

Summary
The glucocorticoid receptor (GR) acts as a ubiquitous cortisol-dependent transcription factor (TF). To identify co-factors, we used protein-fragment complementation assays and found that GR recognizes FLI1 and additional ETS family proteins, TFs relaying proliferation and/or migration signals. Following steroid-dependent translocation of FLI1 and GR to the nucleus, the FLI1-specific domain (FLS) binds with GR and strongly enhances GR’s transcriptional activity. This interaction has functional consequences in Ewing sarcoma (ES), childhood and adolescence bone malignancies driven by fusions between EWSR1 and FLI1. In vitro, GR knockdown inhibited the migration and proliferation of ES cells, and in animal models, antagonizing GR (or lowering cortisol) retarded both tumor growth and metastasis from bone to lung. Taken together, our findings offer mechanistic rationale for repurposing GR-targeting drugs for the treatment of patients with ES.
Keywords: cancer therapy, Ewing sarcoma, glucocorticoid receptor, metastasis, protein-fragment complementation assay
Graphical Abstract
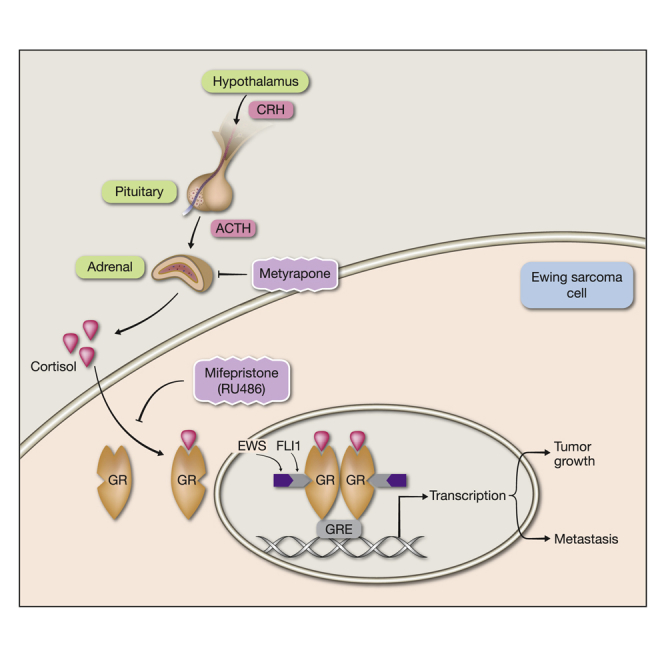
Highlights
-
•
Glucocorticoids activate GR, an inducible TF regulating growth and metabolism
-
•
Protein complementation assays identified FLI1 and ERG as GR binders and activators
-
•
EWS-FLI1, a fusion protein, drives Ewing sarcoma (ES) and binds with activated GR
-
•
Pharmacological inhibition of GR activation retards progression in ES animal models
The single oncogene of Ewing sarcoma (ES), a childhood cancer, encodes a FLI1 fusion protein. Srivastava et al. report physical interactions between the fusion protein and the glucocorticoid receptor. Drug-induced inhibition of these interactions retards the progression of ES in mouse models.
Introduction
Endogenous glucocorticoids (GCs) modulate multiple physiological and cellular processes, including cell proliferation, metabolism, and apoptosis (Lin and Wang, 2016, Ramamoorthy and Cidlowski, 2016). Synthetic GCs like dexamethasone (DEX) have been widely used in the treatment of hematologic malignancies, as cytotoxic agents, and in the treatment of solid tumors to prevent complications associated with cancer therapy (Rutz, 2002). The cellular actions of GCs are mediated by the glucocorticoid receptor (GR). Like other nuclear receptors, the GR resides in the cytoplasm, stabilized by chaperone proteins. Once bound by GCs, the GR translocates into the nucleus to regulate many target genes, either positively or negatively. Positive regulation, a mechanism known as GR-dependent transactivation (TA), entails receptor binding to GC response elements (GREs). A similarly important negative regulation is called GR-dependent transrepression (TR). This is mediated by physical interactions between GR and DNA-bound transcription factors (TFs), such as nuclear factor κB (NF-κB) (Ray and Prefontaine, 1994), Stat5 (Stöcklin et al., 1996), and the activator protein 1 (AP-1) (Petta et al., 2016).
Apart from transrepression, GR can sequester specific TFs, thereby preventing their binding to the respective response elements (Rogatsky et al., 2003). Interestingly, the ability of GR to interfere with NF-κB and AP-1 may lead to mutual inhibition (reviewed in Petta et al., 2016). Despite mutual antagonism, AP-1 has emerged as a key partner in GR-regulated transcription by means of enhancing GR binding to specific sites in the genome (Biddie et al., 2011). Likewise, although recruitment of GR to DNA-bound Stat3 is associated with transcriptional antagonism, the reciprocal recruitment of Stat3 to DNA-bound GR results in transcriptional synergism (Langlais et al., 2012).
Interestingly, GR maintains a crosstalk with the EGFR (epidermal growth factor receptor) pathway (Lauriola et al., 2014). Upon stimulation, EGFR activates a biochemical cascade culminating in the activation of ERK, a mitogen-activated protein kinase (MAPK). In mammary cells, this translates to cell migration, but pre-treatment with DEX strongly inhibits cell migration (Lauriola et al., 2014). Members of the E-twenty-six (ETS) family of TFs are downstream effectors of ERK. Because GR inhibits the EGFR-to-MAPK pathway and a previous report showed that transforming growth factor beta enhances the GC response of the mammary tumor virus promoter through GABP, an ETS factor (Aurrekoetxea-Hernández and Buetti, 2004), we raised the possibility that GR physically interacts with ETS family members. By utilizing the protein-fragment complementation assay (PCA) (Michnick et al., 2007), we discovered that GR interacts with several ETS factors, including FLI1 and ERG. Importantly, gene fusions involving ERG frequently drive prostate cancer (Tomlins et al., 2005). Similarly, Ewing sarcoma (ES), a common bone malignancy in children and young adolescents, is driven by fusions between EWSR1 and either FLI1 or ERG (Arnaldez and Helman, 2014, Kovar, 2014). In line with functional crosstalk, the experiments presented herein indicate that specific ETS proteins can enhance GR-mediated transcription, in analogy to the ability of AP-1 and Stat3 to augment GR-mediated transcription (Biddie et al., 2011, Langlais et al., 2012). We further report that in ES animal models, a GR antagonist or a cortisol-lowering drug retarded tumor growth and metastasis. These findings offer new pharmacological strategies for the treatment of ES.
Results
PCAs Reveal Hormone-Inducible Interactions between GR and Members of the ETS Family
Because transactivation and transrepression by GR involve complex formation with major TFs (Philips et al., 1997), we hypothesized that ETS family factors are similarly controlled. To test this, we used PCA (reviewed in Michnick et al., 2007), which uses two inactive fragments of luciferase, which are fused to two proteins of interest. We used a previously described adaptation of the Gaussia luciferase (Gluc) assay (Gilad et al., 2014). Gluc was split into an amino-terminal fragment, Gluc1, and a carboxyl-terminal fragment, Gluc2 (Figure 1A). A library comprising seventeen ETS factors fused to Gluc1 was constructed. Likewise, Gluc2 was fused to the carboxyl terminus of GR. As a control, we fused Gluc2 to the estrogen receptor alpha (ERα), ERβ, and the mineralocorticoid receptor (MR; Figure S1; Table S1).
Figure 1.

FLI1 and Ligand-Activated GRs Translocate to the Nucleus and Then Physically Interact in Living Cells
(A) Schemes of the Gaussia luciferase protein, an amino-terminal segment (Gluc1) fused to an ETS factor (either ETV7 or FLI1), and a carboxyl terminal segment (Gluc2) fused to GR. Amino acid numbers and luciferase activity are indicated.
(B) HEK293T cells (6 × 103), pre-transfected with combinations of plasmids, Gluc1 (encoding the indicated ETS factor), and Gluc2 (fused GR), were starved overnight and thereafter treated (60 min) with vehicle or DEX (1 μM). Shown are normalized, DEX-induced fold changes in luciferase activity (means of triplicates ± SE; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001).
(C) HEK293T cells pre-transfected (in sextuplicates) with GR-Gluc2, and the indicated Gluc1-ETS plasmid was treated with vehicle, DEX (1 μM), or a combination of DEX and RU486 (1 μM each). Shown are normalized fold changes in luminescence (means ± SE). ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ns, not significant.
(D) Pre-starved monolayers of HEK293T cells were treated with solvent (C), DEX, RU486, or the combination of drugs. Cell extracts were subjected to immunoprecipitation (IP) with an anti-FLI1 or a control antibody (IgG) and immunoblotting (IB) with an anti-GR antibody. Images are representative of three replicates.
(E) HEK293T cells (6 × 103) were transfected with plasmids encoding Gluc1 (fused FLI1 or ERG) and Gluc2 (fused GR), and 24 h later, they were starved overnight and treated with vehicle or DEX (1 μM) for the indicated intervals. Shown are normalized fold changes in luciferase activity. ∗p ≤ 0.05; ∗∗p ≤ 0.01 (n = 3).
