the reaction conditions while irradiating 8Me (for these and several additional control experiments, see the Supporting Information (SI)).
The previously proposed mechanism by Fensterbank and co‐workers employing a photo(redox) catalyst3s proposes a photochemical oxidative addition of the diazonium salt to the AuI complex producing a highly Lewis acidic AuIII intermediate, that provides an open coordination site able to function as a π‐acid. Here, as in many other studies,2i, 3e, 3k, 3ab a vinyl AuIII intermediate is proposed to then form upon reaction with an o‐alkynylphenol substrate and reductive elimination results in the formation of the arylated benzofuran products. Stoichiometric experiments have indeed demonstrated that the photochemical oxidative addition of diazonium salts is feasible both in an inter‐ and intramolecular fashion.3v, 6 In the present case, varying the conditions of the reaction solely by irradiation with light yielded two different products (Table 1, entry 2). We wanted to investigate where this difference originates and if these two reactions have a common intermediate. We probed if oxidative addition of a diazonium salt to the (pre)catalyst is feasible by irradiating solutions of Ph3PAuCl or Ph3PAuNTf2 in the presence of a diazonium salt and base (NaHCO3). During a period of 2 h we did not observe changes to the concentrations of the diazonium salt or Au complexes (see SI). This result suggests that oxidative addition is not feasible in the present case. Together with the observation that azobenzofuran 8Me forms we can speculate that a vinyl AuI complex can form under the reaction conditions. We decided to prepare vinyl AuI complex 9 following a reported procedure4c and treated it with diazonium salt 2H (Scheme 2).
Scheme 2.
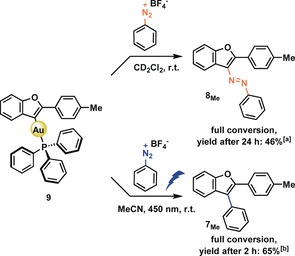
Reactivity of the isolated vinyl AuI complex 9 towards diazonium salt 2H with and without irradiation. Conditions: 60 mm, 1.10 equiv. reactant. [a] Determined by 1H NMR spectroscopy, using benzyl acetate as internal standard. [b] Determined by GC‐MS, using hexamethylbenzene as internal standard.
Without irradiation, azocompound 8Me was obtained in 46 % yield after 24 h, as determined by 1H NMR spectroscopy. Both 9 and 8Me could be characterized by single‐crystal X‐ray structure analysis (Figure 1).
Figure 1.
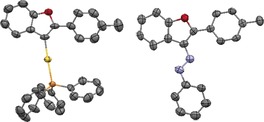
Solid state X‐ray structure of vinyl AuI complex 9 (left) and azobenzofuran 8Me (right). Thermal ellipsoids are shown at 50 % probability, hydrogen atoms are omitted for clarity.
When we carried this reaction out in the presence of a light source, we observed arylated benzofuran 7Me after 2 h in 65 % yield (Scheme 2). We note that both reactions gave full conversion. A by‐product formed during both reactions is 2‐(p‐tolyl)benzofuran 10, which arises from protodeauration. 10 is also formed under catalytic conditions (see SI), where a large excess of acidic 6Me is present, which we suspect to serve as a proton donor. We confirmed this hypothesis by treating 9 with 6Me and PhOH as proton sources which proceeded slowly to 10 (see SI). As irradiation of Ph3PAuCl or Ph3PAuNTf2 in the presence of a diazonium salt and base did not show any reaction in the absence of substrate 6Me, we might postulate that the formation of a vinyl AuI complex precedes potential oxidative addition. The stoichiometric reactions between 9 and diazonium salt 2H also demonstrate the intrinsically nucleophilic nature of these complexes. These experiments clearly demonstrate that vinyl AuI complexes can form under the reaction conditions and may serve as intermediates in these transformations, where they are of key relevance to the photochemical activation required for N2‐extrusion. The mechanistic scenarios we propose are outlined in Scheme 3.
Scheme 3.
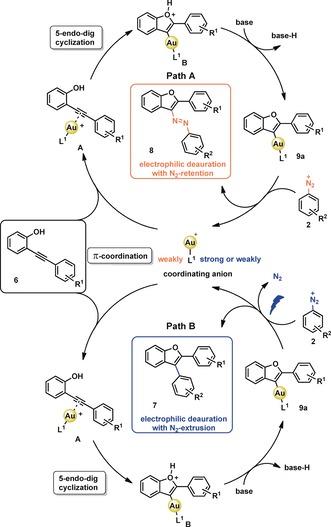
Proposed mechanisms for the divergent Au catalysis.
We propose, that both reaction pathways proceed through π‐coordination of the catalyst A. The subsequent activation of the triple bond leads to intramolecular nucleophilic attack of the oxygen atom and forms intermediate B via a 5‐endo‐dig cyclization (Scheme 3). Deprotonation of intermediate B by base provides vinyl AuI complex 9 a. Once the formation of a vinyl AuI intermediate is complete, the diazonium salt 2 reacts either as a N‐electrophile to form the azobenzofuran product 8 under N2‐retention (Scheme 3 , Path A) or as a C‐electrophile to give arylated benzofuran 7 with concomitant N2‐extrusion (Scheme 3 , Path B). At this point we cannot differentiate if the photochemical activation after formation of the vinyl AuI intermediate 9 a leads to oxidative addition or results in direct C−C bond formation from the vinyl Au complex 9 a due to loss of N2. Interestingly, a related stoichiometric experiment between an isolated vinyl AuI complex and a diazonium salt was reported by Shin and co‐workers.3w In this case, however, a catalytic amount of Ru(bpy)3(PF6)2 was still required to achieve photochemical C−C bond formation, emphasizing the intrinsically different reactivity observed here in the absence of such an additive. Very recently Fensterbank and co‐workers also described the reaction between an isolated vinyl AuI intermediate, essentially the same as the one reported here just differing by the phosphine ligand used, and an iodoalkyne which resulted in C−C bond formation under photolytic conditions.7 However, this reaction also benefitted largely from the presence of an Ir photosensitizer, which the authors attributed to energy transfer from an excited triplet state of the Ir complex to the vinyl AuI intermediate, where the subsequent C−C bond forming reaction takes place on the triplet surface. The reaction we report here does not make use of such an additive and yet efficiently results in C−C bond formation. We may therefore ask how photochemical activation promotes this reaction, especially as both reaction partners are essentially colorless (see Figure 2, top right). A possible scenario is the formation of a donor–acceptor complex between the vinyl AuI complex and the diazonium salt, which we explored computationally (for technical details and a more detailed description see SI). We first fully optimized the geometry of the non‐covalently bound complex I between the Au complex 9 and PhN2 + (see Figure 2, top left) at the TPSS‐D3(BJ)/def2‐SVP/PCM(MeCN) level of theory and then explored excited states through TD‐DFT calculations (CAM‐B3LYP‐D3(BJ)/def2‐SVPD/PCM(MeCN)).8 We found a low‐lying S1 excited state that corresponds to charge transfer from the HOMO, which is located at the electron rich vinyl AuI portion, to the LUMO, which is located on the PhN2 + unit (see Figure 2, middle). This is also very well reflected in the natural transition orbitals which are shown in the SI. Vertical excitation to the computed S1 state lies 45.8 kcal mol−1 above the S0 ground state (Figure 2, bottom left). Notably, we found that the triplet state (T1) within this geometry is energetically lower lying (29.7 kcal mol−1). Optimization of this triplet state II further lowers the energy to 13.2 kcal mol−1 and more interestingly results in elongation of the C−N bond distance in the PhN2 + unit from 1.362 to 1.449 Å with the spin density being distributed over both the vinyl Au and the PhN2 + fragments (Figure 2, bottom right), indicating C−N bond activation. These results show that photochemical activation from donor–acceptor complex I is possible and point us in the direction that subsequent steps may take place on the triplet surface, similar to the proposed pathway by Fensterbank and co‐workers.7 The exact pathway of internal conversion, intersystem crossing and especially the contribution from the heavy element Au remain to be elucidated and should give further insight on how Au‐catalyzed transformations can benefit from photochemical activation. We shall note here, that the non‐covalently bound complex I is also primed for C−N bond formation, the reaction we observe in the absence of a light source. Thus, complex I lies at the heart of the mechanistic divergence between the two pathways described in this paper.
Figure 2.
