

Abstract
To expand the arsenal of industrially applicable oxidative enzymes, fusions of alcohol dehydrogenases with an NADPH‐oxidase were designed. Three different alcohol dehydrogenases (LbADH, TbADH, ADHA) were expressed with a thermostable NADPH‐oxidase fusion partner (PAMO C65D) and purified. The resulting bifunctional biocatalysts retained the catalytic properties of the individual enzymes, and acted essentially like alcohol oxidases: transforming alcohols to ketones by using dioxygen as mild oxidant, while merely requiring a catalytic amount of NADP+. In small‐scale reactions, the purified fusion enzymes show good performances, with 69–99 % conversion, 99 % ee with a racemic substrate, and high cofactor and enzyme total turnover numbers. As the fusion enzymes essentially act as oxidases, we found that commonly used high‐throughput oxidase‐activity screening methods can be used. Therefore, if needed, the fusion enzymes could be easily engineered to tune their properties.
Keywords: alcohol dehydrogenases, biocatalysis, enzyme engineering, fusion enzymes, oxidases
Going self‐sufficient: Alcohol dehydrogenases (ADHs) can catalyze selective alcohol oxidations and can be applied to kinetically resolve racemic alcohols, or to produce carbonyl building blocks. By fusing ADHs with an NADPH oxidase, self‐sufficient biocatalysts can be produced that act like alcohol oxidases. These enzyme fusions can be used for conversions and for activity screening to aid enzyme engineering.
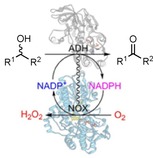
Alcohol oxidations are vital for the synthesis of various carbonyl compounds.1, 2 In particular, a catalyst that features strict enantioselectivity can be used for the kinetic resolution of alcohols.3 Enzymes can catalyze highly selective alcohol oxidations, and such biocatalytic oxidations have a relatively low environmental impact compared to chemically catalyzed oxidations.3, 4, 5, 6
The two main classes of enzymes that catalyze alcohol oxidations are alcohol oxidases (EC 1.1.3) and alcohol dehydrogenases (ADHs, EC 1.1.1.1). Although oxidases are attractive biocatalysts, because they depend on molecular oxygen as electron acceptor, there are not many alcohol oxidases available. On the other hand, there is a large array of characterized ADHs with various substrate specificities. The dehydrogenation reaction that the ADHs catalyze typically involves oxidized nicotinamide adenine dinucleotide (phosphate) (NAD(P)+) as electron acceptor plus an alcohol substrate, and transforms these into NAD(P)H and an aldehyde or ketone product. Inversely, the enzymes can also catalyze ketone reductions by using a reduced nicotinamide cofactor.
A major challenge in applying ADHs for alcohol oxidations is their dependence on the nicotinamide cofactor. Because the cofactor is too expensive to be applied in stoichiometric amounts, a recycling system is necessary to enable alcohol oxidations in an economically feasible manner.6, 7 In addition, alcohol oxidation with NAD(P)+ is thermodynamically less favorable than the reverse reaction, thus efficient recycling is needed to push against the equilibrium. One typical NAD(P)+‐recycling approach is the addition of an excess of a sacrificial ketone substrate, like acetone, that is readily reduced by the same ADH. This approach keeps the system simple, though it has some drawbacks, such as difference in pH optimum for the two reactions, occupation of the active sites by different substrates, which leads to inhibition, and poor atom efficiency due to the excess of sacrificial substrate. Another downside is the inhibition caused by the product from the sacrificial ketone. Alternatively, an NAD(P)H oxidase (NOX, EC 1.6.3) can be used to regenerate NAD(P)+.6, 7 NOXs typically contain a tightly bound flavin cofactor and efficiently oxidize NAD(P)H by using molecular oxygen, thereby forming hydrogen peroxide (type 1 NOX), or water (type 2 NOX).
Another type of flavin‐containing enzyme is the Baeyer–Villiger monooxygenase (BVMO, EC 1.14.13), which can also bind NAD(P)H and oxygen, but which is used to catalyze Baeyer–Villiger oxidations or other oxygenations.8 One property of this class of enzymes is that, after binding of NAD(P)H and oxygen and formation of the peroxyflavin intermediate, an uncoupling reaction can occur. During this reaction, the reactive peroxyflavin shunts back to the oxidized state, H2O2 is formed, and NADP+ is released. A mutant of a BVMO, phenylacetone monooxygenase (PAMO), was found to have a greatly enhanced uncoupling rate, thereby acting as an NADPH oxidase.9 Despite a profound change in activity, the C65D mutant PAMO was found to be as stable as the wild‐type PAMO, which is one of the most thermostable BVMOs characterized (T m=60 °C). With this favorable stability and the more alkaline pH optimum of the mutant (pH 8.0) compared to that of some other natural NOXs,7 PAMO C65D is an attractive biocatalyst to apply for NADP+ recycling.
Considering the combination of an ADH with a NOX for performing alcohol oxidations, we explored the approach of fusing these two enzymes together (Scheme 1). With this approach, the enzymes can be produced and purified in one go, and, as the enzymes are colocalized, the NOX could support the rapid regeneration of NADP+. In recent years, the possibilities and advantages of enzyme fusions have been explored for various enzyme types, including fusions of redox enzymes.10, 11, 12 For instance, to enable NADPH‐dependent enzymes to be recycled, various BVMOs and a P450 monooxygenase were fused to phosphite dehydrogenase.13, 14, 15 Some studies showed that, rather than using a sacrificial substrate like phosphite, it is possible to fuse ADHs with a cyclohexanone monooxygenase (CHMO) to enable cascade reactions from alcohols to esters,16 or from cyclohexanol to caprolactone,17 for which the fusions were more efficient than the separate enzymes. Another recent example of enzyme fusions is the combination of oxidases with a peroxidase to produce fusions that can be used for cascade reactions, as the hydrogen peroxide that is produced by the oxidase can be used directly by the fused peroxidase.1
Scheme 1.
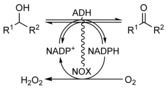
Alcohol dehydrogenases (ADHs) can catalyze alcohol oxidations and ketone reductions. By fusing an ADH with a NOX enzyme, which can oxidize the reduced nicotinamide cofactor NADPH by using oxygen, the equilibrium is driven toward catalyzing alcohol oxidations. In essence, the fusion of the two enzymes acts like an alcohol oxidase: an alcohol substrate is converted at the cost of oxygen, and hydrogen peroxide is produced.
The aim of this study is to investigate whether fusing an ADH with a NOX produces a bifunctional enzyme that can be applied for dioxygen‐driven alcohol oxidations by facilitating the regeneration of NADP+ (Scheme 1). Fusions were made by pairing the PAMO C65D mutant (NOX) with three NADP+dependent alcohol dehydrogenases: LbADH (R‐selective) from Lactobacillus brevis, TbADH from Thermoanaerobacter brockii, and a commercial ADH (ADHA).
The organization of the fusions was inspired by our previous study on ADH/CHMO fusions, in which we found a clear difference in ADH activity depending on the orientation: ADH–CHMO or CHMO–ADH.17 The findings from that study indicate that short‐chain dehydrogenases/reductases (SDRs) lose activity as N‐terminal fusions (ADH–BVMO), possibly through perturbation of the dimer/tetramer formation of the SDRs. Although LbADH was not investigated in that study, other studies found that a C‐terminal His tag was detrimental to the activity.19 For ADHA, we have similar evidence (unpublished results). Considering their classification as SDRs, we presumed that they would be active as C‐terminal fusions (BVMO–ADH). Therefore, we designed the fusion enzymes NOX‐A, NOX‐L, and T‐NOX (Table 1), each with an N‐terminal His tag.
Table 1.
Alcohol dehydrogenase–NADPH‐oxidase fusions produced.
|
Enzyme |
N‐terminal |
Linker |
C‐terminal |
M W [kDa] |
|---|---|---|---|---|
|
NOX‐A |
PAMO C65D |
SGSAAG |
ADHA |
90.3 |
|
NOX‐L |
PAMO C65D |
SGSAAG |
LbADH |
90.5 |
|
T‐NOX |
TbADH |
SGSAAG |
PAMO C65D |
101.4 |
A: ADHA, L: LbADH from L. brevis; T: TbADH from T. brockii, NOX: PAMO mutant C65D.
The three fusion constructs were first cloned and then transformed into Escherichia coli for recombinant expression. The expression levels were found to be similar to those of the individual ADH and NOX enzymes, based on SDS‐PAGE with samples from the cell‐free extracts (Figures S1 and S2 in the Supporting Information) and the amount of purified enzyme. After affinity chromatography purification, 40–150 mg of fusion enzyme per liter culture could be obtained. For wild‐type PAMO, 40 mg of purified enzyme per liter culture was reported.20
The UV/Vis absorbance spectra of the fusion enzymes were different from the spectrum of the single NOX. In a typical spectrum of oxidized FAD in NOX, the two absorbance maxima at 350–385 nm and at 440–460 nm have roughly the same height; for the fusions the 350–385 nm peak was more pronounced. This an indication of the presence of some fully reduced and/or semiquinone flavin. It is difficult to pinpoint the cause of this change; possibly the cells experienced more oxidative stress during the expression of these fusions, and this affected the oxidation state of the NOX. To fully oxidize the flavin cofactor in the purified fusion enzymes, they were incubated overnight with 10 mm potassium ferricyanide (K3[Fe(CN)6]), which was subsequently removed by gel‐filtration chromatography. After this treatment, the spectra from the fusion enzymes resembled the spectrum of the individual NOX, thus indicating that the FAD is in oxidized state (Figure S3).
As mentioned before, the fusion of a protein to the N or C terminus of an enzyme can greatly influence the activity of that enzyme. Therefore, we determined the kinetic parameters of the ADH–NOX fusions and the single enzymes (Table 2). Cyclohexanol was chosen as model substrate for the alcohol oxidation activity, as it is a known substrate for each of the three ADHs. The alcohol oxidation results show minor differences in k cat and K M values between the fused and nonfused ADH enzymes; at most 1.5‐ to 2‐fold differences in kinetic parameters. This indicates that the activity of the ADH enzymes was unaffected by the fusion. For the NOX activity, the differences were in a similarly small range, though there was a remarkable increase in K M for NADPH of the NOX‐A fusion. Still, the K M,NADPH for NOX‐A is in the micromolar range. Overall, we found that the fused enzymes largely retained their catalytic properties, and proceeded to investigate their utility as biocatalysts.
Table 2.
Alcohol and NADPH oxidation kinetics of the fusion enzymes.[a]
|
Enzyme |
Cyclohexanol oxidation |
NADPH oxidation |
||||
|---|---|---|---|---|---|---|
|
|
k cat |
K M |
k cat /K M |
k cat |
K M |
k cat /K M |
|
|
[s−1] |
[mm] |
[s−1 m −1] |
[s−1] |
[μm] |
[s−1 mm −1] |
|
NOX |
– |
– |
– |
5.0[b] |
3.5[b] |
1400 |
|
ADHA |
0.26 |
19 |
14 |
– |
– |
– |
|
NOX‐A |
0.56 |
10 |
56 |
5.1 |
27 |
190 |
|
LbADH |
2.2 |
31 |
71 |
– |
– |
– |
|
NOX‐L |
2.0 |
29 |
69 |
4.4 |
5.8 |
760 |
|
TbADH |
8.3 |
3.7 |
2200 |
– |
– |
– |
|
T‐NOX |
5.7 |
5.8 |
980 |
2.8 |
5.7 |
490 |
[a] The kinetics were determined by measuring the change in absorbance at 340 nm at various concentrations of substrate (5–10 different concentrations, in duplicate or triplicate, Figures S4 and S5). The alcohol oxidation rates were measured in 20 mm KPO4 (pH 7.5), and NADPH oxidation was measured in 50 mm Tris⋅HCl (pH 8.0), both at 25 °C. [b] Data taken from ref. 9 (in 50 mm Tris⋅HCl (pH 7.5)).
To assess the applicability of the fusion enzymes for alcohol oxidations, the conversion of cyclohexanol by NOX‐A was tested. The reaction mixtures consisted of fusion enzyme, buffer, substrate, and NADP+. Initially, only moderate conversions could be achieved, ranging from 32 to 76 % (data not shown). Possibly, the hydrogen peroxide that is formed during the reaction was inactivating one or both enzymes. Another complication could be the loss of FAD from the NOX fusion. To evaluate whether the addition of catalase or FAD could improve the level of conversion, each additive was added separately (Table 3). Both additives significantly improved the level of conversion. For the catalase, the reasons for the improvement are fairly straightforward: removing the hydrogen peroxide prevents damage to the enzymes while it regenerates oxygen that can be used by the NOX. The beneficial effect of additional FAD might lie in the stabilizing effect of forcing the enzyme to remain in its holo (FAD‐bound) state. A recent study found that the addition of FAD indeed improves the stability of FAD‐dependent monooxygenases.22 In that study, superoxide dismutase (SOD) was also found to improve biocatalyst performance. Yet, when we added 40 U of SOD, no improvement was found.
Table 3.
Effect of additives on conversion by NOX‐A.
|
Enzyme |
Additive |
Conversion [%] |
TTN[a] (enzyme) |
TTN[a] (cofactor) |
|---|---|---|---|---|
|
NOX‐A |
– |
75 |
39 500 |
395 |
|
NOX‐A |
FAD |
93 |
46 500 |
465 |
|
NOX‐A |
catalase |
89 |
44 500 |
445 |
|
NOX‐A |
FAD+catalase |
95 |
47 500 |
475 |
[a] TTN: total turnover number (amount of substrate converted per fusion enzyme). Reaction conditions: 50 mm cyclohexanol with 1 μm of NOX‐A in 50 mm Tris⋅HCl (pH 8.5), 100 μm NADP+, 64 h at 24 °C, 500 rpm (ThermoMixer Eppendorf); 10 μm FAD, 1000 U catalase. Experiments were performed in duplicate.
Based on the results, FAD and catalase were included in all subsequent conversions of cyclohexanol and 1‐phenylethanol. We also included 1‐phenylethanol as an additional test substrate to explore kinetic resolutions of this chiral alcohol (Scheme 2). As TbADH is not active towards 1‐phenylethanol, the Tb‐NOX fusion was not tested for this substrate.
Scheme 2.
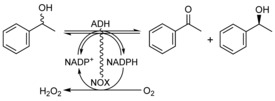
Kinetic resolution of rac‐1‐phenylethanol with the NOX‐ADH fusions. The reaction would ideally yield 50 % acetophenone, and 50 % of 99 % ee (R)‐ or (S)‐1‐phenylethanol.
The results clearly show that the fusions can be used for effective alcohol oxidations, with high total turnover numbers (TTN) for both the enzyme and the cofactor (Table 4). Both metrics are of interest from an industrial perspective, as the biocatalyst and the cofactor have high cost contributions.7 However, the performance of T‐NOX was considerably poorer than those of the other two fusion enzymes. Even though T‐NOX displayed the best kinetic parameters for cyclohexanol oxidation (Table 2), it showed the worst conversion (69 %) of the three fusions enzymes, with only 23 000 turnovers (Table 4). Still, the conversion was significantly better than the conversion without FAD and catalase (only 51 % conversion), thus suggesting that this fusion in particular suffered from the formation of hydrogen peroxide. Despite the addition of a substantial amount of catalase, some hydrogen peroxide can still accumulate because the affinity of catalase towards hydrogen peroxide is rather poor (K M=>10 mm). TbADH is a medium‐chain dehydrogenase that features a cysteine‐coordinated zinc that is involved in catalysis, which could make it more sensitive to peroxide‐induced inactivation.
Table 4.
Conversions of cyclohexanol and racemic 1‐phenylethanol by ADH/NOX fusion enzymes.
|
Substrate |
Enzyme |
Conversion |
ee [a] |
TTN |
TTN |
|---|---|---|---|---|---|
|
|
|
[%] |
[%] |
(enzyme) |
(cofactor) |
|
cyclohexanol |
NOX‐A |
95 |
n.a. |
31 666 |
475 |
|
|
NOX‐L |
99 |
n.a. |
33 000 |
495 |
|
|
T‐NOX |
69 |
n.a. |
23 000 |
345 |
|
rac‐1‐phenylethanol |
NOX‐A |
94 |
99 (R) |
31 333 |
470 |
|
|
NOX‐A |
56[b] |
99 (R) |
28 000 |
140 |
|
|
NOX‐L |
50 |
99 (S) |
16 666 |
250 |
Reaction conditions: 50 mm substrate with 1.5 μm of fusion enzyme in 50 mm Tris⋅HCl (pH 8.5), 100 μm NADP+, 64 h at 24 °C, 500 rpm (ThermoMixer Eppendorf); 10 μm FAD, 1000 U catalase. Reactions with rac‐1‐phenylethanol included 2 % DMSO. [a] Enantiomeric excess of the remaining alcohol substrate (Figure S6), [b] 0.5 μm NOX‐A, 25 mm substrate, 100 μm NADP+, 50 mm N‐cyclohexyl‐2‐aminoethanesulfonic acid (CHES; pH 9.0) for 24 h at 24 °C.
For the conversions of 1‐phenylethanol, it was gratifying to note that NOX‐L retained the strict enantioselectivity of the native LbADH, with 50 % conversion to acetophenone and yielding 99 % ee of the (S)‐1‐phenylethanol. On the other hand, NOX‐A showed a strong preference for the (S)‐substrate. Depending on the duration of the reaction, one could achieve (44 %) (R)‐1‐phenylethanol of 99 % ee, or primarily acetophenone (94 %).
With the developed fusion approach, dioxygen‐driven self‐sufficient alcohol dehydrogenases were generated that could be applied for selective alcohol oxidations. Essentially, as the overall reaction only consumes molecular oxygen and produces hydrogen peroxide, these fusion enzymes can be regarded as artificial alcohol oxidases. With that in mind, we were interested to see whether such “alcohol oxidases” are also suitable for established oxidase‐based, activity‐screening methods. One commonly used method for screening activity with oxidases is the horseradish peroxidase (HRP)‐coupled assay.21 When the oxidases transform alcohols and concomitantly produce hydrogen peroxide, the HRP can use the peroxide to oxidize fluoro‐ or chromogenic substrates, by which an easily detectable product is formed.
First, it was tested whether oxidase activity could be measured by means of a commonly used HRP‐based method. The assay included 4‐aminoantipyrine (AAP) and 3,5‐dichloro‐2hydroxybenzenesulfonic acid (DCHBS) which, upon peroxidase‐catalyzed oxidation, form a stable pink product. When using cell‐free extract (after growth and expression of NOX‐A) and 30 mm cyclohexanol, the reaction mixture turned pink after a few minutes, whereas the control reactions, which excluded one of the components, remained colorless (Figure 1). It should be made clear that no NADP+/NADPH was added to the assay mixture, this shows that the amount of nicotinamide cofactor in the extract was enough to support catalysis by NOX‐A. E. coli cells contain roughly 100 μm of NADP+/ NADPH.23 Even though ADH activity can be monitored by detecting NAD(P)H formation, this peroxidase‐based assay offers a cheap (no cofactor needed), facile and rapid method to measure or detect ADH/NOX oxidase activity.
Figure 1.

With an HRP‐coupled assay, the alcohol oxidation activity of the NOX‐A fusion can be detected without any addition of NADP+ (3). The reaction mixture included buffer (50 mm Tris⋅HCl pH 7.5), HRP (0.8 U), AAP (0.1 mm) and DCHBS (1 mm), cell‐free extract containing NOX‐A (10 % v/v) and 30 mm cyclohexanol. Controls: 1) no substrate and 2) no cell‐free extract.
Aside from screening the activity of cell‐free extracts, another peroxidase‐based method has been developed that allows the screening of oxidase activity in colonies.24 Such a high‐throughput approach is extremely well suited to enzyme engineering strategies. This approach is highly appealing for making large mutant libraries, as positive mutants can be selected based on the color of the colonies. We explored this option with the NOX‐A fusion. After the cells had been transformed with a construct for expression of the NOX‐A fusion, they were plated onto a permeable membrane that was on top of a layer of LB agar containing arabinose for expression. After growing for 24 h at 30 °C, the membrane with the colonies was transferred to an empty plate, frozen at −20 °C, thawed for partial lysis, and submerged in an assay mix. Colonies that expressed the NOX‐A fusion quickly turned an intense blue, but not the colonies that express only NOX (Figure 2) or the ADH (not shown). The blue colonies could be picked and used for growth and plasmid isolation, relying on the cells that survived the freeze–thawing step. This approach could be valuable when engineering an ADH/NOX towards a new substrate, as colonies would only turn blue when it has activity toward the substrate that is in the assay mix. In previous studies that used this method for screening of oxidase mutants, it was shown to be extremely useful for identifying improved variants, for example, in the case of engineering amine oxidase variants.24
Figure 2.
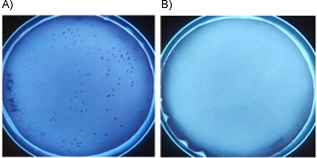
The fusion of an ADH with a NOX enables the detection of alcohol oxidation activity in colonies. The plates contain colonies that expressed A) NOX‐A or B) NOX. Only the colonies that produce the fusion enzyme turn dark blue after addition of the assay mix.
The designed NOX–ADH fusions presented in this study are not only suitable for performing alcohol oxidations with high total turnover numbers, the fusion of the two enzymes also enabled oxidase‐based activity screening. All three ADH enzymes were active in fusion with a NOX, and each enzyme retained its catalytic properties and level of expression. Although the hydrogen peroxide‐forming NOX might not be suitable for some ADHs, very good conversions could be attained with addition of catalase. In this regard, ADH fusions with a water‐forming NOX could be more appealing from an industrial perspective.7 An alternative approach could be a triple fusion: adding a catalase as a fusion partner. However, so far catalase fusion proteins have been found to be problematic, as they could not be expressed (unpublished results). Considering that current screening of ADH mutants is quite labor intensive and costly, it is worth noting that the NOX fusion partner opens doors to a rapid and cheap qualitative activity screening, and a convenient qualitative colony‐based screening. The NOX–ADH fusion approach could be a valuable tool for the development of useful and robust biocatalysts.
Experimental Section
Chemicals, reagents, enzymes and strains: Chemicals, medium components, and reagents were obtained from Sigma–Aldrich, Merck, Acros Organics, Alfa Aesar, Thermo Fisher, and Fisher Scientific. Oligonucleotides, superoxide dismutase (bovine SOD, recombinant), catalase (Micrococcus lysodeikticus), and HRP were obtained from Sigma. T4 ligase and the restriction enzyme BsaI were ordered from New England Biolabs. The PfuUltra Hotstart PCR master mix was purchased from Agilent Technologies. E. coli NEB 10‐beta (New England Biolabs) chemically competent cells were used as host for cloning the recombinant plasmids, and for protein expression. Precultures were grown in glass tubes with lysogeny broth (LB); for the subsequent main culture terrific broth (TB) was used in baffled flasks.
Golden Gate cloning: All fusion constructs were cloned with the Golden Gate cloning approach. Before commencing the amplification and assembly of the genes into the vector, the pamo gene (TFU_RS07 375) was mutated through QuikChange to remove a BsaI site, and to introduce the C65D mutation. Henceforth, the mutated pamo gene will be referred to as nox. Primers were designed that contained a flanking region coding for the BsaI restriction site. These primers were used for PCR to amplify the nox and three alcohol dehydrogenase genes: Tbadh (X64841.1), Lbadh (AJ544275.1), and adhA. In addition to the BsaI sites, linker regions were added to reverse primers of the first gene and the forward primer of the second gene of a construct. These introduced linker regions together code for a short peptide linker (SGSAAG) after ligation of the PCR products. The primers were designed such that the PCR products from one adh gene and the nox gene could be inserted into a pBAD vector. The pBAD vector contains two BsaI restriction sites, with an upstream region coding for an N‐terminal His6 tag, an AraC promoter, and an ampicillin resistance gene. The fusion constructs were produced by incubating together two PCR products (an adh gene and nox), Golden Gate pBAD vector, BsaI restriction enzyme, T4 ligase, ligation buffer, and sterile Milli‐Q water. The incubation temperature alternated between 16 °C (for 5 min) and 37 °C (for 10 min) for 30 cycles, was then set to 55 °C for 10 min, and finally to 80 °C for 20 min to inactivate the enzymes. To transform host cells with the fusion constructs, the Golden Gate reaction mixture (3 μL) was added to chemically competent E. coli cells, and a heat shock (42 °C) was applied for 30 s. After overnight growth on an LB agar plate with ampicillin, colonies were picked and grown in liquid LB, then the plasmids were isolated and sent for sequencing (GATC, Germany) to confirm correct ligation of the genes.
Culture growth and protein purification: Fusion enzymes were produced and purified as described previously.17 The E. coli cells harboring the plasmids were grown in LB (5 mL) with ampicillin (50 μg mL−1; 37 °C, 135 rpm, 16–24 h). From this preculture, an aliquot (2 mL, 4 %, v/v) was used to inoculate TB (50 mL), which contained ampicillin (50 μg mL−1) and 0.02 % filtered l‐arabinose. The cells were grown in a 250‐mL baffled flask (Sigma) at 24 °C and 135 rpm for 40 h. After harvesting by centrifugation (3000 g, 4 °C, 20 min), the cells were stored at −20 °C for several days. To purify the enzymes, the cells were first resuspended in buffer (10 mL; 50 mm Tris⋅HCl pH 8, 5 mm imidazole) supplemented with FAD (10 μm) and PMSF (phenylmethylsulfonyl fluoride, 100 μm). Then the cell suspension was cooled in ice water and subjected to sonication (5 s on/off, 10 min); subsequently the lysate was centrifuged for 45 min at 18 514 g and 4 °C (Eppendorf F‐34‐6‐38 rotor in 5810 R centrifuge). The supernatant was filtered (pore size 0.45 μm) into a gravity flow column containing Ni2+ Sepharose resin (1 mL; GE Healthcare), which was then closed and incubated for 60 min at 4 °C on a rocking table. The flow‐through was collected, and the resin was washed with five column volumes of two solutions consisting of buffer (Tris⋅HCl; 50 mm, pH 8.0) and imidazole 10 and 20 mm. Subsequently, the bound proteins were eluted by applying imidazole (500 mm) in Tris⋅HCl (50 mm, pH 8.0). The obtained yellow elute was then incubated with KFe(CN)6 (100 μm) for 16 h at 4 °C on a rocking table to oxidize any reduced FAD. The yellow solution was applied to a PG‐10 desalting column that was pre‐equilibrated with Tris⋅HCl (50 mm, pH 8.0), and the fusion enzyme was eluted with the same buffer. Purified protein was analyzed by spectrophotometry (200–700 nm) and SDS‐PAGE.
SDS‐PAGE and UV/Vis spectra: During the purification, small samples were taken before each step. SDS loading dye was added to these samples, and the mixture was incubated at 95 °C for 5 min, then centrifuged at 13 000 g for 1 min. The samples were loaded onto a precast SDS‐PAGE gel (GenScript, USA), and the gels were run in a Mini‐PROTEAN Tetra Vertical Electrophoresis Cell (Bio‐Rad) with the current being applied by using a PowerPac HC High‐Current Power Supply (Bio‐Rad) set at 120 V. When the blue front of the loading dye reached the bottom of the gel, after about 70 min, the gel was removed from the chamber, and stained with Coomassie InstantBlue (Expedeon, US). An UV/Vis absorption spectrum from 200 to 700 nm (V‐330 Spectrophotometer, JASCO) was taken of each purified fusion protein diluted 1:10 in buffer (50 mm Tris⋅HCl, pH 8) in a quartz cuvette (Hellma Analytics, Germany). The protein concentration (ϵ 441=12.4 mm −1 cm−1)20 was calculated by using the values at 441 nm.
Activity measurements and determination of kinetic parameters: Kinetic measurements were made by following the formation or depletion of NADPH at 340 nm. After the enzyme (≤0.1 μm) had been mixed with substrate in buffer (50 mm Tris⋅HCl pH 8.0), NADP or NADPH (100 μm) was added, briefly mixed in a cuvette, and then the reaction was followed (V‐330 Spectrophotometer, JASCO). For ADH activity, cyclohexanol was used as substrate; for the NOX activity, only NADPH was used, although in different concentrations. The slopes of the initial 20 s were used to calculate the activity rates. The obtained slope value is expressed in absorption change per minute [Abs min−1]. This value was then divided by the extinction coefficient of NADPH (ϵ 340=6.22 mm −1 cm−1) in accordance with the Beer–Lambert law to give a value in mm per minute. With that value and the enzyme concentration, the activity rate was calculated. All measurements were made in duplicate or triplicate. Activity data were fitted with GraphPad Prism 6.0 (GraphPad; Figures S4 and S5).
Conversions: Small‐scale biotransformations were performed in 2 mL Eppendorf tubes, with 0.5 mL of reaction mixture. The mixture consisted of fusion enzyme (1–5 μm), cofactor (200 μm NADP+), substrate, and buffer (50 mm Tris⋅HCl, pH 8.5, unless stated otherwise). A control without fusion enzyme was run in parallel. The tubes with the reaction mixtures were incubated (24 °C, 600 rpm, ThermoMixer C, Eppendorf), the the samples were extracted three times with an equivalent volume of ethyl acetate. The pooled extract (1.5 mL) was dried over magnesium sulfate, and then analyzed by using GC‐MS (HP‐5 column, injection temperature: 250 °C, oven temperature gradient: 40–30 °C, 5 °C min−1) or by using chiral GC (Hydrodex β‐TBDAc column (Aurora Borealis, The Netherlands), injection temperature: 250 °C, oven temperature gradient: 60–90 °C, 5 °C min−1).
Oxidase activity assay: After expression of the NOX‐A in E. coli, the cells were harvested, dissolved in buffer, and lysed by sonication. Only a small volume (e.g., 500 μL) of solubilized cells is needed for this assay. The insoluble fraction was separated by centrifugation (18 514 g, Eppendorf F‐34‐6‐38 rotor in 5810 R centrifuge, 30 min, 4 °C). In a well of a 96‐well plate, the soluble fraction after lysis (20 μL), AAP (1.0 mm, 20 μL), DCHBS (10 mm, 20 μL), HRP (4 U, 4 μL), substrate, and buffer to a total volume of 200 μL were combined. As the lysate will contain NADP+/NADPH, it is not necessary to add the cofactor. The formation of a pink product (as a result of peroxide production) can be followed over time to obtain a reaction rate (ϵ 515=26 mm −1 cm−1).
Colony‐screening assay: The assay was performed as described previously,24 with some adjustments. Cells harboring the plasmid for expression of NOX‐A were plated onto a porous membrane (nitrocellulose, Amersham, UK) on an LB agar plate that contained ampicillin (50 μg mL−1) and arabinose (0.02 %). After 40 h of growth at 30 °C, or when the colony‐size large enough to pick multiple times, the membrane was transferred to an empty Petri dish and incubated at −20 °C for 1 h. After the membrane with the colonies had been thawed at room temperature for 1 h, the assay mix (20 mL), which contained melted agarose (10 mL, 2 % w/v), potassium phosphate buffer (10 mL, 100 mm, pH 7.5), HRP (100 U), 4‐chloro‐1‐naphthol (2 mm), cyclohexanol (50 mm; substrate), NADP+ (100 μm), was gently poured onto the membrane. The plate was incubated at 30 °C, and after minutes/hours, the colonies with active “oxidases” developed a dark blue color.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research received funding from the European Union (EU) project ROBOX (grant agreement no. 635734) under the EU′s Horizon 2020 Program Research and Innovation actions H2020‐LEIT BIO‐2014‐1. The views and opinions expressed in this article are only those of the authors and do not necessarily reflect those of the European Union Research Agency. The European Union is not liable for any use that may be made of the information contained herein.
F. S. Aalbers, M. W. Fraaije, ChemBioChem 2019, 20, 51.
References
- 1. Mallat T., Baiker A., Chem. Rev. 2004, 104, 3037–3058. [DOI] [PubMed] [Google Scholar]
- 2. Sheldon R. A., Catal. Today 2015, 247, 4–13. [Google Scholar]
- 3.
- 3a. Hollmann F., Arends I. W. C. E., Buehler K., Schallmey A., Bühler B., Green Chem. 2011, 13, 226–265; [Google Scholar]
- 3b. Dong J., Fernández-Fueyo E., Hollmann F., Paul C. E., Pesic M., Schmidt S., Wang Y., Younes S., Zhang W., Angew. Chem. Int. Ed. 2018, 57, 9238–9261; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 9380–9404. [Google Scholar]
- 4. Schmid R. D., Urlacher V. in Modern Biooxidation: Enzymes, Reactions and Applications (Eds.: R. D. Schmid, V. Urlacher), Wiley-VCH, Weinheim, 2007, pp. XIII–XIV. [Google Scholar]
- 5. Pickl M., Fuchs M., Glueck S. M., Faber K., Appl. Microbiol. Biotechnol. 2015, 99, 6617–6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu J., Wu S., Li Z., Curr. Opin. Chem. Biol. 2018, 43, 77–86. [DOI] [PubMed] [Google Scholar]
- 7. Rehn G., Pedersen A. T., Woodley J. M., J. Mol. Catal. B 2016, 134, 331–339. [Google Scholar]
- 8. Torres Pazmiño D. E., Dudek H. M., Fraaije M. W., Curr. Opin. Chem. Biol. 2010, 14, 138–144. [DOI] [PubMed] [Google Scholar]
- 9. Brondani P. B., Dudek H. M., Martinoli C., Mattevi A., Fraaije M. W., J. Am. Chem. Soc. 2014, 136, 16966–16969. [DOI] [PubMed] [Google Scholar]
- 10. Conrado R. J., Varner J. D., DeLisa M. P., Curr. Opin. Biotechnol. 2008, 19, 492–499. [DOI] [PubMed] [Google Scholar]
- 11. Elleuche S., Appl. Microbiol. Biotechnol. 2015, 99, 1545–1556. [DOI] [PubMed] [Google Scholar]
- 12. Yang H., Liu L., Xu F., Appl. Microbiol. Biotechnol. 2016, 100, 8273–8281. [DOI] [PubMed] [Google Scholar]
- 13. Torres Pazmiño D. E., Snajdrova R., Baas B.-J., Ghobrial M., Mihovilovic M. D., Fraaije M. W., Angew. Chem. Int. Ed. 2008, 47, 2275–2278; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 2307–2310. [Google Scholar]
- 14. Torres Pazmiño D. E., Riebel A., de Lange J., Rudroff F., Fraaije M. D. Mihovilovic. M. W., ChemBioChem 2009, 10, 2595–2598. [DOI] [PubMed] [Google Scholar]
- 15. Beyer N., Kulig J. K., Bartsch A., Hayes M. A., Janssen D. B., Fraaije M. W., Appl. Microbiol. Biotechnol. 2017, 101, 2319–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jeon E. Y., Baek A. H., Bornscheuer U. T., Park J. B., Appl. Microbiol. Biotechnol. 2015, 99, 6267–6275. [DOI] [PubMed] [Google Scholar]
- 17. Aalbers F. S., Fraaije M. W., Appl. Microbiol. Biotechnol. 2017, 101, 7557–7565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Colpa D. I., Lončar N., Schmidt M., Fraaije M. W., ChemBioChem 2017, 18, 2226–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Peters C., Rudroff F., Mihovilovic M. D., Bornscheuer U. T., Biol. Chem. 2017, 398, 31–37. [DOI] [PubMed] [Google Scholar]
- 20. Fraaije M. W., Wu J., Heuts D. P. H. M., van Hellemond E. W., Spelberg J. H. L., Janssen D. B., Appl. Microbiol. Biotechnol. 2005, 66, 393–400. [DOI] [PubMed] [Google Scholar]
- 21. Ferrari A. R., Gaber Y., Fraaije M. W., Biotechnol. Biofuels 2014, 7, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goncalves L. C. P., Kracher D., Milker S., Fink M. J., Rudroff F., Ludwig R., Bommarius A. S., Mihovilovic M. D., Adv. Synth. Catal. 2017, 359, 2121–2131. [Google Scholar]
- 23. Bennett B. D., Kimball E. H., Gao M., Osterhout R., Van Dien S. J., Rabinowitz J. D., Nat. Chem. Biol. 2009, 5, 593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alexeeva M., Enright A., Dawson M. J., Mahmoudian M., Turner N. J., Angew. Chem. Int. Ed. 2002, 41, 3177–3180; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 3309–3312. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary