

Abstract
The orphan receptor GPR125 (ADGRA3) belongs to subgroup III of the adhesion G protein−coupled receptor (aGPCR) family. aGPCRs, also known as class B2 GPCRs, share basic structural and functional properties with other GPCRs. Many of them couple to G proteins and activate G protein−dependent and −independent signaling pathways, but little is known about aGPCR internalization and β‐arrestin recruitment. GPR125 was originally described as a spermatogonial stem cell marker and studied for its role in Wnt signaling and cell polarity. Here, using cell‐based assays and confocal microscopy, we show that GPR125 is expressed on the cell surface and undergoes constitutive endocytosis in a β‐arrestin−independent, but clathrin‐dependent manner, as indicated by colocalization with transferrin receptor 1, an early endosome marker. These data support that the constitutive internalization of GPR125 contributes to its biological functions by controlling receptor surface expression and accessibility for ligands. Our study sheds light on a new property of aGPCRs, namely internalization; a property described to be important for signal propagation, signal termination, and desensitization of class A (rhodopsin‐like) and B1 (VIP/secretin) GPCRs.
Keywords: adhesion GPCR, GPR125, ADGRA3, internalization, endocytosis, β‐arrestin
Here, using cell‐based assays and confocal microscopy, we show that GPR125 is expressed on the cell surface and undergoes constitutive endocytosis in a β‐arrestin−independent, but clathrin‐dependent manner, as indicated by colocalization with transferrin receptor 1, an early endosome marker. These data support that the constitutive internalization of GPR125 contributes to its biological functions by controlling receptor surface expression and accessibility for ligands.

Introduction
G protein−coupled receptors (GPCRs) are versatile, seven‐transmembrane domain (7TM) proteins that regulate a variety of intracellular signaling cascades in response to extracellular stimuli, such as hormones, neurotransmitters, ions, photons, and odorants.1 They are among the most intensively studied drug targets due to their substantial involvement in human pathophysiology and pharmacological tractability2 with up to 35% of all marketed drugs acting via GPCRs.3 Ligand‐mediated activation of GPCRs initiates signaling through heterotrimeric G proteins, as well as through G protein−independent pathways. Moreover, some GPCRs also show constitutive activity that can be further modulated by ligands of different efficacies.1
Class B2 of the superfamily of GPCRs consists of 33 adhesion GPCRs (aGPCRs) in humans. Like other GPCRs, aGPCRs are membrane‐located proteins that possess the signature 7TM domain, with a structural hallmark containing a large extracellular N‐terminus with adhesion properties. Many aGPCRs are autoproteolytically cleaved at the GPCR proteolytic site (GPS), which lies within the GPCR autoproteolysis−inducing domain. This results in the N‐terminal fragment (NTF) that is noncovalently linked to the C‐terminal fragment (CTF) containing the 7TM domain.4, 5 Multiple molecular mechanisms of G protein activation have been proposed for aGPCRs, involving the positioning of the NTF relative to the CTF, which enables the Stachel (also called stalk) located downstream of the GPS motif to act as tethered agonist.6, 7
In contrast to class A rhodopsin−like receptors, where several G protein−recognizing motifs have been identified,8 there is so far no conserved aGPCR domain that would determine whether the receptor interacts with G proteins. Despite this, ligand‐mediated9, 10, 11, 12, 13, 14, 15 and constitutive16 G protein signaling have been described for several aGPCRs involving activation of all four major subclasses of G proteins (Gαs, Gαi, Gαq, and Gα12) and a variety of intracellular signaling second messengers leading to many different functional endpoints.16, 17, 18, 19, 20, 21, 22, 23, 24
After internalization, the receptors are either targeted for degradation or recycled back to the cell surface where they can be stimulated again by their ligands.25 To our knowledge, little is known about intracellular trafficking of aGPCRs compared with class A, B1, and C receptors, apart from certain aGPCR's interaction with β‐arrestin and endosomal proteins.17, 18, 26, 27, 28 The arrestin family consists of four members: arrestin‐1 and arrestin‐4, also known as visual arrestins, are exclusively expressed in the retina,29 whereas the two β‐arrestin isoforms, β‐arrestin‐1 and β‐arrestin‐2 (also named arrestin‐2 and arrestin‐3, respectively), are ubiquitously expressed in most tissues.30
GPR125 (ADGRA3) belongs to subfamily III of aGPCRs (Fig. 1), also denoted as ADGRA,31 and was initially identified as a spermatogonial stem cell marker.32 The receptor is broadly expressed in various tissues with a particularly high expression reported in the murine choroid plexus, where it is upregulated after brain injury.33 GPR125 has been suggested to be essential during development as a modulator of planar cell polarity (PCP) in zebrafish.34 Using genetically manipulated zebrafish, it has been shown that GPR125 regulates noncanonical Wnt signaling by recruiting Dishevelled (Dvl) proteins to the cell surface, and thereby directing cellular orientation during convergence and extension movements in gastrulating zebrafish.32, 34 GPR125 has also been studied in the context of cancer. Specifically, its ability to modulate Wnt signaling leads to increased survival of patients with colorectal cancer,35 thereby suggesting that GPR125 is acting as a tumor suppressor in it. However, in myeloid sarcoma, GPR125 seems to have an oncogenic effect as its upregulation contributes to tumor formation.36
Figure 1.
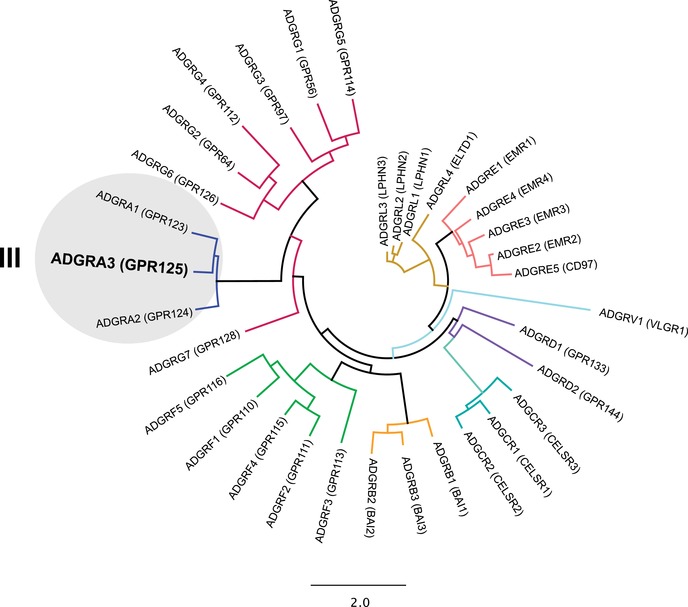
Cladogram of class B2 adhesion GPCRs (aGPCRs). aGPCR amino acid sequences were aligned using MAFFT and the cladogram was constructed using PhyML; Likelihood‐joining. Class B2 receptors are divided into nine different subfamilies marked by the different colors of the branches. GPR125 (ADGRA3) belongs to subfamily III, together with GPR123 (ADGRA1) and GPR124 (ADGRA2).
Inspired by the regulation of GPR125 expression in cancer, we decided to investigate the cellular distribution of this receptor and how β‐arrestin and clathrin are involved in the regulation of GPR125 expression on the cellular surface.
Materials and methods
Phylogenetic analysis
For phylogenetic analysis of the 33 human aGPCRs (Table S1, online only), amino acids were aligned using MAFFT plug‐in Geneious Pro Version 10.2.6 (Biomatters Ltd). The phylogenetic tree was constructed with the neighbor‐joining module, and the reliability of the tree was analyzed by bootstrap analysis (100‐fold resampling).
Constructs and cloning
GPR125 (codon‐optimized for mammalian cell expression) with N‐terminal flag‐tag in pcDNA3.1(+) vector was obtained from GenScript.
Cells and culture conditions and transfection
HEK293 cells were grown in Dulbecco's modified Eagle's medium with 10% fetal bovine serum (FBS), 180 units/mL penicillin, and 45 µg/mL streptomycin. The clustered regularly interspaced short palindromic repeats (CRISPR)‐modified β‐arrestin‐1/2 gene knockout (KO) and parental HEK293 cells were grown as previously described.37 The stable clone of inducible GPR125 HEK293 cells was generated as previously described38 and grown the same as parental HEK293 cells. GPR125 knockdown (KD) HEK293 cells (see below for the generation of these cells) were grown the same as parental HEK293 cells in the medium supplemented with 5 µg/mL puromycin. CHO‐K1 EA‐arrestin2 cells were grown in Ham's F‐12 medium containing 10% FBS, 2 mM glutamine, 180 units/mL penicillin, 45 µg/mL streptomycin, and 250 µg/mL hygromycin. HEK293 cells were transfected using Lipofectamine® 2000 (Invitrogen) according to the manufacturer's recommendation for the enzyme‐linked immunosorbent assay (ELISA) and the β‐arrestin‐1 and β‐arrestin‐2 recruitment bioluminescence resonance energy transfer (BRET) experiments. The calcium phosphate method for transfection was used in the β‐arrestin‐1 recruitment BRET assay in GPR125 KD cells, as previously described.37 For immunohistochemistry and PathHunter® GPCR β‐arrestin‐2 experiments, HEK293 and CHO‐K1 EA cells were transfected using the FuGENE® transfection reagent (Promega) according to the manufacturer's recommendation. All experiments were performed 24 h after transfection, unless otherwise stated.
Generation of GPR125 KD HEK293 cells
HEK293 cells were seeded in a 96‐well plate. The next day, the media was changed and hexadimethrine bromide (at a final concentration of 8 µg/mL) was added to each well (100 µL total volume). The cells were transduced with lentivirus transduction particles—shRNA human GPR125 (clone ID NM‐145290.2_3261s1c1; Sigma) according to the manufacturer's recommendation. Treatment with puromycin (5 µg/mL) was initiated 2 days post transduction and continued until control cells (nontransduced HEK293 cells) were dead. The receptor KD was confirmed by real‐time qPCR. To reintroduce GPR125 expression, stable GPR125 KD cells were transfected with codon‐optimized GPR125 plasmid DNA that could not be targeted by the shRNA due to an altered nucleotide sequence.
Real‐time qPCR on cell lysates
RNA was extracted using the RNeasy® Micro Kit (Qiagen). Approximately 1 µg of total RNA was reverse transcribed with the High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). qPCR was performed using the QuantStudioTM 6 Flex Real‐Time PCR System (Thermo Fisher Scientific) and the PowerUP SYBR® Green Master Mix (Thermo Fisher Scientific). Cycle threshold (Ct) values were obtained using the QuantStudio Real‐time PCR Software, and the Δ–ΔCt method was used to calculate the relative fold change of cDNA levels compared with a reference gene (glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH)). Sequences of the forward and reverse GPR125 primers were CTGGCACATGCTTGTGAACT and GGCAAGGGTGGAATAGTGAA, respectively.
Surface expression determined by ELISA
Transiently transfected HEK293 cells were fixed for 10 min with 3.7% formaldehyde, washed with TBS/CaCl2, and blocked for 30 min at room temperature (RT) with 2% BSA/TBS/CaCl2. For total GPR125 cell content, cells were permeabilized with 0.2% saponin (Sigma) after fixation. Cells were incubated with primary antibody (mouse M1 anti‐flag 1:2250, Sigma) in 1% BSA/TBS/CaCl2 for 1 h at RT, washed with TBS/CaCl2, and incubated with secondary antibody (goat anti‐mouse HRP 1:1000, Thermo Fisher Scientific) in 1% BSA/TBS/CaCl2 for 1 h at RT. They were subsequently exposed to 3,3′,5,5′‐tetramethylbenzidine for 5 min, and the reaction stopped with 0.2M H2SO4. Absorbance at 450 nm was measured with the EnVision® plate reader (PerkinElmer). The experiment was performed in triplicate at least three times independently. Data were analyzed using GraphPad Prism (GraphPad Software, San Diego, CA).
Internalization
HEK293 cells were seeded and transiently transfected. Twenty‐four hours posttransfection, cells were incubated in cold DMEM containing primary antibody (mouse M1 anti‐flag 1:2250, Sigma) for 1 h at 4 °C. After three washes with cold DMEM, cells were either immediately fixed (t = 0) or incubated with prewarmed DMEM at 37 °C for various periods of time (t = 10, 20, 30, 40, and 50 min) to induce internalization before being fixed at the given time points. From there on, the procedure followed that of the ELISA described above. The experiment was performed in quadruplicate at least three times independently. Data were analyzed in GraphPad Prism.
Western blots using HEK293 cell lysates
HEK293 cells were lysed using 150 µL RIPA lysis buffer (Milipore), containing cOmplete® Mini 20 protease inhibitor cocktail (Roche) and phosphatase inhibitor cocktail 3 (Sigma) and 50 µL 4× Laemmli's sample buffer containing 50 mg/mL dithiothreitol. The cell lysates were sonicated and centrifuged at 13,000 × g for 10 min at 4 °C.
The samples were incubated at 30 °C for 30 min before running on an SDS gel (4−15% TGX Stain‐Free Precast Gels (Bio‐Rad)). The proteins were blotted on a polyvinylidene difluoride membrane using the Trans‐Blot turbo transfer system (Bio‐Rad). For the antibody staining, the membrane was first blocked with 2× Odyssey blocking buffer (Li‐Cor) for 30 min, incubated with primary antibody (mouse anti‐FLAG M1 1:2000, Sigma) in 1× blocking buffer/PBS/CaCl2 for 1 h at RT, washed with PBS + 0.01% Tween 20 (Sigma), and incubated with secondary antibody (goat anti‐mouse IRDye 800CW Li‐COR) in 1× blocking buffer/PBS/CaCl2 for 1 h at RT. After washing twice with PBS + 0.01% Tween 20 (Sigma) and once with PBS, the membrane was imaged using a Li‐Cor scanner and analyzed with the Image StudioTM software. The experiment was performed at least three times independently.
Immunocytochemistry and confocal microscopy
Inducible HEK293 cells were cultured on glass coverslips placed in 24‐well plates, and receptor expression was induced with tetracycline (0.25 µg/mL). Cells were fixed 24 h after tetracycline induction with 3.7% formaldehyde, washed with PBS, permeabilized in 0.1% TritonTM X‐100 for 10 min at RT, blocked in PBS with 0.1% Tween 20 and 2% bovine serum albumin for 1 h at RT, and then incubated with primary antibody (mouse M1 anti‐FLAG, 1:2250, Sigma) for 1 h at RT. After washing with PBS, the cells were incubated with secondary antibody (donkey anti‐mouse conjugated to Alexa Fluor 568, 1:500, Life Technologies) for 1 h at RT. Cells were nuclear stained with 4′,6‐diamidino‐2‐phenylindole (DAPI) and mounted in Mowiol® (Sigma) before imaging on a confocal microscope (LSM 700, Zeiss) with a 63× oil immersion plan‐apochromat objective.
Antibody feeding experiments
Fibronectin‐coated glass slides were placed in 24‐well plates and HEK293 cells or β‐arrestin‐1 and β‐arrestin‐2 KO cells were allowed to adhere before transfection the next day. Twenty‐four hours posttransfection, the cells were incubated with primary antibody (mouse M1 anti‐FLAG, 1:2250, Sigma) in media at 4 °C for 1 hour. They were then fixed immediately (t = 0) or incubated in normal media for 30 min at 37 °C to induce internalization (t = 30) before fixation. All samples were blocked in 2% BSA before incubation with Alexa Fluor 488−conjugated goat anti‐mouse antibody (1:500, Invitrogen) for 1 h at RT. Cells were then permeabilized with 0.2% saponin (Sigma) and incubated with Alexa Fluor 568−conjugated donkey anti‐mouse antibody (1:500, Life Technologies) for 1 h at RT. The glass slides were removed from the 24‐well plate and mounted on slides, sealed, and imaged as described above. To study colocalization, GPR125 was cotransfected with GFP‐tagged plasmids containing either transferrin receptor 1 (TfR1) or lysosomal‐associated membrane protein 1 (LAMP1). Here, GPR125 was detected by Alexa Fluor 568−conjugated antibodies but otherwise, the procedure was the same as described above. CoLocalizer Pro 5.4.3 (CoLocalization Research Software) was used to detect colocalization of the red and green fluorescence emitted by GPR125 (red) and TfR1 (green) or LAMP1 (green), and to calculate Pearson's correlation coefficients (Pr). To quantify the amount of internalized receptor in parental HEK and in β‐arrestin‐1 and β‐arrestin‐2 KO cells after 30 min, 10 images were taken, respectively, using a confocal microscope (LSM 710, Zeiss) and a plan apochromat 63×/1.40 Oil DIC M27 objective. For the red fluorescence signal quantification, the following settings were used: 8.21 airy units, optical section 7‐micron, 561 nm laser at 2%, and averaging four times (fixed for all quantified images). For the green fluorescence signal quantification, the following settings were used: 9.59 airy units, optical section 7‐micron, 488 nm laser at 1%, and averaging four times (fixed for all quantified images). Data analysis was performed using the Zen software (Zeiss).
Real‐time ‐arrestin‐1 and ‐arrestin‐2 recruitment experiments
The BRET experiments were carried out as previously described.29, 39 Briefly, 1 day after seeding cells in a 6‐well plate, 60–80% confluent parental HEK293 cells and stably transduced GPR125 KD cells were transiently transfected with the following plasmids: GPR125, the human glucagon‐like peptide‐1 (GLP‐1) receptor (GLP‐1R, positive control), or empty pcDNA3.1(+) vector (negative control), in combination with the BRET donors Renilla luciferase−fused arrestins RLuc8–arrestin‐2–Sp1 or RLuc8–arrestin‐3–Sp1, the BRET acceptor mem‐linker‐citrine‐SH3, and GPCR kinases 2 or 6 (GRK2 or GRK6) to facilitate β‐arrestin‐1 and β‐arrestin‐2 recruitment. One day (for Lipofectamine‐transfected cells) or 2 days (for cells transfected with the calcium‐phoshate method, i.e., GPR125 KD cells) later, the cells were washed with PBS and resuspended in PBS with 5 mmol/L glucose. We added 85 µL of cell suspension to each well on a 96‐well isoplate followed by the addition of PBS with 5 µmol/L coelenterazine h (final concentration). After 10 min at RT, increasing concentrations of GLP‐1 were added to the positive control cells. Luminescence was measured by EnVision plate reader (PerkinElmer) (RLuc8 at 485 ± 40 nm and YFP at 530 ± 25 nm). The experiment was performed in triplicate at least three times independently.
PathHunter GPCR ‐arrestin‐2 experiments
The recruitment of β‐arrestin‐2 was measured using the PathHunter β‐arrestin assay as described previously.29 Briefly, cDNA encoding GPR125 and US28 (positive control) was fused to the ProLinkTM (PK) C‐terminal protein tag and the small fragment of β‐galactose (β‐gal) and cloned into the PK2 vector. The experiments were performed using the CHO‐K1 EA arrestin cell line with the stable expression of β‐arrestin coupled to the β‐gal large fragment. One day after seeding the cells (96‐well plate), they were transfected at a cell density of 60–80% with FuGENE reagent using different amounts of cDNA (GPR125, US28, and PK2 (vector control)). β‐Arrestin‐2 recruitment was detected as β‐gal activity using the PathHunter detection kit (DiscoverX). Chemiluminescent substrate composed of Galacton‐StarTM substrate, Emerald‐IITM solution, and PathHunter cell assay buffer in a ratio of 1:5:19, respectively, was added to the cells (50 µL/well). The luminescent signal was determined after 60 min incubation using the EnVision Multilabel Plate Reader (PerkinElmer).
Statistical analysis
Data were analyzed using a paired Student's t‐test or a one‐way ANOVA (multiple comparisons). P values < 0.05 were considered statistically significant.
Results
GPR125 is expressed on the cell surface and undergoes constitutive internalization
To determine the localization pattern of GPR125, we used an N‐terminally flag‐tagged construct (Fig. 2A). GPR125 surface expression was determined by ELISA in transiently transfected HEK293 cells (Fig. 2B) and the overall cellular localization was observed in inducible GPR125 HEK293 cells by microscopy (Fig. 2C and D). We found that GPR125 is mainly expressed on the cell surface of HEK293 cells, as the total amount of GPR125 expressed in these cells was comparable with surface‐expressed GPR125 (Fig. 2B). This was also the case for the positive control, GPR3940 (Fig. 2B). The internalization of GPR125 was determined using an antibody feeding approach, in which primary antibodies were directed against the N‐terminal flag‐tag of GPR125. Differently labeled secondary antibodies distinguished surface expression (green) and intracellularly located receptors (red). This is done by application before (green, t = 0 min) and after (red, t = 30 min) a temperature shift that induced the internalization event, followed by cell permeabilization to allow intracellular staining (Fig. 3A and B). Consistent with the surface expression determined by ELISA and microscopy (Fig. 2B and C), GPR125 was initially detected on the cell surface at 4 °C (Fig. 3A and B). After 30 min at 37 °C to allow internalization, GPR125 was mainly located in intracellular vesicles (Fig. 3C and D). These data were corroborated in transiently transfected nonpermeablized HEK293 cells, by measuring GPR125 cell surface expression over time after a temperature shift from 4 to 37 °C. After 30 min, the cell surface expression of GPR125 had decreased to 48% of the initial level, and after 1 h only 29% GPR125 remained on the cell surface (Fig. 3E). The class A receptor GPR183 (also known as EBI‐2) was included as a positive control. This receptor is predominantly cell surface−expressed41 and internalizes constitutively as well as ligand dependently (after addition of the endogenous agonist 7α,25‐dihydroxycholesterol (7α,25‐OHC)).42, 43 Consistent with a previous report,43 23.5% of surface‐expressed GPR183 was constitutively internalized after 30 min (Fig. 3F), whereas the addition of 7α,25‐OHC enhanced the internalization to 43% (Fig. 3G), that is, to comparable levels with the constitutive internalization of GPR125 (Fig. 3E). In summary, GPR125 is mainly expressed on the cell surface and undergoes constitutive internalization under physiological temperatures to the same degree as the agonist‐induced GPR183.
Figure 2.
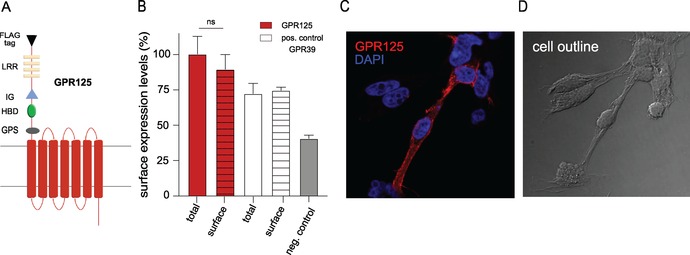
Cell surface expression of GPR125. (A) Diagram of the GPR125 receptor construct with the indicated motifs: GPS, 7TM proteolytic site; HBD, hormone‐binding domain; IG, immunoglobulin domain; and LRR, leucine‐rich region. (B) Cell surface expression of GPR125 (red bar) in transiently transfected HEK293 cells determined by ELISA against the N‐terminal flag‐tag, using pcDNA vector (gray bar) as baseline. The total amount of the receptors was measured by permeabilizing the cell membrane (striped bars). GPR39 was included as a positive control (white bar). (C) GPR125 is mainly expressed on the cell surface (red) of stably transfected HEK293 cells (pool clones) 24 h after induction with tetracycline. The nucleus is stained with DAPI (blue) and the cell outline is shown in D. Error bars in B indicate SEM for at least three independent biological replicates.
Figure 3.
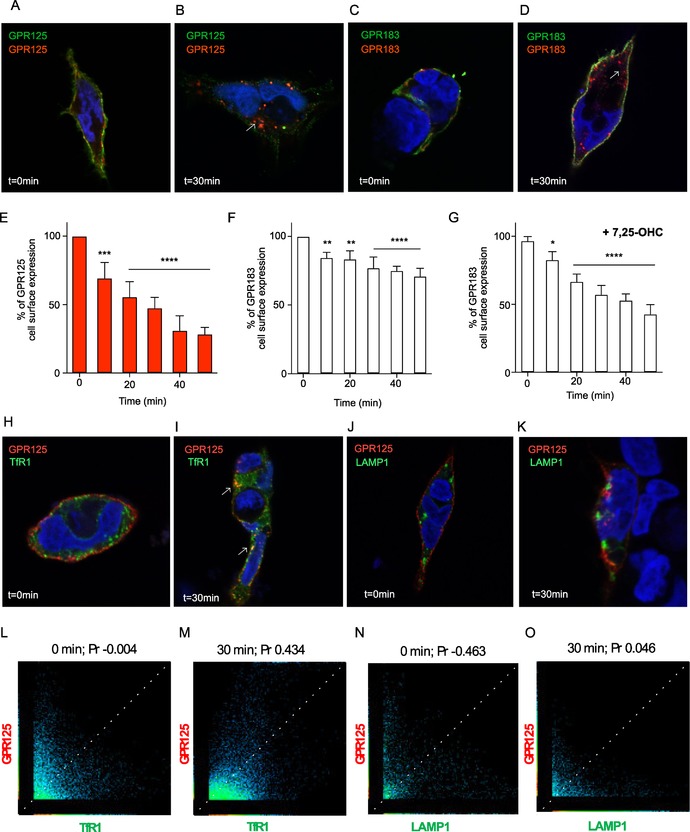
Constitutive endocytosis of GPR125 and colocalization with early and late endosomal markers. (A) Cell surface localization of GPR125 before (green) and (B) after induction of internalization, (red). (C and D) GPR183 included as a positive control. (E) Cell surface expression of GPR125 and GPR183 (F and G) after inducing receptor internalization (+/–ligand). The expression levels were normalized to each receptor at t = 0 minute. (H and I) Colocalization of TfR1 fused to GFP expressed together with GPR125 detected before, H, and after, I, inducing receptor endocytosis (white arrows). (J and K) Colocalization of LAMP1 fused to GFP expressed together with GPR125 detected before and after induction of endocytosis. (L−P) Scatterplots of the correlation between GPR125 and TfR1 or LAMP1 in panels E−H. (M and N) Determination of Pearson's correlation coefficient (Pr) between GPR125 and TfR1. (O and P) Pr of GPR125 and LAMP1. Error bars in E–G indicate SEM for three independent biological replicates. * P < 0.05; ** P < 0.01 *** P < 0.001; and **** P < 0.0001, using one‐way ANOVA analyses.
GPR125 colocalizes with the early endosome marker TfR1 but not with the late endosome marker LAMP1
Having established that GPR125 is constitutively internalized, we cotransfected GPR125 with GFP‐tagged marker of early endosomes (TfR1) and late endosomes/early lysosomes (LAMP1) to determine the route taken by internalized receptors. Using this approach, we would thus be able to describe whether GPR125 is internalized via clathrin‐mediated endocytosis as TfR144 (colocalization of GPR125 with TfR1) or following lysosomal protein degradation pathways (colocalization of GPR125 with LAMP139). Consistent with the surface expression of GPR125, the colocalization between GPR125 and the endosomal markers TfR1 and LAMP1 was weak at 4 °C (Fig. 3H and J, respectively). For TfR1, a Pearson's correlation coefficient (Pr) of –0.043 was obtained (Fig. 3L), whereas the Pr of LAMP1 was –0.463 (Fig. 3N). After 30 min at 37 °C, GPR125 moderately colocalized with TfR1 (Fig. 3I) with a Pr of 0.463 (Fig. 3M), but only weakly with LAMP1 (Fig. 3K) with a Pr of 0.046 (Fig. 3O). In conclusion, GPR125 colocalized with TfR1 after internalization, indicating that GPR125 is predominantly endocytozed in a clathrin‐mediated manner and possibly recycled similarly to what is observed with TfR1,45 as the receptor does not follow protein degradation pathways within the time frames analyzed in these experiments.
‐Arrestin‐1 and ‐arrestin‐2 recruitment to GPR125
Given the robust internalization of GPR125 (Fig. 3), we wanted to determine if GPR125 recruits β‐arrestin to initiate internalization. To this aim, we used a BRET‐based real‐time β‐arrestin recruitment assay, which gives a luminescence signal, when RLuc8‐fused β‐arrestin (RLuc8–arrestin‐2–Sp1 or RLuc8–arrestin‐3–Sp1, BRET donors) is recruited to the cell membrane in proximity to the membrane‐anchored citrine (mem‐linker‐citrine‐SH3, BRET acceptor).39 Following cotransfection with GPR125, we did not observe any increase in β‐arrestin‐1 and β‐arrestin‐2 recruitment to the cell membrane (Fig. 4A and B). In fact, a decreased recruitment signal was observed in both β‐arrestin‐1 (Fig. 4C) and β‐arrestin‐2 (Fig. 4D) upon cotransfection with GRK2 or GRK6 kinases that are both ubiquitously expressed in human cells. GPR125 expression in cells was confirmed by western blotting and its surface expression by ELISA (Fig. 4G and H). Given that endogenous expression of GPR125 in HEK293 cells (Fig. S1, online only) may be affecting this experiment, we wanted to confirm the effect observed upon reintroduction of GPR125 in GPR125 KD cells. We used a stable lentivirus‐transduced HEK293 cell line, where endogenous GPR125 expression was knocked down by GPR125‐specific short‐hairpin RNA (shRNA). The KD of GPR125 was confirmed by real‐time qPCR (Fig. S1A, online only). GPR125 was reintroduced in a concentration‐dependent manner by transfection with engineered GPR125 plasmid DNA untargetable by the shRNA (Fig. S1B, online only). Under these settings, the lack of β‐arrestin‐1 recruitment was confirmed and again a decrease in β‐arrestin‐1 was detected upon cotransfection with GRK2 and GRK6, although these results did not reach statistical significance (Fig. S1C, online only). As a positive control, we included the GLP‐1R stimulated with increasing concentrations of the endogenous agonist GLP‐1 and observed a ligand‐dependent β‐arrestin recruitment in parental HEK293 and GPR125 KD cells (Fig. 4E and F and Fig. S1D, online only). To further determine if there is a direct interaction of β‐arrestin with GPR125, we used transiently transfected cells, coexpressing the PK‐tagged GPR125 and the enzyme acceptor (EA)‐tagged β‐arrestin‐2. In the case of β‐arrestin‐2 interacting with the receptor, the β‐galactosidase enzyme fragments (EA and PK) will reconstitute a functional enzyme and a chemiluminescent signal can be detected. We determined a moderate interaction of GPR125 with β‐arrestin‐2 at 60 min, but this recruitment of β‐arrestin‐2 was not comparable with the interaction observed with our positive control US28 (Fig. S1E, online only).
Figure 4.
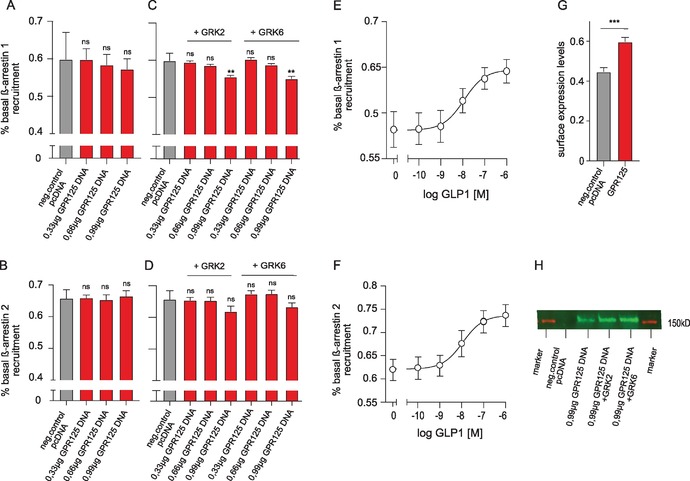
β‐Arrestin recruitment by GPR125. (A and B) β‐Arrestin‐1 and β‐arrestin‐2 recruitment was determined by performing BRET experiments in HEK293 cells. (C and D) β‐Arrestin‐1 and β‐arrestin‐2 recruitment of GPR125 after cotransfection with GRK2 and GRK6 in HEK293 cells. (E and F) GLP‐1R−mediated β‐arrestin‐1 and β‐arrestin‐2 recruitment in a ligand‐dependent manner was included as a positive control. (G and H) GPR125 expression was confirmed by western blotting and ELISA. Error bars indicate SEM for at least three independent biological replicates. ** P < 0.01 and *** P < 0.001, using one‐way ANOVA analyses.
To confirm that GPR125 internalization was independent of β‐arrestins, the antibody feeding experiments were performed in parental HEK293 and β‐arrestin‐1 and β‐arrestin‐2 KO cells. GPR125 got internalized in both cell lines (Fig. 5A−D). Red fluorescence intensities were measured to quantify the amount of internalized receptors; no difference was found in the amount of internalized GPR125, comparing parental HEK293 and β‐arrestin‐1 and β‐arrestin‐2 KO cells (Fig. 5E). Moreover, the amount of remaining surface‐expressed GPR125 (green fluorescence signal) was also equivalent in both cell lines (Fig. 5E).
Figure 5.
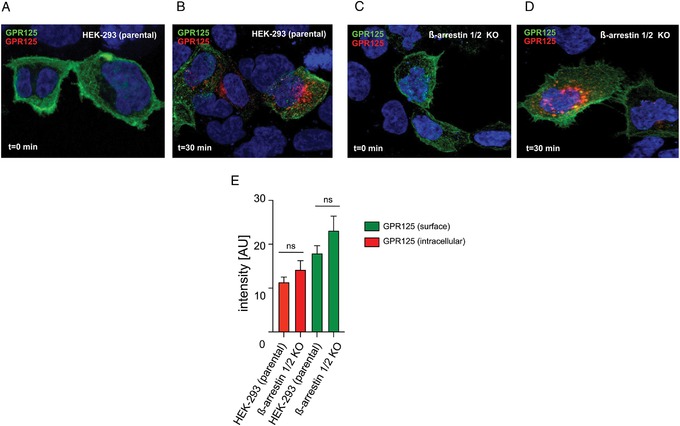
Constitutive endocytosis of GPR125 in parental HEK293 and β‐arrestin‐1 and β‐arrestin‐2 KO cells. (A) Cell surface localization of GPR125 before (green) and (B) after induction of internalization, (red) in parental HEK293 cells and (C and D) β‐arrestin‐1 and β‐arrestin‐2 KO cells. (E) Quantification of internalized GPR125 (red fluorescence signal) and remaining cell surface GPR125 (green fluorescence signal) in parental HEK293 and β‐arrestin‐1 and β‐arrestin‐2 KO cells. Error bars indicate SEM.
Discussion
In the present study, we show that GPR125 is expressed on the cell surface and that it is internalized in a constitutive manner at a speed corresponding to that of an agonist‐induced class A receptor (GPR183). Moreover, we show that the internalization of GPR125 happens in a β‐arrestin−independent, but TfR1 colocalizing/clathrin‐dependent manner. These findings are important amendments to the current knowledge of the biology of GPR125. No ligands have yet been described for GPR125, and it, therefore, still remains an orphan, like most aGPCRs.46 Moreover, its cleavage status is not known and based on its atypical GPS‐motif (SLS) and not the usual (HLS/HLT), it is possibly not cleaved via an autoproteolytic process. Finally, no G protein−mediated signaling has been described for GPR125. In this manner, it resembles the two other adhesion receptors in the subfamily III (GPR123 and GPR124), with which it also currently shares the status of being an orphan47 (Fig. 1).
Receptor endocytosis is a common mechanism to regulate receptor function
β‐arrestin−mediated endocytosis is a common and well‐described desensitization mechanism for class A and B1 receptors.37, 48 Also, β‐arrestin−independent internalization, as observed here for GPR125 (Fig. 4), has been described before as not all receptors rely on arrestins for their internalization. Clathrin‐mediated endocytosis is the major pathway for uptake of molecules and is essentially regulated by the GTPase dynamin.45 There is a growing evidence that GPCRs use clathrin‐mediated internalization as an alternative route, independent of phosphorylation and arrestin recruitement.48 For instance, this has been described for the class A melanocortin‐4 receptor (MC4R)49 and the two chemokine receptors, CXCR4 (a human CXC‐chemokine receptor)50 and US2851 (encoded by the human cytomegalovirus, HCMV). For CXCR4, it has been suggested that the constitutive and agonist‐induced receptor endocytosis are defined by distinct endocytic machineries: the constitutive internalization initiated by CXCR4 phosphorylation by protein kinase C, leading to dynamin‐dependent internalization, whereas the ligand‐induced internalization relies on arrestin recruitment.50, 52, 53 For US28, the internalization can take place in the absence of arrestins, yet US28 also recruits arrestins and is able to use them as an internalization route.51 Of particular note, US28 internalization is an intrinsic function of this receptor, as it scavenges chemokines as an immune escape mechanism for HCMV.54
We observed a relatively fast constitutive internalization of GPR125 (Fig. 3E). In contrast to the comprehensive literature on ligand‐mediated receptor endocytosis, less is known about constitutive internalization and how it regulates GPCR function and biology,55 except for those cases where it functions as a ligand scavenger (like for US2854 and atypical chemokine receptors56) or where constitutive internalization (and recycling) ensures sufficient receptor expression during prolonged exposure to the agonist (as for GPRC6A).57 The constitutive endocytosis of GPR125 could be a consequence of basal receptor activity, as described for the MC4R49 and since no G protein signaling has been described for GPR125, this remains to be determined in the future. Moreover, emerging evidence suggests that GPCRs are capable of signaling intracellularly from endosomes, as shown for the vasopressin receptor 2 (V2R; class A) GLP‐1 (class B1).58, 59 It is, therefore, possible that internalized GPR125 located in early endosomes could signal intracellularly. Since it does not colocalize with the late endosome marker LAMP1 (Fig. 3K), it is likely that GPR125 gets recycled back to the cell surface as it has been described for the β2‐adrenergic receptor60 and the incretin receptors.37
Arrestin recruitment and the following downstream signaling cascade
Interaction of β‐arrestins with activated GPCRs was initially considered to be the stop signal of receptor activation and subsequent initiation of receptor endocytosis.61 Recruitment of arrestins to initiate endocytosis has been vastly documented for class A and B1 receptors and most GPCRs seem to get internalized in an arrestin‐dependent manner.25 However, the role of arrestins solely as signaling stoppers and receptor endocytosis initiators has expanded to include the capacity to act as signaling molecules on their own, independently of G proteins.62 There have been examples of arrestin‐initiated signaling cascades, that is, induced by ERK1/2 MAP‐kinases.63 Recent results though showed for a broad panel of class A GPCRs that β‐arrestins are nonessential for driving ERK/MAPK signaling.64, 65 The acknowledgement of G protein−independent signaling has fostered another phenomenon among GPCRs, namely biased signaling, describing pathway‐dependent signaling.66 In class A receptors, it is most often seen that endogenous ligands are unbiased with similar potencies for G protein activation and β‐arrestin recruitment, although exceptions exist, such as CCR7.67 By contrast, in class B1 receptors (VIP/secretin receptors), there is a general bias toward higher potency for G protein signaling than for arrestin recruitment.37 However, for aGPCRs, very little is known about their use of arrestins and here, signaling bias remains to be described. Some aGPCRs (BAI1/ADGRB1, BAI3/ADGRB3, GPR56/ADGRG1, and GPR64/ADGRG2) have been shown to interact with β‐arrestins.17, 26, 27, 68 For BAI1, BAI3, and GPR56, only receptor interaction with β‐arrestin was detected,17, 68 whereas the interaction of β‐arrestin‐1 with GPR64/Gαq/CFTR (an ion channel) was shown to initiate signaling.26 For GPR125, where only the modulation of Wnt signaling has been described as the sole signaling,35 we observed a suppression of β‐arrestin recruitment to the receptor when measured in real time; the effect that was increased upon cotransfecting with GRK2 and GRK6.
Is the endocytosis of GPR125 a part of its biological function?
Receptor‐mediated endocytosis enables cells to take up molecular complexes, such as ligand−receptor complexes. Uptake of transferrin by TfR1 defines clathrin‐dependent internalization.61 We show that GPR125 gets internalized in a clathrin‐dependent manner, determined by its colocalization with the transferrin receptor in our confocal microscopy studies. It is still unclear how endocytosis and Wnt signaling are intertwined, but it is known that clathrin‐dependent endocytosis promotes PCP.61 GPR125 recruits a subset of PCP components into the membrane subdomains, which suggests that GPR125 modulates the composition of the Wnt/PCP membrane complexes.34 Therefore, GPR125‐induced clathrin‐dependent endocytosis could be important for the modulation of Wnt signaling and subsequently, PCP. Another receptor of the same subgroup III, GPR124 has also been suggested to be a potential candidate for stabilization of ligand–receptor interactions and thereby essential for Wnt‐induced internalization.51 It will be of interest to investigate whether internalization of GPR125 (and of its sibling GPR124) is directly linked to the modulation of Wnt signaling. Our study paves the way to a better understanding of aGPCR internalization and contributes to the knowledge about their biological function.
Author contributions
K.S. designed the research study, conducted the experiments, analyzed the data, and wrote the manuscript. S.O.B and T.L.J. designed the research study, conducted the experiments, analyzed the data, and revised the manuscript. K.R., W.A.L., K.J.M., V.D., J.K.H.R., M.S.K., and G.M.H contributed to the experimental work, analyzed the data, and revised the manuscript. M.M.R. designed the research study, analyzed the data, and wrote the manuscript. All authors commented on and approved the final manuscript.
Competing interests
The authors declare no competing interests.
Supporting information
Figure S1. β‐Arrestin recruitment by GPR125. (A) Real‐time qPCR analysis of GPR125 in HEK293 and GPR125 knockdown (KD) HEK293 cells relative to the expression of the control gene, GAPDH. (B) β‐Arrestin‐1 recruitment was determined by performing BRET experiments in HEK293 GPR125 KD cells, (C) after cotransfection with GRK2 and GRK6 as in the parental HEK293 cells. GLP‐1 receptor−mediated β‐arrestin‐1 recruitment in a ligand‐dependent manner was included as a positive control in HEK293 GPR125 KD cells. (D) β‐Arrestin‐2 recruitment to GPR125 using the PathHunter DiscoverX system. Error bars indicate SEM for at least three independent biological replicates. * P < 0.05; ** P < 0.01 using Student's t‐test (E).
Table S1. aGPCR genes and corresponding NCBI accession numbers.
Acknowledgments
We thank Frederik Vilhardt (University of Copenhagen, Denmark) for kindly supplying the constructs for TfR1 and LAMP1 fused to GFP and Asuka Inoue (Tohoku University, Japan) for supplying β‐arrestin‐1 and β‐arrestin‐2 KO cells. We further thank Sussi Mørkeberg Kristoffersen for her technical support. This work was supported by the European Research Council: VIREX Grant agreement 682549, Call ERC‐2105‐CoG, and the Lundbeck Foundation.
References
- 1. Hilger, D. , Masureel M. & Kobilka B.K.. 2018. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 25: 4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Petersen, S.C. et al 2015. The adhesion GPCR GPR126 has distinct, domain‐dependent functions in Schwann cell development mediated by interaction with laminin‐211. Neuron 85: 755–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hauser, A.S. et al 2018. Pharmacogenomics of GPCR drug targets. Cell 172: 41–54.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lin, H.‐H. et al 2004. Autocatalytic cleavage of the EMR2 receptor occurs at a conserved G protein‐coupled receptor proteolytic site motif. J. Biol. Chem. 279: 31823–31832. [DOI] [PubMed] [Google Scholar]
- 5. Nieberler, M. , Kittel R.J., Petrenko A.G., et al 2016. Control of adhesion GPCR function through proteolytic processing In Adhesion G Protein‐Coupled Receptors. Handbook of Experimental Pharmacology. Vol. 234 Langenhan T. & Schöneberg T., Eds.: 83–109. Cham: Springer. [DOI] [PubMed] [Google Scholar]
- 6. Liebscher, I. et al 2014. A tethered agonist within the ectodomain activates the adhesion G protein‐coupled receptors GPR126 and GPR133. Cell Rep. 9: 2018–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stoveken, H.M. , Hajduczok A.G., Xu L. & Tall G.G.. 2015. Adhesion G protein‐coupled receptors are activated by exposure of a cryptic tethered agonist. Proc. Natl. Acad. Sci. USA 112: 6194–6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schwartz, T.W. , Frimurer T.M., Holst B., et al 2006. Molecular mechanism of 7TM receptor activation—a global toggle switch model. Annu. Rev. Pharmacol. Toxicol. 46: 481–519. [DOI] [PubMed] [Google Scholar]
- 9. Jeong, S.‐J. , Luo R., Li S., et al 2012. Characterization of G protein‐coupled receptor 56 protein expression in the mouse developing neocortex. J. Comp. Neurol. 520: 2930–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Luo, R. et al 2011. G protein‐coupled receptor 56 and collagen III, a receptor–ligand pair, regulates cortical development and lamination. Proc. Natl. Acad. Sci. USA 108: 12925–12930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li, S. et al 2008. GPR56 regulates pial basement membrane integrity and cortical lamination. J. Neurosci. 28: 5817–5826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Iguchi, T. et al 2008. Orphan G protein‐coupled receptor GPR56 regulates neural progenitor cell migration via a G alpha 12/13 and Rho pathway. J. Biol. Chem. 283: 14469–14478. [DOI] [PubMed] [Google Scholar]
- 13. Silva, J.‐P. et al 2011. Latrophilin 1 and its endogenous ligand Lasso/teneurin‐2 form a high‐affinity transsynaptic receptor pair with signaling capabilities. Proc. Natl. Acad. Sci. USA 108: 12113–12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Little, K.D. , Hemler M.E. & Stipp C.S.. 2004. Dynamic regulation of a GPCR‐tetraspanin‐G protein complex on intact cells: central role of CD81 in facilitating GPR56‐Galpha q/11 association. Mol. Biol. Cell 15: 2375–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lelianova, V.G. et al 1997. Alpha‐latrotoxin receptor, latrophilin, is a novel member of the secretin family of G protein‐coupled receptors. J. Biol. Chem. 272: 21504–21508. [DOI] [PubMed] [Google Scholar]
- 16. Peeters, M.C. et al 2015. The adhesion G protein‐coupled receptor G2 (ADGRG2/GPR64) constitutively activates SRE and NFκB and is involved in cell adhesion and migration. Cell. Signal. 27: 2579–2588. [DOI] [PubMed] [Google Scholar]
- 17. Kishore, A. , Purcell R.H., Nassiri‐Toosi Z. & Hall R.A.. 2016. Stalk‐dependent and stalk‐independent signaling by the adhesion G protein‐coupled receptors GPR56 (ADGRG1) and BAI1 (ADGRB1). J. Biol. Chem. 291: 3385–3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stephenson, J.R. et al 2013. Brain‐specific angiogenesis inhibitor‐1 signaling, regulation, and enrichment in the postsynaptic density. J. Biol. Chem. 288: 22248–22256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Araç, D. et al 2012. Dissecting signaling and functions of adhesion G protein‐coupled receptors. Ann. N.Y. Acad. Sci. 1276: 1–25. [DOI] [PubMed] [Google Scholar]
- 20. Gupte, J. et al 2012. Signaling property study of adhesion G‐protein‐coupled receptors. FEBS Lett. 586: 1214–1219. [DOI] [PubMed] [Google Scholar]
- 21. Demberg, L.M. et al 2017. Activation of adhesion G protein‐coupled receptors: agonist specificity of Stachel sequence‐derived peptides. J. Biol. Chem. 292: 4383–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bohnekamp, J. & Schöneberg T.. 2011. Cell adhesion receptor GPR133 couples to Gs protein. J. Biol. Chem. 286: 41912–41916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liebscher, I. et al 2015. A tethered agonist within the ectodomain activates the adhesion G protein‐coupled receptors GPR126 and GPR133. Cell Rep. 10: 1021. [DOI] [PubMed] [Google Scholar]
- 24. Schöneberg, T. , Liebscher I., Luo R., et al 2015. Tethered agonists: a new mechanism underlying adhesion G protein‐coupled receptor activation. J. Recept. Signal Transduct. 35: 220–223. [DOI] [PubMed] [Google Scholar]
- 25. Pavlos, N.J. & Friedman P.A.. 2017. GPCR signaling and trafficking: the long and short of it. Trends Endocrinol. Metab. 28: 213–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang, D.‐L. et al 2018. Gq activity‐ and β‐arrestin‐1 scaffolding‐mediated ADGRG2/CFTR coupling are required for male fertility. elife 7: e33432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Southern, C. et al 2013. Screening β‐arrestin recruitment for the identification of natural ligands for orphan G‐protein–coupled receptors. J. Biomol. Screen. 18: 599–609. [DOI] [PubMed] [Google Scholar]
- 28. Purcell, R.H. , Toro C., Gahl W.A. & Hall R.A.. 2017. A disease‐associated mutation in the adhesion GPCR BAI2 (ADGRB2) increases receptor signaling activity. Hum. Mutat. 38: 1751–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gabe, M.B.N. et al 2019. Enhanced agonist residence time, internalization rate and signalling of the GIP receptor variant [E354Q] facilitate receptor desensitization and long‐term impairment of the GIP system. Basic Clin. Pharmacol. Toxicol. 10.1111/bcpt.13289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Reiter, E. & Lefkowitz R.J.. 2006. GRKs and β‐arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol. Metab. 17: 159–165. [DOI] [PubMed] [Google Scholar]
- 31. Hamann, J. et al 2015. International Union of Basic and Clinical Pharmacology. XCIV. Adhesion G protein‐coupled receptors. Pharmacol. Rev. 67: 338–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Waheeb, R. & Hofmann M.‐C.. 2011. Human spermatogonial stem cells: a possible origin for spermatocytic seminoma. Int. J. Androl. 34: e296–e305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pickering, C. et al 2008. The Adhesion GPCR GPR125 is specifically expressed in the choroid plexus and is upregulated following brain injury. BMC Neurosci. 9: 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li, X. et al 2013. Gpr125 modulates Dishevelled distribution and planar cell polarity signaling. Development 140: 3028–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu, Y. et al 2018. Elevated G‐protein receptor 125 (GPR125) expression predicts good outcomes in colorectal cancer and inhibits wnt/β‐catenin signaling pathway. Med. Sci. Monit. 24: 6608–6616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fu, J.‐F. et al 2013. Involvement of Gpr125 in the myeloid sarcoma formation induced by cooperating MLL/AF10(OM‐LZ) and oncogenic KRAS in a mouse bone marrow transplantation model. Int. J. Cancer 133: 1792–1802. [DOI] [PubMed] [Google Scholar]
- 37. Gabe, M.B.N. et al 2018. Human GIP(3‐30)NH 2 inhibits G protein‐dependent as well as G protein‐independent signaling and is selective for the GIP receptor with high‐affinity binding to primate but not rodent GIP receptors. Biochem. Pharmacol. 150: 97–107. [DOI] [PubMed] [Google Scholar]
- 38. Hjortø, G.M. , Kiilerich‐Pedersen K., Selmeczi D., et al 2013. Human cytomegalovirus chemokine receptor US28 induces migration of cells on a CX3CL1‐presenting surface. J. Gen. Virol. 94: 1111–1120. [DOI] [PubMed] [Google Scholar]
- 39. Donthamsetti, P. , Quejada J.R., Javitch J.A., et al 2015. Using bioluminescence resonance energy transfer (BRET) to characterize agonist‐induced arrestin recruitment to modified and unmodified G protein‐coupled receptors. Curr. Protoc. Pharmacol. 70 10.1002/0471141755.ph0214s70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shimizu, Y. , Koyama R. & Kawamoto T.. 2017. Rho kinase‐dependent desensitization of GPR39; a unique mechanism of GPCR downregulation. Biochem. Pharmacol. 140: 105–114. [DOI] [PubMed] [Google Scholar]
- 41. Rosenkilde, M.M. et al 2006. Molecular pharmacological phenotyping of EBI2. An orphan seven‐transmembrane receptor with constitutive activity. J. Biol. Chem. 281: 13199–13208. [DOI] [PubMed] [Google Scholar]
- 42. Benned‐Jensen, T. et al 2011. Ligand modulation of the Epstein–Barr virus‐induced seven‐transmembrane receptor EBI2. J. Biol. Chem. 286: 29292–29302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Daugvilaite, V. et al 2017. Biased agonism and allosteric modulation of G protein‐coupled receptor 183 — a 7TM receptor also known as Epstein–Barr virus‐induced gene 2. Br. J. Pharmacol. 174: 2031–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mayle, K.M. , Le A.M. & Kamei D.T.. 2012. The intracellular trafficking pathway of transferrin. Biochim. Biophys. 1820: 264–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. van Dam, E.M. & Stoorvogel W.. 2002. Dynamin‐dependent transferrin receptor recycling by endosome‐derived clathrin‐coated vesicles. Mol. Biol. Cell 13: 169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Langenhan, T. 2019. Adhesion G protein–coupled receptors—candidate metabotropic mechanosensors and novel drug targets. Basic Clin. Pharmacol. Toxicol. 10.1111/bcpt.13223. [DOI] [PubMed] [Google Scholar]
- 47. Vallon, M. & Essler M.. 2006. Proteolytically processed soluble tumor endothelial marker (TEM) 5 mediates endothelial cell survival during angiogenesis by linking integrin αvβ3 to glycosaminoglycans. J. Biol. Chem. 281: 34179–34188. [DOI] [PubMed] [Google Scholar]
- 48. Wolfe, B.L. & Trejo J.. 2007. Clathrin‐dependent mechanisms of G protein‐coupled receptor endocytosis. Traffic 8: 462–470. [DOI] [PubMed] [Google Scholar]
- 49. Mohammad, S. et al 2007. Constitutive traffic of melanocortin‐4 receptor in Neuro2A cells and immortalized hypothalamic neurons. J. Biol. Chem. 282: 4963–4974. [DOI] [PubMed] [Google Scholar]
- 50. Signoret, N. et al 1998. Differential regulation of CXCR4 and CCR5 endocytosis. J. Cell Sci. 111: 2819–2830. [DOI] [PubMed] [Google Scholar]
- 51. Fraile‐Ramos, A. , Kohout T.A., Waldhoer M. & Marsh M.. 2003. Endocytosis of the viral chemokine receptor US28 does not require beta‐arrestins but is dependent on the clathrin‐mediated pathway. Traffic 4: 243–253. [DOI] [PubMed] [Google Scholar]
- 52. Orsini, M.J. , Parent J.L., Mundell S.J., et al 1999. Trafficking of the HIV coreceptor CXCR4. Role of arrestins and identification of residues in the c‐terminal tail that mediate receptor internalization. J. Biol. Chem. 274: 31076–31086. [DOI] [PubMed] [Google Scholar]
- 53. Cheng, Z.J. et al 2000. Beta‐arrestin differentially regulates the chemokine receptor CXCR4‐mediated signaling and receptor internalization, and this implicates multiple interaction sites between beta‐arrestin and CXCR4. J. Biol. Chem. 275: 2479–2485. [DOI] [PubMed] [Google Scholar]
- 54. Kledal, T.N. , Rosenkilde M.M. & Schwartz T.W.. 1998. Selective recognition of the membrane‐bound CX3C chemokine, fractalkine, by the human cytomegalovirus‐encoded broad‐spectrum receptor US28. FEBS Lett. 441: 209–214. [DOI] [PubMed] [Google Scholar]
- 55. Scarselli, M. & Donaldson J.G.. 2009. Constitutive internalization of G protein‐coupled receptors and G proteins via clathrin‐independent endocytosis. J. Biol. Chem. 284: 3577–3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Graham, G.J. , Locati M., Mantovani A., et al 2012. The biochemistry and biology of the atypical chemokine receptors. Immunol. Lett. 145: 30–38. [DOI] [PubMed] [Google Scholar]
- 57. Jacobsen, S.E. et al 2017. The GPRC6A receptor displays constitutive internalization and sorting to the slow recycling pathway. J. Biol. Chem. 292: 6910–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bünemann, M. & Hosey M.M.. 1999. G‐protein coupled receptor kinases as modulators of G‐protein signalling. J. Physiol. 517: 5–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Irannejad, R. et al 2013. Conformational biosensors reveal GPCR signalling from endosomes. Nature 495: 534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Seachrist, J.L. & Ferguson S.S.. 2003. Regulation of G protein‐coupled receptor endocytosis and trafficking by Rab GTPases. Life Sci. 74: 225–235. [DOI] [PubMed] [Google Scholar]
- 61. Thomsen, A.R.B. et al 2016. GPCR‐G protein‐β‐arrestin super‐complex mediates sustained G protein signaling. Cell 166: 907–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cahill, T.J. et al 2017. Distinct conformations of GPCR–β‐arrestin complexes mediate desensitization, signaling, and endocytosis. Proc. Natl. Acad. Sci. USA 114: 2562–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Luttrell, L.M. & Lefkowitz R.J.. 2002. The role of β‐arrestins in the termination and transduction of G‐protein‐coupled receptor signals. J. Cell Sci. 115(Pt 3): 455–465. [DOI] [PubMed] [Google Scholar]
- 64. Alvarez‐Curto, E. et al 2016. Targeted elimination of G proteins and arrestins defines their specific contributions to both intensity and duration of G protein‐coupled receptor signaling. J. Biol. Chem. 291: 27147–27159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Grundmann, M. et al 2018. Lack of beta‐arrestin signaling in the absence of active G proteins. Nat. Commun. 9: 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Steen, A. , Larsen O., Thiele S. & Rosenkilde M.M.. 2014. Biased and g protein‐independent signaling of chemokine receptors. Front. Immunol. 5: 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jørgensen, A.S. , Rosenkilde M.M. & Hjortø G.M.. 2018. Biased signaling of G protein‐coupled receptors – from a chemokine receptor CCR7 perspective. Gen. Comp. Endocrinol. 258: 4–14. [DOI] [PubMed] [Google Scholar]
- 68. Hamoud, N. et al 2018. Spatiotemporal regulation of the GPCR activity of BAI3 by C1qL4 and Stabilin‐2 controls myoblast fusion. Nat. Commun. 9: 4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. β‐Arrestin recruitment by GPR125. (A) Real‐time qPCR analysis of GPR125 in HEK293 and GPR125 knockdown (KD) HEK293 cells relative to the expression of the control gene, GAPDH. (B) β‐Arrestin‐1 recruitment was determined by performing BRET experiments in HEK293 GPR125 KD cells, (C) after cotransfection with GRK2 and GRK6 as in the parental HEK293 cells. GLP‐1 receptor−mediated β‐arrestin‐1 recruitment in a ligand‐dependent manner was included as a positive control in HEK293 GPR125 KD cells. (D) β‐Arrestin‐2 recruitment to GPR125 using the PathHunter DiscoverX system. Error bars indicate SEM for at least three independent biological replicates. * P < 0.05; ** P < 0.01 using Student's t‐test (E).
Table S1. aGPCR genes and corresponding NCBI accession numbers.