

Abstract
Transformation of β‐oxoesters with PhI(OCOCF3)2 leads to α‐(ortho‐iodophenyl)‐β‐oxoesters. These materials are the starting point for the synthesis of 6‐carboxybenzo[b]azocin‐2‐ones by a sequence of aryl amination and ring transformation. This reaction sequence starts with copper‐catalyzed formation of N‐alkyl anilines from the iodoarenes and primary amines in the presence of K3PO4 as stoichiometric base. The intermediate products underwent ring transformation by addition of the nitrogen into the carbonyl group of the cycloalkanone, furnishing benzo‐annulated eight‐membered ring lactams. Under the same reaction conditions, the cyclohexanone and cycloheptanone derivatives gave no aminated products, but ring‐transformed to benzofuran derivatives. The title compounds of this investigation contain two points for further diversification (the lactam nitrogen and the carboxylate function), thus, the suitability of this compound class as a scaffold was proven by appropriate functionalizations. The first series of derivatizations of the scaffold was initiated by hydrogenolytic debenzylation of N‐benzyl derivative to provide the NH‐congener, which could be deprotonated with LDA and alkylated at nitrogen to give further examples of this compound class. Secondly, the ester function was submitted to saponification and the resulting carboxylic acid could be amidated using HATU as coupling reagent to furnish different amides.
Keywords: aryl amination, lactams, medium sized rings, ring expansion, scaffolds
Expanding rings: A sequence of aryl amination of α‐(iodophenyl)‐β‐oxoesters followed by ring transformation is the key to accessing hexahydrobenzo[b]azocin‐2‐on‐6‐carboxylic esters.
Introduction
Organic compounds that contain a benzannulated eight‐membered lactam ring (i.e. benzazocin‐2‐ones)1 have been considered as potential drugs, for example as inhibitors of the angiotensin converting enzyme (ACE).2 Furthermore, they show affinity as ligands for the dopamine D3 3 or the GABAA receptor.4 Moreover, some natural products possess this structural motif (Figure 1): Decursivine (1), an antimalarial indole alkaloid from Rhaphidophora decursiva,5 sulpinine C (2), an antiinsectan metabolite from Aspergillus sulphureus,6 the tryptamine derived balasubramide (3) from Clausena indica 7 and asporyzin A (4) from the fungus Aspergillus oryzae associated with the red alga Heterosiphonia japonica.8 Benzazocinones possess two chiral boat‐like conformations and the phenylene ring in them defines an element of planar chirality; the inversion barrier has been studied by NMR investigations and DFT calculations to be in the range of 30–100 kJ mol−1.9, 10
Figure 1.
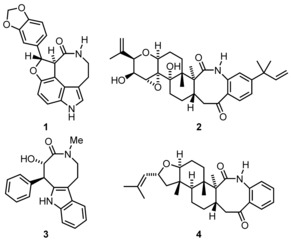
Four naturally occurring benzazocinone derivatives.
Synthetic routes to the azocane ring system were recently reviewed by Voskressensky.11, 12 An obvious synthetic access to hexahydrobenzo[b]azocin‐2‐one derivatives is provided by Beckmann rearrangement of oximes from benzosuberones.13 A less evident, though very efficient access to the target structure is achieved by oxidative cleavage of cyclopenta[b]indole derivatives with periodate.14 Not necessarily most effective, but very interesting routes to eight‐membered ring lactams involve ring expanding transformations.15 For example, Tan et al.16 accessed the target structure by ring expansion of indanones in a reaction sequence, which started with an aldol reaction with the ester enolate of ethyl acetate followed by Weinreb‐amide formation. The ring expanding transformation was then initiated by oxidation of the aromatic ring with PIFA, which led to intramolecular ipso‐substitution with trans‐annular C−C‐bond cleavage. Very similar was the route published by Liu et al.,17 who have replaced the PIFA‐oxidation step by photocatalysis with a Ru complex. A formal [6+2] cyclization of silyloxy alkynes and vinylazetidines leading to monocyclic azocanones was very recently reported by Wu et al.18
We have reported an access to eight‐membered ring lactams by ring transformation of ten different β‐oxoesters 5 with 1,4‐dicarbonyl motif (Scheme 1). Bi‐catalyzed conversion with 25 primary amines R2‐NH2 via azabicyclo[3.3.0]‐intermediates 8 furnished a library of about 250 hexahydroazocinones 9.19 This transformation was then applied to pyrrolidine 6 and tetrahydrothiophene derivatives 7 to furnish diazocanes 10 20 and thiazocanes 11.21 Furthermore, benzo‐ (products 12),10 pyrido‐ (products 13 and two regioisomers)22 and thienoannulated congeners 14 (and two regioisomers)23 were prepared.
Scheme 1.
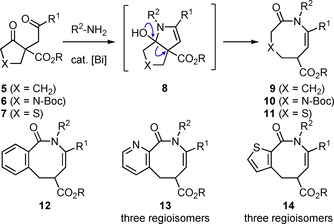
Preparation of eight‐membered ring lactams 9–14 by Bi‐catalyzed ring transformation of 1,4‐diketones 5–7 with primary amines; [Bi]=Bi(NO3)3⋅5 H2O.
A very elegant, asymmetric organocatalytic approach to benzo[b]azocinones 16 was recently published by Rodrigues, Coquerel and co‐workers, who ring‐expanded cyclobutanone derivatives.24 An illustrative example is given in Scheme 2. Cyclobutanoncarboxamide 15 was converted in an organocatalyzed Michael addition with ortho‐(Boc‐amino)‐ω‐nitrostyrene to furnish the lactam 16 with good yield and remarkable stereoselectivity. The transformation proceeded via the product of the conjugated addition, compound 17, which underwent ring transformation via an azabicyclo[4.2.0]intermediate after addition of the carbamate nitrogen to the carbonyl group within the four‐membered ring.
Scheme 2.
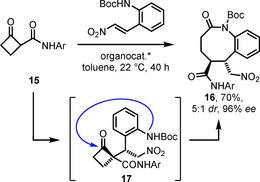
Ring transformation after organocatalyzed Michael addition; Ar=4‐C6H4CO2Et.
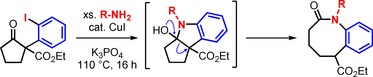
In the present work we propose the preparation of hexahydrobenzo[b]azocin‐2‐on‐6‐carboxylates 18 by ring transformation of β‐oxoesters 20 with an α‐(ortho‐iodophenyl)‐residue (Scheme 3). Our plan is to perform aryl amination with primary amines R‐NH2. Expected products would undergo cyclization to azabicyclo[3.3.0]‐intermedates 19 similar to intermediates 8 in Scheme 1. The project is actually based on the availability of compounds 20, which can be conveniently accessed by iodophenylation of a β‐oxoester with PhI(OCOCF3)2 [PIFA, phenyliodobis(trifluoroacetate)], which was recently reported by Shafir and co‐workers.25
Scheme 3.
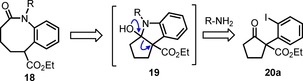
Preparation of hexahydrobenzo[b]azocin‐2‐on‐6‐carboxylates 18 from β‐oxoesters 20 with an α‐(ortho‐iodophenyl)‐residue.
Results and Discussion
The starting materials of this study, α‐(ortho‐iodophenyl)‐β‐oxoesters 20 a–20 c were accessed from the β‐oxoesters 21 a–21 c following the original report25 with stoichiometric amount of PIFA and TFAA (trifluoroacetic anhydride) in a mixture of MeCN and TFA (trifluoroacetic acid). In our hands, the yields in the range of 47–63 % were a little bit better compared to the literature (Scheme 4). The product constitution is proposed to result from a [3,3]‐sigmatropic rearrangement (“ioda‐Claisen reaction”) of an intermediate 22 which was formed by substitution of a trifluoroacetate residue by the enol tautomer of the oxoesters 21 a–21 c at the hypervalent iodine atom. This rearrangement is followed by rearomatization by tautomerization and reductive elimination of TFA from a hypervalent iodine species.
Scheme 4.
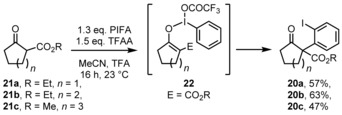
Literature known preparation of starting materials 20 a–20 c from oxoesters 20 a–20 c and PIFA.
We have chosen the Buchwald–Hartwig26 coupling reaction27 for our first efforts for aryl amination of compound 20 a with the primary amine BnNH2. As precatalysts we have chosen Pd2(dba)3 with BINAP, DPPF, and Xantphos as ligands and Cs2CO3, KHMDS and NaOtBu as bases in refluxing toluene, however, acyclic product 24 a was observed as the only isolable and unique compound together with several unspecified decomposition products. Compound 24 a results from two processes: Pd‐catalyzed reductive deiodination and retro‐Claisen reaction induced by intermolecular nucleophilic attack of the amine to the endocyclic carbonyl group. Therefore, we turned to Ullmann‐type28 condensations29 with catalytic amounts of CuI and Cs2CO3 (with or without phenanthroline as ligand) in solvents like 1,4‐dioxane, DMF, and acetonitrile, and we were indeed able to detect the target structure 18 a in the reaction mixture. Finally, inspired by reports of Buchwald et al.,30 we used K3PO4 as base, and the amount of product 18 a increased (Scheme 5). After screening of reaction temperature and solvent, we ultimately identified the following optimal reaction conditions for the formation of compound 18 a: 0.15 equivalents CuI and 2 equivalents K3PO4 in neat BnNH2 at 110 °C for 16 h gave 56 % yield of product 18 a. Cyclopenta[b]benzofurane derivative 23 a was formed as a byproduct and could be isolated in 9 % yield, which results presumably from Cu‐mediated carbon–oxygen coupling and subsequent elimination of water from an intermediate hemiacetal. Furthermore, deiodinated and ring‐opened byproduct 24 a was isolated in 16 %. We then submitted various primary amines to the conversion with oxoester 18 a under the optimized conditions and were able to isolate further five lactams 18 b–18 f together with varying amounts of benzofuran 23 a as well as acyclic products 24 and 25 with (X=H) or without (X=I) reductive deiodination as byproducts (see Table 1). For alkylamines (R=nBu, nHex, Cy and allyl) the products 18 b–18 e were obtained in ca. 50 % yield. For 2‐ethoxyethylamine, the yield was slightly lower (product 18 f in 38 % yield). Table 1 lists the yields of the major products 18 a–18 f as well as the yields of byproducts 23 a, 24 a, 24 c–24 e, 25 b, and 25 c.
Scheme 5.
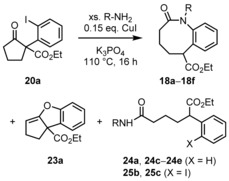
Benzazocinone formation after Ullmann‐type aryl amination, for residues R and X as well as yields see Table 1.
Table 1.
Residues R, X and yields of benzoazocinones 18 and byproducts 23 a, 24 and 25.
|
R |
Product 18 |
Byproduct 23 a |
Byproduct 24 |
Byproduct 25 |
|---|---|---|---|---|
|
Bn |
56 % (18 a) |
9 % |
16 % (24 a) |
0 % |
|
nBu |
51 % (18 b) |
0 % |
0 % |
15 % (25 b) |
|
nHex |
50 % (18 c) |
4 % |
16 % (24 c) |
2 % (25 c) |
|
Cy |
50 % (18 d) |
11 % |
13 % (24 d) |
0 % |
|
allyl |
49 % (18 e) |
0 % |
20 % (24 e) |
0 % |
|
CH2CH2OEt |
38 % (18 f) |
0 % |
0 % |
0 % |
Conversion of the congeners 20 b and 20 c under the respective reaction conditions with benzyl‐ or butylamine did not give lactams as products, but the dibenzofuran and cyclohepta[b]benzofuran derivatives 23 b and 23 c were isolated (in 43 and 18 % yield, respectively; Scheme 6).
Scheme 6.
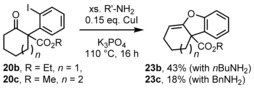
Formation of dibenzofurane and cyclohepta[b]benzofuran derivatives 23 b and 23 c.
Compound 18 b was obtained as a crystalline material suitable for single crystal X‐ray structure determination.31 In Figure 2, a representation of the molecular structure is given. Being a carboxamide, the nitrogen atom N1 is planar (angles C2‐N1‐C10a 123.26°, C2‐N1‐C1′ 120.30°, C1′‐N1‐C10a 116.12°, sum 359.68°) and the C2−N1 bond with a length of 1.3658 Å is rather a double bond. The bond N1−C10a with 1.4289 Å is a single bond. The eight and six membered rings are almost perpendicular at their junction (dihedral angles C2‐N1‐C10a‐C6a 60.75° and C4‐C5‐C6‐C6a 86.16°). Therefore, there seems to be no electronic influence of the amide group towards the aromatic ring, which is also reflected by the chemical shifts of the four aromatic protons in the 1H NMR spectrum (δ 7.21–7.35 ppm).
Figure 2.
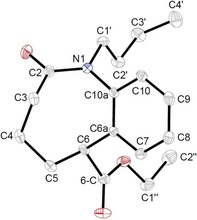
The ORTEP‐representation of the molecular structure of compound 18 b in the solid state proves the constitution.
In order to prove the versatility of the benzoazocinones 18 as new heterocyclic scaffolds, we envisioned diversifying transformations at the lactam‐nitrogen and the exocyclic carboxyl function. First of all, the benzyl group of compound 18 a was removed with H2/Pd/C to furnish compound 18 g (79 %, Scheme 7). In order to achieve full conversion, the temperature had to be raised to 50 °C, upon which the aromatic ring of part of the starting material was hydrogenated to furnish the N‐(cyclohexylmethyl) congener 18 h (10 %). After NH deprotonation with LDA, it was reacted with various alkyl bromides. First of all, the N‐allyl compound 18 e was isolated in surprisingly low yield (24 %, 46 % brsm) together with some starting material 18 g. On the other hand, the prenylation proceeded straightforwardly without allylic inversion (75 % of product 18 i). With methyl bromoacetate, compound 18 j was obtained in 79 % yield. Introducing some steric hindrance with the secondary halide ethyl α‐bromopropionate gave again lower yield (product 18 k in 34 %, 53 % brsm) together with recovered starting materials 18 g. Interestingly, this compound was isolated as two diastereoisomers with 87:13 dr, which is rather a remarkable selectivity considering the 1,5‐distance of the two stereocenters.
Scheme 7.
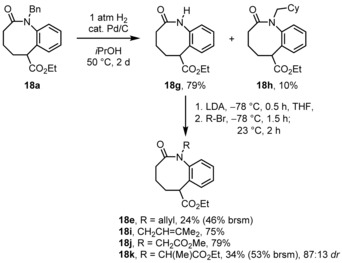
Hydrogenolytic debenzylation of the lactam‐nitrogen followed by alkylation reaction.
For the second diversifying strategy we first submitted compound 18 a to ester saponification yielding compound 26 in 81 % yield (Scheme 8). It was then coupled with the HATU–DIPEA protocol32 [HATU=O‐(7‐azabenzotriazol‐1‐yl)‐N,N,N′,N′‐tetramethyluronium hexafluorophosphate), DIPEA=ethyldiisopropylamine] with the ethyl esters of aminoisobutyric acid and β‐alanine to give the amides 27 a and 27 b in good yield (87 % and 85 %, respectively). By application of the same reaction conditions, trifluoroethylamine could be coupled to furnish compound 27 c with 88 % yield.
Scheme 8.
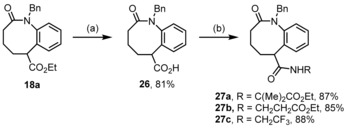
Ester saponification and amide coupling. Reagents and conditions: (a) NaOH, H2O‐EtOH, 80 °C, 16 h; (b) HATU, DIPEA, 1.5 equiv R‐NH2, CH2Cl2, 23 °C, 16 h.
Furthermore, we intended to prepare the 6‐amino derivative of the scaffold by Hofmann degradation of the carboxylate function in compound 26. We relied on a literature procedure applying the hypervalent iodine reagent PIDA [PhI(OAc)2] (Scheme 9).20 First of all, the parent unsubstituted amide 27 d was prepared in 70 % yield by activation of the acid 26 with Boc2O and conversion of the mixed anhydride with hartshorn salt (ammonium carbonate). The degradation proceeded with PIDA and the intermediate isocyanate was removed with MeOH to furnish the carbamate 28, however, the yield was moderate.
Scheme 9.
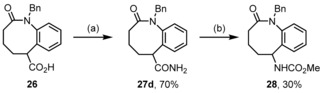
Preparation and Hofmann degradation of amide 27 d. Reagents and conditions: (a) 1. 1.5 equiv Boc2O, 1.8 equiv pyridine, 1,4‐dioxane, 23 °C, 0.5 h; 2. 2.8 equiv (NH4)2CO3, 23 °C, 16 h; (b) 1.0 equiv PIDA, 2.5 equiv KOH, MeOH, CH2Cl2, 0 °C→23 °C, 16 h.
