

Abstract
Soybean cyst nematode (SCN, Heterodera glycines) is a major pest of soybean that is spreading across major soybean production regions worldwide. Increased SCN virulence has recently been observed in both the United States and China. However, no study has reported a genome assembly for H. glycines at the chromosome scale. Herein, the first chromosome‐level reference genome of X12, an unusual SCN race with high infection ability, is presented. Using whole‐genome shotgun (WGS) sequencing, Pacific Biosciences (PacBio) sequencing, Illumina paired‐end sequencing, 10X Genomics linked reads and high‐throughput chromatin conformation capture (Hi‐C) genome scaffolding techniques, a 141.01‐megabase (Mb) assembled genome was obtained with scaffold and contig N50 sizes of 16.27 Mb and 330.54 kilobases (kb), respectively. The assembly showed high integrity and quality, with over 90% of Illumina reads mapped to the genome. The assembly quality was evaluated using Core Eukaryotic Genes Mapping Approach and Benchmarking Universal Single‐Copy Orthologs. A total of 11,882 genes were predicted using de novo, homolog and RNAseq data generated from eggs, second‐stage juveniles (J2), third‐stage juveniles (J3) and fourth‐stage juveniles (J4) of X12, and 79.0% of homologous sequences were annotated in the genome. These high‐quality X12 genome data will provide valuable resources for research in a broad range of areas, including fundamental nematode biology, SCN–plant interactions and co‐evolution, and also contribute to the development of technology for overall SCN management.
Keywords: chromosome scale, evolution, genome assembly, Heterodera glycines, Soybean cyst nematode, X12
1. INTRODUCTION
Soybean cyst nematode (SCN) has become a major pest in soybean (Glycine max Merr.) worldwide, posing a serious threat to the sustainability of the soybean industry (Kim et al., 2016; Koenning & Wrather, 2010; Woo et al., 2014). SCN is estimated to cause annual yield losses of more than $1.2 billion in the United States (Koenning & Wrather, 2010) and more than $120 million in China (Wang, Zhao, & Chu, 2015). Moreover, increased virulence of SCN has been observed, and the dominant races appear to be shifting (Howland, Monnig, Mathesius, Nathan, & Mitchum, 2018; Hua et al., 2018; Lian et al., 2017). To some extent, this indicates that the ecological environment has caused evolution of nematode virulence, which is a serious threat to agro‐ecology and agricultural production. Classifying SCN populations by the published race scheme (Riggs & Schmitt, 1988) or the HG type test (Niblack et al., 2002) based on their virulence phenotype involves assessing the reproductive potential of a given population on a set of soybean indicator lines. Currently, planting SCN‐resistant cultivars is the primary method of controlling the nematode (Mitchum, 2016; Mitchum, Wrather, Heinz, Shannon, & Danekas, 2007; Niblack, Colgrove, Colgrove, & Bond, 2008). Because SCN‐resistant cultivars can invoke a defence against SCN population, a solid understanding of SCN genome information is the basis for analysing the mechanisms underlying pathogenicity and breeding for new SCN‐resistant cultivars (Gardner, Heinz, Wang, & Mitchum, 2017; Kadam et al., 2016; Patil et al., 2019).
Race X12 was isolated from a soybean field heavily infected by SCN in Shanxi Province, China. To date, this race is able to successfully parasitize all resistant soybean germplasm tested, including the four indicator lines of the race scheme (Peking, Pickett, PI88788 and PI90763) (Riggs & Schmitt, 1988), the seven indicator lines of the HG type test (Peking, PI88788, PI90763, PI437654, PI 209332, PI 89772 and PI548316) (Niblack et al., 2002) and ZDD2315, the most promising elite resistant germplasm from China. Indeed, ZDD2315 is resistant to all SCN populations identified thus far, except for the newly identified race X12 (Lian et al., 2017). PI437654 is another elite resistant germplasm from the United States that is vulnerable to few natural SCN populations (Donald & Young, 2004; Jiao et al., 2015). Accordingly, X12 is thought to express additional or new virulence factors compared with other races, and it constitutes a potentially serious threat to soybean production, especially in China. Overall, genetic and genomic information for X12 is crucial for understanding the evolution of SCN parasitism genes and breeding additional resistant cultivars. Genome sequencing of the free‐living nematode Caenorhabditis elegans (The C. elegans sequencing consortium, 1998) and the parasitic nematodes Meloidogyne hapla (Opperman et al., 2008) and Globodera rostochiensis (Akker et al., 2016) has provided reference genomes that can be utilized for comparison with parasitic nematodes. The genetic map of SCN was reported in 2005 with 10 linkage groups (Atibalentja et al., 2005). Although a draft genome sequence for SCN was recently published with 738 contigs in the genome, these contigs were not successfully assembled into chromosomes (Masonbrink et al., 2019). Genome sequencing of the X12 race is extremely important for our understanding of SCN virulence genes.
In this study, PacBio sequencing, 10X Genomics sequencing and Hi‐C were applied to assemble the genome of the newly reported SCN race X12. The sequence information provided in this study combined with other published nematode genomes will allow for comparative genomic approaches to study fundamental nematode biology, gene function, nematode parasitism and evolution. As plant–parasitic nematodes are among the most damaging and difficult‐to‐control agricultural pests, available genome sequences will help scientists meet the current and future worldwide demands for food and bioenergy by providing powerful information for the development of new control paradigms and by minimizing crop losses.
2. MATERIALS AND METHODS
2.1. Selection of individuals for sequencing
The genomes of Heterodera glycines are challenging to sequence and assemble because these animals are dioecious with exceptionally high levels of population heterozygosity (Masonbrink et al., 2019; reviewed by Jones et al., 2013). To assemble this highly heterozygous population, H. glycines race X12 was first purified using ZDD2315, an elite resistant soybean germplasm in China (Lu, Gai, Zheng, & Li, 2006) with high resistance to all races detected in the SCN survey in Huang‐Huai Valleys (Lian et al., 2016), except for race X12, to which it is highly susceptible. X12 (Hg type 1.2.3.4.5.6.7) was grown on ZDD2315 in a greenhouse at HeNan Academy of Agricultural Science. The starting culture was a single cyst selected from the X12 population, which was bulked for eight generations on ZDD2315 planted in steam‐pasteurized soil and grown with approximately 16 daylight hours at 28°C. The X12 cysts used for genome sequencing were cultivated from soil infected with the 8th‐generation purified X12 population.
2.2. DNA/RNA isolation
The forms of H. glycines include cysts (Figure 1a) and early (Figure 1b) (second‐stage juveniles, J2), middle (third‐stage juveniles, J3) and late (fourth‐stage juveniles, J4) life stages (as reviewed by Jones et al., 2013). Cyst nematodes moult to J2 in the egg; J2 is the dormant stage of the life cycle. J2, J3 and J4 are similar in morphology. Specimens for the four life stages of SCN were isolated from the X12 population. J2 were collected, followed by collection of J3 and J4 at 3, 8 and 15 days postinfection according to standard nematological methods (De Boer, Yan, Smant, Davis, & Baum, 1998). The samples were purified by sucrose gradient centrifugation (De Boer et al., 1996). Genomic DNA was extracted from approximately 20,000 eggs using MasterPure Complete DNA Purification Kit, and total RNA was extracted from 10,000 eggs or 300 J2/ J3/ J4 using Exiqon miRCURY RNA Isolation Kit.
Figure 1.

The combination of cysts on soybean roots (a) and micrograph of Heterodera glycines (soybean cyst nematode) second‐stage juvenile (J2) (b) [Colour figure can be viewed at http://wileyonlinelibrary.com]
2.3. Genome sequencing
The DNA extracted from H. glycines was used for genome sequencing and sheared with a sonication device (Bioruptor Pico) for paired‐end library construction. Libraries with 350 base pair (bp) insert sizes were produced according to the instructions provided in the ×TEN Illumina Library Preparation Kit. The Illumina HiSeq ×TEN platform was used to generate 13.65‐gigabyte (Gb) whole‐genome sequencing data, and the clean reads obtained from this process were employed for subsequent analyses. Construction of a 10X Genomics library produced 31.32‐Gb sequencing data. For PacBio library construction, H. glycines genomic DNA was sheared to ~20 kb, and filtered fragments were converted into the proprietary 9 SMRTbell library using PacBio DNA Template Preparation Kit. In total, 28.48 Gb of quality‐filtered data was obtained from PacBio sequencing.
2.4. Genome assembly and quality control
The genome size was estimated based on the k‐mer spectrum of de novo data. All raw reads from the PacBio platform were aligned to each other using ‘daligner’ executed using the mail script of the falcon (v0.7) assembler. Overlapping reads and raw subreads were processed to generate consensus sequences, and error correction of the assembly was performed using the consensus‐calling algorithm Quiver (smrtlink_5.0.7). The paired‐end clean reads from the Illumina platform were further corrected using Pilon (v1.22), and the reads obtained after strict error correction were further used for the subsequent scaffolding.
The 10X Genomics scaffold extension was performed using fragScaff (v140324.1) software, in which the linked reads generated using the 10X genomic library were aligned to the consensus sequence of the PacBio assembly. To obtain the superscaffold, only the consensus sequence with linked‐read support was used for assembly.
To assess the accuracy of the assembled X12 genome, a small fragment library was selected for comparison of the assembled genome using bwa software (v0.7.8) (Burton et al., 2013; Li & Durbin, 2009; Rao et al., 2014; Yaffe & Tanay, 2011). Core Eukaryotic Genes Mapping Approach (CEGMA: http://korflab.ucdavis.edu/dataseda/cegma/; Parra, Bradnam, & Korf, 2007) analysis was performed to assess the completeness and continuity of the SCN genome (X12 race) assembly, along with six additional published genomes, based on a core eukaryotic gene (CEG) library with 248 conserved genes. In addition, the assembly was evaluated with Benchmarking Universal Single‐Copy Orthologs (BUSCO: http://busco.ezlab.org/; Simao, Waterhouse, Ioannidis, Kriventseva, & Zdobnov, 2015).
2.5. Repeat prediction
LTR_FINDER (v1.0.7), RepeatScout (v1.0.5) and RepeatModeler (v1.0.3) were used for de novo identification of repeat elements and for generating a repeat element database. This database was used in RepeatMasker (v4.07) to predict repeat elements. Putative repeats were further filtered on the basis of copy number.
2.6. Gene structure prediction
For gene structure prediction, both de novo and homology‐based approaches were combined to predict protein‐coding genes in the SCN genome. For the former, gene sets from Ascaris_suum (wormbase. WBPS6), Brugia malayi (ensembl. metazoa.v32), Caenorhabditis_briggsae (ensembl.metazoa.v32), C. elegans (ensembl.metazoa.v32), Drosophila melanogaster (ensembl.metazoa.v32) and Onchocerca volvulus (ensembl.metazoa.v34) were used as queries to search against the SCN genome. For the de novo‐based method, Augustus (v3.2.3), GlimmerHMM (v3.0.4), snap (v2013.11.29) and Genscan (v1.0) were employed as engines to predict gene models. The gene prediction results derived from both methods were merged using GLEAN to generate a consensus gene set.
2.7. Functional annotation of protein‐coding genes
Translated coding sequences were aligned to known databases such as Swiss‐Prot (v20180824), Nr (v20180716), Pfam (v31.0), kegg (v20160503) and InterPro (v5.31‐70.0). We annotated all protein‐coding genes identified in this study by retrieving functional terms according to Swiss‐Prot, Nr, kegg, InterPro, GO and Pfam.
2.8. Chromosome preparation
Chromosome preparation was performed according to the method of Du et al. (2016). Briefly, eggs or J2 were collected and treated with colchicine (0.005 g/L) in 2‐ml tubes for 3–5 hr before fixation in a solution of 3:1 ethanol:acetic acid (v/v) for 2–3 days. The samples were suspended in 45% acetic acid and squashed on a slide. After freezing at −70°C overnight, the slides that contained mitotic chromosomes were dehydrated in 100% ethanol followed by DAPI (4′‐,6‐diamidino‐2‐phenylindole) staining, and the chromosomes were observed and photographed under a fluorescence microscope using 450–490 nm excitation.
2.9. Hi‐C scaffolding of the assembly to the chromosome level
The Hi‐C clean data were aligned to the primary assembly using bwa software. Only read pairs with both reads in the pair aligned to contigs were considered for scaffolding. The scaffolds (greater than 100 bp) were selected by Lachesis (v201701) to scaffold the assembly at the chromosome level.
2.10. Phylogenetic analysis and species divergence time estimation
To investigate the phylogenetic position of H. glycines, orthologous and paralogous groups from 11 species were assigned by OrthoMCL as follows: pig roundworm, B. malayi, C. briggsae, C. elegans, D. melanogaster, Haemonchus contortus, Heterorhabditis bacteriophora, Loa loa, M. hapla, O. volvulus and Trichoplax adhaerens. Orthologous groups that contained only one gene for each species were represented by the gene encoding the longest protein sequence. Genes encoding protein sequences shorter than 50 amino acids were filtered out to exclude putative fragmented genes. All‐against‐all blastP was applied to identify similarities among the filtered protein sequences in these species with an E‐value cut‐off of 1e−5. muscle (Robert, 2004) with default parameters was used to generate a multiple sequence alignment of the protein sequences in each single‐copy family. The alignments of each family were then concatenated to form a superalignment that was used for phylogenetic tree reconstruction using maximum‐likelihood methods (Guindon et al., 2010; Yang & Rannala, 2012). Species divergence time was estimated using mcmctree (http://abacus.gene.ucl.ac.uk/software/paml.html) in the paml software package. The correction time points were T. adhaerens and D. melanogaster (1147–713 million years ago), D. melanogaster and M. hapla (946–551 million years ago), and M. hapla and C. elegans (217.5–190.0 million years ago).
3. RESULTS AND DISCUSSION
3.1. Genome summary
Multiple libraries with different insert sizes were constructed from DNA extracted from the eggs of the purified X12 population. In total, 95.22 Gb of sequencing data was generated, of which 13.65 Gb (96.81X coverage) was produced from Illumina reads, 28.48 Gb (201.97X coverage) from PacBio reads, 31.32 Gb (222.13X coverage) from 10X Genomics linked‐read libraries and 21.77 Gb (154.39X coverage) from the Hi‐C library (Table S1). The assembled genome is estimated to be 141.01 Mb, with scaffold and contig N50 sizes of 16.27 Mb and 330.54 kb, respectively (Figure 2b). In addition, the sequencing results (SCN_Lian) were compared with the newly released sequencing results (SCN_Masonbrink) of 2019 (Masonbrink et al., 2019) and the genomes of the plant–parasitic nematode M. hapla (Opperman et al., 2008) and the free‐living nematode C. elegans (The C. elegans sequencing consortium, 1998) (Table 1). The genome size of SCN_Lian is 141.01 Mb, which is almost identical to that of SCN_Masonbrink, at 123 Mb. Notably, SCN_Masonbrink did not assemble the genome of H. glycines at the chromosome scale, though SCN_Lian did. The BUSCO value of SCN_Lian is 53.4% compared with 72% for SCN_Masonbrink, but the BUSCO value of SCN_Masonbrink is ~54% when analysed using the nematode database and the genomic data supplied by Masonbrink et al. Therefore, there is little difference in assembly quality between the genomes of SCN_Masonbrink and SCN_Lian. The GC content of SCN_Lian (36.89%) is similar to that of C. elegans (35.4%), whereas M. hapla has an unusually low GC content of 27.4%. SCN_Masonbrink annotated 29,769 genes, and SCN_Lian annotated 11,882 genes.
Figure 2.

Chromatin conformation capture‐based improved assembly Heterodera glycines genome. (a) Postclustering heat map showing the density of Hi‐C interactions between scaffolds from the proximity‐guided assembly. (b) Statistics of the completeness of the hybrid de novo assembly of X12 race genome. Listed are the assembled genome of ~141 Mb with scaffold and contig N50 size of 16.27 Mb and 330.54 Kb. Also listed in the table are the size and number of N60, N70, N80 and N90 of contigs and scaffolds. (c) Clustering of scaffolds using Hi‐C data into pseudochromosome‐scale scaffolds. Listed are the 258 scaffolds of total length ~12 Mb used for clustering. Also listed in the table are the cluster numbers, the number of contigs and the reference length of contigs [Colour figure can be viewed at http://wileyonlinelibrary.com]
Table 1.
Comparison of Heterodera glycines genome statistics with other plant–parasitic nematodes Meloidogyne hapla and Caenorhabditis elegans
| H. glycines | M. hapla | C. elegans | ||
|---|---|---|---|---|
| SCN‐Masonbrink | SCN‐Lian | |||
| Sequencing material | Inbred population TN10 (Hg type 1.2.6.7) | Natural population X12 (Hg type 1.2.3.4.5.6.7) | ||
| Genome size, Mb | 123.85 | 141.01 | 54 | 100 |
| Contigs, bp | 738 | 889 | 3,452 | N/A |
| Contig N50, Kb | 304,130 | 330,544 | N/A | N/A |
| Scaffolds | N/A | 267 | 1,523 | N/A |
| Scaffolds N50, bp | N/A | 16,265,615 | 83,645 | 17,494,000 |
| Assembled, bp | N/A | 141,354,287 | 53,578,246 | 100,267,623 |
| Sequence coverage, % | N/A | 98.33 | 99.2 | 100 |
| Per cent complete BUSCO, % | 72 | 53.4 | 59 | 99.6 |
| G+C, % | N/A | 36.89 | 27.4 | 35.4 |
| Annotated genes | 29,769 | 11,882 | 14,420 | 20,060 |
| Repeats numbers accounted for the genome, % | 34 | 51.10 | 17 | 16.5 |
| Identified SNPs | 1,619,134 | 247,046 | N/A | N/A |
| Chromosomes | NA | 18 | 16 | 6 |
| Chromosomes‐level assembly | N/A | Yes | N/A | Yes |
Abbreviation: SCN, soybean cyst nematode.
The data quality control results are shown in Tables S2–S4 and Figures S1 and S2. The following was obtained for assessment of polymerase length distribution: read number of 2,080,111, with mean read length of 13,703 and read length N50 of 23,355. Insert size length distribution showed the following: read number of 2,080,111, with mean read length of 9,875 and read length N50 of 14,429. Assessment of subread length distribution revealed that the read number was 3,179,171, with mean read length of 8,948 and read length N50 of 12,988. According to bwa software, the mapping rate of all small fragment reads to the genome was approximately 90.72%, and the coverage rate was approximately 98.33% (Table S5); thus, the reads show good agreement with the assembled genome. After sorting chromosome coordinates, removing repeated sequences and performing single nucleotide polymorphism (SNP) calling for the BWA comparison results, 247,046 SNPs were obtained, with 0.213% SNP heterozygosity and 0.0024% SNP homozygosity based on SAMtools (http://samtools.sourceforge.net/) (Table S6); therefore, the genome assembly has high single‐base accuracy. In addition, the GC content and average depth of the assembled genome were calculated and mapped using 10k Windows without repeated calculation. The results showed that the GC content is concentrated in a region encompassing 40% of the genome, without apparent separation, which showed that the genome was not contaminated by foreign sources (Table S7 and Figure S3).
The results of CEGMA analysis demonstrated that the assembly was complete, with mapping rate of 86.29% (a total of 214 genes) (Table S8). BUSCO evaluation results also indicated that the assembly result was complete, with 53.4% assembled complete single‐copy genes of 978 homologous single‐copy genes (Table S9). Remarkably, only 53.4% of the genes in the H. glycines assembly are single copy according to the BUSCO analysis, with 3.7% duplicated. For comparison, the BUSCO results for SCN_Masonbrink indicate that 56% of the genes in H. glycines are single copy, with 16% duplicated (Masonbrink et al., 2019).
Results of repeat prediction showed that the X12 genome contains 51.10% repeat sequences. Repetitive sequence statistics and classification results are shown in Tables S10 and S11 and Figure S4. The genome of H. glycines is diploid and consists of repeated sequences with higher nucleotide divergence (19.21%) than the genomes of Meloidogyne species, which are polyploid and consist of duplicated regions with low nucleotide divergence (~8%) (Abad et al., 2008; Blanc‐Mathieu et al., 2017; Sato et al., 2018; Szitenberg et al., 2017).
Gene structure prediction was performed, and 11,882 protein‐coding genes were predicted, with a mean of 1,233.92 bp of coding sequence (CDS) and 8.3 exons per gene (Table S12 and Figure S5). The transcript lengths of genes, CDSs, exons and introns of SCN are comparable to those of the genomes used for homology‐based prediction (Table S13 and Figure S6). In addition, noncoding RNA genes were predicted in the SCN genome, including a total length of 17,688‐bp ribosomal RNA (rRNA), 46,685‐bp transfer RNA (tRNA), 39,375‐bp microRNA (miRNA) and 21,549‐bp snRNA genes (Table S14). Based on functional annotation of protein‐coding genes, 64.5% (7,663), 76.5% (9,093), 60% (7,126), 70.7% (8,405), 49.1% (5,840) and 61.5% (7,303) of genes are annotated in Swiss‐Prot, Nr, kegg, InterPro, GO and Pfam, respectively. The four life stages of SCN were isolated and then mixed before sequencing for genome annotation. In total, 9,383 protein‐coding genes (79.0%) with conserved functional motifs and functional terms were successfully annotated (Table S15 and Figure S7). The distribution of genes, GC contents, long terminal repeats (LTRs), long interspersed nuclear elements (LINEs), short interspersed nuclear elements (SINEs), tRNAs, miRNAs, snRNAs and rRNAs in X12’s chromosomes are shown in Figure 3.
Figure 3.

The genome characteristics of Heterodera glycines. Circos plot showing the genomic features. 1u = 40 kb, small scale means 5u and large scale means 25u. From outer to inner circles: Track a: nine chromosomes of the genome; Track b: gene distribution in nine chromosomes; Track c: GC content distribution in nine chromosomes; Track d: LTR distribution in nine chromosomes; Track e: LINE distribution in nine chromosomes; Track f: SINE distribution in nine chromosomes; Track g: tRNA located on chromosomes; Track h: miRNA located on chromosomes; Track i: snRNA located on chromosomes; Track j: rRNA located on chromosomes [Colour figure can be viewed at http://wileyonlinelibrary.com]
There are some differences regarding the results for SCN_Lian and SCN_Masonbrink, such as the number of annotated genes. The possible reasons are as follows. First, there were differences in the sequencing technologies used. For SCN_Masonbrink, PacBio long‐read technology was mainly used, whereas combined Illumina short‐read and PacBio long‐read technologies were employed for SCN_Lian. Second, there were differences in the materials sequenced. The inbred population TN10 (Hg type 1.2.6.7) was used for SCN_Masonbrink, but the natural population X12 (Hg type 1.2.3.4.5.6.7), which is the most virulent SCN population identified to date, was utilized for SCN_Lian. The differences in the pathogenicity of these populations may also be judged from the differential proportions of S genes (50.4% and 45.3% in SCN_Lian and SCN_Masonbrink, respectively) and D genes (2.3% and 8.7% in SCN_Lian and SCN_Masonbrink, respectively) in the BUSCO results (Table 2). Third, different annotation methods were applied. Gene annotations were performed using Braker for SCN_Masonbrink with an unmasked assembly, which annotated 29,769 genes including 12,357 expressed repetitive elements and showed that the H. glycines genome has a significant number of repeats, at 34% of the genome. To prevent the number of genes from being too high, which can be caused by false positives from repeats during gene annotation, repeat masking before structure annotation was performed for SCN_Lian, as also conducted in many other studies (Xu et al., 2013; Zhang et al., 2019). To obtain more comprehensive and accurate repeat sequences, homologous sequence alignment and ab initio prediction were performed. Ultimately, 11,882 annotated genes and 51.10% nonredundant repeat sequences were obtained.
Table 2.
Genomic statistics and Benchmarking Universal Single‐Copy Orthologs analysis using nematode database
| Scientific name | Version | Genome size | Gene number | BUSCO genome |
|---|---|---|---|---|
| Caenorhabditis_elegans | ensembl.metazoa.v32 | 98M | C:98.6% (S:98.0%, D0.6%), F:0.8%, M:0.6%, n:982 | |
| Caenorhabditis_briggsae | ensembl.metazoa.v32 | 106M | C:97.7% (S:97.0%, D0.7%), F:1.5%, M:0.8%, n:982 | |
| Ascaris_suum | ensembl.metazoa.v32 | 265M | C:89.8% (S:88.0%, D1.8%), F:6.6%, M:3.6%, n:982 | |
| Brugia_malayi | wormbase.WBPS6 | 93M | C:96.6% (S:96.0%, D0.6%), F:2.4%, M:1.0%, n:982 | |
| Onchocerca_volvulus | ensembl.metazoa.v32 | 94M | C:97.6% (S:97.3%, D0.3%), F:1.7%, M:0.7%, n:982 | |
| Meloidogyne hapla | 54M | 14420 | C:59.9% (S:58.7%, D1.2%), F:9.4%, M:30.7%, n:982 | |
| Meloidogyne incognita | 184M | 43718/45351 | C:61.8% (S:25.8%, D36.0%), F:8.1%, M:30.1%, n:982 | |
| Heterodera glycines (SCN‐Lian) | 135M | 11882 | C:52.7% (S:50.4%, D2.3%), F:9.6%, M:37.7%, n:982 | |
| H. glycines (SCN‐Masonbrink) | 129M | 29769 | C:54.0% (S:45.3%, D8.7%), F:10.4%, M:35.6%, n:982 |
Abbreviation: SCN, soybean cyst nematode.
3.2. Chromosome observation and Hi‐C scaffolding
The chromosome number of H. glycines during meiosis was observed under a fluorescence microscope using 450–490 nm excitation (2n = 18) (Figure 4). The Illumina‐based Hi‐C data were remapped to the PacBio assembly, clustering into nine pseudomolecules using the Proximo Hi‐C scaffolding pipeline (Figure 2a). The Hi‐C scaffolding was able to anchor and order with high confidence all of the 258 scaffolds into nine pseudomolecules. The scaffold sizes ranged from 7.6 to 185 Mb with an N50 of 16.3 Mb (Figure 2c). The overall scaffolding rate was 91.2% (Table S16).
Figure 4.

Observation of the chromosome of Heterodera glycines in meiosis under fluorescence microscope with 450–490 nm excitation light (2n = 18) [Colour figure can be viewed at http://wileyonlinelibrary.com]
3.3. Evolutionary analysis
A total of 25,535 gene family clusters were constructed. The genes used for gene family clustering in each species are shown in Table S17. In total, 482 single‐copy gene families are common to all 12 species. The distribution of single‐copy orthologs, multiple‐copy orthologs, genes unique to H. glycines and other orthologs in different species is shown in Table S18. Protein sequences from the 482 single‐copy gene families were used for phylogenetic tree reconstruction, and the estimation of divergence time was performed (Figure 5) with mcmctree software. Synteny diminished as phylogenetic relatedness declined, and our results showed that the divergence time between H. glycines and M. hapla is approximately 143.6 million years. Thus, the divergence of H. glycines preceded that of the model nematode C. elegans. Moreover, because plant parasitism is a lifestyle found in three different clades in the nematode tree of life, plant parasitism appeared at least three times independently during the evolution of nematodes (Danchin & Perfus‐Barbeoch, 2009). It was also inferred that plant parasites evolved from fungus‐feeding nematodes, according to previous results that showed consistent coclustering of plant parasites with fungivorous species (Holterman et al., 2006).
Figure 5.
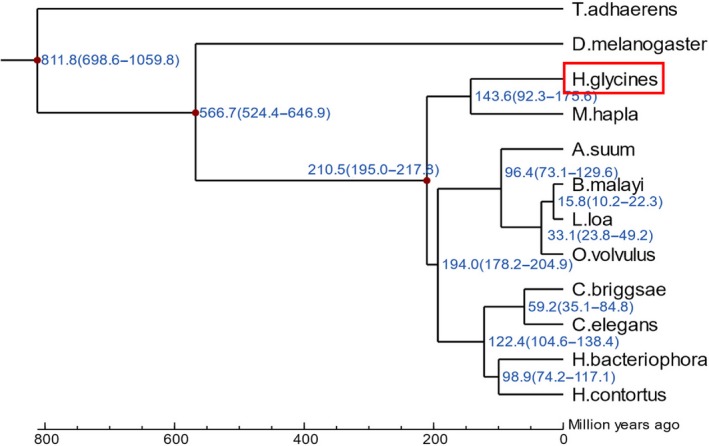
Phylogenetic relationships of species related to Heterodera glycines. Phylogenetic tree of the single‐copy gene families coexistent in the 12 species representing the relatedness of each species. Red box surrounded denotes the sequenced species H. glycines. Node labels represent node ages [Colour figure can be viewed at http://wileyonlinelibrary.com]
4. CONCLUSIONS
The chromosome‐level reference genome sequence of X12, a notable race of the SCN H. glycines, has immediate and important implications for research on plant nematodes and also for broader biological studies. In total, approximately 95.22 Gb of sequencing data were generated using a combination of long‐ and short‐read techniques. The assembled genome contains 267 scaffolds, with an N50 scaffold length of 16.27 Mb and a total length of 141.01 Mb. The assembly was estimated to contain 86.29% of core genes according to CEGMA analysis and 53.4% of complete single‐copy genes. The assembled genome is more contiguous than is the previously published H. glycines genome (Masonbrink et al., 2019), with 1.2‐fold more contigs and a 1.09‐fold greater N50 contig length. The mapping rate for reads back to the assembled genome is approximately 90.72%. A total of 11,882 genes were predicted, assisted by RNA sequencing data, and 79.0% homologous sequences were annotated in the genome. This high‐quality genome assembly of H. glycines will help to enable the identification of virulence‐related genes.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
AUTHOR CONTRIBUTIONS
Yun Lian and Weiguo Lu conceived and designed the experiments. He Wei, Chenfang Lei and Jinying Li collected the cyst samples and managed the data. Jinshe Wang performed the data analysis. Haichao Li collected the sequencing data. Jianqiu Guo collected the soil sample containing X12 from the soybean field. Pei Du participated in the chromosome preparation. Yongkang Wu, Shufeng Wang, Hui Zhang and Tingfeng Wang revised the manuscript.
Supporting information
ACKNOWLEDGEMENTS
We thank all the members of the soybean research group for helpful comments and discussions.
Lian Y, Wei H, Wang J, et al. Chromosome‐level reference genome of X12, a highly virulent race of the soybean cyst nematode Heterodera glycines . Mol Ecol Resour. 2019;19:1637–1646. 10.1111/1755-0998.13068
Funding information
This research was supported by the National Natural Science Foundation of China (31371652), Central Plains Science and Technology Innovation leading Talent Project (194200510003) and National Key Research and Development Program of China (2017YFD0101400).
DATA AVAILABILITY STATEMENT
The genomic (SRA917187) and transcriptomic sequence reads (SRA 917415) were deposited at the NCBI (National Center for Biotechnology Information) with BioProject Accession no.: PRJNA535374. The assembled genome and annotation results were available at the Dryad Digital Repository: https://doi.org/10.5061/dryad.5b2m501. In addition, the supplementary figures and tables are provided in Supporting Information.
REFERENCES
- Abad, P. , Gouzy, J. , Aury, J. M. , Castagnone‐Sereno, P. , Danchin, E. G. , Deleury, E. , … Wincker, P. (2008). Genome sequence of the metazoan plant‐parasitic nematode Meloidogyne incognita . Nature Biotechnology, 26, 909–915. [DOI] [PubMed] [Google Scholar]
- Akker, S. E. D. A. , Laetsch, D. R. , Thorpe, P. , Lilley, C. J. , Danchin, E. G. J. , Rocha, M. D. , … Jones, J. T. (2016). The genome of the yellow potato cyst nematode, Globodera rostochiensis, reveals insights into the basis of parasitism and virulence. Genome Biology, 17, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atibalentja, N. , Bekal, S. , Domier, L. L. , Niblack, T. L. , Noel, G. R. , & Lambert, K. N. (2005). A genetic linkage map of the soybean cyst nematode Heterodera glycines . Molecular Genetics and Genomics, 273, 273–281. [DOI] [PubMed] [Google Scholar]
- Blanc‐Mathieu, R. , Perfus‐Barbeoch, L. , Aury, J. M. , Da Rocha, M. , Gouzy, J. , Sallet, E. , … Danchin, E. G. J. (2017). Hybridization and polyploidy enable genomic plasticity without sex in the most devastating plant‐parasitic nematodes. PLoS Genetics, 13, e1006777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton, J. N. , Adey, A. , Patwardhan, R. P. , Qiu, R. , Kitzman, J. O. , & Shendure, J. (2013). Chromosome‐scale scaffolding of de novo genome assemblies based on chromatin interactions. Nature Biotechnology, 31(12), 1119–1125. 10.1038/nbt.2727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danchin, E. G. J. , & Perfus‐Barbeoch, L. (2009). The genome sequence of Meloidogyne incognita unveils mechanisms of adaptation to plant–parasitism in Metazoa In Pierre P. (Ed.), Evolutionary biology (Chapter 17, pp. 287–302. Berlin, Heidelberg: Springer. [Google Scholar]
- De Boer, J. M. , Overmars, H. A. , Pomp, H. , Davis, E. L. , Zilverentant, J. F. , Goverse, A. , … Schots, A. (1996). Production and characterization of monoclonal antibodies to antigens from second‐stage juveniles of the potato cyst nematode, Globodera rosto‐chiensis . Fundamental and Applied Nematology, 19, 545–554. [Google Scholar]
- De Boer, J. , Yan, Y. , Smant, G. , Davis, E. L. , & Baum, T. J. (1998). In‐situ hybridization to messenger RNA in Heterodera glycines . Journal of Nematology, 30, 309. [PMC free article] [PubMed] [Google Scholar]
- Donald, P. , & Young, L. (2004). Characterization of two soybean cyst nematode populations that reproduce on PI 437654 source of resistance. Journal of Nematology, 36, 315–316. [Google Scholar]
- Du, P. , Li, L. N. , Zhang, Z. X. , Liu, H. , Qin, L. I. , Huang, B. Y. , … Zhang, X. Y. (2016). Chromosome painting of telomeric repeats reveals new evidence for genome evolution in peanut. Journal of Integrative Agriculture, 15(11), 2488–2496. [Google Scholar]
- Gardner, M. , Heinz, R. , Wang, J. , & Mitchum, M. G. (2017). Genetics and adaptation of soybean cyst nematode to broad spectrum soybean resistance. G3 (Bethesda), 7(3), 835–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon, S. , Dufayard, J. F. , Lefort, V. , Anisimova, M. , Hordijk, W. , & Gascuel, O. (2010). New algorithms and methods to estimate maximum‐likelihood phylogenies: Assessing the performance of PhyML 3.0. Systematic Biology, 59, 307–321. [DOI] [PubMed] [Google Scholar]
- Holterman, M. , van der Wurff, A. , van den Elsen, S. , van Megen, H. , Bongers, T. , Holovachov, O. , … Helder, J. (2006). Phylum‐wide analysis of SSU rDNA reveals deep phylogenetic relationships among nematodes and accelerated evolution toward crown Clades. Molecular Biology and Evolution, 23(9), 1792–1800. [DOI] [PubMed] [Google Scholar]
- Howland, A. , Monnig, N. , Mathesius, J. , Nathan, M. , & Mitchum, M. G. (2018). Survey of Heterodera glycines population densities and virulence phenotypes during 2015–2016 in Missouri. Plant Disease, 102(12), 2407–2410. [DOI] [PubMed] [Google Scholar]
- Hua, C. , Li, C. , Hu, Y. , Mao, Y. , You, J. , Wang, M. , … Wang, C. (2018). Identification of HG types of soybean cyst nematode Heterodera glycines and resistance screening on soybean genotypes in Northeast China. Journal of Nematology, 50(1), 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao, Y. Q. , Vuong, T. D. , Liu, Y. , Meinhardt, C. , Liu, Y. , Joshi, T. , … Nguyen, H. T. (2015). Identification and evaluation of quantitative trait loci underlying resistance to multiple HG types of soybean cyst nematode in soybean PI 437655. TAG. Theoretical and Applied Genetics., 128, 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, J. T. , Haegeman, A. , Danchin, E. J. , Gaur, H. S. , Helder, J. , Jones, M. G. K. , … Perry, R. N. (2013). Top 10 plant‐parasitic nematodes in molecular plant pathology. Molecular Plant Pathology, 14(9), 946–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadam, S. , Vuong, T. D. , Qiu, D. , Meinhardt, C. G. , Song, L. , Deshmukh, R. , … Nguyen, H. T. (2016). Genomic‐assisted phylogenetic analysis and marker development for next generation soybean cyst nematode resistance breeding. Plant Science, 242, 342–350. [DOI] [PubMed] [Google Scholar]
- Kim, K. S. , Vuong, T. D. , Qiu, D. , Robbins, R. T. , Grover Shannon, J. , Li, Z. , & Nguyen, H. T. (2016). Advancements in breeding, genetics, and genomics for resistance to three nematode species in soybean. Theoretical and Applied Genetics, 129(12), 2295–2311. [DOI] [PubMed] [Google Scholar]
- Koenning, S. R. , & Wrather, J. A. (2010). Suppression of soybean yield potential in the continental United States by plant diseases from 2006 to 2009. Plant Health Progress, 11(1), 5 10.1094/PHP-2010-1122-01-RS [DOI] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25(14), 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian, Y. , Guo, J. Q. , Li, H. C. , Wu, Y. K. , Wei, H. , Wang, J. S. , … Lu, W. G. (2017). A new race (X12) of soybean cyst nematode in China. Journal of Nematology, 49(3), 321–326. [PMC free article] [PubMed] [Google Scholar]
- Lian, Y. , Wang, J. S. , Li, H. C. , Wei, H. , Li, J. Y. , Wu, Y. K. , … Lu, W. G. (2016). Race distribution of soybean cyst nematode in the main soybean producing area of Huang‐Huai Rivers Valley. Acta Agronomica Sinica, 42(10), 1479–1486. [Google Scholar]
- Lu, W. G. , Gai, J. Y. , Zheng, Y. Z. , & Li, W. D. (2006). Construction of a soybean genetic linkage map and mapping QTLs resistant to soybean cyst nematode (Heterodera glycines Ichinohe). Acta Agronomica Sinica, 32(9), 1272–1279. [Google Scholar]
- Masonbrink, R. E. , Maier, T. R. , Muppirala, U. , Seetharam, A. S. , Lord, E. , Juvale, P. S. , … Baum, T. J. (2019). The genome of the soybean cyst nematode (Heterodera glycines) reveals complex patterns of duplications involved in the evolution of parasitism genes. BMC Genomics, 20, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchum, M. G. (2016). Soybean resistance to the soybean cyst nematode Heterodera glycines: An update. Phytopathology, 106(12), 1444–1450. [DOI] [PubMed] [Google Scholar]
- Mitchum, M. G. , Wrather, J. A. , Heinz, R. D. , Shannon, J. G. , & Danekas, G. (2007). Variability in distribution and virulence phenotypes of Heterodera glycines in Missouri during 2005. Plant Disease, 91, 1473–1476. [DOI] [PubMed] [Google Scholar]
- Niblack, T. L. , Arelli, P. R. , Noel, G. R. , Opperman, C. H. , Orf, J. H. , Schmitt, D. P. , … Tylka, G. L. (2002). A revised classification scheme for genetically diverse populations of Heterodera glycines . Journal of Nematology, 34, 279–288. [PMC free article] [PubMed] [Google Scholar]
- Niblack, T. L. , Colgrove, A. L. , Colgrove, K. , & Bond, J. P. (2008). Shift in virulence of soybean cyst nematode is associated with use of resistance from PI 88788. Plant Health Progress, 9(1), 29– 10.1094/PHP-2008-0118-01-RS [DOI] [Google Scholar]
- Opperman, C. H. , Bird, D. M. , Williamson, V. M. , Rokhsar, D. S. , Burke, M. , Cohn, J. , … Windham, E. (2008). Sequence and genetic map of Meloidogyne hapla: A compact nematode genome for plant parasitism. Proceedings of the National Academy of Sciences, 105, 14802–14807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra, G. , Bradnam, K. , & Korf, I. (2007). CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics, 23, 1061–1067. [DOI] [PubMed] [Google Scholar]
- Patil, G. B. , Lakhssassi, N. , Wan, J. , Song, L. , Zhou, Z. , Klepadlo, M. , … Nguyen, H. T. (2019). Whole genome re‐sequencing reveals the impact of the interaction of copy number variants of the rhg‐1 and Rhg4 genes on broad‐based resistance to soybean cyst nematode. Plant Biotechnology Journal, 17(8), 1595–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao, S. S. P. , Huntley, M. H. , Durand, N. C. , Stamenova, E. K. , Bochkov, I. D. , Robinson, J. T. , … Aiden, E. L. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell, 159(7), 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs, R. D. , & Schmitt, D. P. (1988). Complete characterization of the race scheme for Heterodera glycines . Journal of Nematology, 20, 392–395. [PMC free article] [PubMed] [Google Scholar]
- Robert, C. E. (2004). MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato, K. , Kadota, Y. , Gan, P. , Bino, T. , Uehara, T. , Yamaguchi, K. , … Shirasu, K. (2018). High‐quality genome sequence of the root‐knot nematode Meloidogyne arenaria genotype A2‐O. Genome Announcements, 6(26), e00519‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simao, F. A. , Waterhouse, R. M. , Ioannidis, P. , Kriventseva, E. V. , & Zdobnov, E. M. (2015). BUSCO: Assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- Szitenberg, A. , Salazar‐Jaramillo, L. , Blok, V. C. , Laetsch, D. R. , Joseph, S. , Williamson, V. M. , … Lunt, D. H. (2017). Comparative genomics of apomictic root‐knot nematodes: Hybridization, ploidy, and dynamic genome change. Genome Biology and Evolution, 9, 2844–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The C. elegans sequencing consortium . (1998). Genome sequence of the nematode C. elegans : A platform for investigating biology. Science, 282, 2012–2018. [DOI] [PubMed] [Google Scholar]
- Wang, H. M. , Zhao, H. H. , & Chu, D. (2015). Genetic structure analysis of populations of the soybean cyst nematode, Heterodera glycines, from north China. Nematology, 17, 591–600. [Google Scholar]
- Woo, M. O. , Beard, H. , MacDonald, M. H. , Brewer, E. P. , Youssef, R. M. , Kim, H. , & Matthews, B. F. (2014). Manipulation of two α‐endo‐β‐1,4‐glucanase genes, AtCel6 and GmCel7, reduces susceptibility to Heterodera glycines in soybean roots. Molecular Plant Pathology, 15(9), 927–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Q. , Chen, L. L. , Ruan, X. A. , Chen, D. J. , Zhu, A. D. , Chen, C. L. , … Yijun Ruan, Y. J. (2013). The draft genome of sweet orange (Citrus sinensis). Nature Genetics, 45(1), 59–67. [DOI] [PubMed] [Google Scholar]
- Yaffe, E. , & Tanay, A. (2011). Probabilistic modeling of Hi‐C contact maps eliminates systematic biases to characterize global chromosomal architecture. Nature Genetics, 43(11), 1059–1065. [DOI] [PubMed] [Google Scholar]
- Yang, Z. , & Rannala, B. (2012). Molecular phylogenetics: Principles and practice. Nature Reviews Genetics, 13(5), 303–314. 10.1038/nrg3186 [DOI] [PubMed] [Google Scholar]
- Zhang, X. J. , Jianbo Yuan, J. B. , Sun, Y. M. , Li, S. H. , Gao, Y. , Yu, Y. , … Jianhai Xiang, J. H. (2019). Penaeid shrimp genome provides insights into benthic adaptation and frequent molting. Nature Communications, 10, 356 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The genomic (SRA917187) and transcriptomic sequence reads (SRA 917415) were deposited at the NCBI (National Center for Biotechnology Information) with BioProject Accession no.: PRJNA535374. The assembled genome and annotation results were available at the Dryad Digital Repository: https://doi.org/10.5061/dryad.5b2m501. In addition, the supplementary figures and tables are provided in Supporting Information.