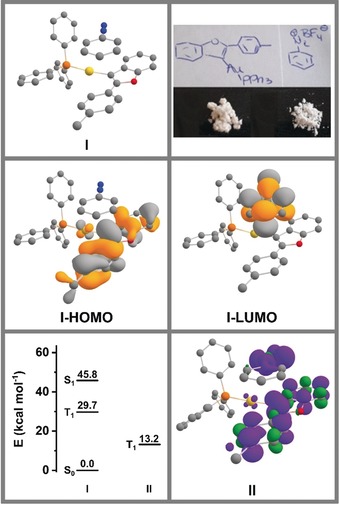
Top: Proposed donor–acceptor complex I relevant for photochemical activation and photos of 9 and PhN2BF4; Middle: depictions of the HOMO and LUMO of the donor–acceptor complex I; Bottom: Energy profile of the excited states for I and II and spin density plot for II. All data shown was obtained at the CAM‐B3LYP‐D3(BJ)/def2‐SVPD/PCM(MeCN)//TPSS‐D3(BJ)/def2‐SVP/PCM(MeCN) level of theory.
In both reaction paths shown in Scheme 3 the Au center has to be readily available to act as a π‐acid, a pathway that is favored when using a weakly coordinating anion, such as Tf2N− The photoreaction using Ph3PAuCl might therefore require the replacement of the counter anion (chloride) leading to a cationic species. As Table 1 entry 1 demonstrates that Ph3PAuCl even leads to small amounts of product 8Me, which we judge to fully rely on the π‐acidic nature of AuI, we believe that the generation of a cationic AuI intermediate is possible under the reaction conditions. Our study therefore demonstrates that the presence or absence of light can completely change the outcome of Au‐catalyzed reactions at the stage of a common vinyl AuI intermediate 9 a and that no additional photo(redox) catalyst is required.
With this mechanistic scenario in mind, we briefly probed the reaction conditions for a small set of substrates. We first continued our efforts to optimize the formation of azocompounds and replaced Ph3PAuNTf2 with the NHC‐Au complex IMesAuNTf2, which has also been shown previously to lead to stable vinyl AuI complexes,4c, 9 an intermediate that is key to this reaction. Following a brief screening we identified that the more soluble base DTBP in combination with IMesAuNTf2 resulted in improved yields (for screening, see SI). We then briefly investigated the scope of the reaction towards electron donating and electron withdrawing groups at the para‐position of the diazonium salt and the alkyne, respectively (Table 2).
Table 2.
Substrate scope for the formation of substituted azobenzofurans and arylated benzofurans.
|
Entry |
|
R1 |
R2 |
Time [h] |
Isolated yield [%] No irradiation |
|---|---|---|---|---|---|
|
1 |
8OMe |
OMe |
H |
16 |
88 |
|
2 |
8Me |
Me |
H |
27 |
78 |
|
3 |
8H |
H |
H |
168 |
22 |
|
4 |
8F |
F |
H |
21 d |
33 |
|
5 |
8Me,OMe |
Me |
OMe |
48 |
39 |
|
6 |
8Me,Me |
Me |
Me |
27 |
69 |
|
7 |
8Me,F |
Me |
F |
48 |
59 |
|
Entry |
|
R1 |
R2 |
Time [h] |
Isolated yield [%] Irradiation |
|---|---|---|---|---|---|
|
8 |
7Me |
Me |
H |
2 |
51 (64[a]) |
|
9 |
7Me,Me |
Me |
Me |
4 |
46 |
|
10 |
7Me,F |
Me |
F |
2 |
53 |
|
11 |
7Me,NO2 |
Me |
NO2 |
2 |
32 (34[b]) |
[a] 10 mol % [Au]. [b] Reaction time: 30 minutes.
The formation of the azocompound is favored if the aryl group on the alkyne contains an electron donating substituent, resulting in yields up to 88 % (Table 2, entry 1). The reaction time increases significantly and yields decrease as the substituent does not contain an electron donating group (entries 3–4). The substitution pattern of the diazonium salt also influences the reactivity. 2‐(p‐Tolylethynyl)phenol 6Me reacts in moderate yields with electron donating and withdrawing groups at the diazonium salt (entries 5–7). For the photochemical gold‐catalyzed C−C bond formation we probed reactivity using a set of diazonium salts with varying electronic properties and obtained the desired products in moderate yields (entries 8–11). In particular, we find that the yield for the diazonium salt with the most electron withdrawing group gave a surprisingly low yield, which is in contrast to the previous finding reported by Fensterbank and co‐workers which found this to be particularly high‐yielding.3s Upon closer inspection we noticed that the product was in fact partially consumed by the excess of diazonium salt used under photolytic conditions (see SI). A reduction of the reaction time to 30 min unfortunately did not increase the yield of product 7Me,NO2, yet resulted in full conversion of the starting material. One possible scenario for this more rapid reaction could be a more efficient photochemical activation from the proposed donor–acceptor complex I (see above). In addition, we found that for the more electron rich diazonium salt 2Me product formation was retarded (entry 9).
In summary, we have demonstrated that the mechanistic pathway of Au‐catalyzed reactions using diazonium salts can be altered through the application of a light source. If the reaction is carried out under blue‐LED irradiation, arylated benzofurans are obtained with N2‐extrusion from the diazonium salt under aerobic conditions without the need for an additional photo(redox) catalyst in moderate yields. If the reaction is carried out in the absence of a light source, the N2‐unit from the diazonium salt is retained and azobenzofurans are formed in moderate to high yields. Stoichiometric experiments demonstrate that these products are accessible from the same vinyl AuI intermediate and lead us to propose that the often suggested photochemical oxidative addition3a–3c, 3d–3l, 3o–3x, 3y–3ae either occurs after the formation of a vinyl AuI species or may even not involve oxidation state changes.
We expect our findings to further fuel the research area of Au‐catalyzed photoredox catalysis and inspire new strategies for transformations using light in combination with Au catalysts.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Prof. Dr. A. J. Minnaard (University of Groningen) for providing access to a photoreactor. S.T. is grateful for a Ph.D. scholarship from the German Academic Exchange Service (DAAD). J.E.M.N.K. acknowledges access to computational facilities at the Peregrine high‐performance computing cluster (University of Groningen) and access to the national computing facilities provided by the Netherlands Organisation for Scientific Research (NWO project number 17664). J.E.M.N.K. acknowledges funding from the Netherlands Organisation for Scientific Research (NWO START‐UP grant). A.S.K.H. is grateful for support by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy (2082/1–390761711).
S. Taschinski, R. Döpp, M. Ackermann, F. Rominger, F. de Vries, M. F. S. J. Menger, M. Rudolph, A. S. K. Hashmi, J. E. M. N. Klein, Angew. Chem. Int. Ed. 2019, 58, 16988.
Contributor Information
Prof. Dr. A. Stephen K. Hashmi, Email: hashmi@hashmi.de.
Dr. Johannes E. M. N. Klein, Email: j.e.m.n.klein@rug.nl.
References
- 1.For selected reviews on photoredox catalysis, see:
- 1a. McAtee R. C., McClain E. J., Stephenson C. R. J., Trends Chem. 2019, 1, 111–125; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Marzo L., Pagire S. K., Reiser O., König B., Angew. Chem. Int. Ed. 2018, 57, 10034–10072; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10188–10228; [Google Scholar]
- 1c. Revathi L., Ravindar L., Fang W. Y., Rakesh K. P., Qin H. L., Adv. Synth. Catal. 2018, 360, 4652–4698; [Google Scholar]
- 1d. Twilton J., Le C., Zhang P., Shaw M. H., Evans R. W., MacMillan D. W. C., Nat. Rev. Chem. 2017, 1, 0052; [Google Scholar]
- 1e. Menigaux D., Belmont P., Brachet E., Eur. J. Org. Chem. 2017, 2008–2055; [Google Scholar]
- 1f. Prier C. K., Rankic D. A., MacMillan D. W. C., Chem. Rev. 2013, 113, 5322–5363; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1g. Hari D. P., König B., Angew. Chem. Int. Ed. 2013, 52, 4734–4743; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 4832–4842. [Google Scholar]
- 2.For selected reviews on Au catalysis, see:
- 2a. Pflästerer D., Hashmi A. S. K., Chem. Soc. Rev. 2016, 45, 1331–1367; [DOI] [PubMed] [Google Scholar]
- 2b. Dorel R., Echavarren A. M., Chem. Rev. 2015, 115, 9028–9072; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Zhang L., Acc. Chem. Res. 2014, 47, 877–888; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2d. Ohno H., Isr. J. Chem. 2013, 53, 869–882; [Google Scholar]
- 2e. Braun I., Asiri A. M., Hashmi A. S. K., ACS Catal. 2013, 3, 1902–1907; [Google Scholar]
- 2f. Liu L.-P., Hammond G. B., Chem. Soc. Rev. 2012, 41, 3129–3139; For recent reviews on Au catalysis involving photochemical activation, see: [DOI] [PubMed] [Google Scholar]
- 2g. Zidan M., Rohe S., McCallum T., Barriault L., Catal. Sci. Technol. 2018, 8, 6019–6028; [Google Scholar]
- 2h. McCallum T., Rohe S., Barriault L., Synlett 2017, 28, 289–305; [Google Scholar]
- 2i. Zhang M., Zhu C., Ye L.-W., Synthesis 2017, 49, 1150–1157; [Google Scholar]
- 2j. Levin M. D., Kim S., Toste F. D., ACS Cent. Sci. 2016, 2, 293–301; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2k. Hopkinson M. N., Tlahuext-Aca A., Glorius F., Acc. Chem. Res. 2016, 49, 2261–2272. [DOI] [PubMed] [Google Scholar]
- 3.For selected examples on photoredox Au catalysis involving diazonium salts, see:
- 3a. Akram M. O., Banerjee S., Saswade S. S., Bedi V., Patil N. T., Chem. Commun. 2018, 54, 11069–11083; [DOI] [PubMed] [Google Scholar]
- 3b. Witzel S., Sekine K., Rudolph M., Hashmi A. S. K., Chem. Commun. 2018, 54, 13802–13804; [DOI] [PubMed] [Google Scholar]
- 3c. Xie J., Sekine K., Witzel S., Krämer P., Rudolph M., Rominger F., Hashmi A. S. K., Angew. Chem. Int. Ed. 2018, 57, 16648–16653; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 16890–16895; [Google Scholar]
- 3d. Tabey A., Berlande M., Hermange P., Fouquet E., Chem. Commun. 2018, 54, 12867–12870; [DOI] [PubMed] [Google Scholar]
- 3e. Chakrabarty I., Akram M. O., Biswas S., Patil N. T., Chem. Commun. 2018, 54, 7223–7226; [DOI] [PubMed] [Google Scholar]
- 3f. Dong B., Peng H., Motika S. E., Shi X., Chem. Eur. J. 2017, 23, 11093–11099; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3g. Bansode A. H., Shaikh S. R., Gonnade R. G., Patil N. T., Chem. Commun. 2017, 53, 9081–9084; [DOI] [PubMed] [Google Scholar]
- 3h. Witzel S., Xie J., Rudolph M., Hashmi A. S. K., Adv. Synth. Catal. 2017, 359, 1522–1528; [Google Scholar]
- 3i. Sauer C., Liu Y., De Nisi A., Protti S., Fagnoni M., Bandini M., ChemCatChem 2017, 9, 4456–4459; [Google Scholar]