Finally, the N‐allyl group of compound 18 e seemed to be perfectly suited for further transformations, for example, olefin cross‐metathesis. Therefore, we converted it with an excess of methyl acrylate in the presence of one of Evonik's catMETium RF catalysts33 (Scheme 10). The internal olefin 18 l was obtained exclusively as trans‐diastereoisomer together with some unreacted starting material (44 % yield, 59 % brsm).
Scheme 10.
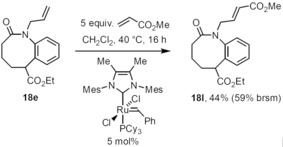
Olefin cross‐metathesis of allylic amide 18 e; Mes=2,4,6‐Me3C6H2.
Conclusions
A novel synthesis of benzo[b]azocin‐2‐ones by a sequence of aryl amination and ring transformation of ethyl 1‐(ortho‐iodophenyl)‐2‐oxocyclopentancarboxylate 20 a was introduced. Additionally, the nitrogen atom and the carboxylate function define two points for further diversification, thus, the suitability of this compound class as a scaffold was proven by appropriate functionalization. Starting point of this investigation was the preparation of α‐(ortho‐iodophenyl)‐β‐oxoesters 20 a–20 c by transformation of β‐oxoesters 21 a–21 c with PhI(OCOCF3)2 (PIFA). After aryl amination of the cyclopentanone congener 20 a with six primary amines, which was accomplished with catalytic amounts of CuI and K3PO4 as stoichiometric base, the intermediate N‐alkyl anilines underwent ring transformation by addition of the nitrogen into the carbonyl group of the cycloalkanone, furnishing the benzo‐annulated eight‐membered ring lactams 18 a–18 f (38–56 % yield). Under the same reaction conditions, the cyclohexanone and cycloheptanone derivatives 20 b and 20 c gave no aminated products, but ring‐transformed to benzofuran derivatives 23 b and 23 c. The first series of derivatizations of the scaffold was initiated by hydrogenolytic debenzylation of N‐benzyl derivative 18 a to provide the NH‐congener 18 g, which could be deprotonated with LDA and alkylated at nitrogen to give further examples 18 i–18 k of this compound class. Another representative (product 18 l) was obtained by olefin cross‐metathesis of N‐allyl lactam 18 e with methyl acrylate. Secondly, the ester function of compound 18 a was submitted to saponification (81 % yield) and the resulting carboxylic acid 26 could be amidated using HATU as coupling reagent to furnish three different amides 27 a–27 c (85–88 % yield). The N‐unsubstituted parent amide 27 d was obtained by amidation with (NH4)2CO3 and could be further transformed by Hofmann degradation using PhI(OAc)2 (PIDA) and MeOH to give carbamate 28 (30 % yield).
Experimental Section
General: Preparative column chromatography was carried out using Merck SiO2 (35–70 μm, type 60A) with hexanes (mixture of isomers, bp. 64–71 °C), tert‐butyl methyl ether (MTBE), EtOAc, and MeOH as eluents. TLC was performed on aluminum plates coated with SiO2 F254. 1H, 19F and 13C NMR spectra were recorded on Bruker Avance DRX 500 and 300 instruments. Multiplicities of carbon signals were determined with DEPT experiments. MS and HRMS spectra of products were obtained with a Waters Q‐TOF Premier (ESI, pos. mode) or Thermo Scientific DFS (EI) spectrometers. IR spectra were recorded on a Bruker Tensor 27 spectrometer equipped with a diamond ATR unit. Compounds 20 a–20 c were literature known and prepared accordingly.25 All other starting materials were commercially available.
General procedure A (GPA) for the α‐arylation of β‐oxoesters 21 a–21 c:25 Under exclusion of air and moisture (nitrogen atmosphere), TFAA (1.5 equiv) was added dropwise to a stirred solution of PIFA (1.3 equiv) and TFA (1.5 L mol−1 PIFA) in MeCN (1.5 L mol−1 PIFA) and the resulting mixture was stirred at ambient temperature for 15 min. Then β‐oxoester 21 (1.0 equiv) was added and the resulting mixture was further stirred at ambient temperature for 16 h. The solvent was removed in vacuum and the residue was purified by column chromatography to yield arylated β‐oxoesters 20 a–20 c.
Ethyl 1‐(2‐iodophenyl)‐2‐oxocyclopentane‐1‐carboxylate (20 a):25 According to GPA, TFAA (2.52 g, 12.0 mmol), PIFA (4.47 g, 10.4 mmol) and β‐oxoester 21 a (1.25 g, 8.00 mmol) were converted in TFA (16 mL) and MeCN (16 mL) to furnish the title compound 20 a (1.64 g, 4.58 mmol, 57 %) after chromatography (SiO2, hexanes/MTBE 3:1, R f=0.30) as a colorless solid. M.p. 74 °C. 1H NMR (300 MHz, CDCl3): δ=1.24 (t, J=7.1 Hz, 3 H), 1.63–1.80 (m, 1 H), 2.03–2.15 (m, 1 H), 2.44–2.57 (m, 3 H), 3.20 (ddd, J=13.5 Hz, J=9.7 Hz, J=7.0 Hz, 1 H), 4.14–4.30 (m, 2 H), 6.92–6.97 (m, 2 H), 7.28 (td, J=7.8 Hz, J=1.3 Hz, 1 H), 7.93 (dd, J=8.3 Hz, J=1.2 Hz, 1 H) ppm. All spectroscopic data are in accordance with the literature.25 C14H15IO3 (358.18 g mol−1).
For the preparation of compounds 20 b and 20 c see the Supporting Information.
General procedure B (GPB) for the Ullmann type coupling of β‐oxoesters 20 a–20 c with amines: Under exclusion of air and moisture (nitrogen atmosphere), a Schlenk tube was charged with α‐arylated β‐oxoester 20 (1.0 equiv), K3PO4 (2.0–3.0 equiv) and CuI (15 mol %), three times evacuated and flushed with nitrogen. The amine (1‐1.8 L mol−1) was then added and the tube was tightly closed. The resulting mixture was stirred at 110 °C for 16 h and subsequently cooled to ambient temperature. The mixture was diluted with MTBE (20 L mol−1), water (20 L mol−1) and sat. aqueous NH4Cl solution (2 L mol−1) and the layers were separated. The aqueous layer was extracted with MTBE (2×20 L mol−1). The combined organic layers were dried (MgSO4), filtered and the solvent was removed in vacuo. The crude product was purified by column chromatography to furnish benzazocinones 18 together with byproducts 23, 24, and 25.
Conversion of β‐oxoester 20 a with benzylamine: According to GPB, β‐oxoester 20 a (179 mg, 500 μmol), K3PO4 (212 mg, 1.00 mmol) and CuI (14 mg, 75 μmol) were converted with benzylamine (0.5 mL). The crude product was purified by column chromatography (SiO2, hexanes/MTBE 1:2) to yield the benzofuran 23 a (10 mg, 43 μmol, 9 %, R f=0.65) as a pale yellow oil. Secondly, the benzazocinone 18 a (95 mg, 0.28 mmol, 56 %, R f=0.30) was eluted as a pale yellow solid. M.p. 60–63 °C. As the third fraction, the acyclic amide 24 a (28 mg, 82 μmol, 16 %, R f=0.12) was obtained as a pale yellow oil.
Ethyl 1‐benzyl‐2‐oxo‐1,2,3,4,5,6‐hexahydrobenzo[b]azocine‐6‐carboxylate (18 a): 1H NMR (500 MHz, CDCl3): δ=1.10 (t, J=7.1 Hz, 3 H), 1.49 (dddd, J=14.1 Hz, J=12.7 Hz, J=11.2 Hz, J=5.6 Hz, 1 H), 1.76–1.85 (m, 1 H), 1.88–1.96 (m, 2 H), 2.30–2.34 (m, 1 H), 2.38 (dd, J=14.1 Hz, J=5.0 Hz, 1 H), 3.20 (dd, J=11.2 Hz, J=0.9 Hz, 1 H), 3.97–4.03 (m, 2 H), 4.68 (d, J=14.0 Hz, 1 H), 5.33 (d, J=14.0 Hz, 1 H), 7.17–7.19 (m, 1 H), 7.22–7.30 (m, 8 H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ=14.06 (CH3), 24.42 (CH2), 32.28 (CH2), 33.13 (CH2), 44.69 (CH), 52.69 (CH2), 60.59 (CH2), 125.95 (CH), 127.31 (CH), 127.40 (CH), 127.92 (CH), 128.42 (2 CH), 128.49 (CH), 129.26 (2 CH), 136.74 (C), 139.01 (C), 140.62 (C), 173.68 (C), 173.99 (C) ppm. IR (ATR): ṽ=2941 (w), 2928 (w), 1728 (vs), 1651 (vs), 1493 (m), 1453 (m), 1393 (m), 1296 (m), 1225 (m), 1185 (vs), 1148 (m), 1027 (m), 759 (m), 733 (m), 701 (s) cm−1. HR‐MS (EI, 70 eV): calcd 337.1672 (for C21H23NO3 +), found 337.1665 [M +]. C21H23NO3 (337.42 g mol−1).
Ethyl 2,8 b–dihydro‐1H‐cyclopenta[b]benzofuran‐8 b,carboxylate (23 a): 1H NMR (500 MHz, CDCl3): δ=1.16 (t, J=7.1 Hz, 3 H), 2.27 (ddd, J=11.6 Hz, J=9.9 Hz, J=7.9 Hz, 1 H), 2.44 (ddd, J=14.9 Hz, J=7.9 Hz, J=4.0 Hz, 1 H), 2.78 (dddd, J=14.9 Hz, J=9.9 Hz, J=5.3 Hz, J=1.5 Hz, 1 H), 2.85 (dd, J=11.6 Hz, J=5.3 Hz, 1 H), 4.07–4.13 (m, 2 H), 5.24 (dd, J=4.0 Hz, J=1.5 Hz, 1 H), 6.97–7.01 (m, 2 H), 7.23 (td, J=7.9 Hz, J=1.5 Hz, 1 H), 7.33 (dd, J=7.4 Hz, J=1.1 Hz, 1 H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ=13.98 (CH3), 31.32 (CH2), 37.84 (CH2), 61.40 (CH2), 63.00 (C), 101.56 (CH), 110.74 (CH), 122.51 (CH), 124.81 (CH), 129.03 (C), 129.17 (CH), 161.67 (C), 162.25 (C), 171.76 (C) ppm. IR (ATR): ṽ=2958 (m), 2929 (m), 2859 (m), 1727 (vs), 1686 (m), 1607 (m), 1456 (s), 1239 (s), 1152 (s), 1101 (m), 1017 (m), 835 (m), 751 (s) cm−1. HR‐MS (ESI): calcd 237.1097 (for C14H14LiO3 +), found 237.1105 [M+Li+]. C14H14O3 (230.26 g mol−1).
Ethyl 6‐(benzylamino)‐6‐oxo‐2‐phenylhexanoate (24 a): 1H NMR (500 MHz, CDCl3): δ=1.18 (t, J=7.1 Hz, 3 H), 1.54–1.69 (m, 2 H), 1.75–1.82 (m, 1 H), 2.04–2.12 (m, 1 H), 2.15–2.25 (m, 2 H), 3.52 (t, J=7.6 Hz, 1 H), 4.02–4.16 (m, 2 H), 4.40 (d, J=5.8 Hz, 2 H), 5.80 (br s, 1 H), 7.22–7.33 (m, 10 H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ=14.08 (CH3), 23.63 (CH2), 33.01 (CH2), 36.25 (CH2), 43.55 (CH2), 51.52 (CH), 60.77 (CH2), 127.21 (CH), 127.46 (CH), 127.80 (4 CH), 128.59 (2 CH), 128.65 (2 CH), 138.25 (C), 138.89 (C), 172.16 (C), 173.82 (C) ppm. IR (ATR): ṽ=3294 (w), 2931 (w), 1731 (s), 1646 (s), 1546 (m), 1456 (m), 1174 (m), 1150 (s), 1030 (m), 734 (m), 699 (vs) cm−1. HR‐MS (EI, 70 eV): calcd 339.1829 (for C21H25NO3 +), found 339.1817 [M +]. C21H25NO3 (339.44 g mol−1).
For the conversion of β‐oxoester 20 a with n‐butylamine (products 18 b, 25 b), n‐hexylamine (products 18 c, 24 c, 25 c), cyclohexylamine (products 18 d, 24 d), allylamine (products 18 e, 24 e), and 2‐ethoxyethylamine (product 18 f) see the Supporting Information.
Ethyl 2,3,4,4 a‐tetrahydrodibenzofuran‐4 a‐carboxylate (23 b): According to GPB, β‐oxoester 20 b (105 mg, 279 μmol), K3PO4 (178 mg, 837 μmol) and CuI (8 mg, 0.04 mmol) were converted in n‐butylamine (0.5 mL) to yield the title compound 23 b (29 mg, 0.12 mmol, 43 %) after chromatography (SiO2, hexanes/MTBE 20:1, R f=0.23) as a colorless oil. 1H NMR (500 MHz, CDCl3): δ=1.16 (t, J=7.1 Hz, 3 H), 1.57–1.64 (m, 2 H), 1.86–1.90 (m, 1 H), 2.19–2.31 (m, 2 H), 2.83 (dd, J=8.6 Hz, J=3.1 Hz, 1 H), 4.11 (q, J=7.1 Hz, 2 H), 5.31 (t, J=3.8 Hz, 1 H), 6.91–6.96 (m, 2 H), 7.20 (td, J=7.9 Hz, J=1.2 Hz, 1 H), 7.31 (dd, J=7.4 Hz, J=0.8 Hz, 1 H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ=13.96 (CH3), 19.03 (CH2), 21.97 (CH2), 29.84 (CH2), 54.13 (C), 61.46 (CH2), 99.97 (CH), 109.80 (CH), 121.78 (CH), 124.05 (CH), 128.62 (C), 129.32 (CH), 155.61 (C), 157.87 (C), 171.66 (C) ppm. IR (ATR): ṽ=2981 (w), 2936 (w), 2916 (w), 1728 (vs), 1609 (w), 1596 (w), 1472 (m), 1461 (s), 1223 (vs), 1174 (m), 1157 (m), 1128 (m), 1102 (m), 1087 (vs), 1072 (m), 1022 (m), 751 (s) cm−1. HR‐MS (ESI): calcd 251.1254 (for C15H16LiO3 +), found 251.1251 [M+Li+]. C15H16O3 (244.29 g mol−1).
Methyl 8,9,10,10 a‐tetrahydro‐7H‐cyclohepta[b]benzofuran‐10 a‐carboxylate (23 c): According to GPB, β‐oxoester 20 c (105 mg, 279 μmol), K3PO4 (178 mg, 837 μmol) and CuI (8 mg, 0.04 mmol) were converted in benzylamine (0.5 mL) to yield the title compound 23 c (12 mg, 49 μmol, 18 %) after chromatography (SiO2, hexanes/MTBE 20:1, R f=0.21) as a colorless solid. M.p. 75 °C. 1H NMR (500 MHz, CDCl3): δ=1.38–1.47 (m, 1 H), 1.68–1.80 (m, 3 H), 2.04–2.10 (m, 1 H), 2.12–2.16 (m, 2 H), 2.59–2.63 (m, 1 H), 3.73 (s, 3 H), 5.66 (dd, J=7.5 Hz, J=6.5 Hz, 1 H), 6.85 (d, J=8.0 Hz, 1 H), 6.93 (td, J=7.5 Hz, J=0.9 Hz, 1 H), 7.20 (td, J=8.0 Hz, J=1.4 Hz, 1 H), 7.27 (dd, J=7.2 Hz, J=1.1 Hz, 1 H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ=24.55 (CH2), 27.52 (CH2), 28.85 (CH2), 35.45 (CH2), 52.78 (CH3), 58.75 (C), 104.59 (CH), 109.47 (CH), 121.60 (CH), 123.81 (CH), 129.46 (CH), 129.57 (C), 156.98 (C), 159.98 (C), 171.15 (C) ppm. IR (ATR): ṽ=2926 (m), 2851 (w), 1732 (vs), 1701 (m), 1597 (m), 1476 (s), 1463 (s), 1237 (vs), 1221 (vs), 1158 (m), 1137 (m), 1093 (m), 1073 (m), 1057 (m), 999 (m), 820 (m), 749 (vs) cm−1. HR‐MS (ESI): calcd 251.1254 (for C15H16LiO3 +), found 251.1257 [M+Li+]. C15H16O3 (244.29 g mol−1).
N ‐Debenzylation of benzazocinone 18 a: A suspension of 10 % Pd/C (883 mg, 830 μmol) and benzazocinone 18 a (560 mg, 1.66 mmol) in iPrOH (8 mL) was stirred at 50 °C for 2 d under an atmosphere of hydrogen (1 bar). The mixture was then filtered and the solvent was removed in vacuo. The mixture was submitted to column chromatography (SiO2, hexanes/MTBE 1:5) to yield in the first fraction benzazocinone 18 h (59 mg, 0.17 mmol, 10 %, R f=0.40) as a colorless oil. Secondly, benzazocinone 18 g (324 mg, 1.31 mmol, 79 %, R f=0.16) was obtained as a colorless solid. M.p. 95–100 °C.
Ethyl 2‐oxo‐1,2,3,4,5,6‐hexahydrobenzo[b]azocine‐6‐carboxylate (18 g): 1H NMR (500 MHz, CDCl3): δ=1.18 (t, J=7.1 Hz, 3 H), 1.58–1.67 (m, 1 H), 1.73–1.82 (m, 1 H), 1.93–1.98 (m, 2 H), 2.29–2.33 (m, 1 H), 2.50–2.53 (m, 1 H), 3.71 (d, J=10.8 Hz, 1 H), 4.09–4.19 (m, 2 H), 7.16–7.18 (m, 1 H), 7.26–7.32 (m, 2 H), 7.35–7.36 (m, 1 H), 8.29 (s, 1 H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ=14.00 (CH3), 23.65 (CH2), 32.14 (CH2), 32.51 (CH2), 45.02 (CH), 60.95 (CH2), 125.68 (CH), 127.14 (CH), 127.84 (CH), 128.24 (CH), 135.52 (C), 137.55 (C), 173.92 (C), 176.67 (C) ppm. IR (ATR): ṽ=3189 (w), 2945 (w), 1727 (s), 1659 (vs), 1495 (m), 1443 (m), 1390 (m), 1371 (m), 1301 (m), 1222 (m), 1185 (s), 1142 (m), 1096 (m), 1048 (m), 1017 (m), 764 (s), 734 (m) cm−1. HR‐MS (EI, 70 eV): calcd 247.1203 (for C14H17NO3 +), found 247.1196 [M +]. C14H17NO3 (247.29 g mol−1).
Ethyl 1‐(cyclohexylmethyl)‐2‐oxo‐1,2,3,4,5,6‐hexahydrobenzo[b]azocine‐6‐carboxylate (18 h): 1H NMR (500 MHz, CDCl3): δ=1.05–1.19 (m, 4 H), 1.17 (t, J=7.1 Hz, 3 H), 1.51–1.71 (m, 7 H), 1.80–1.86 (m, 3 H), 1.90–1.96 (m, 1 H), 2.23 (dd, J=11.1 Hz, J=8.1 Hz, 1 H), 2.43–2.46 (m, 1 H), 3.22 (dd, J=13.5 Hz, J=5.2 Hz, 1 H), 3.63 (d, J=10.7 Hz, 1 H), 4.06–4.21 (m, 3 H), 7.20–7.22 (m, 1 H), 7.27–7.31 (m, 2 H), 7.33–7.36 (m, 1 H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ=14.12 (CH3), 24.27 (CH2), 25.80 (CH2), 26.00 (CH2), 26.25 (CH2), 31.51 (CH2), 31.91 (CH2), 32.16 (CH2), 33.26 (CH2), 36.82 (CH), 44.81 (CH), 55.38 (CH2), 60.91 (CH2), 125.52 (CH), 126.99 (CH), 128.02 (CH), 128.20 (CH), 138.68 (C), 141.46 (C), 173.93 (C), 174.43 (C) ppm. IR (ATR): ṽ=2924 (m), 2851 (w), 1732 (vs), 1652 (vs), 1493 (m), 1450 (m), 1395 (m), 1299 (m), 1224 (m), 1183 (m), 1150 (m), 1097 (m), 1048 (m), 1025 (m), 764 (m), 735 (m) cm−1. HR‐MS (EI, 70 eV): calcd 343.2142 (for C21H29NO3 +), found 343.2152 [M +]. C21H29NO3 (343.47 g mol−1).
General procedure C (GPC) for the N ‐alkylation of benzazocinone 18 g: Under exclusion of air and moisture (nitrogen atmosphere) and at −78 °C, nBuLi (2.5 mol L−1 in hexanes, 1.05 equiv) was added dropwise to a stirred solution of diisopropylamine (1.05 equiv) in abs. THF (3 L mol−1). After stirring this mixture for 15 min at −78 °C, a solution of benzazocinone 18 g (1.00 equiv) in abs. THF (2 L mol−1) was added and the resulting mixture was further stirred at −78 °C for 30 min. The alkyl bromide (1.05 equiv) was then added and the resulting mixture was stirred at −78 °C for 1.5 h and for further 2 h at ambient temperature. Subsequently, the mixture was diluted with hydrochloric acid (1 mol L−1, 4 L mol−1) and extracted with MTBE (3×4 L mol−1). The combined organic layers were dried (MgSO4), filtered and the solvent was removed in vacuo. The crude product was purified by column chromatography to yield benzazocinones 18 e, 18 i–18 k.
Ethyl 1‐allyl‐2‐oxo‐1,2,3,4,5,6‐hexahydrobenzo[b]azocine‐6‐carboxylate (18 e): According to GPC, benzazocinone 18 g (124 mg, 500 μmol), nBuLi (0.21 mL, 2.5 mol L−1 in hexanes, 0.53 mmol) and iPr2NH (54 mg, 0.53 mmol) were converted with allyl bromide (64 mg, 0.53 mmol) to yield in the first fraction the title compound 18 e (34 mg, 0.12 mmol, 24 %, R f=0.39) after chromatography (SiO2, hexanes/MTBE 1:5) as a colorless oil. Secondly, starting material 18 g (60 mg, 0.24 mmol, 48 %, R f=0.16) was recovered in another fraction.
Ethyl 2‐oxo‐1‐prenyl‐1,2,3,4,5,6‐hexahydrobenzo[b]azocine‐6‐carboxylate (18 i): According to GPC, benzazocinone 18 g (124 mg, 500 μmol), nBuLi (0.21 mL, 2.5 mol L−1 in hexanes, 0.53 mmol) and iPr2NH (54 mg, 0.53 mmol) were converted with prenyl bromide (79 mg, 0.53 mmol) to yield the title compound 18 i (119 mg, 377 μmol, 75 %) after chromatography (SiO2, hexanes/MTBE 1:5, R f=0.43) as a colorless oil. 1H NMR (500 MHz, CDCl3): δ=1.15 (t, J=7.1 Hz, 3 H), 1.46–1.55 (m, 1 H), 1.49 (s, 3 H), 1.61 (s, 3 H), 1.72–1.91 (m, 3 H), 2.21 (dd, J=11.1 Hz, J=7.9 Hz, 1 H), 2.40 (dd, J=13.5 Hz, J=4.7 Hz, 1 H), 3.51 (d, J=11.2 Hz, 1 H), 3.97 (dd, J=14.4 Hz, J=7.3 Hz, 1 H), 4.11 (q, J=7.1 Hz, 2 H), 4.85 (dd, J=14.4 Hz, J=7.3 Hz, 1 H), 5.24 (t, J=7.3 Hz, 1 H), 7.19–7.22 (m, 1 H), 7.24–7.27 (m, 2 H), 7.29–7.32 (m, 1 H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ=14.08 (CH3), 17.69 (CH3), 24.24 (CH2), 25.58 (CH3), 32.14 (CH2), 32.99 (CH2), 44.75 (CH), 46.51 (CH2), 60.76 (CH2), 118.14 (CH), 126.01 (CH), 126.80 (CH), 127.86 (CH), 128.35 (CH), 136.63 (C), 139.10 (C), 140.55 (C), 173.52 (C), 173.95 (C) ppm. IR (ATR): ṽ=2924 (w), 1733 (s), 1652 (s), 1495 (m), 1456 (m), 1445 (m), 1395 (m), 1297 (m), 1227 (m), 1184 (s), 1149 (m), 1099 (m), 1049 (m), 1027 (m), 769 (m), 738 (m) cm−1. HR‐MS (EI, 70 eV): calcd 315.1829 (for C19H25NO3 +), found 315.1835 [M +]. C19H25NO3 (315.41 g mol−1).
For the preparation of compounds 18 j and 18 k see the Supporting Information.
Ethyl (E)‐1‐[3‐(methoxycarbonyl)‐2‐propenyl]‐2‐oxo‐1,2,3,4,5,6‐hexahydrobenzo[b]azocine‐6‐carboxylate (18 l): Methyl acrylate (215 mg, 2.50 mmol) and catMETium RF {Benzylidenedichloro[4,5‐dimethyl‐1,3‐bis(2,4,6‐trimethylphenyl)‐4‐imidazolin‐2‐ylidene](tricyclohexylphosphano)ruthenium(II)} (25 μmol, 22 mg) were added to a solution of benzazocinone 18 e (144 mg, 501 μmol) in degassed CH2Cl2 (1.5 mL) and the resulting mixture was stirred at 40 °C for 16 h. All volatile materials were evaporated and the crude product was purified by column chromatography (SiO2, hexanes/EtOAc 1:1) to yield the title compound 18 l (76 mg, 0.22 mmol, 44 %, R f=0.27) as a colorless oil. As a second fraction, the starting material 18 e (38 mg, 0.13 mmol, 26 %, R f=0.35) was recovered. 1H NMR (500 MHz, CDCl3): δ=1.15 (t, J=7.1 Hz, 3 H), 1.55 (dddd, J=14.2 Hz, J=12.4 Hz, J=11.1 Hz, J=5.6 Hz, 1 H), 1.77–1.86 (m, 1 H), 1.88–1.97 (m, 2 H), 2.27–2.31 (m, 1 H), 2.46 (dd, J=14.2 Hz, J=4.9 Hz, 1 H), 3.46 (dd, J=11.1 Hz, J=0.8 Hz, 1 H), 3.69 (s, 3 H), 4.11 (q, J=7.1 Hz, 2 H), 4.32 (ddd, J=15.4 Hz, J=6.7 Hz, J=1.1 Hz, 1 H), 4.79 (ddd, J=15.4 Hz, J=6.4 Hz, J=1.4 Hz, 1 H), 5.92 (dt, J=15.7 Hz, J=1.3 Hz, 1 H), 6.97 (dt, J=15.7 Hz, J=6.5 Hz, 1 H), 7.20–7.22 (m, 1 H), 7.28–7.36 (m, 3 H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ=13.93 (CH3), 24.22 (CH2), 31.96 (CH2), 32.90 (CH2), 44.84 (CH), 49.98 (CH2), 51.49 (CH3), 60.98 (CH2), 123.88 (CH), 125.62 (CH), 127.20 (CH), 128.27 (CH), 128.92 (CH), 138.90 (C), 140.20 (C), 141.65 (CH), 166.13 (C), 173.60 (C), 174.10 (C) ppm. IR (ATR): ṽ=2949 (w), 1724 (vs), 1652 (vs), 1494 (m), 1454 (m), 1441 (m), 1390 (m), 1299 (m), 1276 (m), 1225 (m), 1185 (s), 1169 (s), 1150 (m), 1097 (m), 1045 (m), 1022 (m), 996 (m), 972 (m), 765 (m), 740 (m), 718 (m) cm−1. HR‐MS (EI, 70 eV): calcd 345.1571 (for C19H23NO5 +), found 345.1566 [M +]. C19H23NO5 (345.40 g mol−1).
1‐Benzyl‐2‐oxo‐1,2,3,4,5,6‐hexahydrobenzo[b]azocine‐6‐carboxylic acid (26): An aqueous solution of NaOH (0.5 mol L−1, 40 mL) was added to a solution of benzazocinone 18 a (700 mg, 2.07 mmol) in EtOH (2 mL) and the resulting mixture was stirred at 80 °C for 16 h. Subsequently, the mixture was acidified with hydrochloric acid (1 mol L−1, 25 mL) and extracted with CH2Cl2 (3×30 mL). The combined organic layers were dried (MgSO4), filtered and the solvent was removed in vacuo to yield the title compound 26 (519 mg, 1.68 mmol, 81 %) as a colorless solid. M.p. 166–170 °C. 1H NMR (500 MHz, CDCl3): δ=1.51 (dddd, J=14.1 Hz, J=12.6 Hz, J=11.0 Hz, J=5.5 Hz, 1 H), 1.77–1.86 (m, 1 H), 1.90–1.97 (m, 2 H), 2.40 (dd, J=12.0 Hz, J=8.2 Hz, 1 H), 2.45 (dd, J=14.1 Hz, J=4.8 Hz, 1 H), 3.29 (d, J=11.0 Hz, 1 H), 4.86 (d, J=14.1 Hz, 1 H), 5.17 (d, J=14.1 Hz, 1 H), 7.15 (dd, J=7.8 Hz, J=1.0 Hz, 1 H), 7.21–7.30 (m, 6 H), 7.33 (td, J=7.8 Hz, J=1.4 Hz, 1 H), 7.42 (dd, J=7.8 Hz, J=1.3 Hz, 1 H), 10.45 (br s, 1 H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ=24.18 (CH2), 31.90 (CH2), 32.78 (CH2), 44.59 (CH), 52.87 (CH2), 125.86 (CH), 127.46 (CH), 127.50 (CH), 128.02 (CH), 128.46 (2 CH), 128.74 (CH), 129.03 (2 CH), 136.27 (C), 138.57 (C), 140.36 (C), 174.68 (C), 177.26 (C) ppm. IR (ATR): ṽ=3044 (m), 2946 (m), 1728 (vs), 1625 (vs), 1598 (s), 1496 (m), 1456 (m), 1441 (m), 1411 (m), 1287 (m), 1224 (m), 1173 (s), 1145 (m), 781 (m), 761 (m), 722 (m), 701 (s), 681 (m), 640 (m) cm−1. HR‐MS (EI, 70 eV): calcd 309.1359 (for C19H19NO3 +), found 309.1368 [M +]. C19H19NO3 (309.37 g mol−1). The compound was reported in the literature before, but insufficiently characterized.14b
General procedure D (GPD) for the amide coupling of benzazocinone 26: HATU (1.1 equiv) and DIPEA (1.1–2.2 equiv) were added to a stirred solution of benzazocinone 26 (1.0 equiv) and the primary amine (1.5 equiv) in CH2Cl2 (5 L mol−1) and the resulting mixture was stirred at ambient temperature for 16 h. Subsequently, the mixture was washed with water (1×10 L mol−1), sat. aq. NaHCO3 solution (1×10 L mol−1) and brine (1×10 L mol−1). The organic layer was dried (MgSO4), filtered and the solvent was removed in vacuo. The crude product was purified by column chromatography to yield benzazocinones 27 a–27 c.
1‐Benzyl‐2‐oxo‐1,2,3,4,5,6‐hexahydrobenzo[b]azocine‐6‐carboxylic acid N ‐[1‐methyl‐1‐(ethoxycarbonyl)ethyl]amide (27 a): According to GPD, HATU (209 mg, 550 μmol), DIPEA (71 mg, 0.55 mmol) and ethyl 2‐aminoisobutyrate (98 mg, 0.75 mmol) were converted with benzazocinone 26 (154 mg, 500 μmol) to yield the title compound 27 a (183 mg, 433 μmol, 87 %) after chromatography (SiO2, hexanes/MTBE 1:7, R f=0.28) as a colorless oil. 1H NMR (500 MHz, CDCl3): δ=1.14 (s, 3 H), 1.19 (t, J=7.1 Hz, 3 H), 1.22 (s, 3 H), 1.28–1.38 (m, 1 H), 1.69–1.76 (m, 1 H), 1.84 (t, J=12.4 Hz, 1 H), 1.87–1.93 (m, 1 H), 2.27–2.34 (m, 2 H), 2.72 (d, J=10.8 Hz, 1 H), 4.04–4.13 (m, 3 H), 4.16 (d, J=13.9 Hz, 1 H), 5.94 (d, J=13.9 Hz, 1 H), 7.22–7.28 (m, 6 H), 7.32 (d, J=4.0 Hz, 2 H), 7.37 (d, J=7.9 Hz, 1 H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ=13.98 (CH3), 24.38 (CH2), 24.82 (CH3), 24.84 (CH3), 31.94 (CH2), 33.06 (CH2), 44.93 (CH), 51.98 (CH2), 55.64 (C), 60.93 (CH2), 125.60 (CH), 127.85 (CH), 128.07 (CH), 128.13 (CH), 128.52 (CH), 128.64 (2 CH), 129.26 (2 CH), 137.00 (C), 139.48 (C), 139.57 (C), 171.91 (C), 173.55 (C), 173.86 (C) ppm. IR (ATR): ṽ=3410 (w), 2983 (w), 2938 (w), 1737 (s), 1676 (s), 1651 (s), 1493 (s), 1452 (s), 1393 (m), 1383 (m), 1276 (s), 1234 (m), 1214 (m), 1193 (m), 1174 (s), 1148 (vs), 1029 (m), 920 (m), 759 (s), 733 (s), 705 (s), 635 (m) cm−1. HR‐MS (EI, 70 eV): calcd 422.2200 (for C25H30N2O4 +), found 422.2196 [M +]. C25H30N2O4 (422.53 g mol−1).
For the preparation of compounds 27 b and 27 c see the Supporting Information.
1‐Benzyl‐2‐oxo‐1,2,3,4,5,6‐hexahydrobenzo[b]azocine‐6‐carboxamide (27 d): Pyridine (142 mg, 1.80 mmol) and Boc2O (327 mg, 1.50 mmol) were added to a solution of benzazocinone 26 (309 mg, 1.00 mmol) in 1,4‐dioxane (2 mL) and the resulting mixture was stirred at ambient temperature for 30 min. Then (NH4)2CO3 (269 mg, 2.80 mmol) was added and the reaction mixture was stirred at ambient temperature for 16 h. Subsequently, H2O (5 mL) and MTBE (5 mL) were added and the crude product 27 d precipitated. It was filtered off, washed with MTBE (3×5 mL) and dried in vacuum to yield the title compound 27 d (216 mg, 700 μmol, 70 %) as a colorless solid. M.p. 225–227 °C. 1H NMR (500 MHz, CDCl3): δ=1.39 (dddd, J=14.3 Hz, J=12.7 Hz, J=10.8 Hz, J=5.5 Hz, 1 H), 1.72–1.82 (m, 1 H), 1.87–1.96 (m, 2 H), 2.30–2.35 (m, 2 H), 2.72 (d, J=10.8 Hz, 1 H), 3.32 (br s, 1 H), 4.16 (d, J=13.7 Hz, 1 H), 4.89 (br s, 1 H), 6.05 (d, J=13.7 Hz, 1 H), 7.25–7.30 (m, 6 H), 7.36–7.40 (m, 3 H) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ=24.42 (CH2), 31.56 (CH2), 33.12 (CH2), 44.66 (CH), 51.88 (CH2), 125.74 (CH), 127.27 (CH), 127.86 (CH), 128.24 (CH), 128.84 (3 CH), 129.93 (2 CH), 136.82 (C), 139.19 (C), 139.85 (C), 173.77 (C), 175.13 (C) ppm. IR (ATR): ṽ=3318 (m), 3132 (m), 2963 (w), 2928 (w), 1673 (s), 1628 (vs), 1595 (m), 1488 (m), 1450 (m), 1443 (m), 1427 (m), 1411 (m), 1392 (s), 1356 (m), 1333 (m), 1230 (m), 1203 (m), 1157 (m), 1020 (m), 994 (m), 776 (m), 759 (s), 727 (m), 715 (m), 694 (m), 670 (m), 637 (m), 579 (s) cm−1. HR‐MS (EI, 70 eV): calcd 308.1519 (for C19H20N2O2 +), found 308.1511 [M +]. C19H20N2O2 (308.38 g mol−1).
1‐Benzyl‐6‐[(methoxycarbonyl)amino]‐2‐oxo‐1,2,3,4,5,6‐hexahydrobenzo[b]azocine (28): A solution of KOH (68 mg, 1.21 mmol) in MeOH (1 mL) was added at 0 °C to a solution of benzazocinone 27 d (150 mg, 486 μmol) and PhI(OAc)2 (157 mg, 487 μmol) in CH2Cl2 (1 mL). The resulting mixture was stirred at 0 °C for 15 min and for further 16 h at ambient temperature. Subsequently, the reaction mixture was diluted with water (5 mL) and the aqueous layer was extracted with CH2Cl2 (3×5 mL). The combined organic layers were dried (MgSO4), filtered and the solvent was removed in vacuo. The crude product was purified by column chromatography (SiO2, hexanes/EtOAc/MeOH 1:1:0.1) to yield the title compound 28 (50 mg, 0.15 mmol, 30 %, R f=0.30) as a colorless solid. M.p. 114–130 °C. NMR spectra showed doubled and broadened signal sets due to E/Z‐isomers (ratio 1:0.15) at the carbamate C−N‐bond. 1H NMR (500 MHz, CDCl3), major isomer: δ=1.60 (qd, J=12.1 Hz, J=5.6 Hz, 1 H), 1.87–2.07 (m, 3 H), 2.11–2.14 (m, 1 H), 2.29–2.34 (m, 1 H), 3.60 (s, 3 H), 4.38 (br s, 1 H), 4.56 (br d, J=14.5 Hz, 1 H), 5.25 (br d, J=4.6 Hz, 1 H), 5.40 (br s, 1 H), 6.87 (br d, J=6.0 Hz, 1 H), 7.15 (br t, J=7.2 Hz, 1 H), 7.22–7.34 (m, 6 H), 7.39 (dd, J=7.9 Hz, J=1.4 Hz, 1 H) ppm; minor isomer: δ=1.73 (ddt, J=14.3 Hz, J=10.0 Hz, J=4.2 Hz, 1 H), 1.78–1.84 (m, 1 H), 1.87–2.07 (m, 2 H), 2.18 (dd, J=11.9 Hz, J=4.3 Hz, 1 H), 2.29–2.34 (m, 1 H), 3.58 (s, 3 H), 4.80 (d, J=14.2 Hz, 1 H), 4.95–4.99 (m, 1 H), 5.02 (d, J=14.2 Hz, 1 H), 7.01 (dd, J=7.7 Hz, J=1.3 Hz, 1 H), 7.22–7.34 (m, 8 H) ppm; a signal for the NH proton was not observed. 13C{1H} NMR (125 MHz, CDCl3), major isomer: δ=23.65 (CH2), 32.72 (CH2), 36.43 (CH2), 50.51 (CH), 52.06 (CH3), 52.40 (CH2), 125.25 (CH), 126.15 (CH), 127.27 (CH), 127.67 (CH), 128.37 (2 CH), 128.86 (CH), 128.88 (2 CH), 137.53 (C), 139.87 (C), 141.94 (C), 155.78 (C), 174.04 (C) ppm; minor isomer: δ=20.80 (CH2), 31.98 (CH2), 32.16 (CH2), 51.97 (CH3), 52.43 (CH2), 54.86 (CH), 127.32 (CH), 127.87 (CH), 128.33 (CH), 128.57 (CH), 128.68 (2 CH), 129.17 (2 CH), 131.75 (CH), 137.22 (C), 138.94 (C), 140.47 (C), 155.79 (C), 173.43 (C) ppm. IR (ATR): λ−1=3314 (m), 2929 (w), 1717 (s), 1627 (s), 1598 (m), 1521 (m), 1494 (m), 1454 (m), 1447 (m), 1406 (m), 1295 (m), 1247 (s), 1201 (m), 1058 (m), 1025 (m), 911 (m), 906 (m), 759 (m), 734 (s), 702 (s), 626 (m) cm−1. HR‐MS (EI, 70 eV): calcd 338.1625 (for C20H22N2O3 +), found 338.1624 [M +]. C20H22N2O3 (338.41 g mol−1).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
MTBE was obtained as a generous gift from Evonik Industries, Marl, Germany. We are furthermore grateful to Dr. R. Kadyrov (Evonik, Hanau) for providing us with a sample of ruthenium catalyst.
A. Dierks, J. Tönjes, M. Schmidtmann, J. Christoffers, Chem. Eur. J. 2019, 25, 14912.
Contributor Information
Anna Dierks, http://www.uni-oldenburg.de/oc-christoffers/.
Prof. Dr. Jens Christoffers, Email: jens.christoffers@uol.de.
References
- 1. Hussain A., Yousuf S. K., Mukherjee D., RSC Adv. 2014, 4, 43241–43257. [Google Scholar]
- 2.
- 2a. Watthey J. W. H., Gavin T., Desai M., J. Med. Chem. 1984, 27, 816–818; [DOI] [PubMed] [Google Scholar]
- 2b. Watthey J. W. H., Stanton J. L., Desai M., Babiarz J. E., Finn B. M., J. Med. Chem. 1985, 28, 1511–1516. [DOI] [PubMed] [Google Scholar]
- 3. Ortega R., Ravina E., Masaguer C. F., Areias F., Brea J., Loza M. I., Lopez L., Selent J., Pastor M., Sanz F., Bioorg. Med. Chem. Lett. 2009, 19, 1773–1778. [DOI] [PubMed] [Google Scholar]
- 4. Saavedra-Vélez M. V., Correa-Basurto J., Matus M. H., Gasca-Perez E., Bello M., Cuevas-Hernandez R., Garcia-Rodriguez R. V., Trujillo-Ferrara J., Ramos-Morales F. R., J. Comp. Aided Mol. Des. 2014, 28, 1217–1232. [DOI] [PubMed] [Google Scholar]
- 5. Zhang H., Qiu S., Tamez P., Tan G. T., Aydogmus Z., Hung N. V., Cuong N. M., Angerhofer C., Soejarto D. D., Pezzuto J. M., Fong H. H. S., Pharm. Biol. 2002, 40, 221–224. [Google Scholar]
- 6. Laakso J. A., Gloer J. B., Wicklow D. T., Dowd P. F., J. Org. Chem. 1992, 57, 2066–2071. [Google Scholar]
- 7. Riemer B., Hofer O., Greger H., Phytochemistry 1997, 45, 337–341. [DOI] [PubMed] [Google Scholar]
- 8. Qiao M.-F., Ji N.-Y., Liu X.-H., Li K., Zhu Q.-M., Xue Q.-Z., Bioorg. Med. Chem. Lett. 2010, 20, 5677–5680. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Witosińska A., Musielak B., Serda P., Owinska M., Rys B., J. Org. Chem. 2012, 77, 9784–9794; [DOI] [PubMed] [Google Scholar]
- 9b. Musielak B., Holak T. A., Rys B., J. Org. Chem. 2015, 80, 9231–9239. [DOI] [PubMed] [Google Scholar]
- 10. Behler F., Habecker F., Saak W., Klüner T., Christoffers J., Eur. J. Org. Chem. 2011, 4231–4240. [Google Scholar]
- 11. Listratova A. V., Voskressensky L. G., Synthesis 2017, 49, 3801–3834. [Google Scholar]
- 12.Other recent representative methods:
- 12a. Zhou B., Li L., Zhu X.-Q., Yan J.-Z., Guo Y.-L., Ye L.-W., Angew. Chem. Int. Ed. 2017, 56, 4015–4019; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4073–4077; [Google Scholar]
- 12b. Zhao W., Qian H., Li Z., Sun J., Angew. Chem. Int. Ed. 2015, 54, 10005–10008; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10143–10146; [Google Scholar]
- 12c. Shaw M. H., Croft R. A., Whittingham W. G., Bower J. F., J. Am. Chem. Soc. 2015, 137, 8054–8057; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12d. Wu S., Zeng R., Fu C., Yu Y., Zhang X., Ma S., Chem. Sci. 2015, 6, 2275–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Jones D. H., Stephenson G. F., Spray G. W., Wragg W. R., J. Chem. Soc. C 1969, 2176–2181; [Google Scholar]
- 13b. Kenwright J. L., Galloway W. R. J. D., Wortmann L., Spring D. R., Synth. Commun. 2013, 43, 1508–1516. [Google Scholar]
- 14.
- 14a. Gale D. J., Wilshire J. F. K., Austr. J. Chem. 1974, 27, 1295–1308; [Google Scholar]
- 14b. Jones G., Tringham G. T., J. Chem. Soc. Perkin Trans. 1 1975, 1280–1283; [DOI] [PubMed] [Google Scholar]
- 14c. Sigaut F., Levy J., Tetrahedron Lett. 1989, 30, 2937–2940. [Google Scholar]
- 15.Reviews:
- 15a. Donald J. R., Unsworth W. P., Chem. Eur. J. 2017, 23, 8780–8799; [DOI] [PubMed] [Google Scholar]
- 15b. Dowd P., Zhang W., Chem. Rev. 1993, 93, 2091–2115; recent examples: [Google Scholar]
- 15c. Baud L. G., Manning M. A., Arkless H. L., Stephens T. C., Unsworth W. P., Chem. Eur. J. 2017, 23, 2225–2230; [DOI] [PubMed] [Google Scholar]
- 15d. Kitsiou C., Hindes J. J., I′Anson P., Jackson P., Wilson T. C., Daly E. K., Felstead H. R., Hearnshaw P., Unsworth W. P., Angew. Chem. Int. Ed. 2015, 54, 15794–15798; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 16020–16024; [Google Scholar]
- 15e. Hall J. E., Matlock J. V., Ward J. W., Gray K. V., Clayden J., Angew. Chem. Int. Ed. 2016, 55, 11153–11157; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11319–11323; [Google Scholar]
- 15f. Klapars A., Parris S., Anderson K. W., Buchwald S. L., J. Am. Chem. Soc. 2004, 126, 3529–3533. [DOI] [PubMed] [Google Scholar]
- 16. Guney T., Wenderski T. A., Boudreau M. W., Tan D. S., Chem. Eur. J. 2018, 24, 13150–13157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang N., Gu Q.-S., Li Z.-L., Li Z., Guo Y.-L., Guo Z., Liu X.-Y., Angew. Chem. Int. Ed. 2018, 57, 14225–14229; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14421–14425. [Google Scholar]
- 18. Wu A., Feng Q., Sung H. H. Y., Williams I. D., Sun J., Angew. Chem. Int. Ed. 2019, 58, 6776–6780; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 6848–6852. [Google Scholar]
- 19.
- 19a. Rössle M., Christoffers J., Synlett 2006, 106–108; [Google Scholar]
- 19b. Pflantz R., Tielmann P., Rössle M., Hoenke C., Christoffers J., Eur. J. Org. Chem. 2007, 3227–3238. [Google Scholar]
- 20. Penning M., Christoffers J., Eur. J. Org. Chem. 2012, 1809–1818. [Google Scholar]
- 21. Pflantz R., Sluiter J., Kricka M., Saak W., Hoenke C., Christoffers J., Eur. J. Org. Chem. 2009, 5431–5436. [Google Scholar]
- 22. Penning M., Christoffers J., Eur. J. Org. Chem. 2014, 2140–2149. [Google Scholar]
- 23. Penning M., Aeissen E., Christoffers J., Synthesis 2015, 47, 1007–1015. [Google Scholar]
- 24. Zhou Y., Wei Y.-L., Rodriguez J., Coquerel Y., Angew. Chem. Int. Ed. 2019, 58, 456–460; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 466–470. [Google Scholar]
- 25.
- 25a. Jia Z., Galvez E., Sebastian R. M., Pleixats R., Alvarez-Larena A., Martin E., Vallribera A., Shafir A., Angew. Chem. Int. Ed. 2014, 53, 11298–11301; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 11480–11483; [Google Scholar]
- 25b. Wu Y., Arenas I., Broomfield L. M., Martin E., Shafir A., Chem. Eur. J. 2015, 21, 18779–18784. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Louie J., Hartwig J. F., Tetrahedron Lett. 1995, 36, 3609–3612; [Google Scholar]
- 26b. Guram A. S., Rennels R. A., Buchwald S. L., Angew. Chem. Int. Ed. Engl. 1995, 34, 1348–1350; [Google Scholar]; Angew. Chem. 1995, 107, 1456–1459. [Google Scholar]
- 27. Ruiz-Castillo P., Buchwald S. L., Chem. Rev. 2016, 116, 12564–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.
- 28a. Ullmann F., Chem. Ber. 1903, 36, 2382–2384; [Google Scholar]
- 28b. Goldberg I., Chem. Ber. 1906, 39, 1691–1692. [Google Scholar]
- 29.
- 29a. Sambiagio C., Arsden S. P., Blacker A. J., McGowan P. C., Chem. Soc. Rev. 2014, 43, 3525; [DOI] [PubMed] [Google Scholar]
- 29b. Okano K., Tokuyama H., Fukuyama T., Chem. Commun. 2014, 50, 13650–13663. [DOI] [PubMed] [Google Scholar]
- 30.
- 30a. Kwong F. Y., Klapars A., Buchwald S. L., Org. Lett. 2002, 4, 581–584; [DOI] [PubMed] [Google Scholar]
- 30b. Klapars A., Huang X., Buchwald S. L., J. Am. Chem. Soc. 2002, 124, 7421–7428; [DOI] [PubMed] [Google Scholar]
- 30c. Shafir A., Buchwald S. L., J. Am. Chem. Soc. 2006, 128, 8742–8743. [DOI] [PubMed] [Google Scholar]
- 31.CCDC https://www.ccdc.cam.ac.uk/services/strctures?id=doi:10.1002/chem.201903139 (18 b) contains the supplementary crystallographic data for this paper. These data are provided free of charge by http://www.ccdc.cam.ac.uk/.
- 32.
- 32a. Carpino L. A., Imazumi H., El-Faham A., Ferrer F. J., Zhang C., Lee Y., Foxman B. M., Henklein P., Hanay C., Mügge C., Wenschuh H., Klose J., Beyermann M., Bienert M., Angew. Chem. Int. Ed. 2002, 41, 441–445; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 457–461; [Google Scholar]
- 32b. Buschbeck L., Christoffers J., J. Org. Chem. 2018, 83, 4002–4014. [DOI] [PubMed] [Google Scholar]
- 33.
- 33a. Kadyrov R., Rosiak A., Chim. Oggi 2009, 27, 24–26; [Google Scholar]
- 33b. Kadyrov R., Rosiak A., WO 2009/124977 A1.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary