

Abstract
Heightened neuronal plasticity expressed during early postnatal life has been thought to permanently decline once critical periods have ended. For example, monocular deprivation is able to shift ocular dominance in the mouse visual cortex during the first months of life, but this effect is lost later in life. However, various treatments, such as the antidepressant fluoxetine, can reactivate a critical period‐like plasticity in the adult brain. When monocular deprivation is supplemented with chronic fluoxetine administration, a major shift in ocular dominance is produced after the critical period has ended. In the current study, we characterized the temporal patterns of fluoxetine‐induced plasticity in the adult mouse visual cortex, using in vivo optical imaging. We found that artificially induced plasticity in ocular dominance extended beyond the duration of the naturally occurring critical period and continued as long as fluoxetine was administered. However, this fluoxetine‐induced plasticity period ended as soon as the drug was not given. These features of antidepressant‐induced plasticity may be useful when designing treatment strategies involving long‐term antidepressant treatment in humans.
Keywords: experience‐dependent plasticity, mouse brain, neuronal plasticity, optical imaging, visual cortex
Various treatments including antidepressant fluoxetine can reinstate developmental‐like plasticity in adult animals. In the current study, we describe patterns of fluoxetine‐induced plasticity in the mouse visual cortex using in vivo optical imaging.

Abbreviations
- ANOVA
analysis of variance
- BDNF
brain‐derived neurotrophic factor
- MD
monocular deprivation
- NDS
normal donkey serum
- NMDA
N‐methyl‐d‐aspartate
- ODI
ocular dominance index
- OD
ocular dominance
- PNN
perineuronal nets
- PV
parvalbumin
- SSRIs
selective serotonin reuptake inhibitors
- TrkB
tropomyosin receptor kinase B
- WFA
Wisteria floribunda agglutinin
1. INTRODUCTION
The visual cortex has been an established model for plasticity studies for decades (Hubel & Wiesel, 1963). The visual cortex matures during a critical period of increased sensitivity to environmental stimuli, during which developing neuronal networks are shaped based on experience. It was long thought that plasticity declines soon after the critical period ends and remains restricted throughout adulthood.
Previous studies have shown that the antidepressant fluoxetine, when combined with monocular deprivation (MD), can restore developmental‐like plasticity in the visual cortex of rats and mice (Maya Vetencourt et al., 2008; Steinzeig, Molotkov, & Castrén, 2017). The plasticity‐promoting effect of antidepressants is not limited to the visual system, but extends to other brain areas as well, including regions responsible for processing fear and Controlling emotions (Karpova et al., 2011; Mikics et al., 2018).
Additional studies have demonstrated that antidepressants promote neural plasticity on different levels: they enhance hippocampal neurogenesis (Malberg, Eisch, Nestler, & Duman, 2000; Santarelli, 2003) and increase signalling of brain‐derived neurotrophic factor (BDNF) through its receptor TrkB (Björkholm & Monteggia, 2016; Duman & Monteggia, 2006; Nibuya, Morinobu, & Duman, 1995; Saarelainen et al., 2003), independently from serotonin transporter (Rantamäki et al., 2011). Hence, these data suggest that complex processes of neuronal plasticity and structural adaptation may underlie clinical effects of antidepressants.
In the current study, we used the visual cortex as a model to examine characteristics of fluoxetine‐induced plasticity in mice. While a naturally occurring critical period of plasticity lasts for roughly one month in the visual cortex of rodents, it has not been established whether fluoxetine‐induced plasticity is confined to the same timeframe (Hensch, 2005). Here, we have studied whether a window of plasticity, induced by fluoxetine, is dependent on treatment (i.e. remains open for the duration of the treatment, irrespective of time) or on time (i.e. intrinsically closes after a certain period, irrespective of treatment). To answer this question, we extended the standard fluoxetine treatment duration to three months and tested for visual plasticity of ocular dominance.
A novel class of rapid‐acting antidepressants has recently received considerable attention. While the conventional antidepressant drugs (such as fluoxetine) require chronic treatment for their action to begin (Wong & Licinio, 2001), a single dose of the N‐methyl‐d‐aspartate (NMDA) receptor antagonist, ketamine, demonstrates fast antidepressant effects that vastly outlast the presence of ketamine in the body (Zarate et al., 2006). It is not clear whether the plasticity‐inducing actions of fluoxetine also outlast the presence of the drug in the body. To assess the potentially prolonged effects of fluoxetine in mice, we separated the drug treatment and monocular deprivation (MD) by testing the effect of MD after fluoxetine and its metabolites had been cleared from the body.
The structural basis for the plasticity induced by antidepressants remains under investigation. Growing evidence suggests an important role of inhibitory neurons (Hensch, 2005). In particular, perineuronal nets (PNNs)—structures of extracellular matrix formed around parvalbumin (PV)‐containing cells at the end of the critical period—could be potential candidates. Previous findings demonstrated that a reduction in PNNs accompanied plasticity in the visual cortex (Lensjø, Lepperød, Dick, Hafting, & Fyhn, 2017; Pizzorusso, 2002), dentate gyrus (Kobayashi et al., 2010) and amygdala (Karpova et al., 2011). Therefore, we assessed whether reduced PNN expression can contribute to the plasticity effects of fluoxetine.
2. MATERIALS AND METHODS
2.1. Animals
All experiments were performed on adult female C57BL/6J Rcc mice delivered from Envigo (Harlan Laboratories). Animals were housed in standard cages and conditions (temperature 22°C, 12‐hr light/dark cycle), and were provided with food and water ad libitum. Since the previous experiments have shown a period of elevated plasticity in the mouse visual cortex, where longer MD can cause an OD shift up to postnatal day 110, all the animals used in the current study were at least 130 days old at the time of the second imaging session (Lehmann & Löwel, 2008).
2.2. Ethical statement
All experiments were performed according to institutional guidelines and were approved by the County Administrative Board of Southern Finland (Licences number: ESAVI/7551/04.10.07/2013; ESAVI/10300/04.10.07/2016). All the efforts were made to minimize animals suffering and number of animals used in experiments.
2.3. Fluoxetine treatment and experimental groups
Our standard protocol to restore developmental‐like plasticity in the adult mice included four weeks of fluoxetine treatment. Specifically, fluoxetine hydrochloride (Bosche Scientific) was dissolved 80 mg/L in the drinking water for the treatment group (Steinzeig et al., 2017). For the plasticity window experiments, we extended our protocol to 12 weeks (“Long treatment” group), rationalizing that this period significantly exceeds the natural critical period. Treatments longer than this could potentially be toxic, as shown in rats (Inkielewicz‐Stępniak, 2011). For the second group, “Postponed MD”, we used the standard protocol (four weeks of fluoxetine treatment) but postponed the start of MD until one week after the fluoxetine treatment ended. Control groups for all the experiments underwent the same procedures but received regular tap water instead of fluoxetine solution. Water consumption was monitored twice a week in both groups; no decline in drinking was observed in the treatment group (Figure S1).
2.4. Monocular deprivation
To test for cortical plasticity, the mice were monocularly deprived for one week in all groups. The eye contralateral to the hemisphere to be imaged was sutured shut with the silk thread (Ethicon) three mattress sutures under anaesthesia (see below). During the deprivation period, animals were checked daily to exclude those with spontaneous eye reopening. In the Long treatment group, MD occurred during the last week of fluoxetine treatment; in the Postponed MD group, it occurred one week after the last fluoxetine treatment.
2.5. Surgical preparation for optical imaging
To prepare for the optical imaging experiment, mice underwent transparent skull surgery as described earlier (for detailed protocol, see Steinzeig et al., 2017). Briefly, animals were anaesthetized with a mixture of fentanyl (Hameln, Germany) 0.05 mg/kg, midazolam (Hameln, Germany) 5 mg/kg and medetomidine (Orion Pharma) 0.5 mg/kg. Additionally, carprofen 5 mg/kg (ScanVet) was administrated subcutaneously for postsurgery analgesia. During the surgery, animals were fixed to a stereotaxic frame and kept on a heating pad at 37°C to prevent hypothermia. The eyes were protected with eye gel (Viscotears, Alcon). The scalp around the visual cortex was removed, and the skull surface was cleaned of periosteum, blood and debris. A layer of cyanoacrylate glue Loctite 401 (Henkel) was applied, followed by two layers of acryl (EUBECOS) mixed with methyl methacrylate liquid (Dentsply). Finally, a metal bar holder was attached to the surface of the skull and then covered with a mixture of cyanoacrylate glue and dental cement (Dentsply) to guarantee a secure positioning during the optical experiments.
2.6. Optical imaging
We evaluated ocular dominance with an intrinsic signal optical imaging—a haemodynamic‐based method for brain imaging based on the blood oxygenation level and optical properties of the brain tissue underlined neuronal activity.
We utilized continuous‐periodic stimulation with continuous synchronized data acquisition for the processing of the intrinsic signals (Cang, Kalatsky, LöWel, & Stryker, 2005; Kalatsky & Stryker, 2003). Intrinsic signals were recorded with a Dalsa 1M30 CCD camera (Teledyne‐Dalsa) and tandem macro objective (50 × 135 mm, Nikon). The visual stimulus was represented by a drifting thin horizontal bar (2° wide) moving upwards on a high refresh rate stimulus monitor at a temporal frequency of 1 cycle/8.3 s (0.125 Hz) and a spatial frequency of 1/80 deg. The visual stimulus was designed to evoke the response solely in the binocular area of the primary visual cortex. Therefore, the bar was restricted only to the central part of the screen (−15° to +5°). For the imaging experiment, the animals were anaesthetized with 1.2% isoflurane in a mixture of oxygen:air and placed on a heating pad facing the stimulus monitor, 25 cm in front of the test subject. A vertical midline was aligned to the animal's nose. The physiological state of the animals was monitored throughout the experiment with the monitor (Physio Suite, Kent Scientific). The head of the animal was firmly fixed during the recordings, and the eyes remained in a stable position throughout the experiment. First, the vascular maps were collected under green light illumination (540 ± 20 nm). Then, the camera was focused at 600 μm below the cortical surface to record the intrinsic signals under red light (625 ± 10 nm). The frames were collected for 5 min for each eye at a rate of 30 fps and stored as a 512 × 512 pixel image after spatial binning of the camera images. To collect the cortical response for left and right eye stimulation independently, we alternately closed one eye with a patch.
Since the method allows repetitive measurements, we imaged the same animals twice: before (IOS I) and after MD (IOSII).
2.7. IOS data analysis
Cortical maps were computed out of the raw video files using an analysis software package for continuous recording of optical intrinsic signals (VK Imaging). Briefly, to generate primary visual cortex activation maps upon ipsilateral and contralateral stimulation, the software analyses fractional changes in reflectance from the cortical surface on the frequency of stimulation and removes slow haemodynamic artefacts using Fourier transform. To calculate ocular dominance indexes (ODIs), the ipsilateral activity map was smoothened with a low‐pass filter, thresholded at 30% of the peak response to improve signal‐to‐noise ratio and used as a mask to select the binocularly responding region. Then, within the binocularly responding region, the calculations of the ODI were made for each responsive pixel based on the formula (C − I)/(C + I), where “C” refers to the magnitude response of the contralateral eye and “I” refers to that of the ipsilateral eye. For each animal, a final result was obtained by averaging several ODIs. Positive values represent contralateral dominance, while negative values represent ipsilateral dominance.
2.8. Immunohistochemistry
Coronal sections of 40 μm were cut with a vibratome. Staining procedures were performed on free‐floating sections under constant agitation. The lectin Wisteria floribunda agglutinin (WFA) was used to visualize PNNs, as it recognizes N‐acetylgalactosamine‐containing epitopes in the CS‐GAG chains. The sections were quickly rinsed in 1× PBS and blocked with 5% normal donkey serum (NDS) and 0.3% Triton X‐100 (Sigma) in 1× PBS for 1 hr at room temperature. The sections were then incubated for 2 days at 4°C with a primary antibody cocktail of biotin‐conjugated WFA 1:200 (Sigma‐Aldrich, L1516) and guinea pig anti‐parvalbumin 1:1,000 (Synaptic System, 195 004), diluted in 1× PBS with 1% NDS and 0.3% Triton X‐100. After four rounds of 10‐min washings in 1× PBS, the slices were incubated for 2 hr at room temperature with a secondary antibody cocktail composed of Streptavidin Alexa fluorophore 555 conjugate 1:1,000 (Life Technologies, 1600216) and donkey anti‐guinea pig Alexa fluorophore 488 1:200 (Jackson ImmunoResearch Europe Ltd., 706‐545‐148), in 1% NDS and 0.3% Triton X‐100 (Sigma) in 1× PBS. Finally, the slices were washed in 1× PBS four times for 10 min each time, mounted in PB 0.1 M on Super frost slides and cover slipped using fluorescence mounting medium (Dako North America).
2.9. Confocal microscopy and PV‐PNN counting
All slides were coded during the quantitative evaluation of immunostainings to perform the analysis blindly. The primary visual cortex was analysed (areas identical to adult bregma from −2.80 to −3.52) and was determined according to the mouse brain atlas (Paxinos & Franklin, 2nd edition). From each brain, two to three sections were selected and Z‐stack images of 2 μm steps were obtained with a confocal microscope LSM 700 (Carl Zeiss) equipped with a 10× objective lens (10× Plan‐Apochromat 10×/0.45, Carl Zeiss) and imaging Software ZEN 2012 lite (Zeiss). The same microscope and camera settings were used for all of the slides. Image processing was done using ImageJ Software (National Institutes of Health). The region of interest (ROI) was selected in the primary visual cortex and then subdivided into six layers, using the ROI manager of ImageJ software. The minimum filter function of ImageJ with a radius of 0.5 pixel was used for all of the pictures from the Control, Long treatment and Postponed MD groups, in order to decrease the potential influence of random and punctate auto‐fluorescence on the counting procedure. This did not result in a loss of information. This filter uses a greyscale erosion; in particular, by setting the radius at 0.5 pixel, the filter replaces the pixel with the minimum pixel value of the four immediate neighbours. The number of parvalbumin‐positive (PV+), PNN‐positive (PNN+) and co‐localized PV+PNN+ cells was manually counted in each image using the Cell Counter tool of ImageJ. Cells were considered positive and counted if they displayed a PV staining throughout the soma for the PV+, and a clear ring around the soma for the PNN+, and all the neurons fulfilling these criteria were counted as positive regardless of their size or intensity of staining. All cells entirely within the boundaries of the area were counted; those in contact with the top border of each layer were considered in that layer, and those that were in contact with the lower border of the layer were excluded from that layer count.
2.10. PV intensity measurements
To assess the intensity of parvalbumin‐positive interneurons in layers 2–3 and 4 of the visual cortex, maximum intensity pictures were obtained from the original z‐stack pictures. The profiles of the PV cells were selected with the selection tool of ImageJ and a circular ROI where there was no signal was taken as background for each layer. Area mean grey value and integrated density were measured for each selected ROI. To calculate the intensity of fluorescence or CTCF (corrected total cell fluorescence), a formula defined in the ImageJ‐win64 manual was used: CTCF = Integrated density − (Area selected cell × Mean fluorescence of background reading). Then, the averaged CTCF for each layer and picture was calculated and normalized for the number of cells in that layer.
2.11. Statistical analysis
In optical imaging experiments, groups were compared using analysis of variance (ANOVA) and Student's t tests. For post hoc analysis, we performed Sidak's test for multiple comparisons. Data from immunohistochemistry analysis were compared with the non‐parametric Mann–Whitney test, followed by the Dunn's multiple comparison test as post hoc in case of significance. For PV intensity, non‐parametric Kruskal–Wallis test was used. Significance levels were set as *p < .05; **p < .01; and ***p < .001. Data are presented as means ± SEM, with n indicating the number of animals. Statistical analysis was conducted with GraphPad Prism software version 6.00 (GraphPad).
3. RESULTS
3.1. The window of induced plasticity stays open as long as the fluoxetine treatment continues
To determine whether the window of fluoxetine‐induced plasticity remains open as long as the treatment continues, we extended peroral fluoxetine treatment from the basic protocol of 4–12 weeks (Figure 1). Specifically, to test the OD plasticity, we used optical imaging of intrinsic signals (IOS) to assess ocular dominance in the visual cortex after 7 days of MD. The IOS data demonstrated that after 12 weeks of fluoxetine (80 mg/L in the drinking water), closing the eye for 7 days resulted in a shift in OD from 0.2 ± 0.03 to −0.01 ± 0.03 (one‐way ANOVA, Sidak‐corrected). This was indicative of an ipsilateral (open) eye dominance. In the Control group, ODIs remained stable (before MD 0.19 ± 0.02; after MD 0.19 ± 0.02; Figure 2). Analysis of the cortical activation measured by average magnitude of the signal showed significant decrease in the closed eye response in fluoxetine group (Control: 5.1 ± 0.43; fluoxetine: 3.8 ± 0.2 n = 8) and non‐significant open eye increase (Control: 4.2 ± 0.30; fluoxetine: 4.6 ± 0.17; n = 8; ANOVA, Sidak‐corrected; Figure 3). Therefore, after 12 weeks of continuous fluoxetine treatment, MD retains robust effects on binocularity, indicating that continuous fluoxetine treatment keeps the window of critical period‐like plasticity open for at least three months—much longer than the naturally occurring critical period.
Figure 1.
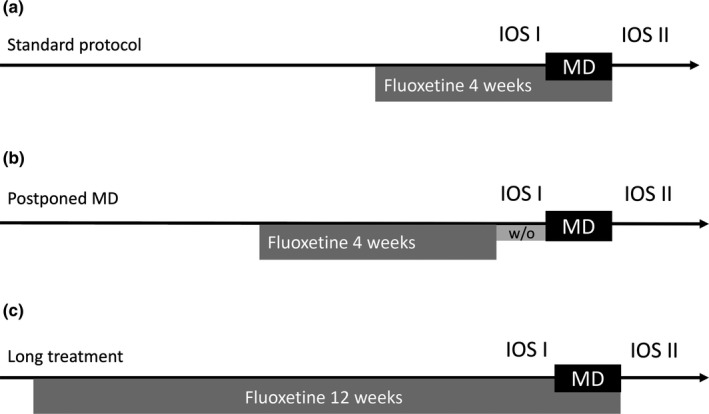
Experimental design. (a) Standard protocol for fluoxetine‐induced plasticity, four‐week treatment with fluoxetine 80 mg/kg dissolved in the drinking water, combined with monocular deprivation during the last week. (b) “Postponed monocular deprivation” protocol, after 4 weeks of fluoxetine treatment, one‐week period of the drug wash‐out was added before the eye closure. (c) “Long treatment” protocol, fluoxetine treatment was expanded to 12 weeks and monocular deprivation applied during the last week. For each group, first imaging session took place before monocular deprivation and the second one just after the eye reopening
Figure 2.

Monocular deprivation leads to a robust shift in ocular dominance after long‐term treatment with fluoxetine (12 weeks). (a) Ocular dominance indexes of fluoxetine‐treated (IOSI 0.2 ± 0.03, IOSII −0.01 ± 0.03; n = 8) and Control groups (IOSI 0.19 ± 0.02, IOSII 0.19 ± 0.02; n = 5; one‐way ANOVA, Sidak‐corrected). (b–d) Representative optical imaging data: optical maps of cortical activity in the binocular part of the primary visual cortex, evoked by a drifting bar stimulus. The Control animals’ ipsilateral patch of activity is smaller, whereas in fluoxetine‐treated animals almost indistinguishable from contralateral patch. That shows the reduction in the response of the closed eye after monocular deprivation and corresponds to lower OD indexes in these animals. b: Control group; c and d: fluoxetine‐treated. Colour maps reflect relative retinotopy; black and white—magnitude of the optical signal. The grey scale below the magnitude maps: fractional change in reflection ×104. The grey circle shows brain axes. Scale bar = 1 mm [Colour figure can be viewed at http://www.wileyonlinelibrary.com/]
Figure 3.

Primary visual cortex activation caused by stimulation of the contralateral and ipsilateral eye after MD. In fluoxetine‐treated group, OD shift was mediated by a significant decrease of deprived contralateral eye response (Control: 5.1 ± 0.43; fluoxetine: 3.8 ± 0.2; n = 8), and only insignificant increase in the open ipsilateral eye (Control: 4.2 ± 0.30; fluoxetine: 4.6 ± 0.17; n = 8; ANOVA, Sidak‐corrected)
3.2. Fluoxetine promotes changes in OD plasticity only during the treatment
To determine whether fluoxetine treatment could evoke long‐lasting changes in brain plasticity after the drug is cleared from the body, we first measured serum concentration of fluoxetine and its active metabolite norfluoxetine after a 4‐week treatment. We found that serum levels of fluoxetine declined quickly within 72 hr and were below detection limit after 7 days following drug withdrawal (Table S1). At 24 hr, the serum concentration of norfluoxetine was 10‐fold higher than that of fluoxetine, but at 7 days, only trace levels were detectable. Thus, for this set of experiments, we Postponed the MD to begin 7 days after the last fluoxetine treatment (Figure 1). OD plasticity in the primary visual cortex was tested after 7 days of MD (Postponed MD, Figure 1). The average ODIs for the Postponed MD group were not statistically different from that of the Control group: ODIs before MD 0.19 ± 0.02; after MD 0.21 ± 0.03 (paired t test; Figure 4). Thus, the OD plasticity window in response to MD is only open when fluoxetine is in the body; after fluoxetine and norfluoxetine were eliminated from the serum, the plasticity window is closed.
Figure 4.
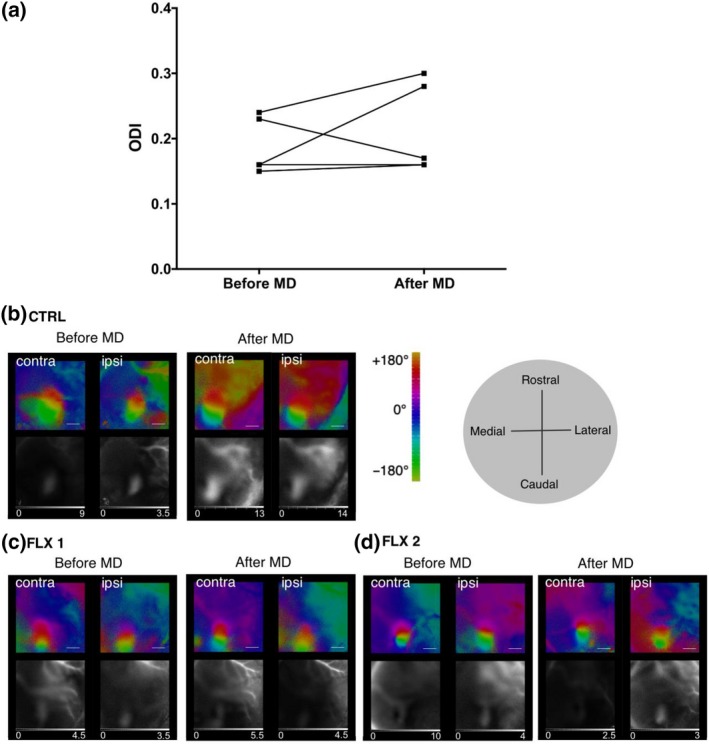
Effects of fluoxetine on plasticity are absent when the monocular deprivation was postponed for one week after the end of the treatment. (a) Ocular dominance indexes before (0.19 ± 0.02) and after (0.21 ± 0.03) monocular deprivation (paired t test, n = 5); (b–d) representative optical imaging data: optical maps of cortical activity in the binocular part of the primary visual cortex, evoked by a drifting bar stimulus. b: Control group; c and d: fluoxetine‐treated. Colour maps reflect relative retinotopy; black and white—magnitude of the optical signal. The grey scale below the magnitude maps: fractional change in reflection ×104. The grey circle shows brain axes. Scale bar = 1 mm [Colour figure can be viewed at http://www.wileyonlinelibrary.com/]
3.3. Fluoxetine‐induced OD shift was not accompanied by a reduction in parvalbumin or perineuronal nets
To examine whether a change in PNNs around PV cells might serve as a source for enhanced plasticity, we investigated PNNs with confocal microscopy. We counted the PNN+ (WFA positive), parvalbumin‐positive (PV+) cells and co‐localization of these two signals (PV+PNN+) in brain slices (Figure 5a). As an additional parameter, we also calculated the ratio of PV+PNN+ neurons to PV+ cells (i.e. the percentage of PV+ cells surrounded with PNN out of all PV+ cells; Balmer, Carels, Frisch, & Nick, 2009). The immunostaining demonstrated no significant difference either in the total amount of PNN+s, PV+s, nor in PV+PNN+s: (PNN+: Control = 43 ± 3, Long treatment = 44 ± 2, Postponed MD = 36 ± 4; p > .05; PV+: Control = 98 ± 6, Long treatment = 91 ± 4, Postponed MD = 73 ± 8; p > .05; PV+PNN+: Control = 36 ± 2, Long treatment = 37 ± 2, Postposed MD = 30 ± 5; p > .05, n = 4 animals per group, Kruskal–Wallis test; Figure 5b). Furthermore, no changes in PV+PNN+/PV+ ratio were observed (Control = 41 ± 3%; Long treatment = 40 ± 2%; Postponed MD = 40 ± 2%; n = 4 animals per group, Kruskal–Wallis test; Figure 5c). In addition, when these parameters were analysed separately for each layer, we only observed a significant reduction in PV+ cell number in the fourth cortical layer in the Postponed MD group; the PV+PNN+/PV ratio remained stable (Figure 5d–g). Consequently, there was no correlation between fluoxetine‐induced plasticity and number of PV+, PNN+, or PV+PNN+ double positive cells, or in their ratio. Additionally, we have analysed intensity of the PV cells, since different PV content was shown to indicate the plasticity level in the hippocampus (Donato, Rompani, & Caroni, 2013). However, our brain samples demonstrated no significant changes in PV intensity (Figure S2).
Figure 5.
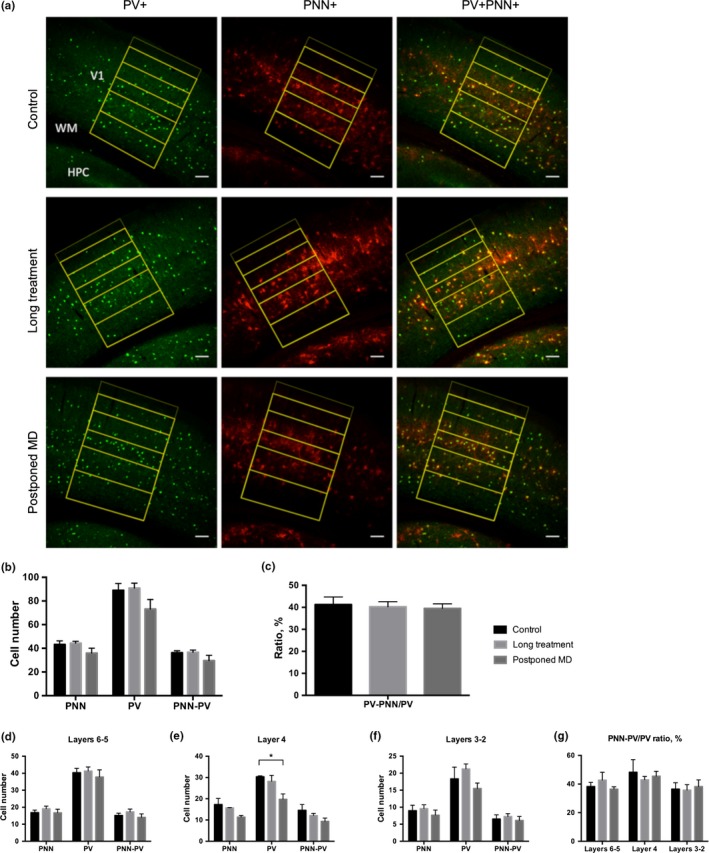
Number of parvalbumin‐positive cells (PV+) or surrounding them perineuronal nets (PNN+) in the primary visual cortex was unaffected after 12‐week chronic fluoxetine treatment. (a) Representative confocal images. Parvalbumin (green) and WFA (red) immunoreactivity. The layers borders are drawn in yellow. Scale bar = 100 μm. (b) Counting results in the primary visual cortex. (PNN+: Control = 43 ± 3, Long treatment = 44 ± 2, Postponed MD = 36 ± 4—p > .05; PV+: Control = 98 ± 6, Long treatment = 91 ± 4, Postponed MD = 73 ± 8—p > .05; PV+PNN+: Control = 36 ± 2, Long treatment = 37 ± 2, Postposed MD = 30 ± 5—p > .05, n = 4 animals per group, Kruskal–Wallis test) (c). Percentage of PV cells surrounded with PNN in the primary visual cortex (Control = 41 ± 3%; Long treatment = 40 ± 2%; Postponed MD = 40 ± 2%—p > .05, n = 4 animals per group, Kruskal–Wallis test). (d) In the layers 6–5 of the primary visual cortex, no change was detected (PNN: Control = 17 ± 1; Long treatment = 19 ± 1; Postponed MD = 17 ± 2; p > .05; PV: Control = 40 ± 2; Long treatment = 41 ± 2, Postponed MD = 38 ± 4; p > .05; PNN‐PV: Control = 15 ± 1; Long treatment = 17 ± 2; Postponed MD = 14 ± 2; p > .05; n = 4 animals per group, Kruskal–Wallis test). (e) In the fourth layer of the primary visual cortex, no change in the amount of perineuronal nets surrounding parvalbumin‐positive interneurons was detected; however, a significant reduction in PV+ cell number was observed in the Postponed MD group (PNN: Control = 17 ± 3; Long treatment = 16; Postponed MD = 11 ± 1; p > .05; PV: Control = 30; Long treatment = 28 ± 3; Postponed MD = 20 ± 2; Control versus Long treatment p > .05; Control versus Postponed MD *p = .028; PNN‐PV: Control = 15 ± 3; Long treatment = 12 ± 1; Postponed MD = 9 ± 1; p > .05; Kruskal–Wallis test and Dunn's test for multiple comparisons). (f) In the layers 3–2 of the primary visual cortex, no change was observed (PNN: Control = 9 ± 2; Long treatment = 10 ± 1; Postponed MD = 8 ± 1; p > .05; PV: Control = 18 ± 3; Long treatment = 1 ± 1; Postponed MD = 15 ± 2; p > .05; PNN‐PV: Control = 6 ± 1; Long treatment = 7 ± 1; Postponed MD = 6 ± 1; p > .05; n = 4 animals per group, Kruskal–Wallis test). (g) Percentage of PV cells surrounded with PNN in the different layers of the primary visual cortex (Layers 6–5: Control = 38 ± 3; Long treatment 43 ± 5; Postponed MD = 36 ± 1; p > .05. Layer 4: Control = 48 ± 1; Long treatment = 43 ± 2; Postponed MD 45 ± 3; p > .05. Layers 3/2: Control = 36 ± 4; Long treatment = 36 ± 4; Postponed MD = 38 ± 5; p > .05. Kruskal–Wallis test) [Colour figure can be viewed at http://www.wileyonlinelibrary.com/]
4. DISCUSSION
The present study shows that the antidepressant fluoxetine restores plasticity in the adult mouse brain, and the window of this plasticity remains open as long as the treatment continues. Interestingly, the window of induced plasticity exceeded the natural critical period of plasticity in the rodent visual cortex. These data are consistent with previous findings on the plasticity‐enhancing effects of enriched environments on ocular dominance (Greifzu, Kalogeraki, & Löwel, 2016; Greifzu et al., 2014). Thus, these findings confirm the view that cortical plasticity is not restricted to early development but can be reactivated and maintained in adulthood.
Previous studies using visually evoked potentials have shown that MD during fluoxetine treatment in adult rodents leads to a reduction in the responses to the closed eye without any significant strengthening of open eye responses, which is typically observed with MD responses during the critical period (Maya Vetencourt, Tiraboschi, Spolidoro, Castrén, & Maffei, 2011; Maya Vetencourt et al., 2008). Furthermore, fluoxetine has no effects on vision in the absence of MD, as assessed with either evoked potentials or imaging of intrinsic signals (Maya Vetencourt et al., 2008; Steinzeig et al., 2017), and actually improves vision in an amblyopic eye in adulthood, when the better eye is patched during fluoxetine treatment (Maya Vetencourt et al., 2008). In agreement with the previous data on the character of the fluoxetine‐induced plasticity, analysis of the visual cortical activity revealed decrease in the deprived eye response and a slight increase in the non‐deprived eye—a pattern typical for juvenile‐like OD plasticity.
We also tested whether fluoxetine affects visual cortex plasticity only while present in the body. We found that once the drug and its active metabolite have been cleared, monocular deprivation did not produce any shift in ocular dominance. While this finding is expected from a pharmacological perspective, the antidepressant effects of ketamine have been found to vastly outlast its presence in the body; therefore, we tested whether fluoxetine induces similar lasting effects on OD plasticity. It is, however, possible that even longer duration of the fluoxetine treatment before the drug withdrawal might produce longer‐lasting effects on plasticity.
Although it has been considered that once reduced at the end of critical periods, plasticity remains restricted the rest of the lifetime, and it has recently been shown that a number of different treatments can reactivate critical period‐like plasticity in the adult brain, including environmental enrichment, disruption of PNNs, genetic manipulations as well as several drug treatments, such as fluoxetine, valproate and inhibitors of acetylcholinesterase (Bavelier, Levi, Li, Dan, & Hensch, 2010; Morishita & Hensch, 2008; Sale, Berardi, & Maffei, 2014; Umemori, Winkel, Didio, Llach Pou, & Castrén, 2018). However, it is not clear whether the different treatments that induce adult plasticity are sharing a common mechanistic pathway or whether there are several parallel mechanisms that independently can lead to a similar induction of critical period‐like plasticity in the adult brain.
The exact mechanisms through which fluoxetine acts to reactivate plasticity remain also unclear. It has been shown that the activation of serotonin 1A receptors and BDNF signalling through TrkB receptor is required (Maya Vetencourt et al., 2008, 2011), but how these signalling pathways are related to each other and through what cell types these effects are mediated are not known. Ongoing studies in our laboratory are addressing these questions through transcriptomic and proteomic analyses as well as electrophysiological recordings, and we expect to be able to shine light on these mechanisms in the near future.
Maturation of PV+‐ interneurons and PNN formation takes place towards the end of the naturally occurring critical period in the visual cortex (Huang et al., 1999), and PV and PNN densities are well‐established markers of the critical period closure (Pizzorusso, 2002). Enzymatic removal of PNN by chondroitinase ABC injection is reopens the critical period in adult rodents in the same way as fluoxetine does (Lensjø et al., 2017; Pizzorusso, 2002). Several studies have demonstrated that chronic fluoxetine treatment in adult mice leads to a reduction in PNN+, PV+ or PV+ cells surrounded with PNN in different brain areas, such as the basolateral amygdala, medial frontal cortex and hippocampus (Karpova et al., 2011; Ohira, Takeuchi, Iwanaga, & Miyakawa, 2013). It has therefore been suggested that fluoxetine‐induced OD plasticity could, at least in part, involve the reduction in PNNs surrounding PV+ interneuron cells. However, the effect of fluoxetine on PNNs appears to be dependent on the treatment protocol and is moderate in size (Ohira et al., 2013). Despite these previous indications that PV and PNN density might be related to visual plasticity during the critical period as well as to plasticity in other brain regions during adulthood, we did not observe such changes in any of these parameters tested after 12 weeks of fluoxetine treatment. As our treatment lasted longer than that in the previous studies, it is possible that compensatory mechanisms take place during the long treatment, thereby restoring PNNs back to normal levels. Of note, PNN changes can sometimes only be appreciated in terms of their intensity; however, the intensity measurements were unfortunately not unavailable for this study. Nevertheless, our findings suggest that changes in PV or PNN density are not necessary for fluoxetine‐induced plasticity in the adult visual cortex.
In the Postponed MD group, a small decline of PV+ cells was observed in the layer four of the primary visual cortex. A reduced number of PV+ neurons might reflect either a reduction in PV‐containing cells or a decrease in parvalbumin expression below detectable levels. The latter could be a sign of reduced inhibition by interneurons, although we did not see any accompanying changes in OD plasticity. Based on a concept proposed by Donato and colleagues, PV‐cell network in the hippocampus can be presented by two states: low or high PV. When low PV state possibly reflects elevated plasticity, high PV is associated with memory consolidation. We thus tested PV cells intensity in the current study, however, found no detectable difference between the groups (Donato et al., 2013).
Our results emphasize the importance of combining fluoxetine with MD for promoting plasticity, since neither treatment alone brings about an OD shift. Clinically, it is known that a combination of antidepressant treatment together with psychotherapy works better than pharmacotherapy alone (Pampallona, Bollini, Tibaldi, Kupelnick, & Munizza, 2004; Vittengl, Clark, Dunn, & Jarrett, 2007), which might reflect psychoterapy‐induced alterations in mood‐relevant circutis rendered plastic by fluoxetine treatment (Castrén, 2005, 2013; Castrén & Antila, 2017). However, treatment of depression is typically initiated by antidepressant treatment alone and psychotherapy is added later. It was therefore important to investigate whether enhanced plasticity induced by fluoxetine spontaneously closes or whether it stays open as long as the treatment lasts. Our data now suggest that a spontaneous closure of plasticity period does not take place, which is consistent with finding using enriched environment (Greifzu et al., 2014, 2016). Therefore, if enhanced plasticity within mood‐related circuits took place during fluoxetine treatment in humans, environmental guidance such as psychotherapy should be beneficial also after an extended treatment with antidepressants alone.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
E.C and A.S. formulated and planned the experiments. A.S. and C.C carried out the experiments, samples collection and data analysis; A.S. prepared the manuscript with help from E.C. and C.C. E.C supervised the project.
Supporting information
ACKNOWLEDGEMENTS
The authors would like to thank Frederike Winkel, Outi Nikkilä and Sulo Kolehmainen for technical help. Funding for this study was provided by ERC Grant No 322742—iPLASTICITY, EU Joint Programme—Neurodegenerative Disease Research (JPND) CircProt project #301225, Sigrid Juselius foundation and Academy of Finland grants #294710 and #307416.
Steinzeig A, Cannarozzo C, Castrén E. Fluoxetine‐induced plasticity in the visual cortex outlasts the duration of the naturally occurring critical period. Eur J Neurosci. 2019;50:3663–3673. 10.1111/ejn.14512
Edited by Patricia Gaspar.
DATA AVAILABILITY STATEMENT
The data are available without restriction. https://doi.org/10.6084/m9.figshare.8058275
REFERENCES
- Balmer, T. S. , Carels, V. M. , Frisch, J. L. , & Nick, T. A. (2009). Modulation of perineuronal nets and parvalbumin with developmental song learning. Journal of Neuroscience, 29, 12878–12885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavelier, D. , Levi, D. M. , Li, R. W. , Dan, Y. , & Hensch, T. K. (2010). Removing brakes on adult brain plasticity: From molecular to behavioral interventions. Journal of Neuroscience, 30, 14964–14971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björkholm, C. , & Monteggia, L. M. (2016). BDNF – A key transducer of antidepressant effects. Neuropharmacology, 102, 72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cang, J. , Kalatsky, V. A. , LöWel, S. , & Stryker, M. P. (2005). Optical imaging of the intrinsic signal as a measure of cortical plasticity in the mouse. Visual Neuroscience, 22, 685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castrén, E. (2005). Is mood chemistry? Nature Reviews Neuroscience, 6, 241–246. [DOI] [PubMed] [Google Scholar]
- Castrén, E. (2013). Neuronal network plasticity and recovery from depression. JAMA Psychiatry, 70, 983. [DOI] [PubMed] [Google Scholar]
- Castrén, E. , & Antila, H. (2017). Neuronal plasticity and neurotrophic factors in drug responses. Molecular Psychiatry, 22, 1085–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato, F. , Rompani, S. B. , & Caroni, P. (2013). Parvalbumin‐expressing basket‐cell network plasticity induced by experience regulates adult learning. Nature, 504, 272–276. [DOI] [PubMed] [Google Scholar]
- Duman, R. S. , & Monteggia, L. M. (2006). A neurotrophic model for stress‐related mood disorders. Biological Psychiatry, 59, 1116–1127. [DOI] [PubMed] [Google Scholar]
- Greifzu, F. , Kalogeraki, E. , & Löwel, S. (2016). Environmental enrichment preserved lifelong ocular dominance plasticity, but did not improve visual abilities. Neurobiology of Aging, 41, 130–137. [DOI] [PubMed] [Google Scholar]
- Greifzu, F. , Pielecka‐Fortuna, J. , Kalogeraki, E. , Krempler, K. , Favaro, P. D. , Schlüter, O. M. , & Löwel, S. (2014). Environmental enrichment extends ocular dominance plasticity into adulthood and protects from stroke‐induced impairments of plasticity. Proceedings of the National Academy of Sciences, 111, 1150–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensch, T. K. (2005). Critical period plasticity in local cortical circuits. Nature Reviews Neuroscience, 6, 877–888. [DOI] [PubMed] [Google Scholar]
- Huang, Z. J. , Kirkwood, A. , Pizzorusso, T. , Porciatti, V. , Morales, B. , Bear, M. F. , … Tonegawa, S. (1999). BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell, 98, 739–755. [DOI] [PubMed] [Google Scholar]
- Hubel, D. H. , & Wiesel, T. N. (1963). Shape and arrangement of columns in cat's striate cortex. Journal of Physiology, 165, 559–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inkielewicz‐Stępniak, I. (2011). Impact of fluoxetine on liver damage in rats. Pharmacological Reports, 63, 441–447. [DOI] [PubMed] [Google Scholar]
- Kalatsky, V. A. , & Stryker, M. P. (2003). New paradigm for optical imaging: Temporally encoded maps of intrinsic signal. Neuron, 38, 529–545. [DOI] [PubMed] [Google Scholar]
- Karpova, N. N. , Pickenhagen, A. , Lindholm, J. , Tiraboschi, E. , Kulesskaya, N. , Agustsdottir, A. , … Castren, E. (2011). Fear erasure in mice requires synergy between antidepressant drugs and extinction training. Science, 334, 1731–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi, K. , Ikeda, Y. , Sakai, A. , Yamasaki, N. , Haneda, E. , Miyakawa, T. , & Suzuki, H. (2010). Reversal of hippocampal neuronal maturation by serotonergic antidepressants. Proceedings of the National Academy of Sciences, 107, 8434–8439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann, K. , & Löwel, S. (2008). Age‐dependent ocular dominance plasticity in adult mice. PLoS ONE, 3, e3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lensjø, K. K. , Lepperød, M. E. , Dick, G. , Hafting, T. , & Fyhn, M. (2017). Removal of perineuronal nets unlocks juvenile plasticity through network mechanisms of decreased inhibition and increased gamma activity. Journal of Neuroscience, 37, 1269–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malberg, J. E. , Eisch, A. J. , Nestler, E. J. , & Duman, R. S. (2000). Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. Journal of Neuroscience, 20, 9104–9110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maya Vetencourt, J. F. , Sale, A. , Viegi, A. , Baroncelli, L. , De Pasquale, R. , F. O'Leary, O. , … Maffei, L. (2008). The antidepressant fluoxetine restores plasticity in the adult visual cortex. Science, 320, 385–388. [DOI] [PubMed] [Google Scholar]
- Maya Vetencourt, J. F. , Tiraboschi, E. , Spolidoro, M. , Castrén, E. , & Maffei, L. (2011). Serotonin triggers a transient epigenetic mechanism that reinstates adult visual cortex plasticity in rats: Epigenetics of serotonin‐induced adult cortical plasticity. European Journal of Neuroscience, 33, 49–57. [DOI] [PubMed] [Google Scholar]
- Mikics, É. , Guirado, R. , Umemori, J. , Tóth, M. , Biró, L. , Miskolczi, C. , … Karpova, N. N. (2018). Social learning requires plasticity enhanced by fluoxetine through prefrontal Bdnf‐TrkB signaling to limit aggression induced by post‐weaning social isolation. Neuropsychopharmacology, 43, 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita, H. , & Hensch, T. K. (2008). Critical period revisited: Impact on vision. Current Opinion in Neurobiology, 18, 101–107. [DOI] [PubMed] [Google Scholar]
- Nibuya, M. , Morinobu, S. , & Duman, R. (1995). Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. Journal of Neuroscience, 15, 7539–7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohira, K. , Takeuchi, R. , Iwanaga, T. , & Miyakawa, T. (2013). Chronic fluoxetine treatment reduces parvalbumin expression and perineuronal nets in gamma‐aminobutyric acidergic interneurons of the frontal cortex in adult mice. Molecular Brain, 6, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pampallona, S. , Bollini, P. , Tibaldi, G. , Kupelnick, B. , & Munizza, C. (2004). Combined Pharmacotherapy and Psychological Treatment for Depression. Arch Gen Psychiatry, 61. [DOI] [PubMed] [Google Scholar]
- Pizzorusso, T. (2002). Reactivation of ocular dominance plasticity in the adult visual cortex. Science, 298, 1248–1251. [DOI] [PubMed] [Google Scholar]
- Rantamäki, T. , Vesa, L. , Antila, H. , Di Lieto, A. , Tammela, P. , Schmitt, A. , … Castrén, E. (2011). Antidepressant drugs transactivate TrkB neurotrophin receptors in the adult rodent brain independently of BDNF and monoamine transporter blockade. PLoS ONE, 6, e20567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saarelainen, T. , Hendolin, P. , Lucas, G. , Koponen, E. , Sairanen, M. , MacDonald, E. , … Castrén, E. (2003). Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant‐induced behavioral effects. Journal of Neuroscience, 23, 349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sale, A. , Berardi, N. , & Maffei, L. (2014). Environment and brain plasticity: Towards an endogenous pharmacotherapy. Physiological Reviews, 94, 189–234. [DOI] [PubMed] [Google Scholar]
- Santarelli, L. (2003). Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science, 301, 805–809. [DOI] [PubMed] [Google Scholar]
- Steinzeig, A. , Molotkov, D. , & Castrén, E. (2017). Chronic imaging through “transparent skull” in mice. PLoS ONE, 12, e0181788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umemori, J. , Winkel, F. , Didio, G. , Llach Pou, M. , & Castrén, E. (2018). iPlasticity: Induced juvenile‐like plasticity in the adult brain as a mechanism of antidepressants: Antidepressant‐induced plasticity. Psychiatry and Clinical Neurosciences, 72, 633–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vittengl, J. R. , Clark, L. A. , Dunn, T. W. , & Jarrett, R. B. (2007). Reducing relapse and recurrence in unipolar depression: A comparative meta‐analysis of cognitive‐behavioral therapy's effects. Journal of Consulting and Clinical Psychology, 75, 475–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, M.‐L. , & Licinio, J. (2001). Research and treatment approaches to depression. Nature Reviews Neuroscience, 2, 343–351. [DOI] [PubMed] [Google Scholar]
- Zarate, C. A. , Singh, J. B. , Carlson, P. J. , Brutsche, N. E. , Ameli, R. , Luckenbaugh, D. A. , … Manji, H. K. (2006). A randomized trial of an N‐methyl‐D‐aspartate antagonist in treatment‐resistant major depression. Archives of General Psychiatry, 63, 856–864. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data are available without restriction. https://doi.org/10.6084/m9.figshare.8058275