

Summary
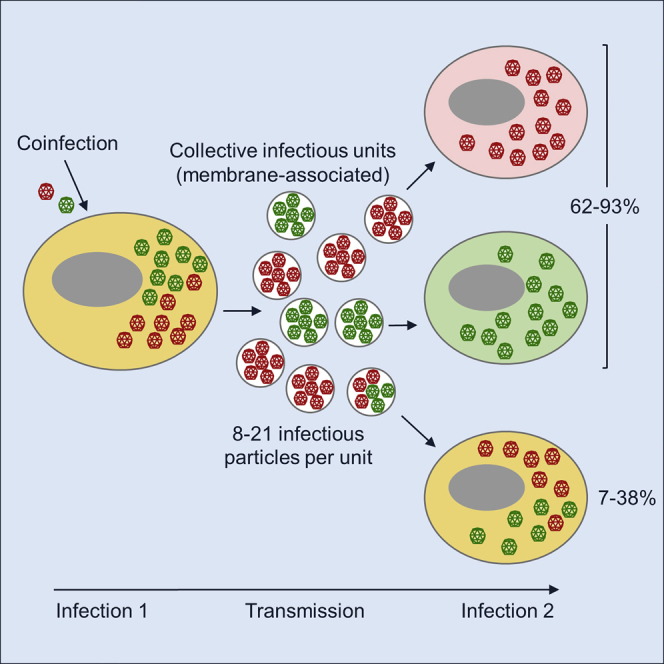
Some viruses are released from cells as pools of membrane-associated virions. By increasing the multiplicity of infection (MOI), this type of collective dispersal could favor viral cooperation, but also the emergence of cheater-like viruses such as defective interfering particles. To better understand this process, we examined the genetic diversity of membrane-associated coxsackievirus infectious units. We find that infected cells release membranous structures (including vesicles) that contain 8–21 infectious particles on average. However, in most cases (62%–93%), these structures do not promote the co-transmission of different viral genetic variants present in a cell. Furthermore, collective dispersal has no effect on viral population sequence diversity. Our results indicate that membrane-associated collective infectious units typically contain viral particles derived from the same parental genome. Hence, if cooperation occurs, it should probably involve sibling viral particles rather than different variants. As shown by social evolution theory, cooperation among siblings should be robust against cheater invasion.
Keywords: enterovirus, viral transmission, collective infectious unit, extracellular vesicles, social evolution
Graphical Abstract
Highlights
-
•
Pools of membrane-associated virions are shed from coxsackievirus-infected cells
-
•
These pools mediate the collective spread of 8–21 infectious particles on average
-
•
Coxsackievirus collective spread typically involves sibling viral genomes
-
•
This spread mode prevents interactions among different virus genetic variants
Enteroviruses can be transmitted as pools of membrane-associated virions. Bou et al. show that these pools tend to be constituted by sibling viral genomes, preventing the widespread mixing of different virus variants.
Introduction
Many viruses remodel endoplasmic reticulum membranes to form distinct organelles or viral factories used for viral replication and assembly (Altan-Bonnet, 2017, Belov, 2016, Fernández de Castro et al., 2016, Hsu et al., 2010, Shulla and Randall, 2016, Romero-Brey and Bartenschlager, 2016, Ravindran et al., 2016). Viral release from cells can also take place in a membrane-associated manner. Notably, hepatitis A virus (Feng et al., 2013), enteroviruses (Robinson et al., 2014, Bird et al., 2014, Chen et al., 2015), noroviruses (Santiana et al., 2018), rotaviruses (Santiana et al., 2018), and Marseilleviruses (Arantes et al., 2016) are secreted from cells as pools of virions inside extracellular vesicles. Such collective dispersal contrasts with a more classical model according to which viral dissemination takes place mainly through free virions (Sanjuán, 2017, Altan-Bonnet, 2016, Sanjuán, 2018, Mutsafi and Altan-Bonnet, 2018). The biological relevance of this type of transmission is supported by several lines of evidence. For instance, enterovirus-containing vesicles are highly infectious and produce large amounts of viral progeny (Chen et al., 2015). Also, rotavirus-containing vesicles were found to be more infectious than free virions via the gastrointestinal entry route in mice (Santiana et al., 2018). However, how membrane-associated spread determines virus-virus interactions and how this impacts viral fitness, genetic diversity, and evolution is still largely unknown.
An important feature of virus collective dispersal is that it should elevate the cellular MOI, defined as the average number of viral genomes that initiate the infection of a cell. Elevating the MOI may increase viral fitness via different processes. For instance, it could reduce the chances that essential viral proteins are missing during the early stages of the infection cycle due to insufficient expression levels, dilution, or degradation. Alternatively, invading a cell with multiple viral genome copies could allow the virus to overcome early infection barriers. This may be achieved, for instance, by overwhelming antiviral factors or by accelerating the infection cycle, which would provide the virus a head start against cellular innate immune responses (Sanjuán and Thoulouze, 2019). A link between collective spread, the MOI, and infection speed has been established for HIV-1 (Boullé et al., 2016) and vesicular stomatitis virus (Andreu-Moreno and Sanjuán, 2018).
Additionally, it has been suggested that increasing the MOI could facilitate genetic complementation among deleterious mutants, enabling a diversity-based type of cooperation, particularly in fast-mutating RNA viruses (Vignuzzi et al., 2006, Bordería et al., 2015, Lauring and Andino, 2010, Domingo and Perales, 2018, Villarreal and Witzany, 2015, Andino and Domingo, 2015, Leeks et al., 2018). Consequently, it has been postulated that collective viral spread through vesicles may increase viral fitness by promoting diversity-based cooperation (Chen et al., 2015, Altan-Bonnet and Chen, 2015, Sanjuán, 2017, Sanjuán and Thoulouze, 2019). A similar argument has been put forward for other types of collective spread such as virion aggregates in poliovirus (Aguilera et al., 2017), polyploid capsids in measles virus (Shirogane et al., 2012), and occlusion bodies in the large DNA baculoviruses (Simón et al., 2013, Clavijo et al., 2010).
On the other hand, it has been amply demonstrated that high MOIs promote the evolution of defective interfering particles and other types of defective viruses that replicate at the expense of fully functional viruses (Marriott and Dimmock, 2010, Rezelj et al., 2018, Manzoni and López, 2018, Brooke, 2017). Because defective viruses behave as social cheaters that benefit from fully functional “helper” viruses without reciprocating, they obtain a competitive advantage that allows them to take over the population at high MOIs, reducing average population fitness severely and increasing extinction risk (Turner and Chao, 1999, Díaz-Muñoz et al., 2017, Chao and Elena, 2017, Grande-Pérez et al., 2005). Thus, collective dispersal might select for defective virus variants.
The evolution of defective viruses, but also diversity-based cooperation, critically require that cells are coinfected with different genetic variants of a virus. However, whether such coinfection is favored by membrane-associated virus intercellular transmission has not been tested. Here, we set out to clarify this using coxsackievirus B3 (CVB3) as a model system. Previous work has shown that CVB3 and other enteroviruses are released from cells inside autophagosome-like, phosphatidylserine-rich vesicles containing multiple virions (Robinson et al., 2014, Chen et al., 2015). We find that, in cells coinfected with two phenotypically marked CVB3 variants, pools of membrane-associated infectious particles released from cells tend to contain only one of the two variants. We also find that ongoing intercellular transmission of these multi-virion structures does not change overall population genetic diversity appreciably, compared with free-virion transmission. Our results suggest that membrane-associated viruses released from infected cells tend to originate from the same parental genome. This may help enteroviruses increase MOIs while keeping genetic relatedness high, preventing invasion by defective viruses. On the other hand, it seems unlikely that this type of collective spread could promote diversity-based cooperation.
