

Abstract
Bioinspired complexes employing the ligands 6‐tert‐butylpyridazine‐3‐thione (SPn) and pyridine‐2‐thione (SPy) were synthesized and fully characterized to mimic the tungstoenzyme acetylene hydratase (AH). The complexes [W(CO)(C2H2)(CHCH‐SPy)(SPy)] (4) and [W(CO)(C2H2)(CHCH‐SPn)(SPn)] (5) were formed by intramolecular nucleophilic attack of the nitrogen donors of the ligand on the coordinated C2H2 molecule. Labelling experiments using C2D2 with the SPy system revealed the insertion reaction proceeding via a bis‐acetylene intermediate. The starting complex [W(CO)(C2H2)(SPy)2] (6) for these studies was accessed by the new acetylene precursor mixture [W(CO)(C2H2)n(MeCN)3−nBr2] (n=1 and 2; 7). All complexes represent rare examples in the field of W−C2H2 chemistry with 4 and 5 being the first of their kind. In the ongoing debate on the enzymatic mechanism, the findings support activation of acetylene by the tungsten center.
Keywords: acetylene hydratases, acetylene ligands, bioinorganic chemistry, tungsten
C2H2 under attack! Bioinspired model complexes for acetylene hydratase demonstrate that nucleophilic attack on a W−C2H2 adduct is feasible. With the insertion of the enzyme's substrate acetylene into the W−N bond of the biomimetic ligand, data, which support the still disputed mechanism involving a W‐η2‐C2H2 intermediate, is provided (see scheme).
Molybdenum and tungsten are found in the active site of a large group of enzymes that catalyze the transfer of an oxygen atom from or to a substrate.1 The structurally related tungstoenzyme acetylene hydratase (AH) is unique among them, because it catalyzes the net hydration reaction of acetylene to acetaldehyde, a nonredox reaction. It is the only enzyme known to accept acetylene as substrate, apart from nitrogenase, which reduces it to ethylene.2 In 2007, structural information obtained by single‐crystal X‐ray diffraction analysis revealed the active site consisting of a six‐coordinate WIV center with a sulfur‐rich coordination sphere derived from two molybdopterin co‐factors and a cysteine residue. The sixth ligand of the distorted octahedron was found to be a water molecule with an aspartate in close proximity.3 The mechanism of the catalytic hydration is still under debate. Four mechanisms have been proposed and evaluated by DFT calculations.3, 4 They can be grouped into two fundamentally different reaction pathways, the major differences being the mode of substrate binding (C2H2 coordinated to W: first shell mechanism; H2O coordinated to W: second shell mechanism, Figure 1). Only the first shell mechanism proposed by Liao and co‐workers, comprising a deprotonated Asp13 and the displacement of H2O by C2H2, revealed a realistic energy barrier. The acetylene is nucleophilically attacked by the liberated H2O supported by concomitant proton transfer to Asp13. The resulting vinyl anion is then protonated by Asp13 to give acetaldehyde after tautomerization. Nonetheless, the second shell mechanism proposed by Einsle and co‐workers cannot be ruled out at present, because the acetylene could also be nucleophilically attacked while still located in the putative substrate pocked above the coordinated water molecule.3
Figure 1.
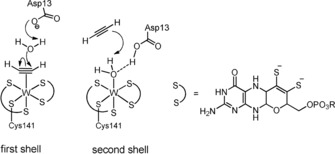
Comparison of first and second shell mechanism (left) and molybdopterin co‐factor (right).
Tungsten acetylene complexes are generally scarce,5 and those qualifying as biomimetic models are even rarer.6, 7, 8 Among the latter, the scorpionate complex [WO(C2H2)Tp′I] (Tp′=hydridotris(3,5‐dimethylpyrazolyl)borate) binds both substrates, C2H2 and H2O, upon halogen abstraction, whereas the recent structural analogue [WO(C2H2)(S‐Phoz)2] (S‐Phoz=2‐(4′4′‐dimethyloxazoline‐2′‐yl)thiophenolate) exhibits reversible C2H2 binding.7, 8 The only structural–functional model [Et4N]2[WO(mnt)2] (mnt=malonitrile) was reported to perform nine catalytic turnovers, whereas no W−C2H2 adduct could be isolated or spectroscopically observed.9, 10 However, recent attempts to reproduce these results failed, which questions the existence of a structural–functional model for AH.11
For the development of tungsten–acetylene complexes capable of nucleophilic attack, we explored two bioinspired ligands with different electronic influence, because biomimetic dithiolene‐type ligands are already well established.12 The use of the electron‐deficient sulfur ligand 6‐tert‐butylpyridazine‐3‐thione and the electron‐richer pyridine‐2‐thione (HSPy) should reveal which electronic properties are needed to gain the desired reactivity on a model complex. In our hand, the electron‐poor SPn system already led to unique reactivities with various other transition metals and thus seemed to be suitable for a model system, in which nucleophilic attack of H2O/OH− to a coordinated acetylene is desired.9, 13
Herein, we report the first intramolecular nucleophilic attack of a W‐bound η2‐C2H2 by the bioinspired S,N‐donor ligands SPn and SPy forming new complexes of the type [W(CO)(C2H2)(CHCH‐L)L].
For the preparation of acetylene complexes with the SPy and SPn ligands, a procedure previously employed for [W(CO)(C2H2)(S‐Phoz)2] was followed.8, 14 Thus, in a first step, previously reported [W2(CO)7Br4]15 was converted to [W(CO)3(MeCN)2Br2],16 which was subsequently reacted with two equivalents of the sodium salt of the respective ligand in dichloromethane. The use of SPy gave the tris‐carbonyl compound [W(CO)3(SPy)2] (1) in 67 % yield.17 The reaction with NaSPn also gave the tris‐carbonyl compound [W(CO)3(SPn)2] (2), as was evidenced by NMR spectroscopy of the crude reaction mixture. However, upon workup, partial dimerization to [W2(CO)4(SPn)4] (3) was observed, which had no influence on subsequent reactivity (Scheme 1). Exposing toluene solutions of the carbonyl compounds 1, 2 and 3 to acetylene atmosphere at room temperature led to highly interesting reactivity. Next to the expected substitution of CO by one molecule of C2H2, a second molecule of acetylene was found in the final product, which had inserted into the tungsten–nitrogen bond forming [W(CO)(C2H2)(CHCH−SPy)(SPy)] (4) and [W(CO)(C2H2)(CHCH−SPn)(SPn)] (5), respectively (Scheme 1). It is noteworthy that for the significant faster formation of 5, the reaction time is crucial, because after two hours, substantial polyacetylene (PA) formation and decomposition was observed.
Scheme 1.
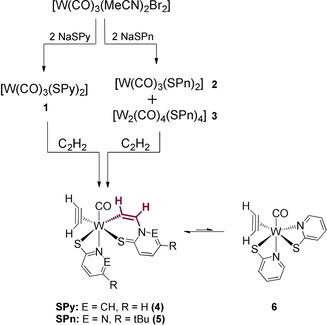
Synthetic strategy for the preparation of 4 and 5.
Characterization by 1H NMR spectroscopy clearly confirmed the two types of acetylene in 4 and 5 with the rotationally hindered η2‐acetylene appearing as two singlets (11.99–12.99 ppm, Table 1) and the protons of the inserted C2H2 as two doublets showing satellites resulting from coupling to the NMR‐active W183 isotope. For complex [W(CO)(C2H2)(CHCH‐SPy)(SPy)] (4), the second η1‐acetylene proton was determined by the COSY cross‐peak, as it is obscured by other ligand signals (see the Supporting Information). Unambiguous evidence delivered single‐crystal X‐ray diffraction analysis. Molecular views are displayed in Figure 3.
Table 1.
Overview of analytical data.
|
|
1H NMR [ppm] |
13C NMR [ppm] |
IR [cm−1] |
X‐ray [Å] |
||||
|---|---|---|---|---|---|---|---|---|
|
|
δ η2‐HC2H |
δ (C≡C) |
ν (C≡O) |
C≡C |
W−C2H2 |
W−CHCHN |
W−CO |
C≡O |
|
6 |
13.77 13.66 12.36 12.24 |
207.58 206.41 |
1911 1900 |
1.316(3) |
2.022(2) 2.045(2) |
– |
1.973(2) |
1.159(3) |
|
5 |
12.99 12.02 |
196.24 191.60 |
1918 |
1.315(7) |
2.036(4) 2.060(4) |
2.079(4) |
1.993(5) |
1.156(6) |
|
4 |
12.93 11.99 |
197.53 192.55 |
1890 |
1.3148(19) |
2.0353(13) 2.0582(13) |
2.0964(13) |
1.9781(12) |
1.1611(15) |
Figure 3.
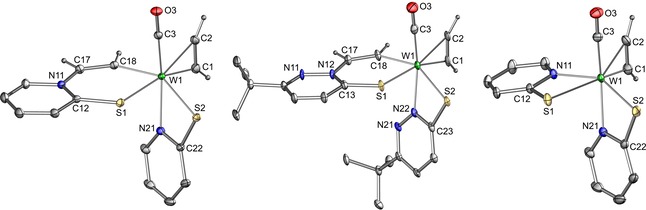
Molecular views of 4 (left), 5 (middle), and 6 (right) showing the atomic numbering Scheme.
With complex 4, the insertion of C2H2 is slower compared to the thiopyridazine system and was further found to be reversible (Scheme 1). Elimination of the inserted acetylene could be observed by 1H NMR spectroscopy revealing the formation of 70 % of complex [W(CO)(C2H2)(SPy)2] (6) within two hours at 45 °C. Under the same condition, no acetylene elimination for the corresponding SPn complex was observed.
The insertion of acetylene is extremely interesting, because it represents an intramolecular, nucleophilic attack on acetylene, which is relevant to the debate on AH mechanism. Furthermore, it has not been observed in [W(CO)(C2H2)(S2CNEt2)2]18 with very similar structural and electronic properties. Therefore, we were curious whether we can prepare the compounds [W(CO)(C2H2)L2] (L=SPy or SPn) without an inserted alkyne to separately investigate the insertion by isotopic labelling experiments. However, selective preparation starting from [W(CO)3L2] with limited amounts of acetylene proved to be futile, because with L=SPn only inserted product and with L=SPy a mixture of the desired [W(CO)(C2H2)(SPy)2] (6) and the inserted compound was observed. We therefore envisioned a procedure by using a tungsten starting material containing only one molecule of acetylene. Subsequent introduction of the S,N‐ligands should allow preparation of [W(CO)(C2H2)L2]. Although compounds of the type [W(CO)(C2R2)LnX2] with substituted alkynes (R=Me, Ph) have been described previously,19 the respective acetylene derivative has been elusive.
Therefore, we exposed an acetonitrile solution of [W(CO)3(MeCN)2Br2] to acetylene atmosphere for 65 minutes forming an olive green precipitate. The poorly soluble material proved to be a mixture of the desired compound with one molecule of acetylene [W(CO)(C2H2)(MeCN)2Br2] (7 a) and with two acetylenes [W(CO)(C2H2)2(MeCN)Br2] (7 b) as shown in Scheme 2. Although we were able to obtain single crystals suitable for X‐ray diffraction analyses confirming their structures (see the Supporting Information, Figures S12 and S14), their separation in bulk was futile, so that the mixture was used for subsequent reactions. The reaction of the mixture 7 (7 a and b) with NaSPy allowed the preparation of the desired tungsten compound [W(CO)(C2H2)(SPy)2] (6) without an inserted acetylene in 29 % yield (Scheme 2). Presumably, NaSPy only reacts with [W(CO)(C2H2)(MeCN)2Br2] (7 a), because [W(C2H2)2(SPy)2] was not observed. This general type of compound is known with substituted alkynes and other bidentate ligands.20 The synthetic procedure allowed also the preparation of deuterium‐labeled [W(CO)(C2D2)(SPy)2] (9) upon using C2D2 (Scheme 2). In contrast, the respective compound with the SPn ligand, [W(CO)(C2H2)(SPn)2], was not accessible, because any isolated product from the reaction of 7 with NaSPn proved to be the inserted complex [W(CO)(C2H2)(CHCH‐SPn)(SPn)] (5).
Scheme 2.
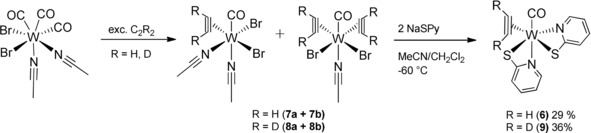
Synthetic procedure of the precursor mixtures 7 a+b and 8 a+b and the labelled complexes 6 and 9.
The 1H NMR spectra of complexes [W(CO)(C2R2)(SPy)2] (R=H 6, D 9) showed a mixture of two dynamic isomers indicated by two sets of acetylene protons (Figure 2 a). We assume the isomeric difference is with respect to the position of the ligands to each other, in which S,S‐, N,N‐ and two S,N‐trans positions are possible and as previously been observed in our S‐Phoz system.8, 14 With compounds 6 and 9, the respective cross‐insertion was investigated: thus, complex 6 was treated with C2D2 and 9 with C2H2 (Scheme 3). The insertion reaction was evaluated by 1H NMR spectroscopy upon comparing the ratio of integrals between a ligand and two acetylene signals (Scheme 3). To obtain comparable data, T 1 of the acetylenic protons of 4 was determined by the inversion recovery experiment (IRE), as these signals gave integrals <1 when recorded with standard T 1 of one second (see the Supporting Information).
Figure 2.
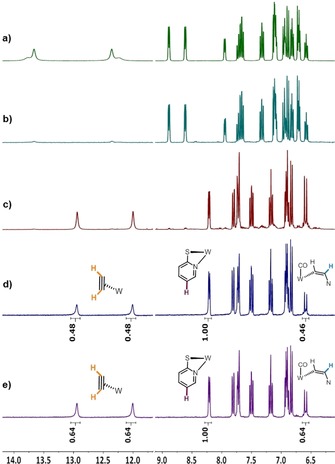
1H NMR spectra in CD2Cl2: a) [W(CO)(C2H2)(SPy)2] (6, two isomers), b) [W(CO)(C2D2)(SPy)2] (9, two isomers), c) [W(CO)(C2H2)(CHCH‐SPy)(SPy)] (4), d) 6+C2D2, and e) 9+C2H2.
Scheme 3.
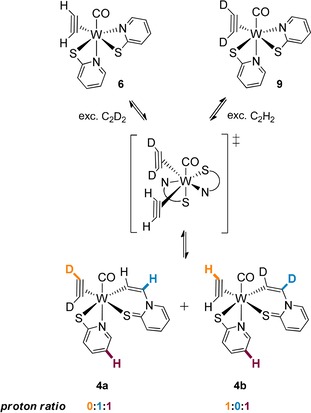
Labelling studies on acetylene insertion: reaction of protonated 6 with C2D2 (left), reaction of deuterated 9 with C2H2 (right).
The ratio of the η2‐C2H2 (11.99 ppm) and η1‐C2H2 (6.58 ppm) protons was determined and referenced to the pyridine ligand proton at 8.20 ppm (Scheme 3 and Figure 2). Upon selective insertion of added C2H2 in 9, formation of [W(CO)(C2D2)(CHCH−SPy)(SPy)] (4 a) with an integral ratio of 0:1:1 (η2:η1:SPy). is expected, whereas a ratio of 1:0:1 is anticipated for the opposite experiment forming [W(CO)(C2H2)(CDCD‐SPy)(SPy)] (4 b). However, the two cross experiments reveal the presence of complex 4 a and b in a 1:1 ratio (Figure 2 d and e), as was indicated by a virtually identical integral of the η2‐C2H2 (11.99 ppm) and the η1‐C2H2 (6.58 ppm) signals.
Equimolar formation of complexes 4 a and b indicates a bis‐acetylene intermediate (Scheme 3), in which insertion of either the deuterated or the protonated acetylene into the cis N−W bond can occur. The bis‐acetylene intermediate is further supported by the observation that the acetylene precursor mixture [W(CO)(C2H2)n(MeCN)3−nBr2] (n=1 (7 a) and 2; (7 b)) reacts exclusively to [W(CO)(C2H2)(CHCH‐SPn)(SPn)] (5) when adding the more reactive SPn‐ligand (see above). Also, integration of the η2‐ and η1‐C2H2 signals, referenced to the pyridine signal at 8.20 ppm of the reaction 9+C2H2, reveals a hydrogen content of 1.3 (Figure 2 e), which is higher than the expected 1. This can be explained by a dynamic exchange of the deuterated acetylene with excess C2H2 in the bis‐acetylene intermediate (Scheme 3) or by impurities of complex 6 in complex 9 (Figure 2 b, residual W−C2H2 signals). The expected hydrogen content of one in the analogous cross‐reaction (Figure 2 d) may originate from less excess of C2D2 due to different gas sources (C2D2 from CaC2 vs. C2H2 from a gas cylinder) and thus less exchange in the intermediate. We rule out exchange in the product as treatment of the fully deuterated complex [W(CO)(C2D2)(CDCD‐SPy)(SPy)] with acetylene for 2.5 hours lead to virtually no changes in the 1H NMR spectrum.
Insertion seems to be facilitated by the more electron‐withdrawing thiopyridazine ligand, which is reflected by the drastic difference in reactivity. Less electron density on the W center leads to weaker π backdonation into the C≡C bond, which makes its LUMO more reactive towards a nucleophilic attack. Furthermore, ring strain reduction of a four‐membered to a six‐membered ring seems not to be the only driving force for the insertion, because only one of the two chelate rings is expanded by acetylene insertion.
Nucleophilic attack on coordinated acetylene in an intramolecular fashion using phosphorous or carbon nucleophiles is known for transition metals, such as Mo or Ir.21, 22 For example, oxidation of [Mo(C2H2)(dppe)2] with [Cp2Fe][BF4] leads to nucleophilic attack of the phosphorus on the acetylenic carbon to form the acetylene‐inserted complex [Mo(CHCH‐PPh2CH2CH2PPh2)(dppe)(MeCN)2][OTf]2.21 In the complex [Ir(C2H2)( )(η5‐C5Me5)][BArF] with the cyclometalated ligand PMe(2,6‐Me2C6H3)2 (= ) the coordinated C2H2 is inserted into the σ Ir−CH2 bond upon thermal activation.22 However, such reactivity has never been observed for a mononuclear W complex. This makes 4 and 5 the first compounds of their kind, which is of particular interest regarding AH, because it represents the first nucleophilic attack on a W‐bound C2H2 and therefore supports the proposed first shell mechanism.
Solid‐state structures of all compounds were obtained by using single‐crystal X‐ray diffraction analysis (Figure 3 and the Supporting Information). Compound [W(CO)(C2H2)(SPy)2] (6) features a S,N‐trans arrangement of the ligands and most likely reflects the minor isomer present in solution. It further displays the typical parallel arrangement of the CO and the C2H2 ligand.7, 23 The structures of 4 and 5 revealed that the N atom of the heterocycle acted as intramolecular nucleophile. The S,N‐trans ligand arrangement is conserved during insertion of acetylene into the W−N bond giving the corresponding S,C‐trans complexes. The W−CHCHN bond length in 5 is shorter than in 4 (2.079(4) versus 2.0964(13) Å), which is in accordance with the more electron‐withdrawing character of the SPn ligand. The thereby formed six‐membered ring is planar and orientated perpendicular to the carbonyl ligand.
In conclusion, the successful synthesis, as well as the structural and spectroscopic characterization of the first nucleophilically attacked W‐bound acetylene, is reported. In this reaction, the coordinated nitrogen atom of the bioinspired ligands 6‐tert‐butylpyridazine‐3‐thione and pyridine‐2‐thione served as intramolecular nucleophile. Acetylene insertion was investigated by using C2D2 and revealed coordination of the second acetylene prior to insertion, as well as reversibility of the insertion reaction. The obtained results show that W−C2H2 adducts are able to react with nucleophiles and therefore support the proposed first shell mechanism of AH.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
C. Vidovič, L. M. Peschel, M. Buchsteiner, F. Belaj, N. C. Mösch-Zanetti, Chem. Eur. J. 2019, 25, 14267.
References
- 1. Bertini I., Gray H. B., Stiefel E. I., Valentine J. S., Biological Inorganic Chemistry: Structure & Reactivity, University Science Books, Sausalito, 2007. [Google Scholar]
- 2. Burgess B. K., Lowe D. J., Chem. Rev. 1996, 96, 2983–3012. [DOI] [PubMed] [Google Scholar]
- 3. Seiffert G. B., Ullmann G. M., Messerschmidt A., Schink B., Kroneck P. M. H., Einsle O., Proc. Natl. Acad. Sci. USA 2007, 104, 3073–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Liao R.-Z., Yu J.-G., Himo F., Proc. Natl. Acad. Sci. USA 2010, 107, 22523–22527; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Vincent M. A., Hillier I. H., Periyasamy G., Burton N. A., Dalton Trans. 2010, 39, 3816–3822; [DOI] [PubMed] [Google Scholar]
- 4c. Antony S., Bayse C. A., Organometallics 2009, 28, 4938–4944. [Google Scholar]
- 5.
- 5a. Umland P., Vahrenkamp H., Chem. Ber. 1982, 115, 3580–3586; [Google Scholar]
- 5b. Williams D. S., Schofield M. H., Schrock R. R., Organometallics 1983, 2, 165; [Google Scholar]
- 5c. Kersting M., El-Kholi A., Mueller U., Dehnicke K., Chem. Ber. 1989, 122, 279–285; [Google Scholar]
- 5d. Nielson A. J., Ware D. C., Polyhedron 1990, 9, 603–610; [Google Scholar]
- 5e. Radius U., Sundermeyer J., Pritzkow H., Chem. Ber. 1994, 127, 1827–1835. [Google Scholar]
- 6. Ricard L., Weiss R., Newton W. E., Chen G. J. J., McDonald J. W., J. Am. Chem. Soc. 1978, 100, 1318–1320. [Google Scholar]
- 7. Crane T. W., White P. S., Templeton J. L., Organometallics 1999, 18, 1897–1903. [Google Scholar]
- 8. Peschel L. M., Belaj F., Mösch-Zanetti N. C., Angew. Chem. Int. Ed. 2015, 54, 13018–13021; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13210–13213. [Google Scholar]
- 9. Yadav J., Das S. K., Sarkar S., J. Am. Chem. Soc. 1997, 119, 4315–4316. [Google Scholar]
- 10. Das S. K., Biswas D., Maiti R., Sarkar S., J. Am. Chem. Soc. 1996, 118, 1387–1397. [Google Scholar]
- 11. Schreyer M., Hintermann L., Beilstein J. Org. Chem. 2017, 13, 2332–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Sugimoto H., Sugimoto K., Inorg. Chem. Commun. 2008, 11, 77–80; [Google Scholar]
- 12b. Sugimoto H., Tarumizu M., Miyake H., Tsukube H., Eur. J. Inorg. Chem. 2007, 4663–4668. [Google Scholar]
- 13.
- 13a. Holler S., Tüchler M., Belaj F., Veiros L. F., Kirchner K., Mösch-Zanetti N. C., Inorg. Chem. 2016, 55, 4980–4991; [DOI] [PubMed] [Google Scholar]
- 13b. Holler S., Tüchler M., Steller B. G., Belaj F., Veiros L. F., Kirchner K., Mösch-Zanetti N. C., Inorg. Chem. 2018, 57, 6921–6931; [DOI] [PubMed] [Google Scholar]
- 13c. Tüchler M., Holler S., Schachner J. A., Belaj F., Mösch-Zanetti N. C., Inorg. Chem. 2017, 56, 8159–8165; [DOI] [PubMed] [Google Scholar]
- 13d. Tüchler M., Gärtner L., Fischer S., Boese A. D., Belaj F., Mösch-Zanetti N. C., Angew. Chem. Int. Ed. 2018, 57, 6906–6909; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 7022–7025; [Google Scholar]
- 13e. Tüchler M., Belaj F., Raber G., Neshchadin D., Mösch-Zanetti N. C., Inorg. Chem. 2015, 54, 8168–8170. [DOI] [PubMed] [Google Scholar]
- 14. Peschel L. M., Vidovič C., Belaj F., Neshchadin D., Mösch-Zanetti N. C., Chem. Eur. J. 2019, 25, 3893–3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Cotton F. A., Falvello L. R., Meadows J. H., Inorg. Chem. 1985, 24, 514–517; [Google Scholar]
- 15b. Peschel L. M., Schachner J. A., Sala C. H., Belaj F., Mösch-Zanetti N. C., Z. anorg. allg. Chem. 2013, 639, 1559–1567. [Google Scholar]
- 16.
- 16a. Braendle A., Vidovič C., Mösch-Zanetti N. C., Niederberger M., Caseri W., Polymer 2018, 10, 881; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b. Baker P. K., Hursthouse M. B., Karaulov A. I., Lavery A. J., Malik K. M. A., Muldoon D. J., Shawcross A., J. Chem. Soc. Dalton Trans. 1994, 3, 3493–3498. [Google Scholar]
- 17.
- 17a. Deeming A. J., Karim M., Powell N. I., J. Chem. Soc. Dalton Trans. 1990, 2321–2324; [Google Scholar]
- 17b. Sukcharoenphon K., Capps K. B., Abboud K. A., Hoff C. D., Inorg. Chem. 2001, 40, 2402–2408. [DOI] [PubMed] [Google Scholar]
- 18. Ward B. C., Templeton J. L., J. Am. Chem. Soc. 1980, 102, 1532–1538. [Google Scholar]
- 19.
- 19a. Templeton J. L. in Advances in Organometallic Chemistry, Vol. 29 (Eds.: F. G. A. Stone, R. West), Academic Press, San Diego, Section l, 1989, pp. 1–100; [Google Scholar]
- 19b. Baker P. K., Muldoon D. J., Lavery A. J., Shawcross A., Polyhedron 1994, 13, 2915–2921; [Google Scholar]
- 19c. Baker P. K., Keys E. M., Polyhedron 1986, 5, 1233–1235. [Google Scholar]
- 20. Baker P. K., Cartwright G. A., Jackson P. D., Flower K. R., Galeotti N., Severs L. M., Polyhedron 1992, 11, 1043–1048. [Google Scholar]
- 21. Ishino H., Kuwata S., Ishii Y., Hidai M., Organometallics 2001, 20, 13–15. [Google Scholar]
- 22. Espada M. F., Poveda M. L., Carmona E., Organometallics 2014, 33, 7164–7175. [Google Scholar]
- 23. Crane T. W., White P. S., Templeton J. L., Inorg. Chem. 2000, 39, 1081–1091. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary