

Summary
Several phage-tail-like nanomachines were shown to play an important role in the interactions between bacteria and their eukaryotic hosts. These apparatuses appear to represent a new injection paradigm. Here, with three verified extracellular contractile injection systems (eCISs), a protein profile and genomic context-based iterative approach was applied to identify 631 eCIS-like loci from the 11,699 publicly available complete bacterial genomes. The eCIS superfamily, which is phylogenetically diverse and sub-divided into six families, is distributed among Gram-negative and -positive bacteria in addition to archaea. Our results show that very few bacteria are seen to possess intact operons of both eCIS and type VI secretion systems (T6SSs). An open access online database of all detected eCIS-like loci is presented to facilitate future studies. The presence of this bacterial injection machine in a multitude of organisms suggests that it may play an important ecological role in the life cycles of many bacteria.
Keywords: Secretion system, bacteria pathogenicity, eCIS, T6SS, database
Graphical Abstract
Highlights
-
•
eCIS loci are widely distributed among bacteria genomes
-
•
eCIS loci encode phage-tail-like proteinaceous machines
-
•
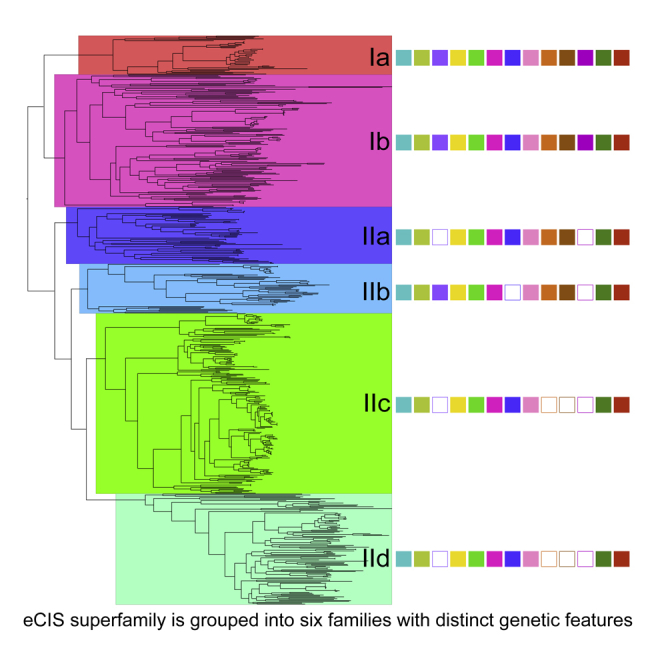
eCIS superfamily is grouped into six families with distinct genetic features
Based on genetic information from three recently identified extracellular contractile injection systems (eCISs), Chen et al. identify 631 eCIS-like loci from 11,699 publicly available complete bacterial genomes, including archaea, and Gram-negative and Gram-positive bacteria. These loci may represent a class of protein delivery systems to serve a variety of roles in these microorganisms.
Introduction
A variety of different bacterial secretion systems have been identified for the transfer of cytoplasmic proteins either out of the cell into the surrounding milieu or directly into the cytoplasm of eukaryotic or bacterial cells (Galán, 2009, Jennings et al., 2017, Russell et al., 2014). The best-described proteinaceous machines fall into the so-called types I-VII secretion systems (Galán and Waksman, 2018, Green and Mecsas, 2016). These secretion systems typically play a key role in the bacterial life cycle, mediating interactions that range from symbiotic to pathogenic relationships. For example, the type VI secretion systems (T6SSs) are capable of targeting both eukaryotic and bacterial cells, either for pathogenesis or to provide a growth advantage against competing bacteria (Jiang et al., 2014, Mougous et al., 2006).
A far less well-characterized phage-tail-like contractile nanomachine was identified in Serratia entomophila, and designated the anti-feeding prophage (AFP). The gut active AFP causes cessation of feeding and death of infected grass grub larvae, the natural host of S. entomophila (Hurst et al., 2004). A similar AFP-like system, the Photorhabdus virulence cassettes (PVCs), was shown to have insecticidal activity against wax moth larvae (Yang et al., 2006). Subsequently, a more distantly related cousin of the AFP/PVC-type devices was identified in the marine bacterium Pseudoalteromonas luteoviolacea. This system, metamorphosis-associated contractile (MAC) structure, triggers metamorphosis of the marine worm Hydroides elegans (Shikuma et al., 2014). Recently, a variant of AFP, AfpX, was identified in S. proteamaculans, which also mediate insect larvae mortality (Hurst et al., 2018). In addition, a Basic Local Alignment Search Tool (BLAST)-based survey listed several putative AFP-like loci in other bacteria termed phage-like-protein translocation structures (PLTSs) (Sarris et al., 2014). All these studies imply that this apparatus may represent a new distinct injection system.
It has been shown that the MAC devices are released by cell lysis. Attachment of the H. elegans free-swimming larvae hypothetically triggers the injection of an effector, inducing metamorphosis of the worm (Shikuma et al., 2014). Likewise, purified AFP and PVC needle complexes can exert a toxic effect, suggesting the effector proteins are pre-loaded into the needle complex before release (Yang et al., 2006).
Visualization of AFP and PVC ultra-structures revealed they are morphologically similar to Pseudomonas aeruginosa R-type bacteriocins (Hurst et al., 2007, Yang et al., 2006). Despite this there are limited genetic similarities, restricted mainly to the contractile tail complex subunits. Furthermore, the R-type bacteriocins do not deliver protein effectors; rather, they create lethal un-gated ion channels in the target bacterial membrane. On the other hand, classical T6SSs are functional in a contact-dependent manner with a minimal set of 13 subunits to form both a bacterial membrane complex and an external bacteriophage-like structure (Records, 2011). Contractile injection systems (CISs), which encode proteins homologous to the phage contractile tails, deliver effectors to mediate bacterial-host interactions. T6SSs are considered as a class of CISs, as their structural components are anchoring to the inner membrane for cytoplasmic localization. Different from the T6SS mode of action, termed “extracellular CISs” (eCISs), the AFP, PVC, and MAC devices resemble headless phages, which are released into the surroundings to bind to and inject into target host cells (Jiang et al., 2019, Leiman and Shneider, 2012, Shikuma et al., 2014). Interestingly, the T6SSiv loci of Amoebophilus asiaticus, which was shown to mediate interactions with host membranes, was reported to be closely related to known eCIS loci in terms of both genetic composition and molecular evolution (Böck et al., 2017). This operon is somewhat of an oddity, comprising aspects of both eCIS and T6SSs. Considering the observed wide distribution of this injection apparatus, it is necessary and timely to analyze and describe the diversity of this system in detail.
Here, based on selected characteristics of three functionally confirmed eCISs (AFP, PVC, and MAC), we describe a protein profile and genomic context-based iterative approach for the large-scale identification of potential eCIS loci. A phylogenetically diverse and broadly distributed superfamily of putative eCIS loci was identified from 11,699 publicly available complete bacterial genomes. The 631 eCIS-like loci cover both Gram-negative and -positive bacteria, as well as archaea. Furthermore, our analysis revealed these loci are sub-divided into six specific subfamilies with distinct patterns in terms of genetic composition and organization. A dedicated online open-access database named dbeCIS summarizing our analysis of these putative eCIS loci has been constructed (http://www.mgc.ac.cn/dbeCIS/). A further comparative analysis of the predicted eCIS and classical T6SS loci in complete genomes provides the evidence that eCIS-like apparatus and classical T6SSs are rarely found encoded in the same genome. With the information from dbeCIS we also identified an additional 143 eCIS positive Salmonella enterica strains by interrogating 178,998 draft Salmonella genomes available in EnteroBase. These apparatuses may represent a new bacterial injection paradigm that likely plays important and diverse roles in the life cycles of many species.
Results
Identification of Putative eCIS Loci
To establish an appropriate pan-genome screening approach for the identification of candidate eCISs, we first sought to accurately define them using information from the three experimentally validated eCISs: AFP, PVC, and MAC (Hurst et al., 2004, Shikuma et al., 2014, Yang et al., 2006). Eleven types of protein are conserved within these eCIS loci (Figure 1A), of which ten components are also present in the close relative of eCIS: T6SSiv (Figure 1B). This implies that these consensus components are important for the function of eCIS-like systems and thus could serve as candidates for our screening process. Moreover, in P. luteoviolacea, single-gene knockouts of macS, macT1/2, or macB (homologs of afp2/3/4, afp1/5, and afp11) abolished the MAC phenotype, indicating these genes are essential for function (Shikuma et al., 2014). Thus, we further narrowed our screening candidates to three subunit types, specifically, homologs of afp2/3/4, afp1/5, and afp11.
Figure 1.

Genetic Composition and Organization of Phage-Tail-Like Systems
(A) Three previously reported eCIS loci and their consensus composition. The 11 consensus components shared by all three loci are summarized below. Experimentally verified essential genes in the MAC are indicated with stars.
(B) The T6SSiv in Amoebophilus asiaticus, which is closely related to eCIS (Böck et al., 2017).
(C) Representatives of classical T6SS and R-type bacteriocin loci. Homologous components carrying the same protein domain as that of eCIS are denoted with the same color. Singleton genes are represented in gray.
(D) Venn diagram of the homologs to the 11 consensus components of eCIS in classical T6SS and R-type bacteriocin. The two key components (afp1/5 and afp11) employed for eCIS screening are indicated.
Although classical T6SS and R-type bacteriocin loci include several genes with predicted protein domains similar to eCIS, they do exhibit distinct differences in overall gene content and organization (Figures 1C and 1D). Well-conserved components of eCIS, absent from classical T6SS, are the Afp11 homologs, which exhibit phage T4 baseplate J-like domains. In addition, while the classical T6SSs and R-type bacteriocins also encode Gp19 domain-like “inner tube” proteins, the eCIS equivalents, Afp1/5-like proteins, can be seen to form a distinct phylogenetic clade (Shikuma et al., 2014).
In the light of the information above, we used the presence of both afp11 and non-phage-like afp1/5 homologs as the criterion to exclude the possible contaminations of classical T6SSs and R-type bacteriocins for the initial large-scale screening of genomes to identify potential members of the eCIS superfamily (Figure 1D).
Hidden Markov model (HMM) profiles of protein alignments can effectively represent the conserved protein families and domains using position-specific scores. Similarity searches based on HMM protein profiles are more sensitive than single-sequence comparison methods (e.g., BLAST) and are commonly used to identify divergent homologs (Pearson and Sierk, 2005). Furthermore, experimentally verified eCIS loci are all encoded in tightly linked gene clusters. Integration of genomic context information can thus effectively suppress false-positive identification and increases the specificity, as previously proposed (Liu et al., 2019). Therefore, we developed a combined protein profile and genomic context-based approach for the identification of eCIS-like loci (Figure 2).
Figure 2.

Identification of eCIS Loci from Publicly Available Complete Bacterial Genomes
The detailed workflow of the HMM profile and genomic-context-based analysis pipeline for the identification of eCIS loci in public bacterial genomes. The list of 11,699 complete bacterial genomes analyzed is available in Table S1.
An iterative screening process was applied to a collection of 11,699 complete bacterial genomes (Table S1), available from GenBank (accessed on June 19, 2018). After careful manual curation, a total of 615 contiguous eCIS loci were identified in 493 genomes (Figure 2), with some genomes encoding more than one eCIS locus. Importantly, even though Photorhabdus genomes also encode both classical T6SS and R-type bacteriocin operons, we note that our results did not misidentify any of these as potential eCISs, which supports the reliability of our approach.
Given the fact that the two key components, afp1/5 and afp11, are in fact encoded by two separated genomic loci in T6SSiv, the close relative of eCIS (Figure 1B), whereas the nanomachine remains functional in A. asiaticus (Böck et al., 2017), we relaxed the screening criteria to allow the report of potential eCIS with homologs of afp1/5 and afp11 encoded in more disparate genomic loci. This was applied only to genomes in which no contiguous eCIS-like loci had previously been identified, also employing extensive manual curation in order to minimize the reporting of false positives. A total of 13 additional genomes were thus identified that appeared to encode what we refer to as “split loci,” that is, eCISs that are encoded across two disparate genomic locations (Figure 2). This approach was validated by the independent detection of the T6SSiv (the close relative of eCIS) of A. asiaticus, as expected.
Finally, more than 85% of the eCIS-like loci we identified encode homologs of at least eight subunit types, including Afp1/5, Afp2/3/4, Afp7, Afp8, Afp9, Afp11, Afp15, and Afp16 (Figure S1). This indicates that not only are these proteins likely to be important for function, as evidenced by their conservation, but also that our screening process effectively excludes operons for other phage-tail-like structures such as prophages, classical T6SSs, and R-type bacteriocins. Therefore, our results imply these eight components can be considered the core backbone of eCIS loci.
Distribution of eCIS Loci
After large-scale screening against complete genomes, along with three reported loci from partially sequenced genomes (AFP, MAC, and AfpX), a total of 631 eCIS-like loci were identified among the available bacterial and archaeal genomes. As depicted in Figure S2, besides the 10 loci encoded within archaeal genomes, 408 loci are found in Gram-negative bacteria with the majority from the Proteobacteria, Cyanobacteria, and Bacteroidetes phyla, including Pseudomonas, Vibrio, Burkholderia, Yersinia, and Salmonella. Also, it is interesting to note, 213 loci were identified in Gram-positive bacteria, mainly from the class Actinobacteria, as previously observed (Sarris et al., 2014).
Due to the intrinsic logic of our screen strategy, Afp11 and Afp1/5 are present in all eCIS-like loci. However, because Afp1/5 homologs are usually in multiple copies in any one eCIS locus, and encode relatively small proteins, it was decided that the Afp11 sequences represent the best candidates for phylogenetic analysis. A maximum likelihood tree was constructed based on the protein sequence of Afp11, to explore the evolutionary relationship between eCISs, which revealed all loci grouped into two distinct phylogenetic clades (Figure 3). The minor clade (lineage I) consists of only Gram-negative bacterial eCIS loci, including all previously reported eCISs with known biological roles, whereas the major clade (lineage II) is mixed and more widely distributed than that of lineage I, containing loci derived from Gram-negative and Gram-positive bacterial and archaeal genomes. While we do see clustering of eCIS loci derived from members of the same genera or species (Figure S2), in general the phylogenetic clades are clearly inconsistent with the taxonomic demarcation at the phylum or class level. It is interesting to note, 94 out of 116 complete Streptomyces genomes encode eCIS loci, which belong to different clades (Figure S2). These results imply that these loci may play important roles in the life cycle of Streptomyces, which warrants further investigation.
Figure 3.

Maximum-Likelihood Tree of 631 Identified eCIS Loci (Root on Midpoint)
Based on Afp11 protein sequences, the tree was constructed by FastTree under Whelan Goldman (WAG) models with gamma optimization. Clades with all members from the same species are collapsed for brevity. Loci derived from archaeal and Gram-positive bacterial genomes are highlighted with red and blue branches, respectively. Previously reported eCIS loci are indicated by solid green circles. Outer strips are color coded by taxonomic groups as indicated by the key. The tree scale represents substitutions per site.
See also Figure S2 for details.
eCIS Loci Segregate into Six Subtypes
The phylogenetic lineages I and II can be sub-divided into two and four subtypes, respectively, which comprise distinct patterns in terms of genetic composition and organization (Figure 4). The two well-characterized examples of eCIS, AFP (and AfpX) and PVC, are members of subtype Ia. All loci from subtype Ia are derived from γ and β Proteobacteria (Figure S2). However, the more recently verified divergent eCISs—MAC and the close relative of eCIS, T6SSiv—belong to subtype Ib, which comprises loci not only from the Proteobacteria but also from other Gram-negative phyla including Bacteroidetes and Cyanobacteria. Nevertheless, in linage I, all components of eCIS-like loci are generally conserved, despite the variation in genomic organization between subtypes Ia and Ib (Figure 4B).
Figure 4.

eCIS Loci Are Divided into Six Families with Different Subtype-Associated Gene Organization
(A) Demarcation of eCIS subtypes based on the same unrooted tree as in Figure 3. Representative genetic compositions of the core components are shown as solid squares color coded as in Figure 1. The previously reported eCIS loci are indicated with red arrows. The support values for major clades are indicated (n = 100).
(B) Representative gene organization of the eCIS loci for each subtype. Known components are color coded with the same schema as in Figure 1. Subtype-specific conserved genes are represented in gray and singletons in white. The percentage above each arrow shows the presence proportion in each subtype. Homologs of the VgrG-like protein Afp8 and the baseplate J-encoding protein Afp11 are highlighted with strips and dots, respectively.
See also Figures S3, S4, S5, and S6 for details.
In contrast, four proteins including Afp6, Afp12, Afp13, and Afp14 are either highly divergent or absent in lineage II eCISs (Figure 4A). Interestingly, though members belonging to each subtype of lineage II are derived from both Gram-negative and Gram-positive bacteria, and even from archaea (for subtypes IIa and IIc), there remains a strong correlation between common gene organization and subtype (Figure S2). Even though all loci of subtype II were identified on the basis of sequence alone, the conserved gene synteny within the different subtypes argues for positive selection based on functional roles.
We further analyzed the conservation of certain subtype-specific genes. In addition to the eight aforementioned core backbone subunits of eCISs, we can also see subtype-specific arrangements and several additional subtype-specific genes (Figures 4B, S3, S4, S5, and S6). For instance, the afp6 gene is absent from all subtypes of lineage II except subtype IIb, whereas the afp10 is solely absent from subtype IIb, which indicates that subtype IIb may employ different regulation and/or target contact mechanisms from other lineage II subtypes. Of note, afp10 encodes a protein carrying potential PAAR-repeat motif, which in T6SSs forms the central spike mounted on VgrG to facilitate membrane puncture (Shneider et al., 2013). Subtypes IIa and IIb encode divergent copies of afp12 and afp13, both of which are absent from most loci of subtypes IIc and IId. Homologs of afp14, which encode putative tape measure proteins in AFP (Rybakova et al., 2015), are generally absent from all subtypes of lineage II. Interestingly, lineage II subtypes carry two or three additional subtype-specific genes, which are generally conserved in the majority (> 50%) of loci within an individual subtype (Figure 4B). However, no known functional domains could be identified in these subtype-specific proteins. Whether these predicted genes serve to compensate the absence of the aforementioned afp gene homologs in these loci, or provide additional features to lineage II subtypes, requires experimental investigations.
Among the conserved afp8 and afp11 genes, variations in gene size and copy number can be observed, correlating with the eCIS subtype (Figure 4B). For example, there is only a single copy of an afp11 homolog in subtypes Ia, Ib, IIb, and IId, although in subtype Ib this gene is notably longer. Conversely, subtypes IIa and IIc encode two copies of this gene. In the case of the T6SS VgrG homolog, afp8, which encodes the equivalent of the phage T4 spike proteins Gp5 and Gp27 (Pukatzki et al., 2007), subtypes Ia, Ib, and IId encode a single contiguous open reading frame (ORF), while in subtypes IIa, IIb, and IIc, the gene is split into two smaller ORFs encoding predicted Gp27 and Gp5 domains, respectively.
Characterization of eCIS Loci and Construction of dbeCIS
On the basis of domain annotation and sequence similarity, a core cluster of eCIS structural genes can now be defined. Table 1 lists the major conserved components of eCIS gene clusters, indicating conserved domains. There are several conserved genes related to baseplate assembly, reminiscent of phage-tail-like contractile structures confirmed by previous studies (Bönemann et al., 2009, Jiang et al., 2019, Kudryashev et al., 2015, Leiman et al., 2009, Liu et al., 2011, Lossi et al., 2013, Planamente et al., 2016, Rybakova et al., 2015, Shneider et al., 2013). Several baseplate structural domains, including LysM, VgrG (Gp27-Gp5), Gp25, Gp6, and Gp7, are remarkably conserved in the majority of eCIS loci, suggesting a baseplate assembly mechanism in common with phage T4 (Taylor et al., 2016). Another subunit conserved in all eCISs is represented by the Afp16-like proteins, which share a domain common with phage T4 Gp3. The Afp16 itself has been shown to regulate sheath formation, providing a mechanism to terminate tube elongation (Rybakova et al., 2013), while Afp14 shares homology with Gp29 and functions as tape measure protein (Rybakova et al., 2015).
Table 1.
Protein Domains Encoded by eCIS Components Compared with Phage and T6SS
| eCIS Gene | Accession Number | Structural or Functional Domain | Ortholog to Phage T4 | Ortholog to T6SS | eCIS Lineage | Reference |
|---|---|---|---|---|---|---|
| Afp1/5 | AAT48338, AAT48342 | phage-tail tube protein | Gp19 | TssD/Hcp | I and II | Leiman et al., 2009 |
| Afp2/3/4 | AAT48339, AAT48340, AAT48341 | phage-tail sheath protein | Gp18 | TssB/TssC | I and II | Lossi et al., 2013 |
| Afp6 | AAT48343 | transcriptional regulator, TETR family | N/A | N/A | I and IIb | Zimmermann et al., 2018 |
| Afp7 | AAT48344 | LysM domain | Gp53 | TssG | I and II | Planamente et al., 2016 |
| Afp8 | AAT48345 | phage-tail spike protein | Gp5/Gp27 | TssI/VgrG | I and II | Leiman et al., 2009 |
| Afp9 | AAT48346 | baseplate wedge protein | Gp25 | TssE | I and II | Kudryashev et al., 2015 |
| Afp10 | AAT48347 | PAAR-repeat motif | Gp5.4 | PAAR-repeat motif | I, IIa, IIc, and IId | Shneider et al., 2013 |
| Afp11 | AAT48348 | baseplate J protein | Gp6 | N/A | I and II | |
| Afp12 | AAT48349 | baseplate protein | Gp6-Gp7 | N/A | I and IIb | |
| Afp13 | AAT48350 | adenovirus fiber protein | Gp12 | N/A | I and IIb | Liu et al., 2011 |
| Afp14 | AAT48351 | tape measure protein | Gp29 | N/A | I | Rybakova et al., 2015 |
| Afp15 | AAT48352 | AAA+ ATPase | N/A | TssH/ClpV | I and II | Bönemann et al., 2009 |
| Afp16 | AAT48353 | phage-tail terminator protein | Gp3 | N/A | I and II | Rybakova et al., 2013 |
Note: eCIS components are represented by AFP locus. Domains are identified by references and HHpred (Zimmermann et al., 2018). N/A, none applicable.
While the majority of eCIS-like loci we detect are encoded as tightly linked contiguous gene clusters, we also identified what we refer to as “split loci” (e.g., T6SSiv, the close relative of eCIS in A. asiaticus), which elaborates phage-tail-like assemblies in the cytoplasm (Böck et al., 2017). These 13 additional split loci were found in both lineages I and II (Table S2), as well as from both Gram-negative and Gram-positive bacteria and archaea.
Interestingly, some bacterial strains encode more than one eCIS locus (Table S3). For example, five eCIS loci were identified in the genome of P. asymbiotica ATCC43949 and six loci in P. luminescens subspecies laumondii TTO1. It should be noted that RNA sequencing (RNA-seq) studies on three Photorhabdus strains (P. luminescens TTO1 and P. asymbiotica strains ATCC43939 and Kingscliff) confirmed transcription of several PVC operons, under certain conditions and growth phases (Mulley et al., 2015). The PVC loci in Photorhabdus are potentially used mainly for combating the insect immune response and, so, are likely to be important for its life cycle (Yang et al., 2006). This may explain the selection for multiple eCISs in this genus.
Based on our findings, we created an open-access and fully searchable database of all eCISs detected, named dbeCIS (http://www.mgc.ac.cn/dbeCIS/). This database integrates all information of the 631 identified eCIS-like loci, including taxonomy, bacterial characteristics, genomic features, sequences, and related publications. Furthermore, dbeCIS provides an interactive linear map for each locus with all known and subtype-specific components color coded and highlighted with clickable links to easily access gene details (Figure S7). Users can easily browse the database by either the hierarchical taxonomic distribution or the aforementioned phylogenetic subtypes of eCIS-like loci. The dbeCIS is also fully searchable via any query text or based on sequence similarities using BLAST.
eCIS Loci in the Genus Salmonella
As described so far, our results are limited to complete genomes. The EnteroBase database (Alikhan et al., 2018) hosts the largest worldwide collection of genomic and metadata of Salmonella, a well-recognized main bacterial agent responsible for foodborne disease. Therefore, in an attempt to further understand eCIS distribution patterns, we extended our search to screening 178,998 draft Salmonella genomes available in EnteroBase. The afp11 gene sequence of S. enteric subspecies diarizonae strain SA20044251, identified in dbeCIS, was aligned to the whole-genome multilocus sequence typing (wgMLST) scheme within EnteroBase. A total of 134 Salmonella genomes were identified to encode an afp11-homolog, and upon further analysis we were able to describe their associated eCIS-like loci (Table S4). Sixty of these genomes belonged to Salmonella collected as human clinical isolates, while the remaining strains were isolated from food, reptiles, and the environment.
To contextualize these genomes within the genus of Salmonella, we defined a dataset that included the 134 eCIS-positive genomes and 926 representative Salmonella genomes previously proposed (Alikhan et al., 2018). From these genomes, we constructed a maximum likelihood phylogeny of concatenated sequences of genes encoding ribosome proteins, as previous described (Jolley et al., 2012). The resulting tree demonstrates that all detected eCIS-positive genomes were most closely related to either S. enterica subspecies salamae or subspecies diarizonae (Figure 5). In addition, we note that based on the afp11 sequence, all eCIS loci from the Salmonella strains presented here are grouped into subtype Ib. Based on eCIS detection in these Salmonella draft genomes, we suggest that afp11 is a good candidate for eCIS loci detection in other draft genomes, and more eCIS loci among various bacterial species would be discovered in this manner.
Figure 5.

Distribution of eCIS within the Salmonella Population
A Grapetree representation of a maximum-likelihood phylogeny of 2,389 SNPs derived from the concatenated sequence of 51 ribosomal encoding genes from 134 eCIS positive Salmonella (Table S4) and 926 representative genomes of S. enterica and S. bongori. Branches greater than 0.01 substitutions per site (dotted) were shortened for clarity. Nodes at the tips were colored by presence of afp11 (a proxy for eCIS), as indicated by the key. Major subspecies were highlighted (key) according to previous results (Alikhan et al., 2018). Node sizes are proportional to the number of genomes that share identical sequences to the node. Tree scale represents frequencies of substitutions per site.
eCIS and T6SS Loci Are Rarely Found Encoded in the Same Genome
T6SS islands comprise 13 core subunits, which form an inverted bacteriophage-like structure on the bacterial cell surface for secretory functions (Silverman et al., 2012). Previous studies show that eCISs also form contractile phage-tail-like structures (Hurst et al., 2004, Jiang et al., 2019, Shikuma et al., 2014, Yang et al., 2006). The homology between classical T6SS and eCIS subunits is a reflection of the shared contractile phage-tail-like mechanism employed by both systems (Figure 6A). Besides an AAA-ATPase subunit, the Afp15-like and TssH proteins, respectively, both eCIS and classical T6SS loci encode several conserved components, including Afp1/5 (TssD), Hcp-like protein; Afp2/3/4 (TssB/C), phage-tail sheath protein; Afp7 (TssG), LysM domain carrying protein; Afp8 (TssI), VgrG-like protein; and Afp9 (TssE), Gp25-like protein. These proteins are components of the phage-tail-like structure, including tube, sheath, and baseplate. It is worth noting that while both eCIS and T6SSi-iii produce contractile phage-tail-like structures, homologs of the proteins encoded by afp11, afp14, and afp16 are not seen in T6SSs, highlighting a fundamental difference in mechanistic deployment. Indeed, a central aspect of the classical T6SSs is that they encode a membrane complex for the synthesis, deployment, anchoring, and recycling of the phage-like external needle. The eCIS loci do not appear to need such a membrane complex, as those that have been experimentally characterized elaborate “single use” freely released needle complexes, as discussed above.
Figure 6.

Relationship of T6SS and eCIS
(A) Comparison between an example T6SS and a subtype Ia eCIS locus. Note, dotted genes, afp11, afp14, and afp16, are encoded in eCIS not classical T6SSi-iii loci.
(B) Venn diagram of shared genomes by the datasets of newly identified eCIS loci and known classical T6SS loci available from the online SecReT6 database. Note, only 15 loci encode all T6SS essential components.
See also Table S5 for details.
Further, we compared the distribution of eCIS and classical T6SS loci among bacterial genomes using our database and the SecReT6 database (Li et al., 2015). Among the 509 strains encoding eCIS-like loci and 498 strains with classical T6SS loci, only 38 bacterial strains had both (Figure 6B). Moreover, of these 38 strains, only 15 encoded all 13 necessary classical T6SS core components (Table S5).
Discussion
Here, we present a protein-profile and genomic context-based iterative method to identify what we propose as a superfamily of phage-tail-based protein injection systems: eCIS. In total, 631 eCIS-like loci were identified, which fall into six distinct phylogenetic subfamilies, distributed among genomes of Gram-negative/positive bacteria and archaea. The widespread distribution of eCIS suggests that these loci may represent essential apparatuses, able to serve a variety of roles in these microorganisms.
Interestingly, our results have revealed that the six subtypes of eCISs represent distinct patterns in terms of genetic composition and organization. It is difficult to speculate at this point whether eCISs within different subtypes have multiple independent origins and evolved by convergent evolution, or whether they have a single common ancestor. Nevertheless, the available evidence suggests that extensive horizontal transfer events might have happened during the spread of eCIS among different microorganisms. Indeed, 25 eCIS loci are carried on plasmids (Table S6), which may have played an important role in the horizontal transfer of eCISs. A good example of this is Calothrix sp. NIES-4101, which carries two subtype IId eCIS loci with the same gene composition and organization encoded on chromosome and plasmid, respectively.
Considering that the T6SS database contains 906 classical T6SS loci in 498 bacterial strains and our database includes 631 eCIS-like loci in 509 bacterial strains, it is a reasonable hypothesis to suggest that eCIS and classical T6SS are in some way “incompatible” or at the very least are not subject to positive selection when both are present. P. asymbiotica ATCC43949 encodes five eCISs and four classical T6SS loci. Our previous RNA-seq results showed that certain eCISs loci could be transcribed under certain conditions, while the T6SS loci were not (Mulley et al., 2015). It is possible that both the eCIS and T6SS apparatus in Photorhabdus require specific host signals or conditions to trigger their expression, as has been seen for the T3SS in other species. Considering that so few examples of bacteria encode both eCIS and T6SS, we speculate that similarity of shared subunit types (i.e., Hcp, VgrG, PAAR, and ATPase domain proteins) could cause misassembly issues in one or both of these multimeric protein machines. It is also possible that, as the two systems may fulfill the same role, it means there is no strong selection pressure to maintain both. Or alternatively, eCIS and T6SS may have a large metabolic cost to produce, and bacteria that have both must have significant evolutionary pressure to retain both structures. While classical T6SSs have been shown to enable contact-dependent delivery of toxins into both eukaryotic and competing bacteria cells, experimental evidence of the three eCISs suggests they are released into the medium to target at eukaryotes. Finally, different from classical T6SS (i-iii), T6SSiv exhibits a closer relationship with eCISs in terms of both genetic composition and phylogeny, as previously revealed (Böck et al., 2017). Nevertheless, T6SSiv lacks many homologs of components that are specific in canonical T6SSs, such as IcmF (Böck et al., 2017), whereas it encodes homologs of the majority components of known eCISs (Figure 1B). The evolutionary relationships between T6SSiv and eCISs would be an interesting focus though they might exhibit different action modes. The 631 loci identified in this study may also cover T6SSiv-like loci and, thus, would be a helpful resource for future studies aimed at addressing this puzzle.
The experimentally verified eCISs are deployed either to manipulate the development or intoxicate invertebrate or single-celled animals. Generally, we do not see them encoded in mammalian host restricted pathogens. So it is likely that many may be involved in commensal, symbiotic, or defensive interactions. In the cases where they are, such as Salmonella, we speculate that they are normally deployed in environmental contexts. We note that the majority of bacteria, which carry eCISs, are found in marine or soil environments, where exposure to simpler animals would be common.
Bacterial secretion systems deliver a plethora of effectors into the environment, a competitor, or a host. To understand the roles these machines play, it is important to elucidate the functions of the effectors. In the case of T6SS, the structural VgrG protein can also serve as an effector. The structural role of VgrG is facilitated by two domains. The N-terminal Gp27 and C-terminal Gp5 domains form a phage-tail spike-like structure that, with assistance from the PAAR protein, allows host cell membrane puncture (Barret et al., 2011). However, certain VgrG homologs, termed as “evolved VgrGs,” have C-terminal extensions with catalytic domains that act as effectors (Ma et al., 2009). We note that none of the eCIS-encoded VgrG homologs have a C-terminal extension, and so appear to be fulfilling a structural role only. To date, only a few eCIS-dependent effectors have been identified in Serratia, Photorhabdus, and Pseudoalteromonas, which are used to interact with eukaryotic cells (Hurst et al., 2004, Rocchi et al., 2019, Yang et al., 2006). However, most of the eCIS-encoding bacteria are not recognized human pathogens, so it is likely that many may be involved in commensal, symbiotic, or defensive interactions. Indeed, in P. luteoviolacea, the eCIS structure is known to trigger metamorphosis of the marine worm H. elegans (Shikuma et al., 2014).
We would like to point out some potential limitations of this study. The current knowledge regarding eCIS function is limited to the three experimentally verified loci (AFP, PVC, and MAC); all of the newly identified eCIS-like loci described here are necessarily based on bioinformatic analysis without experimental support. Therefore, potential false positives and false negatives are inevitable, even though we have balanced the screening approach for sensitivity and specificity. Another limitation is that we have limited our search to completely sequenced and annotated genomes, as our approach uses genomic context information to improve specificity. As many completely sequenced bacterial genomes are of medically important pathogens, this will introduce a certain level of sampling bias, so we suspect the numbers of bacteria using eCISs is being underestimated in the current study. Additional methods to circumvent the incompleteness of whole-genome sequencing (WGS) dataset for accurate eCIS identification in the future are required to further explore the widespread extent of this system in nature. Our screening of > 100,000 genomes of Salmonella WGS data in EnteroBase illustrates the importance and power to extend the systematic analysis to WGS data. Furthermore, according to three functional confirmed eCIS loci (AFP, PVC, and MAC), we performed our screening to identify more than 600 new eCIS loci here, which are only based on conserved protein families and domains. Although these eCIS loci exhibit similar genetic compositions as known eCIS members, more biological experiments are still required to provide further support for these findings.
Here, we described a detailed bioinformatic study of a superfamily of novel bacterial injection apparatuses that utilize phage contractile tail machinery. This system shows fundamentally important differences to the related phage-tail-like systems, such as R-type bacteriocins and classical T6SS. To date, only three of them were experimentally confirmed to be involved in the interaction between bacteria and host, including AFP, PVC, and MAC (Hurst et al., 2004, Shikuma et al., 2014, Yang et al., 2006). Taking into account the taxonomic and genetic diversity of eCISs, we are now able to provide a comprehensive overview of this highly diverse system. In particular, the dbeCIS database offers the community a convenient way to access, query, visualize, compare, and retrieve any related information of potential eCISs identified here. Despite the current limited experimental data regarding eCISs, our results provide insights into the mechanism, function, and evolution of eCISs, and will facilitate further study of what appears to be a very widespread and therefore likely important system.
STAR★Methods
Key Resources Table
Lead Contact and Materials Availability
Further information and requests may be directed to, and will be fulfilled by, the corresponding author Dr. Guowei Yang (yangguowei@hotmail.com). This study did not generate new unique reagents.
Method Details
Data Collection and HMM Protein Profile Building
The initial seed dataset are manually collected from GenBank (Benson et al., 2018) based on previous publications of experimentally verified eCIS loci, including AFP (GenBank accession: AF135182), PVC (GenBank accession: BX470251) and MAC (GenBank accession: KF724687). Homologous groups of each component are identified according to the related literatures followed by BLASTP sequence-similarity confirmation (E-value < 0.1). Afp1 and Afp5 as well as Afp2, Afp3 and Afp4 are two known multiple copy homologous groups in both AFP and PVC, so they are referred as two individual components Afp1/5 and Afp2/3/4 thereafter, respectively. The Clustal-Omega program (Sievers et al., 2011) (v1.2.4) was used to construct multiple protein sequences alignment (MSA) of each homologous group. Then, the MSA files were used to build HMM protein profiles (named seeds profiles hereafter) using the hidden Markov models as implemented in the HMMER3 package (Mistry et al., 2013) (v3.1b2).
The extended dataset include 11,699 completely sequenced and well annotated prokaryotic genomes or chromosomes available from GenBank (accessed at June 19, 2018), including 11,397 Bacteria genomes and 302 Archaea genomes (Table S1). The annotated genome files were downloaded to local system and phrased with BioPerl (Stajich et al., 2002) for further genome wide screen.
Genomic Context-Based Iterative Identification of eCIS
The seeds profiles were used to conduct the first-round screen among the complete genome dataset using the hmmsearch program available from HMMER3 package (Mistry et al., 2013). A deliberately loosened sequence similarity cutoff (hmmsearch E-value < 1e-5) was applied to improve the sensitivity of our approach. However, loosened cutoff will undoubtedly introduced high false positive rate. So in order to increase the specificity, we further designed a genomic-context based criterion: two essential subunit genes homologs, afp11 and afp1/5, are both present in a contiguous genomic region. Here, a contiguous genomic region is defined by a group of identified homologs of aforementioned 13 known components within a given genome that have each adjacent component pair with a maximal genomic distance of 30 kbp. This is an empirical setting to allow our approach to be capable to successfully identify the case of MAC locus.
HMM profiles summarize the evolutionary history of a family by identifying more and less conserved positions within the protein, including more homologs into the profiles improves the sensitivity for similarity searches. But the seeds profiles cover only three known eCIS loci, all derived from Gram-negative Bacteria. To further improve the sensitivity of our approach, based on the results from the first-round screen and the previously report on PLTSs (Sarris et al., 2014), fifteen additional taxonomically diversified representative genomes of both Bacteria and Archaea were further curated and collected. The new dataset was integrated with the seed dataset to form the core dataset for building core protein profiles using the same method mentioned above.
An iterative screening process was then applied to the aforementioned collection of 11,699 genomes, using the new core protein profiles as the initial input for hmmsearch. For each iteration, the newly identified eCIS loci were again included (after curation) to build on the updated protein profiles. These profiles were again used in the next iteration of screening, and so on until no additional eCIS loci were reported (E-value < 1e-5).
Next, given the fact that the two key components, afp1/5 and afp11 are in fact encoded by two separated genomic loci in A. asiaticus (Böck et al., 2017), we relaxed the screening criterion to allow the report of potential eCIS with homologs of afp1/5 and afp11 encoded in more disparate genomic loci. But this was applied only to genomes in which no contiguous eCIS loci had been identified in the previous results. The maximal genomic distance cutoff for each adjacent component pair was retained as 30 kbp for consistence.
Grouping of Subtype-Specific Conserved Genes
Besides known core components many of the newly identified eCIS loci also encode some additional genes, particularly for the loci from lineage II. We therefore conducted a similar protein-profile based identification of potential subtype-specifically conserved gene groups for each lineage II subtype. The software OrthoMCL (Li et al., 2003) (v1.4) was used to identify homologous groups with default parameters among the eCIS loci from each subtype with known core components excluded. The homologous groups produced were individually used to build MSAs and HMM protein profiles as aforementioned. Then, the protein profiles were used as queries for the hmmsearch screen of potential distinct homologs among the dataset of all additional genes within each eCIS locus from the subtype. Only conserved gene groups present in over half of the eCIS loci of the individual subtype were marked as subtype-specific genes.
Phylogenetic Analysis of eCIS Loci
Due to the intrinsic logic of our screen strategy Afp11 and Afp1/5 are present in all eCIS loci. Given that fact that the Afp1/5 homologs are generally present as multiple copies in most eCIS loci and the proteins length are smaller than Afp11, we selected Afp11 as the molecular marker for phylogenetic analysis. For loci with multiple copies of Afp11 only the upstream copy, which is generally longer in length was used for representation. The Clustal-Omega program was used to generate MSA of the 631 Afp11 proteins. FastTree (Price et al., 2010) program (v2.1) was then employed to construct the maximum-likelihood phylogenetic tree under WAG models with gamma optimization. The iTOL (Letunic and Bork, 2016) online server was used to manipulate and present the phylogeny.
Identification and Analysis of eCIS Loci within Salmonella
The gene sequence of afp11 (LFZ55_10775) from S. enterica subspecies diarizonae strain SA20044251 (GenBank accession: CP022135) from dbeCIS (http://www.mgc.ac.cn/cgi-bin/dbeCIS/showecis.cgi?id=A0316) was queried against the whole genome MLST (wgMLST) scheme with the ‘locus search’ feature from EnteroBase (Alikhan et al., 2018) (http://enterobase.warwick.ac.uk/) and identified as NCTC9948_01971 within the wgMLST scheme. Salmonella genomes with apf11 present were combined with a dataset of representative Salmonella previously described (Alikhan et al., 2018). 2,389 SNPs derived from the concatenated alignment of 51 ribosomal encoding genes were called using the EToKi pipeline (https://github.com/zheminzhou/EToKi), with filtering of repetitive genome regions and used to construct a maximum-likelihood tree with RaxML (Stamatakis, 2014) (v8.2.4) using discrete general time reversible (GTRCAT) substitution model with correction for ascertainment bias. Salmonella tree was visualized in GrapeTree (Zhou et al., 2018).
Database Construction
In order to facilitate future studies on eCIS we constructed a publicly accessible online database, named dbeCIS (http://www.mgc.ac.cn/dbeCIS/), to integrate all results generated from previous and current studies. MySQL was used to store all information including genome features, sequence annotations, homologous groups and eCIS typing. The Perl programming language and several modules, such as DBI, GD and CGI, were used to generate dynamic web pages.
Quantification and Statistical Analysis
The statistical analyses are not applicable to this study.
Data and Code Availability
Code Resources
The source code used to identify putative eCIS loci from genomic sequences are available online at: https://github.com/ipb-jianyang/eCIS-screen.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31741008 to G.Y. and 81801980 to N.S.); the CAMS Innovation Fund for Medical Sciences (2017-I2M-3-017 to J.Y.); a Leverhulme Trust Project grant (RPG-2015-194 to N.R.W.); the Biotechnology and Biological Sciences Research Council (BB/L020319/1 to N.-F.A. and Z.Z. and BB/C008367/1 to N.R.W); the Wellcome Trust (202792/Z/16/Z to N.F.-A. and Z.Z.); and a Quadram Institute Bioscience BBSRC-funded Core Capability Grant (BB/CCG1860/1 to N.-F.A.).
Author Contributions
N.R.W., J.Y., and G.Y. conceived the project. L.C., N.S., B.L., N.Z., Y.Z., S.Z., D.Z., M.C., A.H., J.H., N.R.W., J.Y., and G.Y. analyzed the genomic data and constructed the database. N.-F.A. and Z.Z. analyzed the Salmonella WGS data. L.C., N.-F.A., Z.Z., A.H., J.H., N.R.W., J.Y., and G.Y. wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: October 8, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.08.096.
Contributor Information
Nicholas R. Waterfield, Email: n.r.waterfield@warwick.ac.uk.
Jian Yang, Email: yangj@ipbcams.ac.cn.
Guowei Yang, Email: yangguowei@hotmail.com.
Supplemental Information
References
- Alikhan N.F., Zhou Z., Sergeant M.J., Achtman M. A genomic overview of the population structure of Salmonella. PLoS Genet. 2018;14:e1007261. doi: 10.1371/journal.pgen.1007261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barret M., Egan F., Fargier E., Morrissey J.P., O’Gara F. Genomic analysis of the type VI secretion systems in Pseudomonas spp.: novel clusters and putative effectors uncovered. Microbiology. 2011;157:1726–1739. doi: 10.1099/mic.0.048645-0. [DOI] [PubMed] [Google Scholar]
- Benson D.A., Cavanaugh M., Clark K., Karsch-Mizrachi I., Ostell J., Pruitt K.D., Sayers E.W. GenBank. Nucleic Acids Res. 2018;46(D1):D41–D47. doi: 10.1093/nar/gkx1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böck D., Medeiros J.M., Tsao H.F., Penz T., Weiss G.L., Aistleitner K., Horn M., Pilhofer M. In situ architecture, function, and evolution of a contractile injection system. Science. 2017;357:713–717. doi: 10.1126/science.aan7904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bönemann G., Pietrosiuk A., Diemand A., Zentgraf H., Mogk A. Remodelling of VipA/VipB tubules by ClpV-mediated threading is crucial for type VI protein secretion. EMBO J. 2009;28:315–325. doi: 10.1038/emboj.2008.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galán J.E. Common themes in the design and function of bacterial effectors. Cell Host Microbe. 2009;5:571–579. doi: 10.1016/j.chom.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galán J.E., Waksman G. Protein-Injection Machines in Bacteria. Cell. 2018;172:1306–1318. doi: 10.1016/j.cell.2018.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green E.R., Mecsas J. Bacterial Secretion Systems: An Overview. Microbiol. Spectr. 2016;4(1) doi: 10.1128/microbiolspec.VMBF-0012-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst M.R., Glare T.R., Jackson T.A. Cloning Serratia entomophila antifeeding genes--a putative defective prophage active against the grass grub Costelytra zealandica. J. Bacteriol. 2004;186:5116–5128. doi: 10.1128/JB.186.15.5116-5128.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst M.R., Beard S.S., Jackson T.A., Jones S.M. Isolation and characterization of the Serratia entomophila antifeeding prophage. FEMS Microbiol. Lett. 2007;270:42–48. doi: 10.1111/j.1574-6968.2007.00645.x. [DOI] [PubMed] [Google Scholar]
- Hurst M.R.H., Beattie A., Jones S.A., Laugraud A., van Koten C., Harper L. Serratia proteamaculans Strain AGR96X Encodes an Antifeeding Prophage (Tailocin) with Activity against Grass Grub (Costelytra giveni) and Manuka Beetle (Pyronota Species) Larvae. Appl. Environ. Microbiol. 2018;84:e02739-17. doi: 10.1128/AEM.02739-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings E., Thurston T.L.M., Holden D.W. Salmonella SPI-2 Type III Secretion System Effectors: Molecular Mechanisms And Physiological Consequences. Cell Host Microbe. 2017;22:217–231. doi: 10.1016/j.chom.2017.07.009. [DOI] [PubMed] [Google Scholar]
- Jiang F., Waterfield N.R., Yang J., Yang G., Jin Q. A Pseudomonas aeruginosa type VI secretion phospholipase D effector targets both prokaryotic and eukaryotic cells. Cell Host Microbe. 2014;15:600–610. doi: 10.1016/j.chom.2014.04.010. [DOI] [PubMed] [Google Scholar]
- Jiang F., Li N., Wang X., Cheng J., Huang Y., Yang Y., Yang J., Cai B., Wang Y.P., Jin Q. Cryo-EM Structure and Assembly of an Extracellular Contractile Injection System. Cell. 2019;177:370–383.e15. doi: 10.1016/j.cell.2019.02.020. [DOI] [PubMed] [Google Scholar]
- Jolley K.A., Bliss C.M., Bennett J.S., Bratcher H.B., Brehony C., Colles F.M., Wimalarathna H., Harrison O.B., Sheppard S.K., Cody A.J., Maiden M.C. Ribosomal multilocus sequence typing: universal characterization of bacteria from domain to strain. Microbiology. 2012;158:1005–1015. doi: 10.1099/mic.0.055459-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudryashev M., Wang R.Y., Brackmann M., Scherer S., Maier T., Baker D., DiMaio F., Stahlberg H., Egelman E.H., Basler M. Structure of the type VI secretion system contractile sheath. Cell. 2015;160:952–962. doi: 10.1016/j.cell.2015.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiman P.G., Shneider M.M. Contractile tail machines of bacteriophages. Adv. Exp. Med. Biol. 2012;726:93–114. doi: 10.1007/978-1-4614-0980-9_5. [DOI] [PubMed] [Google Scholar]
- Leiman P.G., Basler M., Ramagopal U.A., Bonanno J.B., Sauder J.M., Pukatzki S., Burley S.K., Almo S.C., Mekalanos J.J. Type VI secretion apparatus and phage tail-associated protein complexes share a common evolutionary origin. Proc. Natl. Acad. Sci. USA. 2009;106:4154–4159. doi: 10.1073/pnas.0813360106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I., Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44(W1):W242–W245. doi: 10.1093/nar/gkw290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Stoeckert C.J., Jr., Roos D.S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Yao Y., Xu H.H., Hao L., Deng Z., Rajakumar K., Ou H.Y. SecReT6: a web-based resource for type VI secretion systems found in bacteria. Environ. Microbiol. 2015;17:2196–2202. doi: 10.1111/1462-2920.12794. [DOI] [PubMed] [Google Scholar]
- Liu H., Wu L., Zhou Z.H. Model of the trimeric fiber and its interactions with the pentameric penton base of human adenovirus by cryo-electron microscopy. J. Mol. Biol. 2011;406:764–774. doi: 10.1016/j.jmb.2010.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B., Zheng D., Jin Q., Chen L., Yang J. VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019;47(D1):D687–D692. doi: 10.1093/nar/gky1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossi N.S., Manoli E., Förster A., Dajani R., Pape T., Freemont P., Filloux A. The HsiB1C1 (TssB-TssC) complex of the Pseudomonas aeruginosa type VI secretion system forms a bacteriophage tail sheathlike structure. J. Biol. Chem. 2013;288:7536–7548. doi: 10.1074/jbc.M112.439273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma A.T., McAuley S., Pukatzki S., Mekalanos J.J. Translocation of a Vibrio cholerae type VI secretion effector requires bacterial endocytosis by host cells. Cell Host Microbe. 2009;5:234–243. doi: 10.1016/j.chom.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry J., Finn R.D., Eddy S.R., Bateman A., Punta M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013;41:e121. doi: 10.1093/nar/gkt263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mougous J.D., Cuff M.E., Raunser S., Shen A., Zhou M., Gifford C.A., Goodman A.L., Joachimiak G., Ordoñez C.L., Lory S. A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science. 2006;312:1526–1530. doi: 10.1126/science.1128393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulley G., Beeton M.L., Wilkinson P., Vlisidou I., Ockendon-Powell N., Hapeshi A., Tobias N.J., Nollmann F.I., Bode H.B., van den Elsen J. From Insect to Man: Photorhabdus Sheds Light on the Emergence of Human Pathogenicity. PLoS ONE. 2015;10:e0144937. doi: 10.1371/journal.pone.0144937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson W.R., Sierk M.L. The limits of protein sequence comparison? Curr. Opin. Struct. Biol. 2005;15:254–260. doi: 10.1016/j.sbi.2005.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planamente S., Salih O., Manoli E., Albesa-Jové D., Freemont P.S., Filloux A. TssA forms a gp6-like ring attached to the type VI secretion sheath. EMBO J. 2016;35:1613–1627. doi: 10.15252/embj.201694024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M.N., Dehal P.S., Arkin A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pukatzki S., Ma A.T., Revel A.T., Sturtevant D., Mekalanos J.J. Type VI secretion system translocates a phage tail spike-like protein into target cells where it cross-links actin. Proc. Natl. Acad. Sci. USA. 2007;104:15508–15513. doi: 10.1073/pnas.0706532104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Records A.R. The type VI secretion system: a multipurpose delivery system with a phage-like machinery. Mol. Plant Microbe Interact. 2011;24:751–757. doi: 10.1094/MPMI-11-10-0262. [DOI] [PubMed] [Google Scholar]
- Rocchi I., Ericson C.F., Malter K.E., Zargar S., Eisenstein F., Pilhofer M., Beyhan S., Shikuma N.J. A Bacterial Phage Tail-like Structure Kills Eukaryotic Cells by Injecting a Nuclease Effector. Cell Rep. 2019;28:295–301.e4. doi: 10.1016/j.celrep.2019.06.019. [DOI] [PubMed] [Google Scholar]
- Russell A.B., Peterson S.B., Mougous J.D. Type VI secretion system effectors: poisons with a purpose. Nat. Rev. Microbiol. 2014;12:137–148. doi: 10.1038/nrmicro3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybakova D., Radjainia M., Turner A., Sen A., Mitra A.K., Hurst M.R. Role of antifeeding prophage (Afp) protein Afp16 in terminating the length of the Afp tailocin and stabilizing its sheath. Mol. Microbiol. 2013;89:702–714. doi: 10.1111/mmi.12305. [DOI] [PubMed] [Google Scholar]
- Rybakova D., Schramm P., Mitra A.K., Hurst M.R. Afp14 is involved in regulating the length of Anti-feeding prophage (Afp) Mol. Microbiol. 2015;96:815–826. doi: 10.1111/mmi.12974. [DOI] [PubMed] [Google Scholar]
- Sarris P.F., Ladoukakis E.D., Panopoulos N.J., Scoulica E.V. A phage tail-derived element with wide distribution among both prokaryotic domains: a comparative genomic and phylogenetic study. Genome Biol. Evol. 2014;6:1739–1747. doi: 10.1093/gbe/evu136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shikuma N.J., Pilhofer M., Weiss G.L., Hadfield M.G., Jensen G.J., Newman D.K. Marine tubeworm metamorphosis induced by arrays of bacterial phage tail-like structures. Science. 2014;343:529–533. doi: 10.1126/science.1246794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shneider M.M., Buth S.A., Ho B.T., Basler M., Mekalanos J.J., Leiman P.G. PAAR-repeat proteins sharpen and diversify the type VI secretion system spike. Nature. 2013;500:350–353. doi: 10.1038/nature12453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman J.M., Brunet Y.R., Cascales E., Mougous J.D. Structure and regulation of the type VI secretion system. Annu. Rev. Microbiol. 2012;66:453–472. doi: 10.1146/annurev-micro-121809-151619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stajich J.E., Block D., Boulez K., Brenner S.E., Chervitz S.A., Dagdigian C., Fuellen G., Gilbert J.G., Korf I., Lapp H. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 2002;12:1611–1618. doi: 10.1101/gr.361602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor N.M., Prokhorov N.S., Guerrero-Ferreira R.C., Shneider M.M., Browning C., Goldie K.N., Stahlberg H., Leiman P.G. Structure of the T4 baseplate and its function in triggering sheath contraction. Nature. 2016;533:346–352. doi: 10.1038/nature17971. [DOI] [PubMed] [Google Scholar]
- Yang G., Dowling A.J., Gerike U., ffrench-Constant R.H., Waterfield N.R. Photorhabdus virulence cassettes confer injectable insecticidal activity against the wax moth. J. Bacteriol. 2006;188:2254–2261. doi: 10.1128/JB.188.6.2254-2261.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., Alikhan N.F., Sergeant M.J., Luhmann N., Vaz C., Francisco A.P., Carriço J.A., Achtman M. GrapeTree: visualization of core genomic relationships among 100,000 bacterial pathogens. Genome Res. 2018;28:1395–1404. doi: 10.1101/gr.232397.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann L., Stephens A., Nam S.Z., Rau D., Kübler J., Lozajic M., Gabler F., Söding J., Lupas A.N., Alva V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 2018;430:2237–2243. doi: 10.1016/j.jmb.2017.12.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Code Resources
The source code used to identify putative eCIS loci from genomic sequences are available online at: https://github.com/ipb-jianyang/eCIS-screen.