(F) HEK293T cells were starved overnight for serum factors and then treated with DEX (1 μM) for the indicated intervals. Thereafter, the cells were washed in saline containing Tween 20 (0.1%; PBS-T) and fixed in formaldehyde (4%). This was followed by immunofluorescence staining with 4′,6-diamidino-2-phenylindole (DAPI), along with either a fluorescein isothiocyanate (FITC)-conjugated secondary antibody or a tetramethylrhodamine (TRITC)-labeled antibody (in dark). After washing, cells were mounted, images were captured, and nuclear localization quantified by surveying 180 cells from each condition. Bars, 10 μm.
(G) Starved HEK293T cells were treated with DEX (1 μM) for the indicated intervals and then sedimented prior to fractionation into cytoplasmic and nuclear fractions. Each fraction was resolved using gel electrophoresis and immunoblotting with the indicated antibodies. Images are representative of three biological repeats.
Gluc1-ETS plasmids were expressed in HEK293T cells (Figure S1A). Cells co-expressing GR-Gluc2 and Gluc-ETS plasmids were treated with DEX. As summarized in Figure 1B, ERG and its closest homolog, FLI1, along with PU.1 and ETV4, showed highly significant hormone-induced signals. Next, we confirmed that our protocol could detect the well-established interactions between NF-κB and GR (Figure S1B). To validate inducibility, cells were treated with DEX or a combination of DEX and RU486, a GR antagonist. As shown in Figure 1C, RU486 inhibited DEX-induced interactions between GR and FLI1, PU.1 and ERG, but only a trend was noted with the weaker binder ETV4. To confirm physical interactions in naive cells, we carried out a co-immunoprecipitation assay (Figure 1D), which indicated that endogenous GR physically binds with FLI1.
Notably, MR is sensitive to both mineralocorticoids and GCs, whereas GR responds to GCs but displays insensitivity to mineralocorticoids. To address potential interactions between MR and ETS factors, we utilized the ability of DEX to activate MR. Similarly, we performed PCA with Gluc2 proteins fused to ERα and examined the effects of estradiol (E2). The results established differential specificity of interactions (Figures S1C and S1D). For example, DEX stimulated interactions between MR and both ETV2 and ERG, but no ETS family protein bound with ERα. Still, treatment with E2 reduced the interaction between ERα and ELF3 (Figure S1D). Further studies using co-transfected GR-Gluc2 and Gluc1-ERα detected a significant increase in DEX-induced luciferase activity, but treatment with E2 exerted no marked effect (Figure S1E). This observation implies that GR undergoes ligand-induced alterations that promote GR-ERα interactions, but a similar process may not occur in response to E2. Consistent with this interpretation, it has been reported that endogenous GR physically interacts with ERα (Karmakar et al., 2013). In summary, our assays discovered strong physical interactions between GR and several ETS factors, and control assays established both specificity and inducibility by hormones.
In Living Cells, FLI1 and Ligand-Activated GRs Translocate to the Nucleus to Form a Physical Complex
PCA performed at increasing time intervals demonstrated that GR-FLI1 interactions peaked 60 min after DEX stimulation but decayed thereafter (Figure 1E). Although ERG signals peaked earlier, the signal started decaying 30 min later. Presumably, the delayed and transient interactions are due to subcellular compartmentalization. To test this, we stained GR and FLI1 of HEK293T cells (Figure 1F) and also performed subcellular fractionation (Figure 1G). Evidently, GR translocated to the nucleus within 10 min and remained within nuclei for >120 min. In parallel, FLI1 was basally localized to both the nuclei and cytoplasm. Ten min after stimulation with DEX, it translocated to nuclei, and 30–60 min later, it started returning to the cytosol. These observations corroborated the kinetics of protein-protein interactions determined using PCA (Figure 1E) and indicated that the GR-FLI1 interactions are inducible, transient, and confined to the nucleus.
The DBD-HR-Ligand Binding Domain (LBD) Region of GR Recognizes FLI1’s FLS Domain
To map domains of FLI1 and GR that are engaged upon stimulation with DEX, we constructed Gluc1-FLI1 proteins encompassing two, three, four, or all five FLI1’s domains, which were co-expressed with GR-Gluc2. Subsequently, HEK293 cells co-expressing two constructs were incubated without or with DEX, and luciferase signals were determined. The PCA signals attributed hormone-induced recognition to the FLI1-specific (FLS) region of FLI1, and similar analyses indicated that the DNA binding domain (DBD), along with the flanking hinge region (HR) and ligand-binding domain of GR were essential for DEX-induced recognition of FLI1 (Figures S2A and S2B). Conceivably, LBD is needed for DEX binding, and the proximal DBD and HR domains provide the interface (or folding) needed for FLI1 recognition. Notably, it has been reported that the p65 subunit of NF-κB physically interacts with the DBD of GR (Garside et al., 2004). To map hormone-independent interactions, we performed co-immunoprecipitation assays in the absence of DEX (Figures S2C and S2D). In general, the results were in line with our conclusions: Gluc1-FLI1 proteins containing the FLS region retained hormone-independent binding with endogenous GR, but NTA+5′ETS lost this attribute. Similarly, a GR-Gluc2 protein comprising DBD, HR, and LBD co-precipitated the endogenous FLI1 protein, but the N-terminal domain (NTD), either alone or in combination with the DBD, only weakly recognized FLI1. Notably, although a combination of HR and LBD could not generate DEX-induced signals, this construct strongly recognized FLI1 in the absence of DEX (note weak expression of the HR+LBD construct). The addition of the DBD (DBD+HR+LBD), however, weakened FLI1 recognition. In conclusion, basal (hormone-independent) interactions between GR and FLI1 map to the unique FLS of FLI1 and they require both the HR and LBD of GR. Remarkably, the DBD of GR might inhibit basal interactions with FLI1, but it is essential for DEX-induced enhancement of FLI1 recruitment.
Hormone-Inducible Interactions between FLI1 and DNA-Bound GR Augment Transcriptional Activity of GR
Although the classical mode of GR transactivation involves recognition of the GRE, an alternative mode permits GR to localize to additional genomic sites through binding with other TFs (Whirledge and DeFranco, 2018). To examine tethering, we adopted two reporter systems: (1) the FLI1-binding sequence (BS), and (2) a GRE-driven reporter. Along the classical model, upon co-transfection of each reporter and a plasmid encoding the respective TF, we observed dose-dependent increases in luciferase activity (Figures 2A and S3A). Importantly, increasing the amount of FLI1 induced moderate but reproducible and dose-dependent enhancement of expression from the GRE (Figure 2B), but a reciprocal experiment, which used FLI1-BS and a GR plasmid, achieved no statistical significance (Figure S3B). These observations raised the possibility that FLI1 can activate GRE-bound GRs, but the reciprocal interaction may not occur at the BS.
Figure 2.

DNA Binding and Dimerization Might Be Necessary for the Ability of FLI1 to Enhance Transcription from the GRE
(A–D) HEK293T cells (1.2 × 104) were co-transfected with a reporter plasmid (GRE-luciferase, 2.5 μg) and increasing amounts of a GR expression vector (A), a FLI1-encoding vector (B), or with a combination of a FLI1 vector and a vector encoding GR, wild-type (C), and mutant forms (GRdim-, D). Luciferase activity was determined, in biological triplicates, 48 h later, and normalized. ∗p ≤ 0.05; ∗∗∗p ≤ 0.001.
(E) HEK293T cells were co-transfected with a GRE-luciferase plasmid and the indicated amounts of a FLI1 expression vector. After 24 h, cells were starved overnight and treated for 24 h with DEX, RU486 (each at 1 μM), or with the combination. This was followed by a determination of luciferase activity in triplicates.
(F) A model presenting GR dimers occupying the GRE, either before or after recruiting FLI1 proteins, which enhance the GR’s transcriptional activity.
Consistent with this scenario, the co-expression of GR and FLI1 caused only a small increase in transcription from BS (Figure S3C), but a small amount of FLI1 strongly enhanced GRE activity, especially when GR was co-transfected (Figure 2C). To support a model involving tethering of FLI1 to DNA-bound GR, we used GRdim− (A458T) (Reichardt et al., 1998) because mutagenesis of alanine 458 diminishes binding of GR to DNA and decreases GRE-mediated transactivation (Jewell et al., 2012). Wild-type GR increased transcription by >90-fold, whereas ectopic GRdim− showed a much weaker effect (<4-fold) and lost responsiveness to co-transfected FLI1 (Figure 2D). This observation raised the possibility that intact DNA binding by GR might be important for the ability of FLI1 to transactivate transcription from the GRE. Next, we treated cells with DEX or RU486 to address the endogenous GR (Figure 2E): GRE activity increased by 40-fold in response to DEX and co-treatment with RU486 was inhibitory. The signal further increased when a FLI1 plasmid was introduced. In conclusion, although the reciprocal model cannot be ruled out, our results are consistent with FLI1-mediated transactivation of GR’s target genes. Conceivably, the underlying mechanism entails the recruitment of FLI1 to DNA-bound dimers of GR and results in enhanced transcription from GRE sites (see Figure 2F). This mechanism is analogous to the model allowing Stat3 to cooperate with DNA-bound GR (Langlais et al., 2012).
GR Forms a Transient Complex with the Oncogenic EWS-FLI1 Fusion Protein in Nuclei of ES Cells
Genes encoding ETS family TFs, such as FLI1 and ERG, are frequently deregulated in cancer (Feng et al., 2014). Approximately 50% of all prostate cancers express hybrid ERG genes (Tomlins et al., 2005), and replacement of the N terminus of FLI1 by the transactivation domain of the RNA-binding EWS protein characterizes most ESs (Kovar, 2014). Because the resulting EWS-FLI1 fusion retains the GR binding site, we hypothesized that GR and EWS-FLI1 collaborate in ESs. First, we demonstrated, using immunofluorescence and sub-cellular fractionation, time- and DEX-dependent translocation of GR to nuclei of ES cells, A673 (Figures 3A and 3B). In addition, we verified complex formation between GR and EWS-FLI1 by applying the proximity ligation assay (PLA) (Söderberg et al., 2006). PLA-detected protein-protein interactions were significantly enhanced by DEX and blocked by RU486 (Figure 3C). Congruently, co-immunoprecipitation assays validated interactions between GR and EWS-FLI1 in CHLA9 cells, which express EWS-FLI1 (Figure 4A). As expected, DEX increased the amount of pulled-down fusion protein and RU486 markedly inhibited the interactions. In conclusion, in response to stimulation with DEX, the oncogenic EWS-FLI1 fusion protein forms a complex with GR in the nuclei of ES cells.
Figure 3.

Following DEX-Induced Stimulation of Ewing Sarcoma Cells, GR and EWS-FLI1 Translocate to the Nucleus and Form a Physical Complex
(A) Pre-starved A673 cells were treated with DEX (1 μM) for the indicated intervals. Cells were washed in saline containing Tween 20, fixed in formaldehyde (4%), and permeabilized. The incubation with the primary antibody was followed by an FITC-conjugated secondary antibody and DAPI (45 min in dark). Images were captured and nuclear localization quantified by surveying 180 cells from each condition. Bars, 10 μm.
(B) Pre-starved A673 cells were treated with DEX (1 μM) as indicated. Cell extracts were fractionated into cytoplasmic and nuclear fractions, which were analyzed as in Figure 1F.
(C) A673 cells were probed with antibodies recognizing GR, EWS, and FLI1 and processed for PLA that used a TRITC probe. Counterstaining used DAPI (blue). The squared areas (upper rows) are magnified in the bottom rows. Single antibody controls are shown in the bottom panel, and data quantification is presented in the side panels. Note that the numbers of red dots per nucleus were quantified in at least 6 non-overlapping fields of two independent experiments. ANOVA with Tukey’s multiple comparison tests were used to examine statistical significance. ∗∗p < 0.01; bar, 20 μm.
Figure 4.

Ligand-Induced Activation of GR Associates with Increased Migration and Invasion of Ewing Sarcoma Cells
(A) Serum-starved CHLA9 cells were treated (60 min) with vehicle, DEX (1 μM), and RU486 (1 μM). Extracts were processed for co-immunoprecipitation (IP) and immunoblotting (IB). Images are representative of 3 replicates. Signals were quantified and normalized. IgG, control rabbit antibody.
(B) CHLA9 cells were transfected with either GR-specific or control-scrambled siRNAs (si-C). Knockdown efficiency was tested after 48 h using immunoblotting with antibodies to GR.
(C and D) CHLA9 cells were seeded in Transwell migration chambers or Matrigel-coated invasion chambers. (C) Control siRNAs or siRNAs specific to GR were added 24 h prior to seeding, and both migration and invasion were measured 20 h later or, alternatively, (D) DEX (1 μM) and RU486 (1 μM) were added, and migration and invasion were assayed. ∗p ≤ 0.05, ∗∗p ≤ 0.01. Bars, 500 μm.
(E) CHLA9 cells were transfected with either FLI1-specific or control siRNAs (si-C), and knockdown efficiency was tested 48 h later.
(F) CHLA9 cells were treated with siRNAs as in (E). Twenty-four h later, cells were seeded in Transwell migration chambers and incubated for 20 h with DEX. Thereafter, cell migration was quantified. ∗p ≤ 0.05. Bars, 500 μm.
(G) Plates were pre-coated with fibronectin, and then CHLA9 cells (pre-transfected with si-GR or si-C) were seeded and allowed to attach for 90 min. Adherent cells were stained and optical density (550 nm) was quantified in triplicates. ∗p < 0.05.
(H) A673 cells were incubated for 24 h with either DEX or RU486 and thereafter fixed and stained with DAPI and phalloidin. Bar, 10 μm.
(I) CHLA9 cells were transfected with vectors encoding GR, GRdim− (A458T), or they were un-transfected (UT). Twenty-four h later, we assayed migration and invasion. Signal quantification (means ± SD) and representative fields are presented. ∗p ≤ 0.05; ∗∗∗p ≤ 0.001. Bars, 500 μm (migration) or 100 μm (invasion).
Activation of GR Increases Migration and Invasion of ES Cells
EWS-FLI1 might regulate the cytoskeleton and control TEAD- and YAP-regulated genes (Katschnig et al., 2017), thereby inducing proliferation and migration of ES (Franzetti et al., 2017). Because cell migration can reflect the interaction between GR and EWS-FLI1, we downregulated GR in CHLA9 cells (Figure 4B) and confirmed reduced migration and matrix invasion (Figure 4C). Similarly, pre-treatment with DEX enhanced and RU486 blocked the motility effects (Figure 4D). These effects were confirmed using another Ewing cell line, RD-ES: co-immunoprecipitation confirmed inducible interactions between GR and EWS-FLI1 (Figure S4A), immunoblotting established siRNA-mediated silencing of GR (Figure S4B), and migration and invasion assays (Figures S4C and S4D) duplicated the observations made with CHLA9 cells. In line with functionality, co-treatment of CHLA9 cells with DEX and DO6, a nonsteroidal GR antagonist, inhibited motility (Figure S4E). As a complementary approach, we reduced FLI1 in A673 cells. RNA sequencing confirmed marked effects on a large number of genes involved in growth factor signaling, cell adhesion, and inflammation (Figure S3D; see Table S3). In line with this, when tested in CHLA9 cells, the small interfering RNAs (siRNAs) strongly depleted EWS-FLI1 (Figure 4E) and reduced basal and DEX-induced cell migration (Figure 4F).
Focal adhesion and extracellular matrix proteins are regulated by EWS/FLI1 (Chaturvedi et al., 2014). Accordingly, GR depletion increased both the adhesion of CHLA9 cells to fibronectin (Figure 4G) and the adhesion of two other cell lines, namely, RD-ES and TC-71, to collagen-rich membranes (Figure S4F). Remarkably, when A673 cells were pretreated with DEX, we observed an abundance of actin stress fibers, in contrast to the mostly cortical actin observed with RU486-treated cells (Figure 4H). Consistent with the transactivation model, overexpression of GR increased migration and invasion of CHLA9 cells, but GRdim− was less effective (Figure 4I). In summary, our data are consistent with the following scenario: EWS-FLI1 physically binds with GRs and enhances the transcription of genes able to reduce matrix adhesion, while enhancing the expression of motility genes.
Co-expression of the Interacting Domains of GR and FLI1 Augments Features of Malignancy
To directly link the uncovered GR-to-FLI1 interaction and malignancy, we ectopically expressed in A673 cells the domain mutants presented in Figure S2. As expected, full-length GR and FLI1 strongly increased cell migration, transformed cortical actin into stress fibers, and increased incorporation of radioactive thymidine into DNA (the latter was inhibited by RU486; Figures S4G–S4J). Consistent with the PCA results, which mapped the FLI1 binding site to GR’s DBD+HR+LBD, a mutant containing all three domains displayed as strong activity as full-length GR, whereas mutants unable to bind with FLI1 (i.e., NTD, NTD+DBD, and HR+LBD) displayed reduced activities. Similarly, in line with GR’s binding to the FLI1’s FLS, the deletion mutant NTA+5′ETS+FLS+3′ETS retained all three activities, but shorter mutants (i.e., NTA+5′ETS+FLS and NTA+5′ETS) partially and fully (respectively) lost all three attributes. In the next step, we co-expressed GR and FLI1, along with physically interacting mutants, and challenged cells with RU486. As predicted by our model, co-expressing full-length GR and FLI1 strongly increased migration and stress fiber formation, and both activities were blocked by RU486 (Figures S4K and S4L). Likewise, the combination of GR and the interacting domains of FLI1 were comparably active, and the reciprocal combination (full-length FLI1 combined with GR’s DBD+HR+LBD) was both active and inhibitable by RU486. Finally, combining only the interacting portion of GR (DBD+HR+LBD) and the interacting region of FLI1 (NTA+5′ETS+FLS+3′ETS) exerted similarly strong signals, which were abolished by RU486 but only weakly enhanced by DEX. Taken together, by co-expressing the interacting domains of GR and FLI1, we inferred that features of malignancy of ES cells are closely related to the uncovered physical interaction between these pivotal TFs.
Activation of GR Increases Proliferation and Inhibits Apoptosis of ES Cells
Apparently, EWS-FLI1 promotes cell cycle progression and inhibits apoptosis (Franzetti et al., 2017, Stoll et al., 2013). Hence, we assayed the conversion, by mitochondria, of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) to an insoluble formazan. In line with the proposed molecular collaboration, RU486 and DO6 induced a dose-dependent reduction in the survival of three different ES cell lines (Figures 5A and 5B). Next, we concentrated on A673 cells and depleted FLI1 (Figure 5C). MTT assays confirmed that EWS-FLI1 knockdown reduced cell survival. Consistently, RU486 reduced the fraction of metabolically active cells, but this effect was diminished when the antagonist was combined with the siRNA (Figure 5D). Interestingly, although treatment with DEX almost eliminated the fraction of cells undergoing early apoptosis, RU486 enhanced late apoptosis (Figure 5E), indicating that GRs support cell growth and/or survival. This conclusion was supported by several colony formation assays (Figure 5F). In summary, GR effectively collaborates with EWS-FLI1 by means of supporting cell proliferation, overcoming apoptosis, and promoting various attributes of motile cells.
Figure 5.

Inhibition of GR Decreases Growth and Survival of Ewing Sarcoma Cells
(A) Cell viability assays applied the MTT method on three Ewing sarcoma lines (CHLA9, A673, and RD-ES), which were treated for 24 h with increasing concentrations of RU486.
(B) Increasing concentrations of DO6, a non-steroidal GR antagonist, were incubated with A673 cells as in (A), and an MTT assay was performed in triplicates. ∗p < 0.05.
(C) A673 cells were transfected with either FLI1-specific or control-scrambled siRNAs (si-C). EWS-FLI1 knockdown efficiency was tested after 48 h using immunoblotting.
(D) FLI1-silenced A673 cells (8 × 103) were seeded in 96-well plates and treated with either vehicle or RU486 (10 μM). MTT assays were performed after 48 h in quadruplets. ∗∗p < 0.01; ∗∗∗p < 0.001.
(E) A673 cells were seeded in 100-mm dishes. Thereafter, cells were treated for 48 h with DEX (1 μM), RU486 (10 or 20 μM), or the combination. Shown are results of an apoptosis assay performed using an annexin V/7-AAD kit (BioLegend). Quantification of the fractions of early and late apoptotic cells is shown. The experiment was repeated twice.
(F) The indicated cells were sparsely seeded in 6-well plates. Cells were later treated every other day with either vehicle, DEX (1 μM), RU486 (10 μM), or the combination. Ten days later, cells were fixed and stained with crystal violet. Photos are shown along with bar plots presenting the quantification of colonies in 5 non-overlapping microscope fields. The experiment was repeated twice. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Cortisol Lowering and Treatments with an Antagonist of GR Inhibit ES Xenografts Driven by Either EWS-FLI1 or the Respective ERG Fusion Protein
Next, we asked whether the clinically approved GR agonist (DEX) and antagonist (RU486) can modify tumorigenesis in animal models. Hence, once RD-ES tumors that were pre-implanted in mice became palpable, animals were treated with DEX or RU486. Unlike mice treated with DEX, tumor growth was significantly slower in animals treated with RU486 (Figure 6A). In line with GR-supported tumorigenesis, staining with an antibody to KI67, a proliferation marker, indicated relatively slow rates of cell division in RU486-treated tumors (Figure 6B). Immunoblots of extracts prepared from three tumors of each group are shown in Figure S5A. Despite inter-animal variation, two inhibitors of the cell cycle, p21 and p27, displayed increased abundance in RU486-treated tumors, which also displayed upregulation of the larger isoform of BIM, an initiator of apoptosis. Although we could not detect uniform activation of caspase 3, the data implied that the upstream AKT survival pathway was inhibited by RU-486. Because BIM links apoptosis to autophagy (Luo et al., 2012), we probed for LC3, an autophagy marker, and observed an increased abundance of LC3B-I in RU486-treated tumors (Figure S5A). In conclusion, RU486 inhibits the proliferation of ES tumors, likely through a caspase 3 independent pathway that harnesses both apoptosis and autophagy.
Figure 6.

A GR Antagonist and a Cortisol-Lowering Drug Impede Growth of Xenograft Models of Ewing Sarcoma
(A) RD-ES cells (2 × 106) were implanted subcutaneously in the flanks of SCID mice. Once tumors reached 150 mm3, animals were randomized into three groups (n = 10) that were daily treated intraperitoneally with vehicle, DEX (1 mg/kg), or RU486 (1 mg/kg) and tumorigenic growth was monitored. The means of tumor volumes (+SEM) are shown. Statistical analysis of tumor volumes (day 12) is indicated (RU486 group versus DEX or vehicle).
(B) Representative images of immunofluorescent KI67 staining in paraffin-embedded tumor sections from (A). Scale bars, 100 μm. The scatterplot depicts quantification of KI67 staining in 4 fields of a representative tumor from each group.
(C and D) A673 cells (2 × 106) were implanted in female SCID mice. Once tumors reached 150 mm3, animals were randomized into two groups (n = 9). Each group was daily treated with either vehicle or metyrapone (25 mg/kg).
(C) Actual tumor volumes (±SEM) are presented.
(D) Also shown are tumors harvested from each group of animals; ∗∗∗p < 0.001.
(E and F) STA-ET-11 cells (2 × 106) were implanted subcutaneously in SCID mice. Once tumors reached 150 mm3, animals were randomized into three groups (5 mice per group), which were daily treated intraperitoneally with vehicle, DEX (1 mg/kg), or RU486 (1 mg/kg). (E) The means of tumor volumes (+SEM) are shown along with (F) statistical analysis of tumor volumes measured on day 73.
Assuming that murine cortisol activates GR in xenografts, we tested the effect of cortisol lowering on another ES model, A673. Cells were implanted in severe combined immunodeficiency (SCID) mice and later treated with metyrapone, a clinically approved inhibitor of 11β-hydroxylase, a steroidogenesis enzyme. As expected, a statistically significant decrease in tumor growth rates was associated with the lowered levels of cortisol (Figures 6C and 6D). An analyses of tumor extracts revealed metyrapone-induced effects on BIMEL and LC3B-I, along with elevated γH2AX (Figure S5B). Taken together, our results support a scenario attributing to the hormone-activated form of GR an ability to enhance tumorigenicity of ES.
Predictably, in similarity to EWS-FLI1 expressing tumors, blocking GR might inhibit ES models driven by the EWS-ERG fusion protein. Hence, we tested the STA-ET-11 animal model, which is driven by EWS-ERG. Mice bearing STA-ET-11 tumors were treated with RU486 or with DEX, and tumor volume was monitored for 3 months. The results presented in Figures 6E and 6F are consistent with a model attributing to GR the ability to accelerate the progression of ES: DEX enhanced tumor progression and RU486 significantly decelerated rates of tumor growth. In conclusion, reduced levels of circulating cortisol or blocking GR can inhibit tumor growth in three different animal models of ES.
Inactivation of GR Impedes Bone-to-Lung Metastasis in ES Animal Models
Unlike ES patients presenting localized disease at diagnosis, whose 5-year survival rates approach 70%, patients with metastatic disease have dismal outcomes with 5-year survival rates of 15%–25% (Linabery and Ross, 2008). Because our tests implied that the GR-to-FLI1 interaction controls motility, and both detachment from bone and dissemination of tumor cells are regulated by EWS-FLI1 (Chaturvedi et al., 2012), we examined metastasis in animal models. TC-71 cells pre-engineered to express luciferase were injected into the tibia of SCID mice, which were later treated intraperitoneally with either DEX or RU486. Figures 7A and 7B show whole lungs from representative animals from each group, along with quantified signals from all mice. This analysis indicated that GR activation associated with enhanced bone-to-lung metastasis, whereas blocking GR correlated with fewer lung nodules.
Figure 7.

Pharmacological or Genetic (Inducible) Inhibition of GR Retards Metastasis of Ewing Sarcoma Models
(A and B) Luciferase expressing TC-71 cells (106) were injected into the tibia of female SCID mice (5 weeks old; 7 mice per group). One week later, mice were treated intraperitoneally with DEX or RU486 (1 mg/kg, each). When each tumor reached 10% of body weight, the lungs were excised and examined for metastasis by using bioluminescence imaging. (A) Shown are whole lungs from representative animals and (B) luminescence quantified and presented in a bar graph. ∗∗p ≤ 0.01. The vertical color bar shows level of luminescence.
(C) TC-71 cells were transduced with inducible lentiviral shRNAs specific for GR (from Dharmacon). Two stable cell variants were selected and named inducible sh1 (iSH1) and iSH2. To initiate GR knockdown, cells were grown for 24, 48, or 72 h in culture medium supplemented with doxycycline (1 μg/ml). GR levels were determined using immunoblotting. The abundance of GR, relative to GAPDH, is indicated below each lane. UI, un-induced cells.
(D) Sub-clones iSH1 and iSH2 of TC-71 cells were treated for 48 h with doxycycline (1 μg/ml) and then seeded in Transwell migration chambers. Following 20 h of incubation, cells that reached the lower faces of the chambers were fixed and stained. Shown are representative images of cells that migrated. The bar plots show quantification of areas covered by the cells using ImageJ. ∗p < 0.05 (duplicates). Bars, 500 μm.
(E) Two sub-clones of TC-71 cells (106 cells), which stably express GR-specific inducible shRNAs, were injected into the tibia of 5-week-old female SCID mice (10 mice per group). Once tumors reached 150 mm3, mice were randomly divided into two groups: 4 mice of each group were daily treated (oral gavage) with either saline (un-induced group) or doxycycline (induced group). When each primary tumor reached 10% of body weight, the respective lungs were excised and analyzed for metastasis by using bioluminescence imaging. Shown are whole-lung images corresponding to each mouse in the end of the experiment. The average radiance (photons/s/cm2/sr) is shown for each group.
Next, we used inducible lentiviral short hairpin RNAs (shRNAs) specific to GR. Two different shRNAs were used, namely, inducible sh1 (iSH1) and iSH2. When the corresponding sub-lines of TC-71 were grown in medium supplemented with the inducer doxycycline (DOX), we observed decreases in GR expression (Figure 7C) and lower migration (Figure 7D). Hence, both sub-clones were implanted in the tibia of SCID mice. Once tumors reached a pre-determined volume, mice were treated with either saline or DOX (induced group). When the primary tumor reached 10% of body weight, lungs were analyzed for metastasis. The results presented in Figure 7E indicated that both iSH clones partially lost the ability to dissociate from bones and colonize lungs. Conceivably, GR is essential for the dissemination of metastases, which is in line with a model attributing to EWS-FLI1 the ability to collaborate with GR.
Unlike EWS-ERG-Expressing Cells, Colony-Forming and Migration Abilities of ES Cells Expressing EWS-FEV Are Not Regulated by GR
To directly tie GR to the oncogenic functions of EWS-ETS fusion proteins, we referred to rare Ewing family tumors (EFTs). In addition to EWS-FLI1 and EWS-ERG fusions, which act as transcriptional activators and bind with GR, EFTs include rare cases with fusions of EWS to other ETS family genes. We focused on the FEV transcriptional repressor, which is rarely fused to EWS. The PCA data we generated using Gluc1-FEV indicated that FEV, like ETV6, cannot form physical complexes with GR (Figure S6A). Hence, we obtained STA-ET-10 cells (driven by EWS-FEV), which very weakly give rise to tumors in animals. These cells were compared, in vitro, to STA-ET-11 cells, which are driven by EWS-ERG. As shown in Figures S6B–S6D, STA-ET-11 cells were partly inhibited by RU486, but no inhibition of STA-ET-10 cells could be detected. Similarly, both colony-forming and migratory activities of STA-ET-11 cells were enhanced or inhibited (respectively) following treatment with DEX or RU486, but no effects were detectable in STA-ET-10 cells. Cell survival assays indicated that STA-ET-10 cultures underwent more spontaneous apoptosis than STA-ET-11 cells (Figure S6E). In addition, these experiments confirmed that RU486 exerted no effects on STA-ET-10 cells, unlike STA-ET-11 cells, which were inhibited by the antagonist. In conclusion, our results link the ability of GR to form physical complexes with ETS fusions to the capacity of a GR antagonist to inhibit several attributes of malignancy.
In summary, by using four different EWS-ETS-driven models and examining tumor growth and metastasis, we obtained evidence that strongly associates GR with the progression of ES. Mechanistically, these observations can be explained by the uncovered hormone-dependent physical interaction between GR and FLI1 and the ability of the respective TF complex to enhance transcription from the GC response element. According to our model, when activated by a ligand, GR translocates to the nucleus and forms a complex with EWS-ETS, thereby gaining an augmented ability to alter the transcription of anti- or pro-tumorigenic genes. Beyond the exact molecular mechanisms, our study offers a re-purposing of GR-specific antagonists and cortisol-lowering drugs for the treatment of ES patients.
Discussion
Unlike several other nuclear receptors, the roles for GR in malignancy are not uniform. For instance, GCs have been extensively used in prostate cancer to offset toxicities of chemotherapy and to suppress androgen synthesis (Montgomery et al., 2014). Nevertheless, GCs may promote rather than suppress prostate cancer because GR and AR (androgen receptor) partly share transcriptional programs. Congruently, the overexpression of GR drives resistance to an AR antagonist (Arora et al., 2013). Understanding GR’s action in blood-borne cancer might be simpler: GR mediates extensive apoptosis in leukemia and lymphoma. By discovering and studying strong GR-FLI1 physical interactions, we attribute to EWS-FLI1 an ability to enhance GR’s transcriptional actions and augment ES growth and metastasis. Several lines of previous evidence support the inferred GR-ETS crosstalk: accumulation of PU.1 regulates GC’s effects during erythrocyte differentiation (Gauthier et al., 1993), DEX-mediated transcription of cytochrome P-450c27 involves synergy with ETS2 (Mullick et al., 2001), and another ETS factor, GABPA, influences the responses of a viral promoter to GCs (Aurrekoetxea-Hernández and Buetti, 2004).
FLI1 and other ETS family members serve as ultimate effectors of the MAPK pathway. Mutant forms of the upstream components, such as RAS and RAF, are frequently detected in tumors. According to our results, GR gains enhanced transcriptional activity upon binding with FLI1, independent of the phosphorylation status of the MAPK pathway. It is notable that this pathway regulates both proliferation and cellular migration, and it has been proposed that both outcomes are regulated in ES by the abundance of EWS-FLI1: high levels promote proliferation and fluctuating low levels drive migration (Franzetti et al., 2017). According to another report, the cell-to-cell heterogeneity in expression levels may reprogram the cytoskeleton (Katschnig et al., 2017). In view of the circadian oscillations of GC levels and the herein reported hormone-dependent interactions between GR and EWS-FLI1, it might be speculated that alternate phases of growth and metastasis contribute to ES aggressiveness. Whether the interactions between GR and ERG contribute to aggressiveness of prostate tumors harboring rearranged ERG genes (Tomlins et al., 2005, Tomlins et al., 2007) is a matter for future research.
Similar to the inferred collaboration between EWS-FLI1 and DNA-bound GR, previous studies identified other mechanisms of GR cooperation. For example, cooperative binding of AP-1 and ETS factors to tandem binding sites supports transformation by EWS-ETS (Kim et al., 2006). Mechanistically, AP-1 serves as a pioneer factor that enhances GR binding to select genomic regions (Biddie et al., 2011). In line with this, AP-1 ablation impaired GR-mediated regulation of nearly 50% of the target genes. Similarly, recruitment of GR to DNA-bound Stat3 associates with trans-repression, but recruitment of Stat3 to DNA-bound GR drives transcriptional synergy (Langlais et al., 2012). In analogy, EWS-FLI1 might regulate the transcription of a large fraction of GR-regulated genes involved in tumor progression.
Unlike localized forms of ES, patients with metastatic disease have dismal prognosis. Current research efforts have focused on identifying sensitizers of chemotherapy (Grohar et al., 2014), along with targeting cell surface receptors and histone deacetylases (Arnaldez and Helman, 2014, Mendoza-Naranjo et al., 2013). We offer an alternative strategy that takes advantage of the well-understood pharmacology of GR. Specifically, we tested two strategies, corticosterone lowering using metyrapone and GR antagonism. The latter used RU486 (mifepristone), a clinically approved steroidal antagonist. In light of our findings, the newly developed selective GR modulators (i.e., selective GC receptor modulators [SGRMs] without AR or progesterone receptor [PR] cross-reactivity) (Hunt et al., 2012) and mild corticosterone-lowering drugs might inhibit ES by means of preventing the ability of EWS-FLI1 to synergize with GR.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-FLI1 (immunoblotting) | Abcam | Cat# Ab133485, RRID:AB_2722650 |
| Rabbit polyclonal anti-FLI1 (immunofluorescence) | Santa Cruz Biotechnology | Cat# SC356; RRID:AB_2106116 |
| Mouse monoclonal anti-GR | Santa Cruz Biotechnology | Cat# sc-393232 RRID:AB_2687823 |
| Rabbit polyclonal anti-GR | Santa Cruz Biotechnology | Cat# sc-8992 RRID:AB_2155784 |
| Mouse monoclonal IgG1 anti-EWS | Santa Cruz Biotechnology | Cat# sc-28327, RRID:AB_675526 |
| Mouse monoclonal anti-p27 Kip1 | Cell Signaling Technology | Cat# 3698S, RRID:AB_2077832 |
| Rabbit monoclonal anti-p16 | Abcam | Cat# ab108349, RRID:AB_10858268 |
| Rabbit polyclonal anti-γH2AX (Ser139) | Cell Signaling Technology | Cat# 2577S, RRID:AB_2118010 |
| Mouse monoclonal anti-GAPDH | Merck | Cat# MAB374, RRID:AB_2107445 |
| Anti-Ki67 antibody | Abcam | Cat# ab16667, RRID:AB_302459 |
| Rabbit polyconal anti-Gaussia-luciferase | Nanolight Technology | Cat # 401P, RRID:AB_2572411 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dexamethasone | Sigma-Aldrich | Cat# D4902-25MG |
| RU486 | Sigma-Aldrich | Cat# M8046-100MG |
| Metyrapone | Sigma-Aldrich | Cat# M2696-50MG |
| Coelentrazine | Nanolight Technology | Cat# 303 |
| SYBR Green PCR Master Mix | Thermo Fisher Scientific | Cat# 4309155 |
| Cultrex® RGF BME | R&D System | Cat# 3433-005-02 |
| ITS-G | Invitrogen | Cat# 41400045 |
| Phalloidin | Sigma-Aldrich | Cat# P1951 |
| 3[H]-thymidine | Perkin Elmer, USA | Cat# NET027Z001MC |
| Critical Commercial Assays | ||
| FITC Annexin V Apoptosis Detection Kit I | BD PharMingen™ | Cat# 556547 |
| Dual-Luciferase® Reporter Assay System | Promega | Cat# E1910 |
| Duo link in situ detection kit | Sigma-Aldrich | DUO92008 |
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher Scientific | Cat# 4368814 |
| Dynabeads mRNA DIRECT Purification Kit | Thermo Fisher Scientific | Cat# 61011 |
| Deposited Data | ||
| Raw and processed RNA sequencing data | This Paper | GEO: GSE135229 |
| Experimental Models: Cell Lines | ||
| Human: RD-ES | ATCC | Cat# HTB-166, RRID:CVCL_2169 |
| Human: A673 | ATCC | Cat# CRL-7910, RRID:CVCL_0080 |
| Human: HEK293T | ATCC | CRL-3216, RRID:CVCL_0063 |
| Human: CHLA9 | Gift from Prof. Heinrich Kovar, Children’s Cancer Research Institute, Vienna | N/A |
| Human: STA-ET-11 | Gift from Prof. Peter F. Ambros, Children’s Cancer Research Institute, Vienna | N/A |
| Human: STA-ET-10 | Gift from Prof. Peter F. Ambros, Children’s Cancer Research Institute, Vienna | N/A |
| Human: TC-71 | Gift from Jaffrey Toretsky, Georgetown University, USA | N/A |
| Experimental Models: Organisms/Strains | ||
| Mice: CB17/SCID female | Envigo Israel | N/A |
| Oligonucleotides | ||
| Primers for cloning in Table S2 | This Paper | N/A |
| siRNA-GR | Dharmacon | ON-Target SMART oligonucleotides L-003424-00-0005 |
| siRNA-FLI1 | Dharmacon | siGENOME Human/Mouse/Rat SMARTpool M-003892-00-0005 |
| Human Inducible Lentiviral shRNA for GR | Dharmacon | Cat # V3SH11255 |
| Recombinant DNA | ||
| ETS-Gluc1 fusion plasmids | This paper | N/A |
| Gluc2-plasmids | This paper | N/A |
| Domains of GR and FLI1 plasmids | This paper | N/A |
| GRE-luciferase plasmid | Gift from Prof. Anne Gompel, Paris Descartes University | N/A |
| FLI1 expression vector and FLI1-BS-luciferase plasmid | Gift from Prof. Yaacov Den-David, Chinese Academy of Sciences, Guizhou. | N/A |
| GR expression vector and GRdim mutant plasmid | Gift from Prof. Andrew Cato, Karlsruhe Institute of Technology, Germany | N/A |
| Software and Algorithms | ||
| ImageJ | National Institutes of Health, Bethesda, USA | https://imagej.nih.gov/ij/ |
| Prism 6 | Graph pad | https://www.graphpad.com/scientific-software/prism/ |
| BD FACS Diva software v8.0.1 | BD Biosciences | http://www.bdbiosciences.com/us/instruments/research/software/flow-cytometry-acquisition/bd-facsdiva-software/m/111112/features |
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to the Lead Contact, Yosef Yarden (yosef.yarden@weizmann.ac.il).
Note: This study did not generate new unique reagents.
Experimental Model and Subject Details
Cell Lines
Human embryonic kidney cells, HEK293T, and A673 cells (sex: female) were cultured in DME medium supplemented with fetal bovine serum (10%; FBS). RD-ES cells (sex: male) and TC-71 cells (sex: male) were grown in RPMI supplemented by 15% or 10% FBS, respectively. CHLA9 cells (sex: female) were grown in IMDM supplemented by 20% FBS and a mixture of insulin, transferrin, and selenium. STA-ET-10 cells (sex: female) and STA-ET-11 cells (sex: male) were grown in RPMI supplemented with glutamax and 10% FBS.
Mice
All animal experiments were approved by the Weizmann Institute’s Animal Care and Use Committee. CB17/SCID female mice (5-6 weeks old) were injected subcutaneously into the right dorsal flank with 2.5 million RD-ES, STA-ET-11 or A673 cells in a 0.1 mL suspension in saline. Tumor volume (V/mm3) was estimated using vernier caliper measurements of the longest axis, α/mm, and the perpendicular axis, b/mm. Tumor volume was calculated in accordance with the equation V = (4π/3) x (α/2)2 x (b/2). When the volume of xenografts reached approximately 150 mm3, mice were randomized into groups and treatments initiated. Animals were intraperitoneally treated once per day with DEX, RU486 (both at 1 mg/kg), or metyrapone (25 mg/kg). They were euthanized when tumor size reached 800-900 mm3. For metastasis, mice were anesthetized with ketamine and xylazine. Once animals were fully sedated, we injected into the intratibial (i.t.) region TC-71 cells (2X106) stably expressing GR-specific inducible shRNAs. When tumors reached 100 mm3, mice were randomly divided into two groups, which received (by oral gavage) sterile water, without or with doxycycline (1 mg/ml), until tumor size in the control group reached 10% of body weight. After mice were sacrificed, lungs were extracted and analyzed for metastases using IVIS.
Note: The source and identifier of all the plasmids, cell lines, and mice are listed in the Key Resources Table.
Method Details
Protein Complementation Assays (PCA)
PCA was performed as previously described (Gilad et al., 2014). Briefly, HEK293 cells were reverse transfected in white 96-well tissue culture plates with Gluc-1 and Gluc-2 plasmids (25 ng, each) using the JetPEI reagent. Cells were starved and treated with dexamethasone (1 μM) for 60 minutes, followed by cell lysis and determination of luminescence signals.
Luciferase-Reporter Assay
Cells were co-transfected with a luciferase plasmid containing the consensus glucocorticoid response element (GRE). Additionally, the pGL3-Control vector encoding Renilla luciferase (Promega, Madison, WI) was transfected as a control for transfection efficiency. Luciferase activity was determined using the dual-luciferase reporter assay system, according to the manufacturer’s instructions (Promega). Firefly luciferase luminescence values were normalized to Renilla luminescence and quantified relative to control.
Cell Migration and Invasion Assays
Cells (5X104) were plated in the upper compartment of a 24-well Transwell tray (Corning, Acton, MA). Thereafter, the medium in the lower compartment of the chamber was supplemented with the indicated agents and cells were allowed to migrate for 16 hours at 37°C through the intervening nitrocellulose membrane (8 μm pore size). The filter was later removed and attached cells were fixed for 15 minutes in saline containing paraformaldehyde (3%). Staining with crystal violet followed this step. Cells growing on the upper side of the filter were scraped using a cotton swab; cells located on the bottom side were photographed and counted. Similarly, cell invasion assays were performed using BioCoat Matrigel Invasion Chambers (BD Bioscience, Franklin Lakes, NJ).
Nuclear and Cytoplasmic Fractionation
Cell pellets were lysed in 0.1 mL cytoplasmic lysis buffer (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EGTA, 0.1 mM EDTA, 1 mM DTT and 0.5% NP-40). The cytoplasmic fraction was collected using centrifugation (600 g for 5 minutes). Nuclei were washed and resuspended in nuclear lysis buffer (50 μl; 20 mM HEPES pH 7.9, 0.4 M NaCl, 1 mM EGTA, 1 mM EDTA, 1mM DTT) by repeated freezing and thawing. Supernatants containing the nuclear fraction were collected by centrifugation at 12,000 rpm for 20 minutes.
Cell Lysis, Immunoblotting, and Co-immunoprecipitation Assays
Cell lysates were collected in a mild lysis buffer (50 mM HEPES, pH 7.5, 10% glycerol, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1 mM EGTA, 10 mM NaF and 30 mM β-glycerol phosphate). Proteins were immunoprecipitated from cell lysates using beads conjugated to an antibody. After 2 hours of incubation at 4°C, complexes were washed three times and bound proteins were eluted in 6X Laemmli buffer. Eluates were subjected to electrophoresis and immunoblotting. For immunoblotting, cleared cell lysates were resolved using electrophoresis, followed by electrophoretic transfer to a nitrocellulose membrane. Membranes were blocked with TBS-T (tris-buffered saline containing Tween-20) containing 1% low-fat milk, blotted overnight with a primary antibody, washed three times with TBS-T, incubated for 30 minutes with a secondary antibody linked to horseradish peroxidase, and washed once again with TBS-T. Immunoreactive bands were detected using the ECL reagent (Biorad).
RNA Isolation and Real-Time PCR Analysis
Total RNA was extracted using the PerfectPure RNA Cultured Cell Kit (5-prime, Hamburg) according to the manufacturer’s instructions. Total RNA quantity and quality were determined using the NanoDrop ND-1000 spectrophotometer (Thermo Fischer Scientific, Waltham, MA). Complementary DNA was synthesized using the High Capacity Reverse Transcription kit (Applied Biosystems, Life Technologies, Carlsbad, CA, USA). Real-time qPCR analysis was performed with SYBR Green (Applied Biosystems) and specific primers on the StepOne Plus Real-Time PCR system (Applied Biosystems). qPCR signals (cT) were normalized to beta2-microglobulin (B2M).
Nucleotide Sequencing of RNA
RNA was isolated using Dynabeads mRNA Direct Kit (Thermo Fisher Scientific). NGS libraries were prepared using a modified version of Transeq, as described (Jaitin et al., 2014). In brief, RNA was barcoded and reverse-transcribed using poly-T primers, followed by addition of exonuclease to remove excess RT–PCR primers. Next, the single-stranded cDNA was converted to a double-stranded DNA. The template DNA was then removed using DNase, and the generated RNA was fragmented and ligated to barcoded Illumina adapters. Reverse transcription of this ligation product was performed using primers specific for the Illumina adapters, and libraries of the resulting cDNA were generated and enriched by performing 12–15 PCR cycles. RNA-seq libraries (pooled at equimolar concentrations) were sequenced on an Illumina NextSeq 500 at a median sequencing depth of ∼10 million reads per sample. Sequences were mapped to the human genome, demultiplexed, and filtered.
Immunofluorescence Analyses
Formalin-fixed tumor sections were de-paraffinized and rehydrated. Antigen retrieval was performed in a microwave oven using a citric acid solution (pH 9.0). Slides were blocked in saline containing 20% horse serum, followed by treatment (15 minutes) with a blocking solution, and an overnight incubation with the primary antibody. Thereafter, sections were incubated for 90 minutes with a biotinylated secondary antibody, followed by a Cy3-conjugated Streptavidin. Finally, each slide was examined using a fluorescence microscope. KI67-positive cells were counted using the Image Pro Plus software. To determine localization in cultured cells, cells were washed in saline containing Tween 20 (0.1%; w/v; PBS-T) and fixed in formaldehyde (4%). Next, cells were washed and permeabilized (in saline with 0.1% Triton X-100). Blocking was carried out for 30 minutes using fetal bovine serum (2%; FBS), followed by an incubation with a primary antibody in PBS-T containing FBS (1%). This was followed by FITC-conjugated secondary antibody and DAPI (in dark).
Proximity Ligation Assay (PLA)
A673 cells were serum starved for 16 hours followed by a 60 min stimulus with dexamethasone (1μM) or RU486 (1μM), as indicated. Cells were then fixed with PFA (4%) for 15 minutes, washed with PBS, permeabilized with PBS/Triton X-100 (0.01%) for 15 minutes and hybridized with primary antibodies against FLI1 (1:50) and GR (1:50; overnight). Next, the cells were incubated with secondary antibodies against Rabbit PLUS (DUO92002) and against Mouse MINUS (DUO92002) and processed using the Duolink In Situ Detection Kit (red) containing a tetramethylrhodamine-5-isothiocyanate probe (Sigma-Aldrich). Thereafter, cells were hybridized with phalloidin-FITC and DAPI for counterstaining. Coverslips were washed and placed, cells face down, onto drops of an anti-fade reagent (from Dako). Samples were examined using a widefield fluorescence microscope (Zeiss). Red dots and nuclei were counted and the number of positive stains per cells was calculated from at least 5 non-overlapping microscope fields. One-way ANOVA with Tukey correction was performed.
Apoptosis Assays
Assays were performed using the FITC Annexin V Apoptosis Detection Kit with 7-AAD (from BioLegend) and analyzed using a BD FACSAria Fusion instrument controlled by BD FACS Diva software v8.0.1 (BD Biosciences).
Colony Formation and Adhesion Assays
Cells (150-300) were seeded in 6-well plates. Ten days after treatment, cells were washed, fixed in paraformaldehyde (4%) and then stained for 60 minutes with crystal violet. Cells were then photographed using a binocular microscope and analyzed using ImageJ (NIH, USA). For adhesion tests, plates were coated overnight with Cultrex® RGF BME (R&D Systems) and gently washed thereafter (0.1% albumin in medium). RD-ES and TC-71 cells (30,000 cells/well) were allowed to adhere to the substrate for 8 hours at 37°C. CHLA9 cells were seeded in non-coated plates and allowed to attach for 90 minutes. Unattached cells were removed and adherent cells were rinsed, fixed with paraformaldehyde (4%), and quantified after crystal violet staining (0.1%). The optical density was measured at 550 nm.
Thymidine Incorporation Assay
Cells were plated onto 24-well plates at a density of 5X104 cells/well, followed by plasmid transfection. Sixteen hours later, cells were replaced with fresh serum-free medium containing 3[H]-thymidine (1 μCi). After 48 hours, the reaction was terminated by the addition of ice-cold trichloroacetic acid (5%; TCA). Five minutes later, cells were solubilized at 37°C with in 1N NaOH (for 10 minutes) followed by 1N HCL. Samples were collected into scintillation vials containing scintillation fluid. Radioactivity was determined in a scintillation counter. The results shown are representative of experiments performed in quadruplicates, at least twice.
Quantification and Statistical Analysis
All data were analyzed using the Prism Graph pad software. Significance was assessed using two-way ANOVA followed by Dunnett’s or Sidak’s Multiple Comparison Tests. Some analyses were performed using t test and one-way ANOVA with Tukey’s or Dunnett’s test (∗ p ≤ 0.05; ∗∗ p ≤ 0.01; ∗∗∗ p ≤ 0.001).
Data and Code Availability
The RNA sequencing data generated during this study are available at GEO, accession code GEO: GSE 135229.
Acknowledgments
We thank Tomer Meir Salame and Kira Glatzel for help; Aykut Uren for guidance; Jefferey Toretsky and Peter Ambros for cell lines; and Yaacov Ben-David, Anne Gompel, and Andrew Cato for sharing reagents. This work was performed in the Marvin Tanner Laboratory for Research on Cancer. Y.Y. is the incumbent of the Harold and Zelda Goldenberg Professorial Chair in Molecular Cell Biology. Our studies are supported by the Israel Science Foundation (ISF; 280/15), the European Research Council (ERC; 740469), and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (AMRF; 12).
Author Contributions
S.S. and Y.Y. designed the experiments and wrote the manuscript. S.S., N.B.N., A.S., C.B., S.G., D.D.-G., L.R., D.R., I.M., Y.H., Y.G., and O.M. performed the experiments. R.R., E.D., O.M., D.S., A.Z., Y.T., O.D., and I.A. analyzed statistical and other data. M.L., A.K., I.A., and H.K. reviewed the manuscript.
Declaration of Interests
Yeda, the technology transfer arm of the Weizmann Institute, has submitted a patent application related to the results reported herein.
Published: October 1, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.08.088.
Supplemental Information
References
- Arnaldez F.I., Helman L.J. New strategies in ewing sarcoma: lost in translation? Clin. Cancer Res. 2014;20:3050–3056. doi: 10.1158/1078-0432.CCR-13-0633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora V.K., Schenkein E., Murali R., Subudhi S.K., Wongvipat J., Balbas M.D., Shah N., Cai L., Efstathiou E., Logothetis C. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013;155:1309–1322. doi: 10.1016/j.cell.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurrekoetxea-Hernández K., Buetti E. Transforming growth factor beta enhances the glucocorticoid response of the mouse mammary tumor virus promoter through Smad and GA-binding proteins. J. Virol. 2004;78:2201–2211. doi: 10.1128/JVI.78.5.2201-2211.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biddie S.C., John S., Sabo P.J., Thurman R.E., Johnson T.A., Schiltz R.L., Miranda T.B., Sung M.-H., Trump S., Lightman S.L. Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol. Cell. 2011;43:145–155. doi: 10.1016/j.molcel.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi A., Hoffman L.M., Welm A.L., Lessnick S.L., Beckerle M.C. The EWS/FLI Oncogene Drives Changes in Cellular Morphology, Adhesion, and Migration in Ewing Sarcoma. Genes Cancer. 2012;3:102–116. doi: 10.1177/1947601912457024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi A., Hoffman L.M., Jensen C.C., Lin Y.-C., Grossmann A.H., Randall R.L., Lessnick S.L., Welm A.L., Beckerle M.C. Molecular dissection of the mechanism by which EWS/FLI expression compromises actin cytoskeletal integrity and cell adhesion in Ewing sarcoma. Mol. Biol. Cell. 2014;25:2695–2709. doi: 10.1091/mbc.E14-01-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng F.Y., Brenner J.C., Hussain M., Chinnaiyan A.M. Molecular pathways: targeting ETS gene fusions in cancer. Clin. Cancer Res. 2014;20:4442–4448. doi: 10.1158/1078-0432.CCR-13-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzetti G.A., Laud-Duval K., van der Ent W., Brisac A., Irondelle M., Aubert S., Dirksen U., Bouvier C., de Pinieux G., Snaar-Jagalska E. Cell-to-cell heterogeneity of EWSR1-FLI1 activity determines proliferation/migration choices in Ewing sarcoma cells. Oncogene. 2017;36:3505–3514. doi: 10.1038/onc.2016.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garside H., Stevens A., Farrow S., Normand C., Houle B., Berry A., Maschera B., Ray D. Glucocorticoid ligands specify different interactions with NF-kappaB by allosteric effects on the glucocorticoid receptor DNA binding domain. J. Biol. Chem. 2004;279:50050–50059. doi: 10.1074/jbc.M407309200. [DOI] [PubMed] [Google Scholar]
- Gauthier J.M., Bourachot B., Doucas V., Yaniv M., Moreau-Gachelin F. Functional interference between the Spi-1/PU.1 oncoprotein and steroid hormone or vitamin receptors. EMBO J. 1993;12:5089–5096. doi: 10.1002/j.1460-2075.1993.tb06203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad Y., Shiloh R., Ber Y., Bialik S., Kimchi A. Discovering protein-protein interactions within the programmed cell death network using a protein-fragment complementation screen. Cell Rep. 2014;8:909–921. doi: 10.1016/j.celrep.2014.06.049. [DOI] [PubMed] [Google Scholar]
- Grohar P.J., Segars L.E., Yeung C., Pommier Y., D’Incalci M., Mendoza A., Helman L.J. Dual targeting of EWS-FLI1 activity and the associated DNA damage response with trabectedin and SN38 synergistically inhibits Ewing sarcoma cell growth. Clin. Cancer Res. 2014;20:1190–1203. doi: 10.1158/1078-0432.CCR-13-0901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt H.J., Ray N.C., Hynd G., Sutton J., Sajad M., O’Connor E., Ahmed S., Lockey P., Daly S., Buckley G. Discovery of a novel non-steroidal GR antagonist with in vivo efficacy in the olanzapine-induced weight gain model in the rat. Bioorg. Med. Chem. Lett. 2012;22:7376–7380. doi: 10.1016/j.bmcl.2012.10.074. [DOI] [PubMed] [Google Scholar]
- Jaitin D.A., Kenigsberg E., Keren-Shaul H., Elefant N., Paul F., Zaretsky I., Mildner A., Cohen N., Jung S., Tanay A., Amit I. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science. 2014;343:776–779. doi: 10.1126/science.1247651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewell C.M., Scoltock A.B., Hamel B.L., Yudt M.R., Cidlowski J.A. Complex human glucocorticoid receptor dim mutations define glucocorticoid induced apoptotic resistance in bone cells. Mol. Endocrinol. 2012;26:244–256. doi: 10.1210/me.2011-1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmakar S., Jin Y., Nagaich A.K. Interaction of glucocorticoid receptor (GR) with estrogen receptor (ER) α and activator protein 1 (AP1) in dexamethasone-mediated interference of ERα activity. J. Biol. Chem. 2013;288:24020–24034. doi: 10.1074/jbc.M113.473819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katschnig A.M., Kauer M.O., Schwentner R., Tomazou E.M., Mutz C.N., Linder M., Sibilia M., Alonso J., Aryee D.N.T., Kovar H. EWS-FLI1 perturbs MRTFB/YAP-1/TEAD target gene regulation inhibiting cytoskeletal autoregulatory feedback in Ewing sarcoma. Oncogene. 2017;36:5995–6005. doi: 10.1038/onc.2017.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S., Denny C.T., Wisdom R. Cooperative DNA binding with AP-1 proteins is required for transformation by EWS-Ets fusion proteins. Mol. Cell. Biol. 2006;26:2467–2478. doi: 10.1128/MCB.26.7.2467-2478.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovar H. Blocking the road, stopping the engine or killing the driver? Advances in targeting EWS/FLI-1 fusion in Ewing sarcoma as novel therapy. Expert Opin. Ther. Targets. 2014;18:1315–1328. doi: 10.1517/14728222.2014.947963. [DOI] [PubMed] [Google Scholar]
- Langlais D., Couture C., Balsalobre A., Drouin J. The Stat3/GR interaction code: predictive value of direct/indirect DNA recruitment for transcription outcome. Mol. Cell. 2012;47:38–49. doi: 10.1016/j.molcel.2012.04.021. [DOI] [PubMed] [Google Scholar]
- Lauriola M., Enuka Y., Zeisel A., D’Uva G., Roth L., Sharon-Sevilla M., Lindzen M., Sharma K., Nevo N., Feldman M. Diurnal suppression of EGFR signalling by glucocorticoids and implications for tumour progression and treatment. Nat. Commun. 2014;5:5073. doi: 10.1038/ncomms6073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K.-T., Wang L.-H. New dimension of glucocorticoids in cancer treatment. Steroids. 2016;111:84–88. doi: 10.1016/j.steroids.2016.02.019. [DOI] [PubMed] [Google Scholar]
- Linabery A.M., Ross J.A. Childhood and adolescent cancer survival in the US by race and ethnicity for the diagnostic period 1975-1999. Cancer. 2008;113:2575–2596. doi: 10.1002/cncr.23866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S., Garcia-Arencibia M., Zhao R., Puri C., Toh P.P.C., Sadiq O., Rubinsztein D.C. Bim inhibits autophagy by recruiting Beclin 1 to microtubules. Mol. Cell. 2012;47:359–370. doi: 10.1016/j.molcel.2012.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza-Naranjo A., El-Naggar A., Wai D.H., Mistry P., Lazic N., Ayala F.R.R., da Cunha I.W., Rodriguez-Viciana P., Cheng H., Tavares Guerreiro Fregnani J.H. ERBB4 confers metastatic capacity in Ewing sarcoma. EMBO Mol. Med. 2013;5:1087–1102. doi: 10.1002/emmm.201202343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michnick S.W., Ear P.H., Manderson E.N., Remy I., Stefan E. Universal strategies in research and drug discovery based on protein-fragment complementation assays. Nat. Rev. Drug Discov. 2007;6:569–582. doi: 10.1038/nrd2311. [DOI] [PubMed] [Google Scholar]
- Montgomery B., Cheng H.H., Drechsler J., Mostaghel E.A. Glucocorticoids and prostate cancer treatment: friend or foe? Asian J. Androl. 2014;16:354–358. doi: 10.4103/1008-682X.125392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullick J., Anandatheerthavarada H.K., Amuthan G., Bhagwat S.V., Biswas G., Camasamudram V., Bhat N.K., Reddy S.E., Rao V., Avadhani N.G. Physical interaction and functional synergy between glucocorticoid receptor and Ets2 proteins for transcription activation of the rat cytochrome P-450c27 promoter. J. Biol. Chem. 2001;276:18007–18017. doi: 10.1074/jbc.M100671200. [DOI] [PubMed] [Google Scholar]
- Petta I., Dejager L., Ballegeer M., Lievens S., Tavernier J., De Bosscher K., Libert C. The Interactome of the Glucocorticoid Receptor and Its Influence on the Actions of Glucocorticoids in Combatting Inflammatory and Infectious Diseases. Microbiol. Mol. Biol. Rev. 2016;80:495–522. doi: 10.1128/MMBR.00064-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips A., Maira M., Mullick A., Chamberland M., Lesage S., Hugo P., Drouin J. Antagonism between Nur77 and glucocorticoid receptor for control of transcription. Mol. Cell. Biol. 1997;17:5952–5959. doi: 10.1128/mcb.17.10.5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamoorthy S., Cidlowski J.A. Corticosteroids: Mechanisms of Action in Health and Disease. Rheum. Dis. Clin. North Am. 2016;42:15–31, vii. doi: 10.1016/j.rdc.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A., Prefontaine K.E. Physical association and functional antagonism between the p65 subunit of transcription factor NF-kappa B and the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA. 1994;91:752–756. doi: 10.1073/pnas.91.2.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichardt H.M., Kaestner K.H., Tuckermann J., Kretz O., Wessely O., Bock R., Gass P., Schmid W., Herrlich P., Angel P., Schütz G. DNA binding of the glucocorticoid receptor is not essential for survival. Cell. 1998;93:531–541. doi: 10.1016/s0092-8674(00)81183-6. [DOI] [PubMed] [Google Scholar]
- Rogatsky I., Wang J.-C., Derynck M.K., Nonaka D.F., Khodabakhsh D.B., Haqq C.M., Darimont B.D., Garabedian M.J., Yamamoto K.R. Target-specific utilization of transcriptional regulatory surfaces by the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA. 2003;100:13845–13850. doi: 10.1073/pnas.2336092100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutz H.P. Effects of corticosteroid use on treatment of solid tumours. Lancet. 2002;360:1969–1970. doi: 10.1016/S0140-6736(02)11922-2. [DOI] [PubMed] [Google Scholar]
- Söderberg O., Gullberg M., Jarvius M., Ridderstråle K., Leuchowius K.-J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L.-G., Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- Stöcklin E., Wissler M., Gouilleux F., Groner B. Functional interactions between Stat5 and the glucocorticoid receptor. Nature. 1996;383:726–728. doi: 10.1038/383726a0. [DOI] [PubMed] [Google Scholar]
- Stoll G., Surdez D., Tirode F., Laud K., Barillot E., Zinovyev A., Delattre O. Systems biology of Ewing sarcoma: a network model of EWS-FLI1 effect on proliferation and apoptosis. Nucleic Acids Res. 2013;41:8853–8871. doi: 10.1093/nar/gkt678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlins S.A., Rhodes D.R., Perner S., Dhanasekaran S.M., Mehra R., Sun X.-W., Varambally S., Cao X., Tchinda J., Kuefer R. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- Tomlins S.A., Laxman B., Dhanasekaran S.M., Helgeson B.E., Cao X., Morris D.S., Menon A., Jing X., Cao Q., Han B. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448:595–599. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- Whirledge S., DeFranco D.B. Glucocorticoid Signaling in Health and Disease: Insights From Tissue-Specific GR Knockout Mice. Endocrinology. 2018;159:46–64. doi: 10.1210/en.2017-00728. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA sequencing data generated during this study are available at GEO, accession code GEO: GSE 135229.