Results
Low-Speed Centrifugation Selects for Collective Infectious Units Constituted by Pools of Membrane-Associated Virions
We inoculated confluent HeLa-H1 cells with CVB3 Nancy strain at a viral density of D = 10 plaque-forming units (PFUs) per cell and harvested the culture medium of these virus-producer cells after completion of the infectious cycle (12 h post inoculation [hpi]). Notice that we use the term “viral density” (the ratio of PFUs to cells in a given population or predefined space) to avoid confusion with the cellular MOI, as defined above (the average number of viral genomes that initiate the infection of a cell). We centrifuged the harvested medium at 10,000 × g to sediment large infectious units, including previously described autophagosome-derived vesicles (Robinson et al., 2014, Chen et al., 2015). To more efficiently separate these two subpopulations, we iterated this process three times in total (Figure 1A). We then analyzed the infectivity of each centrifugation fraction by the plaque assay (Figure 1C). The first supernatant (S1) contained (2.4 ± 0.4) × 108 PFUs/mL versus (5.0 ± 0.4) × 106 PFUs/mL in the third resuspended pellet (P3). Filtration of the P3 fraction through 0.1-μm pores reduced its titer by a factor of 15.8 ± 1.0-fold, versus only 1.3 ± 0.1 for the S1 fraction, confirming that the P3 fraction contained large infectious units. We then treated the P3 fraction with Triton X-100 detergent to disrupt membranes. Notably, this increased the titer by a factor of 11.6 ± 1.1, indicating that the large infectious units pelleted by slow-speed centrifugation were collective infectious units (CIUs) constituted by pools of membrane-associated viruses. Based on the above titers and the effect of detergent treatment, these CIUs contained 19.5% ± 1.1% of the total infectious viral progeny at harvest time. This percentage might indeed be higher if some membranous structures were not recovered in the P3 fraction, if detergent treatment did not fully disrupt membranes, or if the S1 fraction already contained infectious virions released from membranes, for instance, due to spontaneous vesicle breakage.
Figure 1.

Fast-Sedimenting CIUs Contain Membrane-Associated Virion Pools
(A) Scheme of the process used to separate CIUs from free virions by low-speed centrifugation. Three serial centrifugation and resuspension steps were carried out in which we separated the supernatant (S) and pellet (P) fractions. P fractions were subjected to Triton detergent to disrupt membranes and release free virions (B fraction).
(B) Transmission electron micrographs of membrane-associated virions obtained from the P3 fraction. The small white bars correspond to 30 nm, the diameter of enterovirus virions. The observed sizes of virion-like structures are close to this size, although often smaller, which is expected if the virion section is not diametrical.
(C–E) Infectivity of the indicated fraction quantified by the plaque assay. The mean and SEM (error bars) titers obtained from three independent assays are shown.
(C) Culture media harvested at 12 hpi.
(D) Culture media harvested at 8 hpi.
(E) Culture media harvested at 12 hpi in which the P2 fraction was purified using annexin V (P2∗ and B2∗).
Inspection of the P3 fraction by transmission electron microscopy confirmed the presence of virion-containing vesicles of different sizes ranging from 100 nm to >1 μm (Figure 1B). Given the small size of enterovirus particles (30 nm), these vesicles should harbor large numbers of virions, potentially increasing the cellular MOI substantially. However, because our protocol simply selected for fast-sedimenting infectious units, the presence of other virion-containing structures could not be discarded. For instance, we might have also selected for replication organelles containing mature viral particles. Western blot analysis of the P3 fraction revealed the presence of LC3, a typical autophagosome marker (Kabeya et al., 2000). However, we also detected the non-processed form of the protein (pro-LC3), which in principle should be associated to the endoplasmic reticulum (Figure S1). Hence, although most of the fast-sedimenting infectious units obtained in the P3 fraction were probably virion-containing vesicles, we henceforth refer to membrane-associated CIUs to conservatively include both virion-containing vesicles and other possible types of lipid-bound viral particles.
Membrane-Associated CIUs Co-transmit CVB3 Genetic Variants Inefficiently
We set out to test whether viral genetic variants present in virus-producer cells were co-transmitted to cells in subsequent rounds of infection (recipient cells). For this, we used CVB3 viruses encoding EGFP or mCherry fluorescent proteins to inoculate HeLa-H1 cells at high viral density (D ≈ 10 PFUs/cell each) with a 1:1 mix of CVB3-EGFP and CVB3-mCherry, hence maximizing coinfection with both variants in producer cells. Flow cytometry confirmed that 99.0% of these cells were fluorescent at 7 hpi, and of these, 95.1% were positive for both EGFP and mCherry (Figure 2A). At 12 hpi, we used slow-speed centrifugation to collect the P3 fraction as above, and used this fraction to inoculate fresh cells at lower density (D ≈ 0.1 PFU/cell) to ensure that most cells received only 1 PFU. Consistent with the expected inoculum density, 9.01% ± 0.10% of the recipient cells were fluorescent at 7 hpi. Among fluorescent cells, 46.6% ± 0.6% were green, 40.4% ± 0.4% were red, and only 13.0% ± 0.5% were doubly fluorescent (see Figure 2B for a representative experiment; Table S1). If cells were infected by free virions independently, the expected percentage of doubly fluorescent cells would be 0.0901 × (0.466 + 0.130) × (0.404 + 0.130) = 2.86%. Hence, the observed percentage of doubly fluorescent cells was 4.5 times higher than expected under free-virion dispersal. However, because the vast majority of producer cells were doubly fluorescent and our high-weight CIUs contained multiple infectious particles, by sheer chance most recipient cells should be infected with both CVB3-EGFP and CVB3-mCherry variants. Yet, this expectation was in sharp contrast with the observed 13.0% ± 0.5% co-transmission frequency, indicating that membrane-associated CIUs typically contained viral progeny derived from the same parental genome.
Figure 2.

Infrequent Co-transmission of EGFP- and mCherry-Encoding CVB3 Viruses through CIUs
(A) Flow cytometry analysis of producer cells inoculated at high density (D ≈ 10 PFUs/cell) with a 1:1 mix of the two variants (culture media harvested at 12 hpi).
(B) Flow cytometry analysis of cells inoculated at lower density (D ≈ 0.1 PFU/cell) with the P3 fraction. Results for one replicate are shown. Cell counts for three replicates are shown in Table S1.
(C) Distribution of the estimated proportion of each variant in individual producer cells (g), rescaling the intensities shown in (A) such that the average rescaled intensities matched the observed proportion of cells infected with each variant (46.4% ± 0.4% CVB3-EGFP versus 53.6% ± 0.4% CVB3-mCherry; Table S1). This was achieved by multiplying all EGFP intensities by 1.82. The resulting coefficients of variation of mCherry and rescaled EGFP intensities were similar (0.72 and 0.68, respectively).
(D) Boxplot of the probability that CIUs containing n infectious particles co-transmitted both variants, assuming random mixing. This probability was calculated as p(n) = 1 − gn − (1 − g)n. The lower and upper limits of the box indicate 25th and 75th percentiles, and the middle line shows the median. Whiskers show the 10th and 90th percentiles, and crosses the 5th and 95th percentiles. Superimposed are shown the n value and observed proportion of coinfected cells in four different cases: P3 fraction harvested at 12 hpi (red), P3 fraction harvested at 12 hpi accounting for possible contamination with free virions (pink), P3 fraction harvested at 8 hpi (brown), and P2∗ fraction harvested at 12 hpi (blue; see text for details). The mean and SEM from three independent assays are shown.
Among-Cell Fluctuations in the Proportion of Each Virus Variant Do Not Explain Low Co-transmission Frequencies
The EGFP- and mCherry-encoding variants were produced at similar overall frequencies, because 46.4% ± 0.4% of recipient cells were EGFP positive versus 53.6% ± 0.4% mCherry positive. However, this proportion may fluctuate among individual cells, leading to potentially large differences in the frequency of each variant in CIUs. To measure this variability, we used the intensity of the EGFP and mCherry signal in each cell as determined by flow cytometry. To better correlate fluorescence intensity with virus abundance, we rescaled the fluorescence signal such that the average signal intensities matched the above percentages of EGFP and mCherry progeny viruses. This allowed us to determine the per-cell statistical distribution of the estimated proportion of EGFP variant to total (EGFP + mCherry) virus in the viral progeny released by producer cells, which we called g (Figure 2C). If the two variants mixed freely, the probability of a CIU containing both variants would be p(n) = 1 − gn − (1 − g)n, where n is the number of infectious particles per CIU. For each n, we calculated the distribution of p(n) based on the distribution of g. This showed that, for n = 12 (the value estimated by detergent-disruption of the P3 fraction), the observed percentage of doubly fluorescent cells (13.0% ± 0.5%) deviated strongly from a model in which the two variants mixed freely (Figure 2D). This deviation was evident even assuming much smaller numbers of particles per CIU, down to n = 2. Hence, unbalances or fluctuations in the proportion of each variant among cells do not explain the low co-transmission frequencies displayed by CIUs.
Low Co-transmission Frequencies Are Not Explained by Contamination with Free Virions
We then set out to explore whether the P3 fraction may contain a substantial proportion of free virions, because this would reduce the chances of coinfection. For this, we repeated the low-speed centrifugation protocol as above and determined the titer of the supernatant fraction, the resuspended pellet, and the detergent-treated resuspended pellet after each centrifugation round (Figure S2). If free virions were efficiently removed by this process, the titer of the supernatant fraction should drop strongly and by a similar factor after each additional centrifugation round. After the first round, the supernatant titer dropped 41.2 ± 8.6 times (S1 versus S2; Figure S2), but in subsequent rounds, the supernatant titer stagnated. This indicates that free viral particles were washed out efficiently by centrifugation, but that they were replenished from the pellet, probably as a result of spontaneous CIU breakage (for instance, vesicle disruption). If such breakage occurred during pellet resuspension, the P3 fraction used for inoculating recipient cells would contain some proportion of free viral particles released from CIUs, potentially affecting interpretation of our results. To quantify this, we developed a mathematical model that specified the titers of each fraction after each centrifugation round as a function of the average number of infectious particles per CIU (n) and the CIU breakage probability (b; Figure S2). Least-squares fitting of the model to the titer data yielded n = 20.3 ± 3.8 and b = 0.117 ± 0.008. This allowed us to estimate the ratio of intact CIUs to total infectious units vp = (1 − b)/(1 − b + nb) = 0.284 ± 0.043. We next calculated what proportion m of all CIUs contained both virus variants (polymorphic CIUs). Among cells receiving only 1 PFU, the observed proportion of doubly to total fluorescent cells (13.0%; see Figure 2B and Table S1) should simply equal the probability of receiving a polymorphic CIU (i.e., mvp = 0.130). However, a non-negligible fraction of fluorescent cells received two PFUs. To account for this, we considered all possible combinations of two PFUs and their associated probabilities (Figure S3). Solving this model yielded m = 0.378 ± 0.016. Hence, after accounting for possible contamination with free virions, we estimated that CIUs contained 20.3 ± 3.8 infectious particles on average, but that only 37.8% ± 1.6% of these CIUs were polymorphic. Albeit higher than the percentage obtained ignoring free-virion contamination, this value was still clearly lower than expected under the null hypothesis of random mixing (Figure 2D).
CIUs Harvested before Lysis Also Exhibit Low Variant Co-transmission Efficiency
As discussed above, our pellet fractions might contain CIUs other than extracellular vesicles, such as, for instance, replication organelles derived from the endoplasmic reticulum and released during lysis. To gain insight into the contribution of extracellular vesicles to the above results, we repeated the low-speed centrifugation protocol using media harvested from producer cells slightly before lysis (8 hpi). Because secretion of virion-containing vesicles precedes lysis (Robinson et al., 2014, Chen et al., 2015) but replication organelles and other possible types of CIUs (for instance, virion aggregates) should be released during lysis, these assays provide a closer view of the ability of extracellular vesicles to promote the co-transmission of different viral genetic variants. We found that the S1 fraction contained (1.9 ± 0.1) × 108 PFUs/mL versus (5.1 ± 0.2) × 106 PFUs/mL in the P3 fraction, and that detergent treatment of the P3 fraction increased titer by a factor of 7.9 ± 0.1 (Figure 1D). These results were similar to those obtained after lysis, suggesting that most CIUs obtained by slow-speed centrifugation were indeed virion-containing vesicles.
We then repeated the flow cytometry analysis. Following inoculation at high density (D ≈ 10 PFUs/cell), 99.7% of producer cells were fluorescent at 7 hpi, of which 95.0% were positive for both EGFP and mCherry. Inoculation of fresh cells with the P3 fraction at a calculated density of D ≈ 0.1 PFUs/cell yielded 9.45% ± 0.23% fluorescent cells, of which 41.0% ± 2.0% were green, 37.2% ± 1.2% were red, and 21.9% ± 1.7% were doubly fluorescent (Table S2). Although the percentage of doubly fluorescent cells was higher than in the assay carried out after lysis, co-transmission still occurred at much lower frequency than expected if the two variants mixed freely (Figure 2D). This strongly suggests that extracellular vesicles are a poor vehicle for the co-transmission of different viral genetic variants.
Phosphatidylserine-Rich CIUs Show Similarly Low Co-transmission Efficiencies
Enterovirus-containing vesicles are rich in phosphatidylserine (Chen et al., 2015), a feature that is shared by other intracellular membranes. To obtain a more purified preparation of membrane-associated viruses, we incubated the second resuspended pellet (P2) with magnetic beads conjugated to annexin V, a protein that binds phosphatidylserine specifically. The annexin-bound fraction (P2∗) contained (0.9 ± 0.1) × 106 PFUs, versus (1.8 ± 0.2) × 108 PFUs in the initial supernatant (Figure 1E). Following detergent treatment of this fraction, the viral titer increased by a factor of 20.6 ± 5.3, confirming that phosphatidylserine-rich membranous structures harbored large numbers of infectious particles and hence were also CIUs. We then harvested the P2∗ fraction at 12 hpi from cells coinfected with the CVB3-EGFP and CVB3-mCherry variants at high density (D ≈ 10 PFUs/cell) and used flow cytometry to assess co-transmission of both virus variants. Inoculation of fresh cells with the P2∗ fraction at a calculated density of D ≈ 0.1 PFU/cell yielded 14.9% ± 1.0% fluorescent cells, of which 55.1% ± 0.2% were green, 17.7% ± 0.3% were red, and 27.2% ± 0.3% were doubly fluorescent (Table S3). Although the proportion of each virus was unbalanced in this assay and although we exceeded 0.1 PFU/cell in recipient cells, co-transmission mediated by these CIUs was again clearly less frequent than expected if the two variants mixed freely (Figure 2D).
Analysis of Infection Foci Shows that the Co-transmission of Different Virus Variants Is Infrequent and Short-Lived
In principle, another possible explanation for the seemingly low efficiency of variant co-transmission is that flow cytometry underestimated the fraction of doubly fluorescent recipient cells. Such an issue could have occurred, for instance, if the timing of expression of the fluorescent proteins was highly variable, although this seemed unlikely because coinfections with both fluorescent viruses were successfully identified in producer cells. To address this possibility, though, we again obtained the P3 fraction from cells inoculated with a mix of CVB3-EGFP and CVB3-mCherry (D ≈ 10 PFUs/cell), and harvested supernatants at 12 hpi. We then inoculated two 96-well plates at limiting dilution, such that most wells received 0 or 1 PFU. At 30 hpi, we examined the resulting infection foci by automated fluorescence microscopy. Because foci contained multiple cells due to secondary rounds of infection, it was unlikely that CVB3-EGFP or CVB3-mCherry was missed. We found infection in 58/192 wells, of which 11 showed 2 foci and the rest showed 1, the total number of examined foci thus being 58 + 11 = 69 (Figure 3). Of these, 29 were positive only for EGFP, 35 were positive only for mCherry, and 5 were doubly fluorescent. Hence only 7.2% (5/69) of the infectious foci showed evidence of co-transmission of the two fluorescently marked viruses. This percentage was even lower than those observed by flow cytometry, thus further supporting the conclusion that CIUs preferentially contained progeny virions originated from the same parental genome.
Figure 3.

Co-transmission of CVB3-EGFP and CVB3-mCherry Viruses Determined by Foci Analysis
Left: virus-producer cells inoculated at high density (10 PFUs/cell) with a 1:1 mix of each variant, shown at 7 hpi. Green and red channels are shown separately, as well as the merged image. Image saturation was maximized so as to reveal the presence or absence of each variant, rather than quantitative information of infection level. Scale bars, 0.3 mm. Right: analysis of infection foci produced in recipient cells inoculated at limiting dilution. A 96-well plate inoculated with the P3 fraction and imaged by whole-well automated microscopy at 30 hpi is shown. Green, red, and yellow regions correspond to EGFP, mCherry, and doubly fluorescent cells, respectively. A magnified view of two wells at an earlier time point is also shown (16 hpi). Top: two independent foci in the same well. Bottom: two variants initially co-transmitted as part of the same infectious unit. This co-transmission was short-lived, as shown by the fact that most cells in the resulting foci were infected with only one variant. Scale bars, 1 mm.
For those cases in which the foci (and thus the initial cell) contained the two variants, we used image analysis to assess whether variant co-transmission was maintained in subsequent infection cycles within foci. The five foci showing a mixture of both variants contained on average 1,024 ± 365 cells at 30 hpi. However, only 4.91% ± 0.96% of these cells were doubly fluorescent (Table 1), indicating that co-transmission of these two variants was short-lived. To a first approximation, the probability of sustained co-transmission of the two variants after k infection cycles should be mk, which rapidly tends to zero. This leads to the conclusion that membrane-associated virus release should not be an efficient vehicle for the ongoing co-transmission of different genetic variants of the virus.
Table 1.
Cell Counts in Five Foci Founded by CIUs Containing Both CVB3-EGFP and CVB3-mCherry Variants
| Cell Count (%) | Total | |||
|---|---|---|---|---|
| EGFP | + | − | + | |
| mCherry | − | + | + | |
| Foci 1 | 228 (46.5) | 227 (46.3) | 35 (7.1) | 490 (100) |
| Foci 2 | 626 (60.7) | 340 (33.0) | 65 (6.3) | 1,031 (100) |
| Foci 3 | 136 (83.4) | 21 (12.9) | 6 (3.7) | 163 (100) |
| Foci 4 | 1,035 (91.1) | 80 (7.0) | 21 (1.8) | 1,136 (100) |
| Foci 5 | 1,323 (57.6) | 847 (36.9) | 128 (5.6) | 2,298 (100) |
| Average | 670 (67.8) | 303 (27.2) | 51 (4.9) | 1,024 (100) |
CIU-Mediated Intercellular Transmission Has Little Effect on Viral Population Genetic Diversity
Because membrane-associated CIUs did not appear to support the long-term co-spread of different variants of CVB3, we did not expect them to have a major impact on overall viral population diversity. Yet, it is conceivable that some co-spreading variants established cooperative interactions, increasing their fitness and, thus, their population frequencies. To test for the effects of collective spread on viral population genetic diversity, we performed 20 serial transfers of CVB3, selecting the P3 fraction after each infection. This experimental evolution protocol was carried out twice in parallel (two evolution lines). As a control, we performed two evolution lines in which we selected the S1 fraction after each transfer. Finally, we also established two lines in which we selected the B3 fraction (detergent-disrupted P3 fraction). After transfer 20, each of the six lines was used for viral RNA extraction, RT-PCR, and Illumina MiSeq sequencing. Compared with the founder virus, the frequency of mutations increased in all evolved populations between 6- and 10-fold (Table 2). However, mutation frequencies were similar among S1, P3, and B3 lines, and genetic diversity was similarly distributed along the genome in the different lines (Figure 4). We therefore conclude that collective intercellular spread of CVB3 in the form of membrane-associated viruses had little effect on the overall accumulation of population genetic diversity.
Table 2.
Illumina Sequence Analysis of Population Genetic Diversity in CVB3 Evolution Lines in which the S1, P3, or B3 Fraction Was Selected after Each Transfer
| Virus Population | Total Bases Read | Sites with Mutations | Total Mutant Bases Read | Mutation Frequencya |
|---|---|---|---|---|
| Founder | 1.04 × 108 | 266 | 11,254 | 1.08 × 10−4 |
| Evolved S1, line 1 | 1.82 × 108 | 1,868 | 160,506 | 8.81 × 10−4 |
| Evolved S1, line 2 | 1.48 × 108 | 1,796 | 150,954 | 10.20 × 10−4 |
| Evolved P3, line 1 | 2.43 × 108 | 2,148 | 256,770 | 10.60 × 10−4 |
| Evolved P3, line 2 | 2.08 × 108 | 2,092 | 188,731 | 9.08 × 10−4 |
| Evolved B3, line 1 | 1.21 × 108 | 2,073 | 89,318 | 7.38 × 10−4 |
| Evolved B3, line 2 | 1.99 × 108 | 2,141 | 172,507 | 8.66 × 10−4 |
Total mutant bases read divided by total bases read.
Figure 4.

Mutation Frequency across the CVB3 Genome
The mutation frequency (mutated/total bases read) per nucleotide site is shown for the founder and one evolution replicate (line 1) for each treatment (S1, P3, and B3). Replicate evolution lines 1 and 2 showed very similar genome-wide diversity patterns (data available from Table S4).
Discussion
We have measured the number of infectious CVB3 particles in membrane-associated CIUs, and we have examined whether these CIUs co-transmit different genetic variants of the virus between cells. In cell cultures harvested after lysis, detergent-based membrane disruption suggested the presence of 11.6 ± 1.1 infectious particles per CIU, but only 13.0% ± 0.5% of these CIUs co-transmitted the two fluorescently marked viruses. When cultures were harvested before lysis, detergent treatment suggested that on average 7.9 ± 0.1 infectious particles per CIU, yet only 21.9% ± 1.7% of these CIUs co-transmitted the two fluorescently marked viruses. The actual CIU size may be higher than estimated by detergent treatment because pellet fractions may already carry free viral particles before detergent treatment, for instance, as a result of spontaneous vesicle breakage. After considering this potential problem, we estimated that CIUs contained on average 20.3 ± 3.8 infectious particles, but that only 37.8% ± 1.6% co-transmitted the two fluorescently marked viruses. We also found that, after annexin V selection of phosphatidylserine-rich particles, detergent treatment increased titer by a factor of 20.6 ± 5.3, again revealing large CIUs, but variant co-transmission frequency was only 27.2% ± 0.3%. In all cases, co-transmission frequencies were much lower than expected under the null hypothesis that these membrane-associated pools of viruses contained a random mix of the two variants present in producer cells.
Hence our results suggest that CVB3 viruses present in the same cell tend to segregate their progenies in different infectious units. A possible explanation for this segregation is that viral genomes are partially compartmentalized inside cells. Enteroviruses such as CVB3 and poliovirus remodel endoplasmic reticulum membranes to form organelles used for viral replication and assembly, a feature that is characteristic of most positive-strand RNA viruses (Altan-Bonnet, 2017, Belov, 2016, Fernández de Castro et al., 2016, Hsu et al., 2010, Shulla and Randall, 2016, Romero-Brey and Bartenschlager, 2016, Ravindran et al., 2016). As a result, groups of membrane-associated viruses released from infected cells would tend to originate from a given parental genome from a given replication organelle. This intracellular spatial structuring may explain why genetic trans-complementation of defective mutants is infrequent for some viral proteins that are tightly associated to viral replication organelles, such as the RNA helicase 2C, the viroporin 2B, or the viral replicase, whereas complementation is more frequent among capsid proteins, which should be more easily interchanged among replication organelles (Bernstein et al., 1986, Kirkegaard and Baltimore, 1986).
There may be additional explanations for the low frequency with which the two virus variants co-dispersed in our experiments, although we believe these are less likely. For instance, CIUs may show large variation in size, such that some may contain tens of virions, but most would contain only very few. However, our statistical analysis of mCherry and EGFP expression levels suggests that, even for CIUs carrying as little as three virions, the observed levels of coinfection were lower than expected under the random mixing hypothesis. Hence CIU size variation could explain our data satisfactorily only if most CIUs carried very few infectious particles. But if this was the case, these structures would fail anyway to support long-lived interactions among different virus variants in the population. Another possible explanation for our results is that most CIUs carry mixes of different variants, but that this diversity is lost in recipient cells due to population bottlenecking occurring at early stages of the infection cycle, which may result from early infection barriers or stochastic losses. However, this is hard to reconcile with the observation that detergent-based membrane disruption released multiple PFUs per CIU.
Our results suggest that collective dispersal of coxsackievirus does not promote cooperation among genetically distinct virus variants, such as, for instance, genetic complementation. On the other hand, our data also suggest that collective spread might be robust against the emergence of defective viruses. Viruses are subject to strong and frequent population bottlenecks associated to dispersal events, including inter-host transmission and intra-host dissemination throughout body sites (Zwart and Elena, 2015, Gutiérrez et al., 2012, McCrone and Lauring, 2018). Whereas under free-virion dispersal the cellular MOI inevitably drops to a single viral genome copy per cell following these bottlenecks, collective viral spread provides means of maintaining cellular MOIs above 1. The risks of cheater invasion might be low, though, if viral genomes present in the same CIU tend to be siblings. Defective viruses might still appear among the progeny of a given virus and, if co-transmitted to the next cell together with their siblings, they would reduce the fitness of that particular lineage. However, our analysis of infection foci showed that variants within a given CIU tend to not be co-transmitted to subsequent infection cycles. Furthermore, even if cheaters succeeded in being co-transmitted with their siblings, they would not strongly interfere with unrelated members of the population, and selection would efficiently purge these cheater-containing lineages. Social evolution theory has established that spatial population structures that help increase genetic relatedness and/or allow for group (multilevel) selection prevent the spread of cheaters and can thus promote the evolution of cooperation (Gardner et al., 2011, West et al., 2006, Nowak, 2006). If interacting viral genomes were unrelated, as is the case under classical high viral density regimens (high PFU/cell ratio), defective interfering particles and other kinds of social cheaters would tend to spread owing to their ability to exploit helper viruses.
Therefore, co-dispersal of pools of related viral particles in the form of membrane-associated viruses might allow for the evolution of cooperation, albeit probably not diversity-based cooperation. Elevating the MOI using CIUs could potentially be a cooperative action in which the fitness benefits would reside in invading cells with a higher number of viral genomes, regardless of their genetic diversity. Such benefits may include accelerating the infection cycle or overwhelming antiviral cellular responses, as we recently suggested for vesicular stomatitis virus (Andreu-Moreno and Sanjuán, 2018). Alternatively, spreading in groups could also afford other types of fitness benefits not related directly to the cellular MOI, such as protection against anti-viral factors including circulating proteases or antibodies (Santiana et al., 2018).
In future work, it would be interesting to investigate whether our results apply to other viruses. Membrane-associated intercellular spread within vesicles has been documented in widely different viruses, but the biogenesis and size of these vesicles are diverse. For instance, norovirus and hepatitis A virus vesicles are likely derived from multivesicular bodies and are smaller than enterovirus vesicles (Feng et al., 2013, Chen et al., 2015, Santiana et al., 2018). These differences may also lead to different levels of mixing among virus variants. However, the types of collective viral spread examined to date have been found to support low levels of mixing between virus variants. For instance, one study used EGFP- and mCherry-encoding HIV-1 to examine the ability of virological synapses to co-transfer both variants from cell to cell (Law et al., 2016). It was found that co-transfer occurred in only 6%–14% of infected cells, which was significantly higher than expected by chance under free-virion infection, yet too low to sustain the joint spread of variants through multiple infection cycles, paralleling our results. Another study found that cell-to-cell spread of tomato mosaic virus through plasmodesmata introduced a strong genetic bottleneck despite allowing for the passage of a large number of viral genomes among cells (Miyashita et al., 2015). Also, unpublished work suggests that most virion-containing vesicles released by cells coinfected with CVB3 and poliovirus are positive for capsids of one virus or the other, but not both (N. Altan-Bonnet, personal communication). This suggests that even capsid precursors are not shared unrestrictedly among viruses present in the same cell, despite their intracellular diffusion being probably less limited than that of viral genomes and non-structural proteins involved in replication.
Overall, these lines of evidence suggest that certain types of collective viral spread do not efficiently support interactions among different virus variants. However, further research is needed to extend our results to other viruses, such as, for instance, noroviruses and rotaviruses spreading through extracellular vesicles, as well as baculoviruses spreading through occlusion bodies.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| CVB3 Nancy infectious clone | Dr. Marco Vignuzzi (Pasteur Institute of Paris, France). | N/A |
| CVB3-Xho-P1-Kpn2I infectious clone | This paper | N/A |
| CVB3-MluI-P2-Kpn2I infectious clone | This paper | N/A |
| CVB3-P1-eGFP infectious clone | This paper | N/A |
| CVB3-P2-mCherry infectious clone | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Kpn2I | ThermoScientific | Cat# FD0534 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 555 | ThermoScientific | Cat# A32732, RRID: AB_2633281 |
| LC3B Polyclonal Antibody | ThermoScientific | Cat# PA535195, RRID: AB_2552505 |
| Anti-BSA antibody | Sigma-MERK | Cat# SAB4301142-100UL |
| Anti-GAPDH, clone 10B13 | Sigma-MERK | Cat# ABS16, RRID: AB_10806772 |
| Goat anti-rabbit IgG-HRP | Santa Cruz Biotechnology | Cat# sc-2004 |
| cOmplete Mini, EDTA-free | Roche | Cat# 04 693 159 001 |
| 4x Laemmli Sample Buffer | BIO-RAD | Cat# 161-0747 |
| Critical Commercial Assays | ||
| Annexin V microbeads kit | Milteny Biotec | Cat# 130-090-201 |
| NEBuilder HiFi DNA assembly mix | New England Biolabs | Cat# E2621L |
| Quick-RNA viral kit | ZymoResearch | Cat# R1035 |
| DNA Clean & Concentrator kit | ZymoResearch | Cat# D4013 |
| Phusion High-Fidelity DNA Polymerase | ThermoScientific | Cat# F530L |
| Pierce ECL Plus Western Blotting Substrate | ThermoScientific | Cat# 32132 |
| AccuScript High-Fidelity 1st Strand cDNA Synthesis Kit | Agilent | Cat# 200820 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE124469 |
| Experimental Models: Cell Lines | ||
| HeLa-H1 | ATCC | ATCC Cat# CRL-1958; RRID:CVCL_3334 |
| Oligonucleotides | ||
| Primer: CVB3 specific oligo-dT: TTTTTTTTTTTTTTCCGCAC | This paper | N/A |
| Primer: CV-1-Forward: TTAAAACAGCCTGTGGGTTGA | This paper | N/A |
| Primer: CV-2143-Reverse: GGCCGAACCACAGAACATAA | This paper | N/A |
| Primer: CV-2045-Forward: TCGAGTGTTTTTAGTCGGACG | This paper | N/A |
| Primer: CV-4923-Reverse: AGCCTTCCCACACACAAGAGG | This paper | N/A |
| Primer: CV-4879-Forward: AACATGCCCATGTCAGTCAAGAC | This paper | N/A |
| Primer: CV-7399-Reverse: CGCACCGAATGCGGAGAA | This paper | N/A |
| Software and Algorithms | ||
| Fiji (ImageJ) | NIH / public domain | https://fiji.sc/ |
| R version 3.x | R Project | https://www.R-project.org/ |
| FastQC 0.11.7 | Babraham Institute | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Cutadapt | https://cutadapt.readthedocs.io/en/stable/ | |
| FASTQ Quality Filter | Cold Spring Harbor Laboratory | http://hannonlab.cshl.edu/fastx_toolkit/index.html |
| Prinseq-lite 0.20.4 | Schmieder and Edwards, 2011 | http://prinseq.sourceforge.net/ |
| ViVan 0.43 | Isakov et al., 2015 | N/A |
Lead Contact and Materials Availability
This study did not generate new unique reagents. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Rafael Sanjuán (rafael.sanjuan@uv.es).
Experimental Model and Subject Details
Virus and cells
HeLa-H1 cells were obtained from the American Type Culture Collection (ATCC, CRL-1958) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), non-essential amino acids, 10 units/mL penicillin, 10 μg/mL streptomycin and 250 ng/mL amphotericin B under standard culturing conditions (37°C and 5% CO2). Cells tested negative for mycoplasma by PCR. The CVB3 Nancy infectious clone was obtained from Dr. Marco Vignuzzi (Pasteur Institute of Paris, France). Viral stocks were produced by inoculating confluent HeLa-H1 cells at 0.1 PFU/cell and incubating them under standard conditions for 24 h. Then, infected cells were subjected to three freeze-thaw cycles, cell debris was removed by centrifugation at 3000 g for 1 min, and the supernatant was collected, aliquoted, and frozen at –80°C.
Method Details
Virus titration by the plaque assay, viral density
Viral density was defined as the number of PFUs per cell in the culture. To adjust the viral density at inoculation, we titrated viral stocks by the plaque assay. For this, we used confluent HeLa-H1 monolayers in 6-well plates. Each well was inoculated with 200 μL for 45 min under standard culturing conditions and then overlaid with DMEM supplemented with 2% FBS, 10 units/mL penicillin, 10 μg/mL streptomycin and 0.8% noble agar. Plaques typically became obvious at 44-48 hpi. Cells were fixed with 10% formaldehyde for 1 h and the agar overlay was removed to stain cells with 2% crystal violet in 10% formaldehyde.
Low-speed centrifugation to select for high-weight infectious units
HeLa-H1 cells were infected with CVB3 in 12-well dishes (2 mL of culture medium) or p100 dishes (9 mL of culture medium) at high viral density (10 PFU/cell) and after 45 min incubation under standard conditions the inoculum was removed, and DMEM supplemented with 2% FBS and 10 units/mL penicillin, 10 μg/mL streptomycin and 250 ng/mL amphotericin B was added. At harvest time (12 or 8 hpi) the extracellular media and the infected cells were collected and centrifuged at 1000 g for 5 min at 4°C. The supernatant was collected and spun again at 10,000 g for 10 min at 4°C if the volume of the culture medium was 2 mL and for 30 min if the volume of the culture medium was 9 mL. The supernatant of this centrifugation was stored until use at 4°C as the S1 fraction, whereas the pellet was resuspended and spun again under the same conditions to remove remaining free viral particles. This step was repeated twice and the third pellet was resuspended and stored until use at 4°C as the P3 fraction. Membranes were disrupted (B3 fraction) by adding 0.16% Triton X-100 and incubating 15 min at 4°C.
Preparation of phosphatidylserine-rich infectious units using annexin V
The second pellet of the above centrifugation-based protocol (P2) was resuspended in 80 μL of binding buffer provided in the Annexin V microbeads kit (Milteny Biotec, CA). Then, 20 μL of microbeads were added and the mix was incubated for 15 min at 4°C. Finally, phosphatidylserine-rich particles were purified following manufacturer’s instructions. Membranes were disrupted as above.
Transmission electron microscopy
The P3 fraction was encased in a 1% agar cone, which was then fixed in 2% paraformaldehyde and 2.5% glutaraldehyde solution in phosphate buffer 0.1 M, and kept for >1 h at room temperature. The fixative was removed by phosphate buffer 0.1 M washes, and preparations were post-fixed and stained with 2% osmium solution, dehydrated in a graded series of ethanol, and stained during dehydration with 2% uranyl acetate in ethanol 70%. Finally, samples were embedded in Durcupan epoxy resin and polymerized for 72 h at 70°C. Semi-thin sections (1.5 μm) were obtained using a Leica EM UC-6 ultra-microtome and stained with 1% toluidine blue at 70°C for optical microscopy. Ultra-thin sections (70-80 nm) were then obtained with a diamond-tipped knife, stained with Reynold’s lead citrate, and imaged by electron microscopy.
SDS-PAGE and western blot
Pellets were resuspended in lysis buffer (50 mM Tris-pH 8, 150 mM NaCl, 5 mM EDTA, 0.5% NP-40) with protease inhibitor and incubated on ice for 30 min, vortexing every 10 min. Then, the proteins were denatured at 95°C for 10 min in reducing Laemmli sample buffer. Denatured proteins were separated on 12.5% sodium dodecyl sulfate polyacrylamide gel by electrophoresis and transferred in transfer buffer (25 mM Tris, 192, glycine, 20% v/v methanol, pH 8.3) to 0.45 μm nitrocellulose membranes by semi-dry transfer in a Novex Semi-Dry Blotter (Invitrogen). The membranes were washed with 0.1% Tween 20 in PBS and then incubated with blocking buffer (5% blotto non-fat dry milk in PBS-tween 0.1%) for 30 min at room temperature. After blocking, the membranes were incubated with primary antibody against LC3B (1:1000, rabbit) overnight at 4°C. Two controls were included: 1:3000 rabbit anti-GAPDH for intracellular content, and 1:2500 rabbit anti-BSA for extracellular culture medium. The excess of primary antibodies was removed washing the membranes 5 times with PBS-Tween 0.1%. Membranes were incubated with a secondary antibody diluted in blocking buffer (goat anti-rabbit IgG-HRP; Santa Cruz Biotechnology) for 90 min at room temperature. Then, membranes were washed as before. Pierce ECL Plus Wester Blotting Substrate was used for detection chemiluminescence on an ImageQuant LAS 500 luminescent image analyzer (GE Healthcare Bio-Sciences AB).
Construction of viruses encoding fluorescent proteins
We generated a CVB3-Xho-P1-Kpn2I construct by site-directed mutagenesis by removing the XhoI site present in P1 and introducing an XhoI site at position 692 as well as a Kpn2I site at position 3314. All mutations in protein-coding regions were synonymous. This plasmid was then linearized with Kpn2I and a synthetic fragment (IDT) encoding eGFP flanked by amino acids that reconstruct an upstream and downstream CVB3 2A cleavage site was ligated using the NEBuilder HiFi DNA assembly mix (New England Biolabs). To introduce mCherry, we generated a CVB3-MluI-P2-Kpn2I construct by site-directed mutagenesis to eliminate the MluI site present in the vector and introduce an MluI site at position 3263 as well as a Kpn2I restriction enzyme site at position 5068. This vector was then linearized with Kpn2I and a synthetic fragment encoding mCherry flanked by the CVB3 3C protease cleavage sites of 2C-3A was ligated using the NEBuilder HiFi DNA assembly mix.
Flow cytometry
HeLa-H1 cells were infected with a 1:1 mix of CVB3-mCherry and CVB3-eGFP at the indicated density to obtain the P3 fraction as detailed above. Confluent HeLa-H1 monolayers in 60 mm dishes were inoculated with this fraction (0.1 PFU/cell) and at 7 hpi the extracellular media was removed, the monolayer was washed with PBS, and the infected cells were collected by detaching them with trypsin-EDTA, resuspended in DMEM containing 10% FBS, washed with PBS by centrifugation at 750 g for 5 min, and resuspended in 4% paraformaldehyde for overnight fixation at 4°C. Fixed cells were centrifuged to remove paraformaldehyde, washed again with PBS, and finally resuspended in 1 mL of PBS. Then, 104 (producer cells) or 105 (recipient cells) events per sample were analyzed in a Becton Dickinson LSRFortessa flow cytometer equipped with 488 nm and 561 nm lasers for eGFP and mCherry excitation, respectively. Controls of non-infected cells, of cells infected only with one variant, and of cells infected with both variants were used to adjust the fluorescent quadrants manually.
Automated fluorescence microscopy
Imaging was performed in an IncuCyte S3 Live-Cell Analysis System (Essen BioScience) housed inside a tissue culture incubator. Images were acquired using phase contrast, green (300-ms exposure) and red (400-ms exposure) channels with a 4X objective. Representative images were selected and used as a reference to define image analysis masks for each acquisition channel. Images were segmented by defining a fluorescence intensity threshold after applying a background correction using the Top-Hat method. Foci with a mixture of both variants were analyzed by manual counting using the Multi-point tool of Fiji (ImageJ).
Experimental evolution
The P3, S1, or B3 fraction was obtained from the culture medium of a founder infection and propagated serially for 20 transfers at an inoculum density of 0.1 PFU/cell, selecting the relevant fraction at 20 hpi after each transfer. Fractions were titrated after each transfer by the plaque assay to keep viral density at inoculation constant.
RNA extraction, RT-PCR and Illumina sequencing
We used 100 μL of supernatant from transfer 20 of each lineage as well as from the founder virus to extract viral RNA using the Quick-RNA viral kit (ZymoResearch), following manufacturer’s instructions. Then, 2 μL of RNA were reverse-transcribed using AccuScript Hi-Fi Reverse Transcriptase (Agilent) with a CVB3-specific oligo-dT (5′TTTTTTTTTTTTTTCCGCAC) and 2.5μL of this reaction were used to amplify the whole genome in three PCR fragments in a total volume of 50 μL using the following pairs of primers: CV-1F (5′TTAAAACAGCCTGTGGGTTGA) and CV-2143-R (5′GGCCGAACCACAGAACATAA) with an annealing temperature of 64°C and an extension time of 1 min 15 s; CV-2045-F (5′TCGAGTGTTTTTAGTCGGACG) and CV-4923-R (5′AGCCTTCCCACACACAAGAGG) with an annealing temperature of 63°C and an extension time of 1 min 40 s; CV-4879-F (5′AACATGCCCATGTCAGTCAAGAC) and CV-7399-R (5′CGCACCGAATGCGGAGAA) with an annealing temperature of 66°C and an extension time of 1 min 30 s. PCRs were carried out with Phusion High-Fidelity DNA Polymerase (Thermo Scientific) under the following thermal profile: an initial denaturation at 98°C for 1 min, 35 cycles of 98°C for 10 s, 20 s at the indicated annealing temperature, and 72°C for the indicated time, followed by 5 min of final extension at 72°C. PCR products were visualized by agarose gel electrophoresis, purified with the DNA Clean & Concentrator kit (ZymoResearch) and quantified by spectrometry (NanoDrop One, Thermo Scientific). Then, an equimolar mix of the three PCR amplicons was used for Illumina sequencing in a MiSeq machine, using paired-end libraries.
Analysis of NGS data
The quality of the FastQ files of NGS sequencing was evaluated with FastQC 0.11.7 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Then, the first 10 nucleotides and the last two nucleotides of each read were cut with Cutadapt (https://cutadapt.readthedocs.io). Reads were then trimmed with FASTQ Quality Filter (http://hannonlab.cshl.edu/fastx_toolkit/) and Prinseq-lite 0.20.4 (Schmieder and Edwards, 2011) by quality (Q30), length (200 nucleotides) and sequencing artifacts (duplications, Ns). The genome of CVB3 Nancy strain (GenBank: JX312064) was used for mapping and SNP calling with ViVan 0.43 (Isakov et al., 2015).
Quantification and Statistical Analysis
All experiments were done in triplicate. Error terms correspond to the standard error of the mean (SEM). Raw flow cytometry files were analyzed using the read.FCS function implemented in R package (https://www.R-project.org) to obtain the intensity values of mCherry and eGFP. The mathematical model used to infer the number of infectious particles per CIU (n), the probability of CIU breakage (b) during pellet resuspension, and the proportion of collective to total infectious units in the P3 fraction (vP) is explained in Figure S2. The probabilistic model used for calculating the proportion of polymorphic to total CIUs (m) from the cytometry counts is explained in Figure S3.
Data and Code Availability
Data Resources
The accession number for raw and analyzed NGS data reported in this paper is GEO: GSE124469.
Acknowledgments
We thank María Durán for help with electron microscopy, Nihal Altan-Bonnet for helpful discussions, and Marco Vignuzzi for the CVB3 infectious clone. R.S. was funded by ERC Consolidator Grant 724519 Vis-a-Vis. J.-V.B. was funded by a Prometeo Grant 2016/122 from the Generalitat Valenciana. R.G. was funded by the Spanish Ramón y Cajal program (RYC-2015-17517) and Ministerio de Ciencia e Innovación (grant BFU2017-86094-R).
Author Contributions
Conceptualization, R.S.; Investigation, J.-V.B.; Resources, R.G.; Writing – Original Draft, R.S.; Writing – Review & Editing, R.S.; Supervision, R.S.; Funding Acquisition, R.S.
Declaration of Interests
The authors declare no competing interests.
Published: October 15, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.09.014.
Supplemental Information
References
- Aguilera E.R., Erickson A.K., Jesudhasan P.R., Robinson C.M., Pfeiffer J.K. Plaques formed by mutagenized viral populations have elevated coinfection frequencies. MBio. 2017;8 doi: 10.1128/mBio.02020-16. e02020–e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altan-Bonnet N. Extracellular vesicles are the Trojan horses of viral infection. Curr. Opin. Microbiol. 2016;32:77–81. doi: 10.1016/j.mib.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altan-Bonnet N. Lipid tales of viral replication and transmission. Trends Cell Biol. 2017;27:201–213. doi: 10.1016/j.tcb.2016.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altan-Bonnet N., Chen Y.H. Intercellular transmission of viral populations with vesicles. J. Virol. 2015;89:12242–12244. doi: 10.1128/JVI.01452-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andino R., Domingo E. Viral quasispecies. Virology. 2015:479–480. doi: 10.1016/j.virol.2015.03.022. 46–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreu-Moreno I., Sanjuán R. Collective infection of cells by viral aggregates promotes early viral proliferation and reveals a cellular-level Allee effect. Curr. Biol. 2018;28:3212–3219.e4. doi: 10.1016/j.cub.2018.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arantes T.S., Rodrigues R.A., Dos Santos Silva L.K., Oliveira G.P., de Souza H.L., Khalil J.Y., de Oliveira D.B., Torres A.A., da Silva L.L., Colson P. The large Marseillevirus explores different entry pathways by forming giant infectious vesicles. J. Virol. 2016;90:5246–5255. doi: 10.1128/JVI.00177-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov G.A. Dynamic lipid landscape of picornavirus replication organelles. Curr. Opin. Virol. 2016;19:1–6. doi: 10.1016/j.coviro.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein H.D., Sarnow P., Baltimore D. Genetic complementation among poliovirus mutants derived from an infectious cDNA clone. J. Virol. 1986;60:1040–1049. doi: 10.1128/jvi.60.3.1040-1049.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird S.W., Maynard N.D., Covert M.W., Kirkegaard K. Nonlytic viral spread enhanced by autophagy components. Proc. Natl. Acad. Sci. USA. 2014;111:13081–13086. doi: 10.1073/pnas.1401437111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordería A.V., Isakov O., Moratorio G., Henningsson R., Agüera-González S., Organtini L., Gnädig N.F., Blanc H., Alcover A., Hafenstein S. Group selection and contribution of minority variants during virus adaptation determines virus fitness and phenotype. PLoS Pathog. 2015;11:e1004838. doi: 10.1371/journal.ppat.1004838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boullé M., Müller T.G., Dähling S., Ganga Y., Jackson L., Mahamed D., Oom L., Lustig G., Neher R.A., Sigal A. HIV Cell-to-Cell spread results in earlier onset of viral gene expression by multiple infections per cell. PLoS Pathog. 2016;12:e1005964. doi: 10.1371/journal.ppat.1005964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooke C.B. Population diversity and collective interactions during influenza virus infection. J. Virol. 2017;91 doi: 10.1128/JVI.01164-17. e01164–e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao L., Elena S.F. Nonlinear trade-offs allow the cooperation game to evolve from Prisoner’s Dilemma to Snowdrift. Proc. Biol. Sci. 2017;284:20170228. doi: 10.1098/rspb.2017.0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.H., Du W., Hagemeijer M.C., Takvorian P.M., Pau C., Cali A., Brantner C.A., Stempinski E.S., Connelly P.S., Ma H.C. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell. 2015;160:619–630. doi: 10.1016/j.cell.2015.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavijo G., Williams T., Muñoz D., Caballero P., López-Ferber M. Mixed genotype transmission bodies and virions contribute to the maintenance of diversity in an insect virus. Proc. Biol. Sci. 2010;277:943–951. doi: 10.1098/rspb.2009.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-Muñoz S.L., Sanjuán R., West S. Sociovirology: conflict, cooperation, and communication among viruses. Cell Host Microbe. 2017;22:437–441. doi: 10.1016/j.chom.2017.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo E., Perales C. Quasispecies and virus. Eur. Biophys. J. 2018;47:443–457. doi: 10.1007/s00249-018-1282-6. [DOI] [PubMed] [Google Scholar]
- Feng Z., Hensley L., McKnight K.L., Hu F., Madden V., Ping L., Jeong S.H., Walker C., Lanford R.E., Lemon S.M. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature. 2013;496:367–371. doi: 10.1038/nature12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández de Castro I., Tenorio R., Risco C. Virus assembly factories in a lipid world. Curr. Opin. Virol. 2016;18:20–26. doi: 10.1016/j.coviro.2016.02.009. [DOI] [PubMed] [Google Scholar]
- Gardner A., West S.A., Wild G. The genetical theory of kin selection. J. Evol. Biol. 2011;24:1020–1043. doi: 10.1111/j.1420-9101.2011.02236.x. [DOI] [PubMed] [Google Scholar]
- Grande-Pérez A., Lázaro E., Lowenstein P., Domingo E., Manrubia S.C. Suppression of viral infectivity through lethal defection. Proc. Natl. Acad. Sci. USA. 2005;102:4448–4452. doi: 10.1073/pnas.0408871102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez S., Michalakis Y., Blanc S. Virus population bottlenecks during within-host progression and host-to-host transmission. Curr. Opin. Virol. 2012;2:546–555. doi: 10.1016/j.coviro.2012.08.001. [DOI] [PubMed] [Google Scholar]
- Hsu N.Y., Ilnytska O., Belov G., Santiana M., Chen Y.H., Takvorian P.M., Pau C., van der Schaar H., Kaushik-Basu N., Balla T. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell. 2010;141:799–811. doi: 10.1016/j.cell.2010.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isakov O., Bordería A.V., Golan D., Hamenahem A., Celniker G., Yoffe L., Blanc H., Vignuzzi M., Shomron N. Deep sequencing analysis of viral infection and evolution allows rapid and detailed characterization of viral mutant spectrum. Bioinformatics. 2015;31:2141–2150. doi: 10.1093/bioinformatics/btv101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkegaard K., Baltimore D. The mechanism of RNA recombination in poliovirus. Cell. 1986;47:433–443. doi: 10.1016/0092-8674(86)90600-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauring A.S., Andino R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 2010;6:e1001005. doi: 10.1371/journal.ppat.1001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law K.M., Komarova N.L., Yewdall A.W., Lee R.K., Herrera O.L., Wodarz D., Chen B.K. In vivo HIV-1 cell-to-cell transmission promotes multicopy micro-compartmentalized infection. Cell Rep. 2016;15:2771–2783. doi: 10.1016/j.celrep.2016.05.059. [DOI] [PubMed] [Google Scholar]
- Leeks A., Segredo-Otero E.A., Sanjuán R., West S.A. Beneficial coinfection can promote within-host viral diversity. Virus Evol. 2018;4:vey028. doi: 10.1093/ve/vey028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoni T.B., López C.B. Defective (interfering) viral genomes re-explored: impact on antiviral immunity and virus persistence. Future Virol. 2018;13:493–503. doi: 10.2217/fvl-2018-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marriott A.C., Dimmock N.J. Defective interfering viruses and their potential as antiviral agents. Rev. Med. Virol. 2010;20:51–62. doi: 10.1002/rmv.641. [DOI] [PubMed] [Google Scholar]
- McCrone J.T., Lauring A.S. Genetic bottlenecks in intraspecies virus transmission. Curr. Opin. Virol. 2018;28:20–25. doi: 10.1016/j.coviro.2017.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita S., Ishibashi K., Kishino H., Ishikawa M. Viruses roll the dice: the stochastic behavior of viral genome molecules accelerates viral adaptation at the cell and tissue levels. PLoS Biol. 2015;13:e1002094. doi: 10.1371/journal.pbio.1002094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutsafi Y., Altan-Bonnet N. Enterovirus transmission by secretory autophagy. Viruses. 2018;10:E139. doi: 10.3390/v10030139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak M.A. Five rules for the evolution of cooperation. Science. 2006;314:1560–1563. doi: 10.1126/science.1133755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindran M.S., Bagchi P., Cunningham C.N., Tsai B. Opportunistic intruders: how viruses orchestrate ER functions to infect cells. Nat. Rev. Microbiol. 2016;14:407–420. doi: 10.1038/nrmicro.2016.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezelj V.V., Levi L.I., Vignuzzi M. The defective component of viral populations. Curr. Opin. Virol. 2018;33:74–80. doi: 10.1016/j.coviro.2018.07.014. [DOI] [PubMed] [Google Scholar]
- Robinson S.M., Tsueng G., Sin J., Mangale V., Rahawi S., McIntyre L.L., Williams W., Kha N., Cruz C., Hancock B.M. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathog. 2014;10:e1004045. doi: 10.1371/journal.ppat.1004045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Brey I., Bartenschlager R. Endoplasmic reticulum: the favorite intracellular niche for viral replication and assembly. Viruses. 2016;8:E160. doi: 10.3390/v8060160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuán R. Collective infectious units in viruses. Trends Microbiol. 2017;25:402–412. doi: 10.1016/j.tim.2017.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuán R. Collective properties of viral infectivity. Curr. Opin. Virol. 2018;33:1–6. doi: 10.1016/j.coviro.2018.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuán R., Thoulouze M.I. Why viruses sometimes disperse in groups†. Virus Evol. 2019;5:vez014. doi: 10.1093/ve/vez025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiana M., Ghosh S., Ho B.A., Rajasekaran V., Du W.L., Mutsafi Y., De Jésus-Diaz D.A., Sosnovtsev S.V., Levenson E.A., Parra G.I. Vesicle-cloaked virus clusters are optimal units for inter-organismal viral transmission. Cell Host Microbe. 2018;24:208–220.e8. doi: 10.1016/j.chom.2018.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder R., Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirogane Y., Watanabe S., Yanagi Y. Cooperation between different RNA virus genomes produces a new phenotype. Nat. Commun. 2012;3:1235. doi: 10.1038/ncomms2252. [DOI] [PubMed] [Google Scholar]
- Shulla A., Randall G. (+) RNA virus replication compartments: a safe home for (most) viral replication. Curr. Opin. Microbiol. 2016;32:82–88. doi: 10.1016/j.mib.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simón O., Williams T., Cerutti M., Caballero P., López-Ferber M. Expression of a peroral infection factor determines pathogenicity and population structure in an insect virus. PLoS ONE. 2013;8:e78834. doi: 10.1371/journal.pone.0078834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner P.E., Chao L. Prisoner’s dilemma in an RNA virus. Nature. 1999;398:441–443. doi: 10.1038/18913. [DOI] [PubMed] [Google Scholar]
- Vignuzzi M., Stone J.K., Arnold J.J., Cameron C.E., Andino R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature. 2006;439:344–348. doi: 10.1038/nature04388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarreal L.P., Witzany G. When competing viruses unify: evolution, conservation, and plasticity of genetic identities. J. Mol. Evol. 2015;80:305–318. doi: 10.1007/s00239-015-9683-y. [DOI] [PubMed] [Google Scholar]
- West S.A., Griffin A.S., Gardner A., Diggle S.P. Social evolution theory for microorganisms. Nat. Rev. Microbiol. 2006;4:597–607. doi: 10.1038/nrmicro1461. [DOI] [PubMed] [Google Scholar]
- Zwart M.P., Elena S.F. Matters of size: genetic bottlenecks in virus infection and their potential impact on evolution. Annu. Rev. Virol. 2015;2:161–179. doi: 10.1146/annurev-virology-100114-055135. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data Resources
The accession number for raw and analyzed NGS data reported in this paper is GEO: GSE124469.