- 3j. Deng J.-R., Chan W.-C., Chun-Him Lai N., Yang B., Tsang C.-S., Chi-Bun Ko B., Lai-Fung Chan S., Wong M.-K., Chem. Sci. 2017, 8, 7537–7544; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3k. Alcaide B., Almendros P., Busto E., Herrera F., Lázaro-Milla C., Luna A., Adv. Synth. Catal. 2017, 359, 2640–2652; [Google Scholar]
- 3l. Alcaide B., Almendros P., Aparicio B., Lázaro-Milla C., Luna A., Faza O. N., Adv. Synth. Catal. 2017, 359, 2789–2800; [Google Scholar]
- 3m. Gauchot V., Sutherland D. R., Lee A. L., Chem. Sci. 2017, 8, 2885–2889; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3n. Tlahuext-Aca A., Hopkinson M. N., Garza-Sanchez R. A., Glorius F., Chem. Eur. J. 2016, 22, 5909–5913; [DOI] [PubMed] [Google Scholar]
- 3o. Cornilleau T., Hermange P., Fouquet E., Chem. Commun. 2016, 52, 10040–10043; [DOI] [PubMed] [Google Scholar]
- 3p. Tlahuext-Aca A., Hopkinson M. N., Sahoo B., Glorius F., Chem. Sci. 2016, 7, 89–93; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3q. Gauchot V., Lee A.-L., Chem. Commun. 2016, 52, 10163; [DOI] [PubMed] [Google Scholar]
- 3r. Kim S., Rojas-Martin J., Toste F. D., Chem. Sci. 2016, 7, 85–88; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3s. Xia Z., Khaled O., Mouriès-Mansuy V., Ollivier C., Fensterbank L., J. Org. Chem. 2016, 81, 7182–7190; [DOI] [PubMed] [Google Scholar]
- 3t. Um J., Yun H., Shin S., Org. Lett. 2016, 18, 484–487; [DOI] [PubMed] [Google Scholar]
- 3u. Alcaide B., Almendros P., Busto E., Luna A., Adv. Synth. Catal. 2016, 358, 1526–1533; [Google Scholar]
- 3v. Huang L., Rudolph M., Rominger F., Hashmi A. S. K., Angew. Chem. Int. Ed. 2016, 55, 4808–4813; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 4888–4893; [Google Scholar]
- 3w. Patil D. V., Yun H., Shin S., Adv. Synth. Catal. 2015, 357, 2622–2628; [Google Scholar]
- 3x. Cai R., Lu M., Aguilera E. Y., Xi Y., Akhmedov N. G., Petersen J. L., Chen H., Shi X., Angew. Chem. Int. Ed. 2015, 54, 8772–8776; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 8896–8900; [Google Scholar]
- 3y. He Y., Wu H., Toste F. D., Chem. Sci. 2015, 6, 1194–1198; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3z. Hopkinson M. N., Sahoo B., Glorius F., Adv. Synth. Catal. 2014, 356, 2794–2800; [Google Scholar]
- 3aa. Shu X.-z., Zhang M., He Y., Frei H., Toste F. D., J. Am. Chem. Soc. 2014, 136, 5844–5847; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3ab. Sahoo B., Hopkinson M. N., Glorius F., J. Am. Chem. Soc. 2013, 135, 5505–5508; For related computational studies, see: [DOI] [PubMed] [Google Scholar]
- 3ac. Bhattacharjee R., Datta A., Chem. Eur. J. 2018, 24, 13636–13646; [DOI] [PubMed] [Google Scholar]
- 3ad. Liu Y., Yang Y., Zhu R., Liu C., Zhang D., Chem. Eur. J. 2018, 24, 14119–14126; [DOI] [PubMed] [Google Scholar]
- 3ae. Zhang Q., Zhang Z.-Q., Fu Y., Yu H.-Z., ACS Catal. 2016, 6, 798–808. [Google Scholar]
- 4.For selected examples of isolated vinyl AuI complexes, see:
- 4a. Wang W., Hammond G. B., Xu B., J. Am. Chem. Soc. 2012, 134, 5697–5705; [DOI] [PubMed] [Google Scholar]
- 4b. Hashmi A. S. K., Angew. Chem. Int. Ed. 2010, 49, 5232–5241; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 5360–5369; [Google Scholar]
- 4c. Hashmi A. S. K., Ramamurthi T. D., Rominger F., Adv. Synth. Catal. 2010, 352, 971–975; [Google Scholar]
- 4d. Hashmi A. S. K., Dondeti Ramamurthi T., Rominger F., J. Organomet. Chem. 2009, 694, 592–597; [Google Scholar]
- 4e. Hashmi A. S. K., Schuster A. M., Rominger F., Angew. Chem. Int. Ed. 2009, 48, 8247–8249; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 8396–8398; [Google Scholar]
- 4f. Liu L. P., Hammond G. B., Chem. Asian J. 2009, 4, 1230–1236; [DOI] [PubMed] [Google Scholar]
- 4g. Liu L.-P., Xu B., Mashuta M. S., Hammond G. B., J. Am. Chem. Soc. 2008, 130, 17642–17643; [DOI] [PubMed] [Google Scholar]
- 4h. Akana J. A., Bhattacharyya K. X., Müller P., Sadighi J. P., J. Am. Chem. Soc. 2007, 129, 7736–7737; [DOI] [PubMed] [Google Scholar]
- 4i. Hashmi A. S. K., Hutchings G. J., Angew. Chem. Int. Ed. 2006, 45, 7896–7936; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 8064–8105. [Google Scholar]
- 5.For selected examples of reactions involving vinyl AuI complexes, see:
- 5a. Zhdanko A., Maier M. E., ACS Catal. 2015, 5, 5994–6004; [Google Scholar]
- 5b. Zhdanko A., Maier M. E., Chem. Eur. J. 2014, 20, 1918–1930; [DOI] [PubMed] [Google Scholar]
- 5c. Hashmi A. S. K., Ramamurthi T. D., Todd M. H., Tsang A. S.-K., Graf K., Aust. J. Chem. 2010, 63, 1619–1626; [Google Scholar]
- 5d. Weyrauch J. P., Hashmi A. S. K., Schuster A., Hengst T., Schetter S., Littmann A., Rudolph M., Hamzic M., Visus J., Rominger F., Frey W., Bats J. W., Chem. Eur. J. 2010, 16, 956–963; [DOI] [PubMed] [Google Scholar]
- 5e. Weber D., Tarselli M. A., Gagné M. R., Angew. Chem. Int. Ed. 2009, 48, 5733–5736; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 5843–5846; [Google Scholar]
- 5f. Wegner H. A., Ahles S., Neuburger M., Chem. Eur. J. 2008, 14, 11310–11313; [DOI] [PubMed] [Google Scholar]
- 5g. Kar A., Mangu N., Kaiser H. M., Beller M., Tse M. K., Chem. Commun. 2008, 386–388; [DOI] [PubMed] [Google Scholar]
- 5h. Yu M., Zhang G., Zhang L., Org. Lett. 2007, 9, 2147–2150; [DOI] [PubMed] [Google Scholar]
- 5i. Worlikar S. A., Kesharwani T., Yao T., Larock R. C., J. Org. Chem. 2007, 72, 1347–1353; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5j. Nakamura I., Yamagishi U., Song D., Konta S., Yamamoto Y., Angew. Chem. Int. Ed. 2007, 46, 2284–2287; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 2334–2337; [Google Scholar]
- 5k. Nakamura I., Sato T., Terada M., Yamamoto Y., Org. Lett. 2007, 9, 4081–4083; [DOI] [PubMed] [Google Scholar]
- 5l. Kirsch S. F., Binder J. T., Crone B., Duschek A., Haug T. T., Liébert C., Menz H., Angew. Chem. Int. Ed. 2007, 46, 2310–2313; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 2360–2363; [Google Scholar]
- 5m. Crone B., Kirsch S. F., J. Org. Chem. 2007, 72, 5435–5438; [DOI] [PubMed] [Google Scholar]
- 5n. Hashmi A. S. K., Rudolph M., Schymura S., Visus J., Frey W., Eur. J. Org. Chem. 2006, 4905–4909. [Google Scholar]
- 6.
- 6a. Kim S., Toste F. D., J. Am. Chem. Soc. 2019, 141, 4308–4315; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Huang L., Rominger F., Rudolph M., Hashmi A. S. K., Chem. Commun. 2016, 52, 6435–6438; [DOI] [PubMed] [Google Scholar]
- 6c. Tlahuext-Aca A., Hopkinson M. N., Daniliuc C. G., Glorius F., Chem. Eur. J. 2016, 22, 11587–11592; [DOI] [PubMed] [Google Scholar]
- 6d. Asomoza-Solís E. O., Rojas-Ocampo J., Toscano R. A., Porcel S., Chem. Commun. 2016, 52, 7295–7298. [DOI] [PubMed] [Google Scholar]
- 7. Xia Z., Corcé V., Zhao F., Przybylski C., Espagne A., Jullien L., Le Saux T., Gimbert Y., Dossmann H., Mouriès-Mansuy V., Ollivier C., Fensterbank L., Nat. Chem. 2019, 11, 797–805. [DOI] [PubMed] [Google Scholar]
- 8.For a complete list of references regarding the computational methods chosen see the Supporting Information.
- 9.
- 9a. Döpp R., Lothschütz C., Wurm T., Pernpointner M., Keller S., Rominger F., Hashmi A. S. K., Organometallics 2011, 30, 5894–5903; [Google Scholar]
- 9b. Roth K. E., Blum S. A., Organometallics 2010, 29, 1712–1716. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary