

Summary
Aims
Classifications of occluding vasculopathies (except vasculitis [1]) may exhibit some difficulties. Firstly, classifications may follow different principles, e.g. clinicopathologic findings, etiology or pathogenesis. Secondly, authors may not distinguish between vasculitis and occluding vasculopathies. Thirdly, occluding vasculopathies are systemic diseases. Organ‐specific variations make morphologic findings difficult to compare. Moreover, subtle changes are recognized in the skin, but may be invisible in other organs. Our aim was to use the skin and subcutis as a tool and clinicopathological correlation as the basic process for classification.
Methods and results
We first differentiate in the skin between small and medium vessel occluding vasculopathies. Here we focus on medium vessel‐occluding vasculopathies. In the second step we differentiate the vessel subtypes. In the final step, we differentiate according to the time point of the coagulation/reorganization process and the involved inflammatory cells/stromal features. By applying the same procedure to the various entities and visualizing the findings in the style of bar codes, the overlaps and differences in the clinical picture as well as the histopathology become more apparent.
Conclusions
Occluding vasculopathies are often not separate entities, but reaction patterns and epiphenomena. Distinguishing them from vasculitides is crucial because of the differences in pathogenesis, therapeutic approach and prognosis.
Introduction
As a continuation of our work on small vessel‐occluding vasculopathies 2, we now focus on medium size vessel‐occluding vasculopathies. Our approach uses the skin and subcutis as a tool and clinicopathological correlation as the basic process for classification. We use an algorithmic approach with pattern analysis, which enables consistent and reliable reporting of histological findings. As was the case with the International Chapel Hill Consensus Conference on the nomenclature of vasculitides from 1994 3, 2012 4 and 2018 5, 6, we first differentiate between small and medium vessel coagulopathies. No true large vessels exist in the skin. According to the Chapel Hill classification of 2012 5, large arteries are confined to the aorta and its large branches, and veins to the vena cava and its large branches. Medium vessels are large organ vessels and small vessels are all vessels smaller than these. According to this classification, we only find small vessels in the skin. However, we differentiate between vessel sizes, because clinical and histopathological appearances vary accordingly. Thus, we use the term “small vessels” when capillaries and postcapillary venules are affected in contrast to medium vessels if primarily arteries (for simplicity also arterioles) or veins at the border of the reticular dermis/subcutis or in the subcutis are involved. In the second step we differentiate either towards arteries or veins 7, 8, 9. In the final step, we differentiate according to the life cycle of the event. Occluding vasculopathies have a characteristic life cycle of histopathologic events. Early stages are dominated by thrombi (fibrin or other materials) without significant inflammation. Reorganization of these thrombi follows, which often leads to “lymphocytic vascular reorganization”, a process dominated by lymphocytes that is accentuated around and within the affected vessel and vessel wall. Finally, there is healing with complete reconstitution of vessels with vessel lumina and/or partial to complete occlusion of vessels by fibroblasts and collagen.
In our algorithmic approach, we use tables and shades to highlight the different features in order to facilitate the comparison between the different manifestations and to grade the importance of certain features (Tables 1, 4, 5, 6, 7, 8, 9, 10). We mark least common denominators in black, prominent characteristic findings in dark grey, variable findings in light grey and missing features in white. By applying the same procedure to the various entities, the overlaps and differences in the clinical picture and in the histopathology become more apparent. We try to visualize these in the style of bar codes (Table 11), which help to simplify the findings and render them comparable.
Table 1.
Livedo reticularis
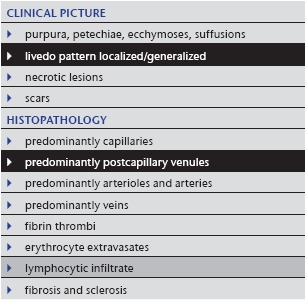
|
Table 4.
Sneddon syndrome
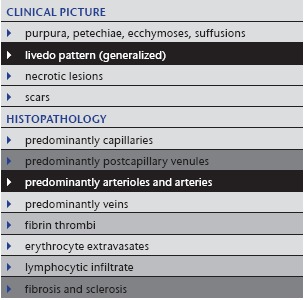
|
Table 5.
Anti‐phospholipid syndrome (Hughes syndrome), systemic lupus erythematosus and malignant atrophic papulosis (Degos disease)
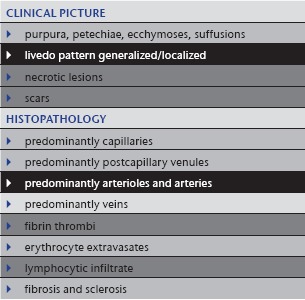
|
Table 6.
Calciphylaxis
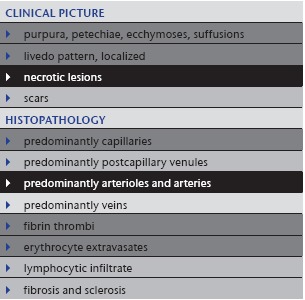
|
Table 7.
Emboli
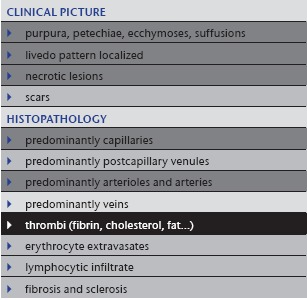
|
Table 8.
Thrombophlebitis
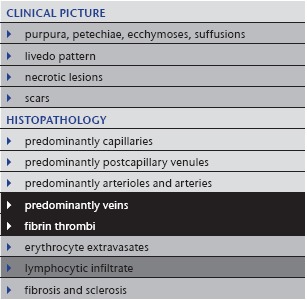
|
Table 9.
Arteriosclerosis and arteriolosclerosis
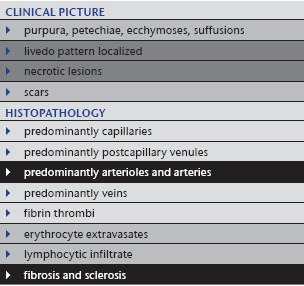
|
Table 10.
Vasculopathy in neurofibromatosis
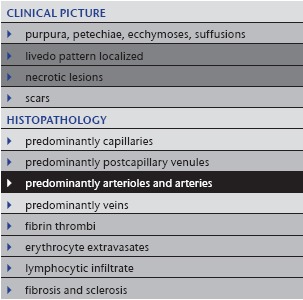
|
Table 11.
Vasculo/coagulopathies – systematic approach and comparison reminiscent of a bar code reader
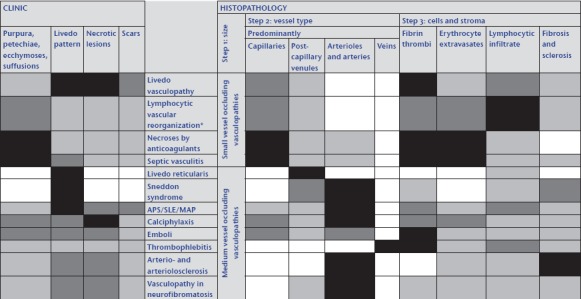
|
Abk.: APS, Antiphospholipid syndrome; SLE, systemic lupus erythematosus; MAP, malignant atrophic papulosis.
*All inflammatory and even proliferative/neoplastic processes may be associated with coagulation disorders and thus with fibrin thrombi; this can lead to lymphocytic vascular reorganization.
Our approach focusses on the clinical picture and histopathology. Etiological and pathogenetic data are not primarily used in our approach. However, pathogenetic evaluation is crucial and laboratory tests and imaging, the latter especially in medium vessel occluding vasculopathies, are obviously necessary and are used to verify or falsify our diagnostic assumptions, based primarily on the clinical pictures and histopathology (see Table 1a, b in part I of the manuscript 2). In addition, other specialties, in particular cardiology, pneumology, nephrology, rheumatology, neurology and ophthalmology, must frequently be included in a multidisciplinary approach in order to achieve optimal patient management.
The characteristic clinical sign of medium vessel pathology, which is also most important to understand from a pathological point of view, is livedo patterning 10, 11, 12. This is not a disease itself, but a condition which indicates irregular blood flow and oxygenation, which can be physiological or a sign of a disease. Livedo patterning occurs in two forms: livedo reticularis and livedo racemosa 13.
I. Livedo reticularis
Under normal circumstances, arteries supply skin from a central artery which drains into a rim of peripheral collecting veins. Neurohumoral or general rheologic disorders generally reduce the blood flow in arteries and veins, the erythrocytes become more deoxygenated than usual, and as a consequence the draining veins become visible as a regular network. Livedo reticularis (Tables 1, 2; Figure 1a) is a bluish discoloration of the skin in a regular, fine and net‐like pattern with closed circles; it usually does not become purpuric and is completely reversible with cessation of the cause. The diameter of the livedo pattern can change according to the anatomical region (small diameter e.g. on feet, large diameter e.g. on thighs). A classic example is cutis marmorata on the thighs of young females. The histological features are mild; postcapillary venules are elongated and dilated (Figure 1b–d), and this affects the upper dermal plexus and the interconnecting veins more than the lower dermal plexus. Endothelial cells are not prominent. Little or no infiltration by lymphocytes can be found. Although the elongated postcapillary venules are the leading histopathological feature, the cause of the livedo pattern is in the larger deep vessels. Neurohumoral constriction or slow/reduced blood flow in instances of rheologic disorders are usually not yet visible in the histopathological sections.
Table 2.
Diseases causing livedo reticularis
| Neurohumoral |
|---|
|
| Rheological |
|---|
|
Figure 1.
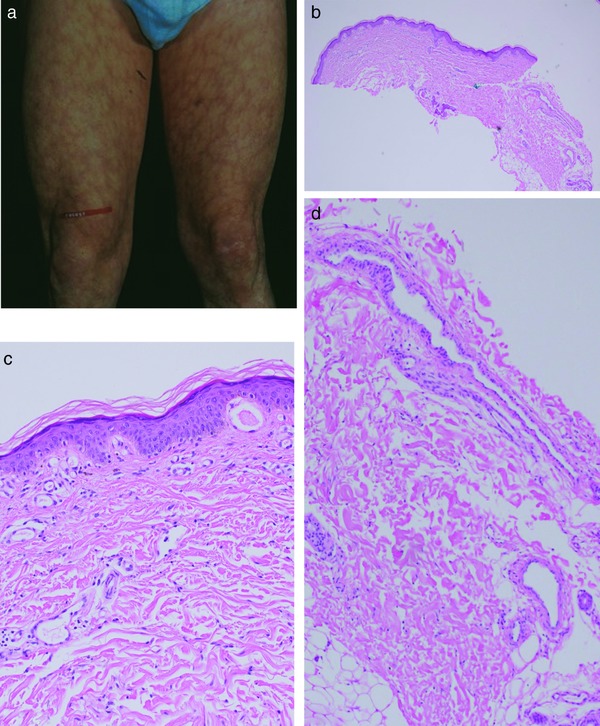
Livedo reticularis. Regular livedo reticularis forming circles to nets on the thighs (a). Prominent dilated and elongated postcapillary venules in the papillary and reticular dermis as well as at the border to the subcutis (b). Prominent postcapillary venules in the papillary dermis (c). Prominent postcapillary venules in the reticular dermis as well as at the border to the subcutis (d).
Depending on the acuity and intensity of a process, there can be fluent crossovers between livedo reticularis and livedo racemosa.
II. Livedo racemosa
By contrast, in livedo racemosa, focal vascular occlusion following vasculitis or thrombus formation/other occlusion will occasionally affect the blood flow and produce an irregular pattern. Livedo racemosa (Table 3; Figure 2a) shows an irregular, lightning or starburst‐like pattern with broad and open circles; the purple discoloration can become purpuric and shows only some fluctuation, but is basically irreversible. Histopathological features of livedo racemosa vary according to the underlying disease and are shown below together with the corresponding diseases.
Table 3.
Diseases causing livedo racemosa
| Affected vessels: capillaries (c), postcapillary venules (pv), arterioles/arteries (a), veins (v) |
|---|
| Vasculitic |
|
| Non‐vasculitic |
|---|
|
Figure 2.
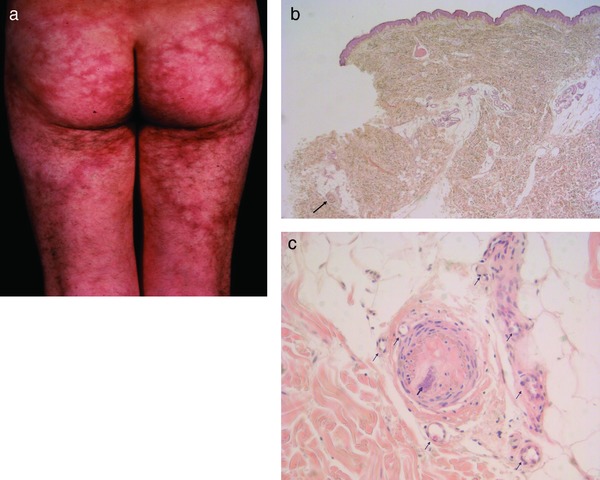
Livedo racemosa/Sneddon syndrome. Irregular livedo racemosa forming open circles to bizarre branches on buttocks (usual area of predilection) and thighs (a). Apparently normal epidermis and dermis. However, higher magnification shows an occluded artery at the dermis‐subcutis border (arrow) (b). Artery completely occluded by fibrosis and early calcification at dermal‐subcutis border (bold arrow); note that the membrana elastica interna of the artery as well as the corona of capillaries (vasa vasorum, thin arrows) are still present, which is unusual in vessels of this size (250–500 µm) and indicates a previous granulation tissue response due to inflammation and/or occluding vasculopathy (c).
1. Sneddon syndrome
Sneddon syndrome (Table 4), which was originally a clinical diagnosis, is probably not a disease but a reaction pattern due to occluding vasculopathies; it primarily affects arterioles and small arteries 15. Various coagulation defects have been associated with patients with Sneddon syndrome 12, 16. The two most often affected organs are the skin, where the process is easily visible, even at a very early stage, and the brain, which is most sensitive to even minor flow disturbances. Signs and symptoms in other organs are usually minor to absent, for example moderate reduction of glomerular filtration rate, minimal changes in electrocardiography, peripheral polyneuropathy and vascular abnormalities in ophthalmoscopy 17. The disorder mainly occurs sporadically, although familial cases and a mutation in CECR1 (cat eye syndrome chromosome region, candidate 1) have been described as a cause of Sneddon syndrome 15, 18.
Clinical picture: Sneddon syndrome mainly affects young adults, women much more often than men (ratio 9 : 1). Buttocks, trunk and dorsal extremities are preferred. Livid, symmetrical and asymptomatic livedo racemosa without purpura is characteristic (Figure 2a). Headache (frequently misinterpreted as migraine) and dizziness are early CNS manifestations 19. Arterial hypertension and cardiac valvulopathy occur in a significant proportion of patients 15.
Histology: Sneddon syndrome characteristically affects arterioles to small arteries in the skin at the border between the dermis and subcutis 17. Rare histological findings of early stages (< 5 % of all biopsies) show fibrin thrombi, swollen endothelial cells, some activated lymphocytes within the lumen and in and around the vessel wall, as well as a rim of granulation tissue with small capillaries around the affected artery (corona of capillaries, vasa vasorum) (Figure 2b, c). In normal arteries of this size (250–500 µm), there is usually no corona. In due course, fibroblasts and myofibroblasts occupy the subendothelial space and narrow the lumen, which may be partially to completely occluded. In such instances the tunica media begins to shrink while the corona of capillaries is still present, although less prominent than before. In the final stage the lumen is either completely occluded by collagen and a few fibroblasts, sometimes even with calcification, or there is recanalization but stenosis of the original lumen. The tunica media is thinned, but some capillaries (approximately 3 to 6) still remind one of the corona of capillaries 20, 21. Postcapillary venules are moderately elongated and dilated. Little or no infiltration by lymphocytes can be found.
2. Anti‐phospholipid syndrome (Hughes syndrome): idiopathic or in association with systemic lupus erythematosus
Anti‐phospholipid syndrome (Table 5) can present in different ways: most frequently similar to Sneddon syndrome but sometimes also with purpuric livedo racemosa, which may ulcerate, as well as a catastrophic variant with rapid hemorrhage and necrosis. The pathological laboratory parameters (anti‐phospholipid antibodies) are characteristic. Anti‐phospholipid syndrome can also appear in association with systemic lupus erythematosus. Usually, typical cutaneous features or manifestations of lupus erythematosus on inner organs and/or positive antinuclear antibodies (ANAs) facilitate this diagnosis. One interesting association with/without lupus erythematosus is malignant atrophic papulosis (Degos disease) 22, which can also show livedo racemosa around characteristic porcelain‐like atrophic papules (own observation and communication) (Figure 3a–c).
Figure 3.
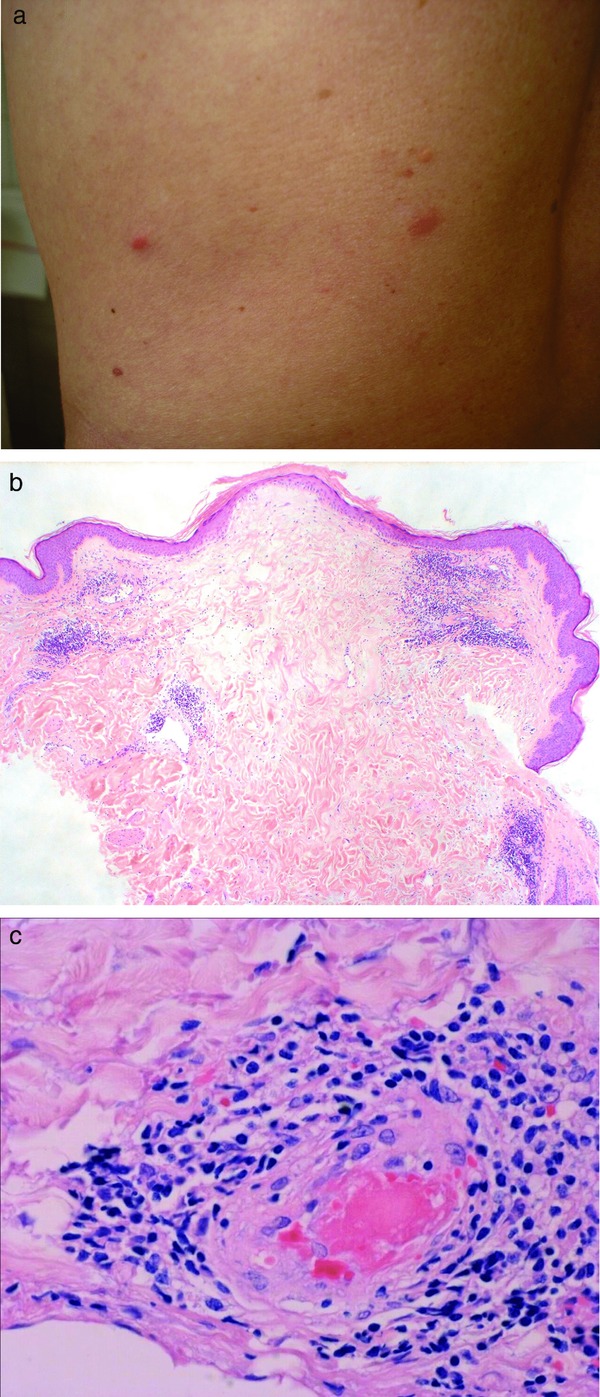
Degos disease. Still reddish (not yet porcelain) papules on the abdomen (a). Wedge‐shaped lesion with hyperkeratotic but atrophic epidermis, increase of mucin and dense lymphocytic infiltrate around dilated and elongated vessels (b). Serial sections with fibrin thrombus occluding the arteriole, which is surrounded by lymphocytes at the bottom of the wedge‐shaped lesion (c).
3. Calciphylaxis
Calciphylaxis (Table 6) or calcific thrombogenic microangiopathy 13 is an occluding vasculopathy affecting small and medium vessels. Patients usually suffer from renal insufficiency with secondary hyperparathyroidism (hyperphosphatemia and hypocalcemia compensated by increased parathormone). Often, additional rheologic defects such as AT‐III deficiency, protein‐C or protein–S deficiency can also be detected.
Clinical picture: Calciphylaxis mainly affects older adults without sexual predilection. Patients suffer from renal insufficiency; they may be on dialysis or post‐transplantation. Extremities are the preferred location. Lesions are symmetrical, discrete to bizarre reticular and confluent. One sees hemorrhagic to necrotic macules, plaques and ulcers (Figure 4a). An acute (often appearing overnight) and extremely painful livedo racemosa/retiform purpura is characteristic. Calciphylaxis has a high mortality, with 75 % of patients dying within three to six months after diagnosis.
Figure 4.
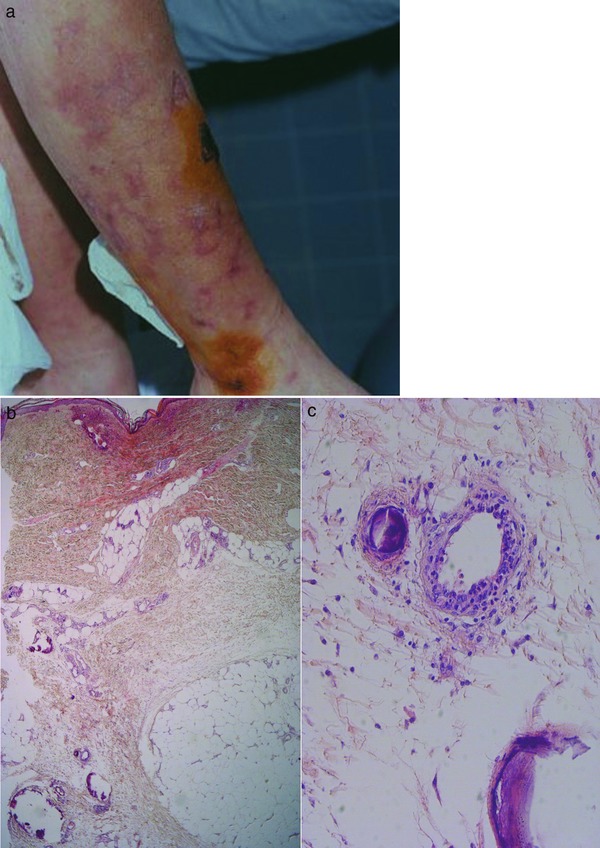
Calciphylaxis. Bizarre livedo racemosa with black necrotic papules to plaques on lower leg (a). Faintly stained specimen due to early necrotic tissue with prominent subepidermal hemorrhage and occluding thrombi as well as subcutaneous calcifications, partially within and around small and medium vessels infiltrated by neutrophils and nuclear dust (b, c).
Histology: Calciphylaxis (Figure 4b, c) shows extensive necrotic lesions with fibrin thrombi and erythrocyte extravasates. In addition, small and medium vessels in the dermis and subcutis show circular perivascular and/or intravascular calcification and a moderate amount of polymorphonuclear neutrophils with leukocytoclasia accentuated around vessels. There are also calcifications in the connective tissue. Additionally, these patients regularly have arteriolosclerosis and arteriosclerosis due to long‐lasting renal disease with secondary arterial hypertension.
4. Emboli
Besides thrombi, a wide variety of emboli (Table 7) may cause acute circulatory problems. According to the size of the emboli, occlusion may affect medium arteries according to the CHCC‐definition (cardiac thrombi/emboli following myocardial infarction or atrial fibrillation; malignancies 23; atrial myxoma 24), small arteries and arterioles (cholesterol emboli following break up of arteriosclerotic plaques; calcium oxalate emboli in association with hyperoxaluria 25) or capillaries (fat, air or gas emboli following extensive bone fractures or decompression sickness [diver's bends]).
Clinical picture: Cholesterol emboli mostly affect dermal arteries/arterioles of older patients with no gender predilection. Acral sites are preferred; arterial puncture for angiography can trigger the process. Lesions range from macules to ulcers 26; they are asymmetrical, with bizarre patterns varying from reticular to confluent (Figure 5a) and appear acutely. Fat particles, on the other hand, predominantly occlude capillaries. Clinically we therefore see purpura and petechiae (Figure 6). The patients mainly have respiratory symptoms caused by fat emboli in the lungs.
Figure 5.
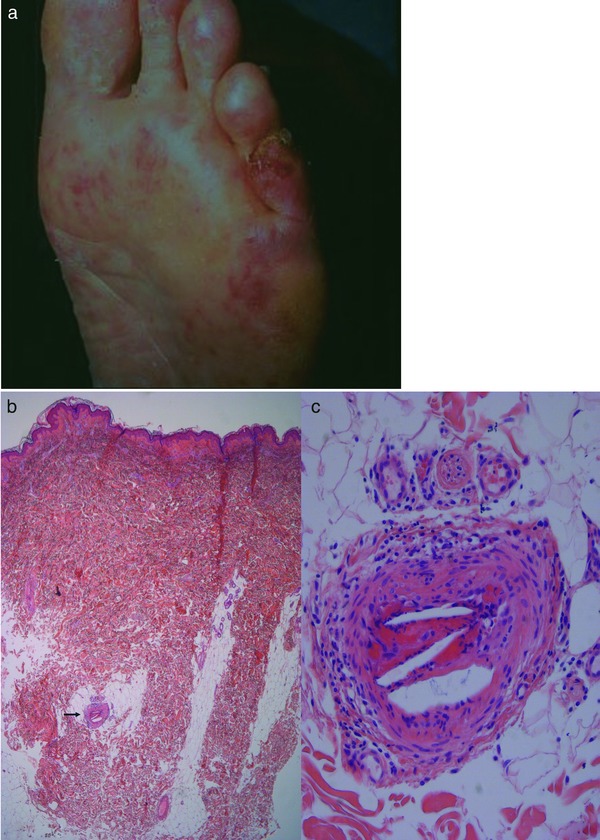
Cholesterol emboli. Bizarre livedo: clinically acute and sometimes painful livedo racemosa with irregular crusted erosions (a). Otherwise normal tissue specimen with abnormal artery at dermis‐subcutis border (b). Artery occluded by fibrin clot with cholesterol clefts in beginning reorganization by granulation tissue with corona of capillaries (c).
Figure 6.
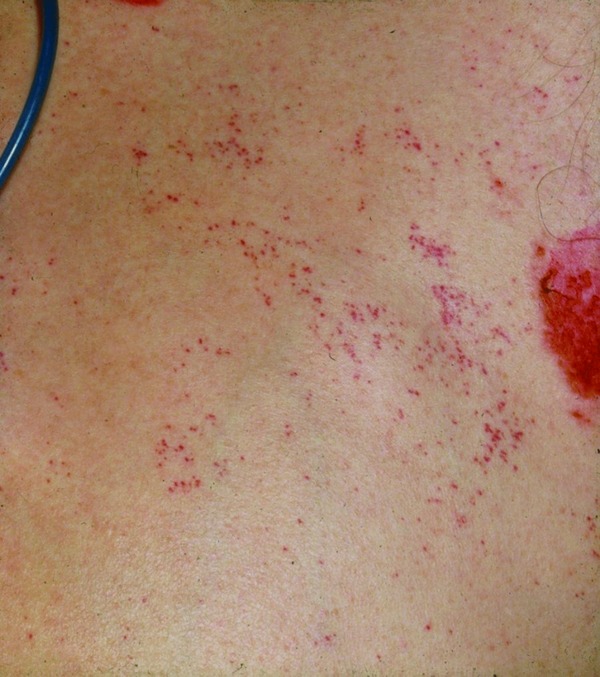
Fat emboli. Purpura and petechiae on the abdomen of a traumatized patient.
Histology: In the early stage, one may find cholesterol emboli (Figure 5b, c) occluding the vessel lumen. Superimposed on this process, we find thrombus formation which may dominate and make detection of cholesterol emboli difficult. Subsequently, lymphocytes appear. In due course, macrophages are attracted and subendothelial proliferation of fibrocytes occurs; at this stage, the cholesterol fragments become surrounded by foamy macrophages and multinucleate giant cells. Frequently, biopsies miss the characteristic features and only show an increase of elongated and dilated postcapillary venules in the upper and mid‐dermis, analogous to findings in livedo reticularis/racemosa. The same pathological process can occur with all other types of emboli.
Comment: Acute ischemia due to cardiac emboli causes a painful, pulseless extremity which is pallid and cool. Fat emboli as well as air and gas emboli cause acute tiny hemorrhages (petechiae), most commonly seen on the trunk (Figure 6). In embolia cutis medicamentosa (Nicolau syndrome) 27 (Figure 7), the intra‐arterial or mostly para‐arterial injection of drugs (agent incorrectly injected into the gluteal musculature) causes acute livedo patterning/retiform purpura and rapid bizarre necrotic lesions. In addition to hemorrhages, septic emboli occasionally show livedo patterning when medium vessels are involved. Finally, metastatic emboli rarely cause a transient and subtle livedo patterning if the disease progresses rapidly. Metastatic emboli usually show no livedo patterning, as the process does not occlude vessels acutely by obstruction, but is an active invasion of endothelium, vasculature and surrounding tissues. Similarly, intravascular proliferations such as reactive angioendotheliomatosis or intravascular lymphoma occlude vessels slowly 13.
Figure 7.
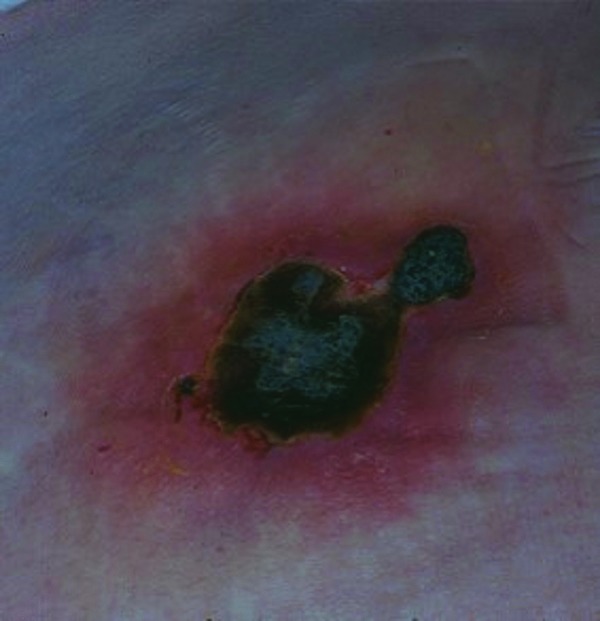
Embolia cutis medicamentosa. Bizarre necrosis surrounded by erythema and livedo racemosa on the buttock.
5. Thrombophlebitis
Iatrogenic thrombophlebitis (Table 8) following vein punctures or indwelling devices is by far the most common problem of occluding vasculopathy in daily practice. Thrombophlebitis may also occur concomitantly with infections, anti‐phospholipid syndrome and/or lupus erythematosus or malignancies.
Clinical picture: While iatrogenic thrombophlebitis is characteristically seen e.g. in the antecubital fossa (Figure 8), other forms tend to occur on the lower extremities. In younger patients, thrombophlebitis may be a sign of anti‐phospholipid syndrome; in older individuals, malignancy is more likely. An edematous and usually painful cord is characteristic. Mondor's disease is a characteristic thrombophlebitis on the lateral trunk associated with breast disease.
Figure 8.
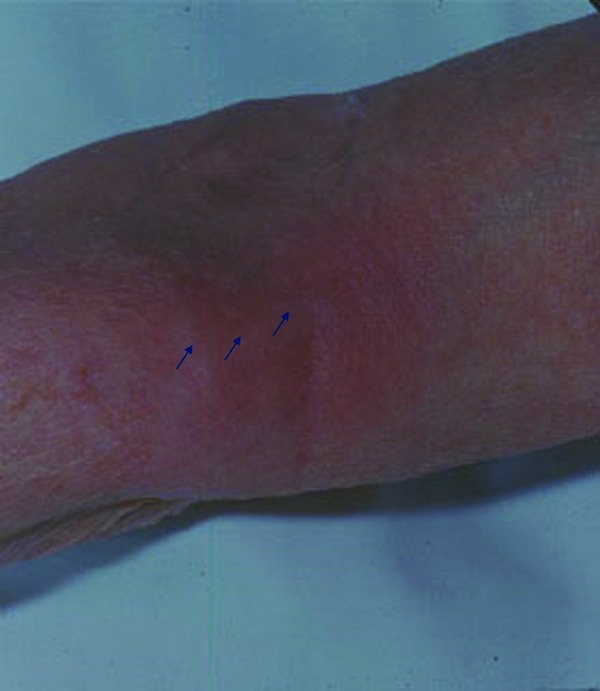
Thrombophlebitis. Iatrogenic thrombophlebitis (arrows indicate area of palpable cord) is mainly seen when indwelling devices are placed above joints.
Histology: Thrombophlebitis, though rarely biopsied, reveals thrombotic occlusion of large subcutaneous veins. Vessels are frequently tortuous and therefore the microtome will cut several lumina at various angles. The vessels are dilated and filled with thrombotic plugs, the endothelium is often missing, the muscle layers are loosened up, and there is an inflammatory infiltrate in and around the vein. The inflammation follows the pattern and time cycle of “lymphocytic vascular reorganization”. Due to subcutaneous tissue necrosis around the vein, one finds micropseudocysts and numerous polymorphonuclear neutrophils in early stages as well as foamy macrophages and foreign body giant cells in later stages of thrombophlebitis. Remarkably, thrombophlebitis is sometimes found unexpectedly in histopathology associated with uncharacteristic clinical appearances such as panniculitis.
We mention the following entities (arteriosclerosis and arteriolosclerosis) in our discussion of occluding vasculopathies, because the livedo pattern is one of the leading clinical features and anticoagulatory treatment is the most important therapeutic option, emphasizing their close relationship.
6. Arteriosclerosis and arteriolosclerosis
Next to thrombophlebitis, arteriosclerosis is the most frequent form of medium vessel vasculopathy in the skin and subcutis (Table 9). Histological changes are often found in skin specimens from the lower extremities as an incidental finding when surgery is performed for other indications. Patients do not develop symptoms if the process happens slowly and does not cause ischemia or necrosis, due to compensation by collateral vascularization. Clinically, patients sometimes show delayed healing after traumata because of impaired circulation. Such patients have frequently suffered from arterial hypertension and/or diabetes for years to decades. However, rapid occlusion of vessels due to arteriosclerosis may hinder the creation of collateral vessels and cause very painful spontaneous ulcers.
Clinical picture: Patients are prone to develop poorly healing post‐traumatic ulcers on the shin (Figure 9a). Such ulcers in combination with arterio/arteriolosclerosis, media sclerosis, arterial hypertension, diabetes and possible trauma are also are known by an eponym (Martorell ulcer) 28 or an acronym (HYTILU; hypertensive ischemic leg ulcers). They are usually solitary, discrete and asymmetrical. Livedo patterning and/or non‐inflammatory retiform purpura are often subtle. The skin itself is cold because of impaired circulation (cold livedo racemosa).
Figure 9.
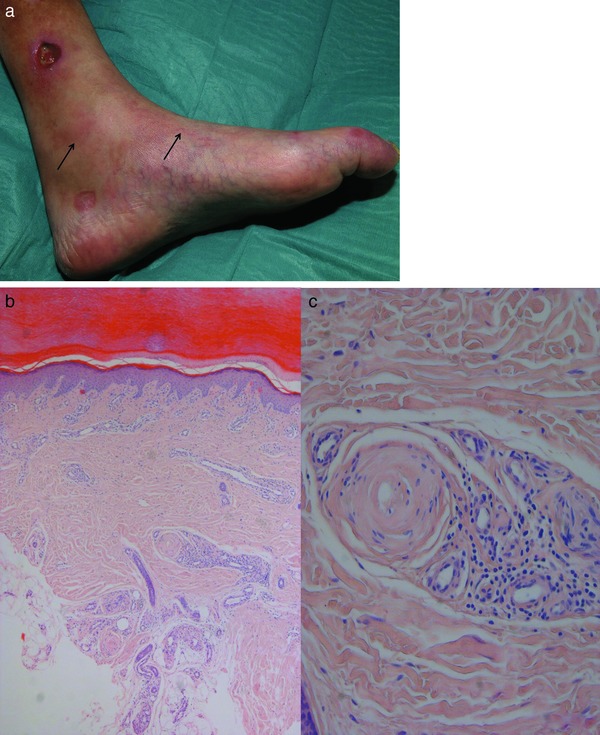
Arteriolosclerosis and arteriosclerosis. Cold (due to ischemia) subtle livedo racemosa (arrows) of lower leg with Martorell/HYTILU hypertensive ischemic leg ulcer (a). Prominent ectatic and elongated capillaries and postcapillary venules, the latter corresponding to livedo patterning, the former to stasis dermatosis; in the deep dermis, there is an occluded artery (b). Characteristic onion‐shaped sclerosis of shrunken artery surrounded by corona of capillaries (c).
Histology: Arteriolo‐ and arteriosclerosis (Figure 9b, c) characteristically show concentric or lamellar fibrosis to sclerosis of arterioles and small arteries in the lower dermis or subcutis. These zones may also show calcifications. The tunica media is frequently atrophic with few to no muscle layers; within the adventitia one finds some small capillaries as vasa vasorum, indicative of an old stage of granulation tissue. This process is analogous to the corona of capillaries seen in Sneddon syndrome. Usually there is no significant inflammatory infiltrate; occasionally one sees macrophages with iron or lipid storage.
7. Vasculopathy in neurofibromatosis
Besides thrombi and emboli, compression of vessels by surrounding tissue may cause livedo patterning (Table 10). Usually this process occurs very slowly, and compensation of blood flow via collaterals does not allow the development of the livedo pattern. If the process is very acute, superimposed by thrombi due to compression of numerous vessels, one will find subtle to moderate livedo racemosa, erosions and ulcerations.
Clinical picture: Vasculopathy in neurofibromatosis is a rare form. Vasculopathy in neurofibromatosis affects young adults, men more commonly than women. The proximal extremities and trunk are the preferred sites. Macules, erosions and ulcers are arranged asymmetrically, producing bizarre reticular to confluent patterns. A subtle, mottled livid livedo racemosa with some erosions or ulcers is characteristic (Figure 10).
Figure 10.
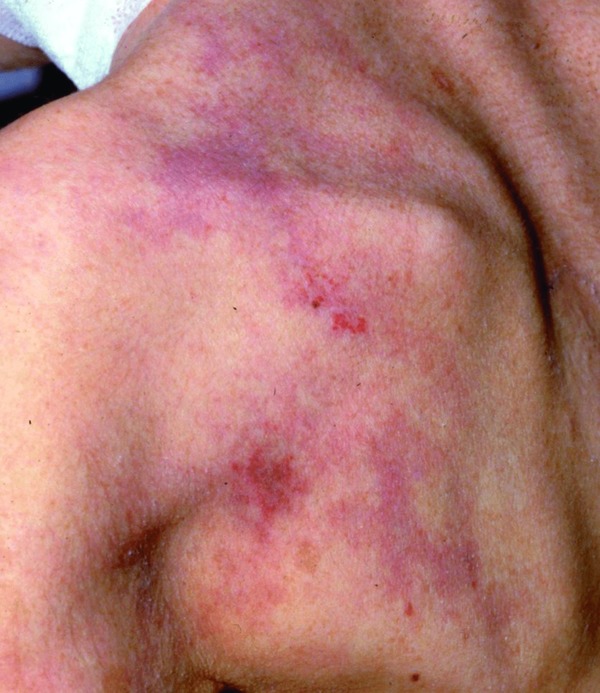
Vasculopathy in neurofibromatosis. Asymmetrical, bizarre macules and erosions surrounded by livedo racemosa.
Histology: First, extensive plexiform or diffuse neurofibromas surround and impinge on the vessels. Second, coagulation phenomena affect the blood flow; reorganization with subendothelial proliferation of fibroblasts and myofibroblasts further impairs the circulation.
Comment: The prognosis of this disease can be grim. We followed a cachectic 36 kg woman with arterial hypertension and extensive gastrointestinal involvement; she died from malabsorption and subsequent septic peritonitis at the age of 28 29.
The mentioned entities in the spectrum of coagulopathies/vasculopathies can benefit from an algorithmic approach. Applying the same questions makes the clinical and histopathological findings become clearer. The style of bar codes (Figure 11; Table 11) simplifies the results and may lead to inaccuracy, because it is usually difficult to put biology into tables. However, it can help to render these entities more understandable and comparable.
Figure 11.
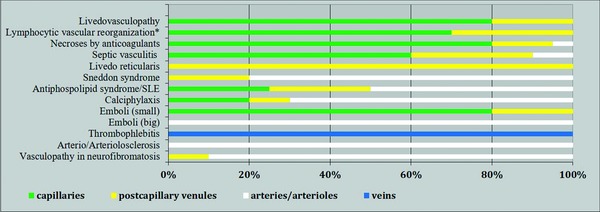
Association of entities/reaction patterns with vessel size. Emboli (small): fat, air, gas; emboli (large): cholesterol, oxalate, embolia cutis medicamentosa.
Conflict of interest
This manuscript was presented in part at National Specialist Dermatopathology EQA Scheme in Warwick, United Kingdom, November 8, 2013.
Acknowledgement
We remember our deceased mentor and friend Prof. Dr. Walter Burgdorf and thank him for his valuable input in the conception of this paper.
References
- 1. Ratzinger G, Zelger BG, Carlson JA et al. Vasculitic wheel – an algorithmic approach to cutaneous vasculitides. J Dtsch Dermatol Ges 2015; 13: 1092–117. [DOI] [PubMed] [Google Scholar]
- 2. Ratzinger G, Zelger BG, Zelger BW. Bar code reader – an algorithmic approach to cutaneous occluding vasculopathies? Part I: small vessel vasculopathies. J Dtsch Dermatol Ges 2019; 17(9): 895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jennette JC, Falk RJ, Andrassy K et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum 1994; 37: 187–92. [DOI] [PubMed] [Google Scholar]
- 4. Jennette JC, Falk RJ, Bacon PA et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013; 65: 1–11. [DOI] [PubMed] [Google Scholar]
- 5. Sunderkotter CH, Zelger B, Chen KR et al. Nomenclature of Cutaneous Vasculitis: Dermatologic Addendum to the 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheumatol 2018; 70: 171–84. [DOI] [PubMed] [Google Scholar]
- 6. Sunderkotter C, Lamprecht P, Mahr A et al. Nomenclature of cutaneous vasculitides – German translation of the dermatologic addendum to the 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. J Dtsch Dermatol Ges 2018; 16: 1425–32. [DOI] [PubMed] [Google Scholar]
- 7. Yus ES, Simon RS, Requena L. Vein, artery, or arteriole? A decisive question in hypodermal pathology. Am J Dermatopathol 2012; 34: 229–32. [DOI] [PubMed] [Google Scholar]
- 8. Chen KR. The misdiagnosis of superficial thrombophlebitis as cutaneous polyarteritis nodosa: features of the internal elastic lamina and the compact concentric muscular layer as diagnostic pitfalls. Am J Dermatopathol 2010; 32: 688–93. [DOI] [PubMed] [Google Scholar]
- 9. Dalton SR, Fillman EP, Ferringer T et al. Smooth muscle pattern is more reliable than the presence or absence of an internal elastic lamina in distinguishing an artery from a vein. J Cutan Pathol 2006; 33: 216–9. [DOI] [PubMed] [Google Scholar]
- 10. Requena L, Kutzner H, Angulo J et al. Generalized livedo reticularis associated with monoclonal cryoglobulinemia and multiple myeloma. J Cutan Pathol 2007; 34: 198–202. [DOI] [PubMed] [Google Scholar]
- 11. In SI, Han JH, Kang HY et al. The histopathological characteristics of livedo reticularis. J Cutan Pathol 2009; 36: 1275–8. [DOI] [PubMed] [Google Scholar]
- 12. Zelger B, Plorer A, Sepp N et al. [Differential livedo syndrome diagnosis]. Hautarzt 1995; 46: 369–79; quiz 77–8. [DOI] [PubMed] [Google Scholar]
- 13. Llamas‐Velasco M, Alegria V, Santos‐Briz A et al. Occlusive nonvasculitic vasculopathy. Am J Dermatopathol 2017; 39: 637–62. [DOI] [PubMed] [Google Scholar]
- 14. Fernandez Armenteros JM, Vea Jodar A, Matas Nadal C et al. Severe and recurrent levamisole‐induced cutaneous vasculopathy. J Cutan Pathol 2018; 45: 309–11. [DOI] [PubMed] [Google Scholar]
- 15. Wu S, Xu Z, Liang H. Sneddon's syndrome: a comprehensive review of the literature. Orphanet J Rare Dis 2014; 9: 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fritsch P, Zelger B. [Livedo vasculitis]. Hautarzt 1995; 46: 215–24; quiz 22–3. [DOI] [PubMed] [Google Scholar]
- 17. Sepp N, Zelger B, Schuler G et al. Sneddon's syndrome – an inflammatory disorder of small arteries followed by smooth muscle proliferation. Immunohistochemical and ultrastructural evidence. Am J Surg Pathol 1995; 19: 448–53. [DOI] [PubMed] [Google Scholar]
- 18. Giannelou A, Zhou Q, Kastner DL. When less is more: primary immunodeficiency with an autoinflammatory kick. Curr Opin Allergy Clin Immunol 2014; 14: 491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stockhammer G, Felber SR, Zelger B et al. Sneddon's syndrome: diagnosis by skin biopsy and MRI in 17 patients. Stroke 1993; 24: 685–90. [DOI] [PubMed] [Google Scholar]
- 20. Zelger B, Sepp N, Stockhammer G et al. Sneddon's syndrome. A long‐term follow‐up of 21 patients. Arch Dermatol 1993; 129: 437–47. [DOI] [PubMed] [Google Scholar]
- 21. Zelger B, Sepp N, Schmid KW et al. Life history of cutaneous vascular lesions in Sneddon's syndrome. Hum Pathol 1992; 23: 668–75. [DOI] [PubMed] [Google Scholar]
- 22. Gibson GE, Su WP, Pittelkow MR. Antiphospholipid syndrome and the skin. J Am Acad Dermatol 1997; 36: 970–82. [DOI] [PubMed] [Google Scholar]
- 23. Marneros AG, Blanco F, Husain S et al. Classification of cutaneous intravascular breast cancer metastases based on immunolabeling for blood and lymph vessels. J Am Acad Dermatol 2009; 60: 633–8. [DOI] [PubMed] [Google Scholar]
- 24. Garcia FVMJ, Sanz‐Sanchez T, Aragues M et al. Cutaneous embolization of cardiac myxoma. Br J Dermatol 2002; 147: 379–82. [DOI] [PubMed] [Google Scholar]
- 25. Lorenz EC, Michet CJ, Milliner DS et al. Update on oxalate crystal disease. Curr Rheumatol Rep 2013; 15: 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Panuncialman J, Falanga V. Unusual causes of cutaneous ulceration. Surg Clin North Am 2010; 90: 1161–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tabor D, Bertram CG, Williams AJK et al. Nicolau Syndrome (Embolia Cutis Medicamentosa): a rare and poorly recognized iatrogenic cause of cutaneous thrombotic vasculopathy. Am J Dermatopathol 2018; 40: 212–5. [DOI] [PubMed] [Google Scholar]
- 28. Hafner J, Nobbe S, Partsch H et al. Martorell hypertensive ischemic leg ulcer: a model of ischemic subcutaneous arteriolosclerosis. Arch Dermatol 2010; 146: 961–8. [DOI] [PubMed] [Google Scholar]
- 29. Obermoser G, Zelger BG, Millonig G et al. Vasculopathy in von Recklinghausen's neurofibromatosis – a diagnostic quandary. J Am Acad Dermatol 2004; 50: S107–9. [DOI] [PubMed] [Google Scholar]