

Summary
Von Willebrand disease (VWD) is the most common inherited bleeding disorder. Most patients with mild and moderate VWD can be treated effectively with desmopressin. The management of severe VWD patients, mostly affected by type 2 and type 3 disease, can be challenging. In this article we review the current diagnosis and treatment of severe VWD patients. We will also discuss the management of severe VWD patients in specific situations, such as pregnancy, delivery, patients developing alloantibodies against von Willebrand factor and VWD patients with recurrent gastrointestinal bleeding. Moreover, we review emerging treatments that may be applied in future management of patients with severe VWD.
Keywords: von Willebrand disease, treatment, severe VWD
Von Willebrand disease (VWD) is the most common inherited bleeding disorder (Leebeek & Eikenboom, 2016). The estimated prevalence of VWD patients with clinically relevant bleeding is 1:10 000 (Sadler et al, 2000). VWD is characterized by mucocutaneous bleeding such as menorrhagia, epistaxis and gum bleeds (de Wee et al, 2012; Leebeek & Eikenboom, 2016). However, other bleeds, such as joint bleeds and gastro‐intestinal bleeds, also occur, especially in more severely affected patients (van Galen et al, 2017). VWD diagnosis is based on personal bleeding history, family history of VWD and a detailed laboratory evaluation (Leebeek & Eikenboom, 2016).
Von Willebrand disease is classified in three subtypes based on a quantitative reduction or a qualitative defect of von Willebrand factor (VWF) (Sadler et al, 2006; Leebeek & Eikenboom, 2016). Type 1 VWD is characterized by a quantitative reduction of VWF, and is the most common (70–80%) form of VWD. The bleeding phenotype of type 1 VWD patients is generally milder than in type 2 and type 3 VWD (de Wee et al, 2012). Type 2 VWD is diagnosed in 20% of VWD patients and is characterized by a qualitative defect of VWF, and the bleeding phenotype is generally more severe than in type 1 VWD patients. Type 3 VWD is the rarest (prevalence <5%) and most severe form of VWD; it is characterized by a complete absence of VWF (Leebeek & Eikenboom, 2016).
The VWF gene, which largely determines VWF levels in circulation, is located at chromosome 12. A large variation of mutations or deletions in the VWF gene are associated with VWD (de Jong & Eikenboom, 2017). The inheritance pattern of VWD is autosomal dominant in the majority of cases (Leebeek & Eikenboom, 2016). In type 1 VWD, most patients have autosomal dominant missense mutations with an incomplete penetrance and a variable expression (Leebeek & Eikenboom, 2016). In type 2 VWD, dominant negative missense mutations of specific VWF gene regions lead to an abnormally functioning VWF. The location of the VWF mutation determines the abnormal VWF function and, subsequently, the subtype of type 2 VWD (de Jong & Eikenboom, 2017). In type 3 VWD, up to 80% of patients have a null allele mutation, which is autosomal recessive and homozygous, or compound heterozygous in most patients (Leebeek & Eikenboom, 2016).
Severe VWD
Unlike in haemophilia, in which the severity of disease is based on factor VIII (FVIII) or factor IX (FIX) levels, there is no uniformly accepted definition for mild, moderate or severe VWD. VWD diagnosis relies on criteria defined by Sadler et al (2006), although that classification did not define severe or moderate VWD.
Federici (2004) classified patients with VWF: Ristocetin cofactor activity (VWF:RCo) levels <10 iu/dl and FVIII coagulant activity (FVIII:C) <20 iu/dl as severe (mainly type 1, 2A, 2B and 3); VWF:RCo 10–30 iu/dl and FVIII:C 20–40 iu/dl (mainly type 1, 2B, 2M and 2N) as moderate and VWF:RCo 30–50 iu/dl and FVIII:C 40–60 iu/dl (mainly type 1) as mild VWD. More recently, it has been suggested that patients with bleeding symptoms and VWF levels between 30–50 iu/dl should not be classified as mild VWD, but as “low VWF” (Leebeek & Eikenboom, 2016; Lavin et al, 2017). In more recently published and ongoing studies on the use of prophylaxis in VWD, severe VWD was defined as VWF:RCo < 20 iu/dl (Abshire et al, 2015). However, the most widely used definition of severe VWD is based on VWF activity levels <10 iu/dl and/or FVIII levels <20 iu/dl. Thus, this includes type 3 VWD patients with a complete absence of VWF and most type 2B VWD patients, who generally also have a severe bleeding phenotype (de Wee et al, 2012). Also patients with type 2A and Type 1 Vicenza, which is characterized by a fast clearance of VWF, fulfil the definition of severe VWD (Leebeek & Eikenboom, 2016). It should be acknowledged that these definitions are not uniformly used and guidelines from Europe or USA may differ on cut‐off values for the diagnosis of VWD and definition of severe VWD (Nichols et al, 2008; Castaman et al, 2013; Laffan et al, 2014).
Therefore, this guideline is intended for a broad range of severe VWD patients with type 1 Vicenza, type 1 with VWF levels <10 iu/dl, type 2A, type 2B and type 3 disease. It is also intended for other individual VWD patients with a severe bleeding phenotype irrespective of their disease type. Additionally, we include recommendations for patients with a high risk for bleeding in specific situations, such as pregnant women with VWD in whom VWF levels do not increase above 50 iu/dl during third trimester, and patients with recurrent gastrointestinal bleeding.
Diagnosis of severe VWD
Von Willebrand disease is characterized by defective platelet adhesion and aggregation. As VWF is a carrier protein for FVIII, thereby preventing FVIII degradation, patients with severe VWD also have reduced FVIII levels. FVIII levels are strongly reduced, particularly in patients with type 3 VWD (Sanders et al, 2015). The laboratory evaluation of an individual with a bleeding phenotype suggestive of VWD, includes measurement of VWF antigen (VWF:Ag), VWF activity [VWF:RCo and VWF: collagen binding activity (VWF:CB)], VWF multimer pattern and FVIII:C (Ng et al, 2015; De Jong & Eikenboom, 2016). The discussion about the cut‐off value of VWF to diagnose VWD is still not completely resolved. According to the European principles of care for VWD, the value was defined as 40 iu/dl whereas others defined VWD based on VWF:Ag or VWF:RCo below 30 iu/dl, which is now also used in most recent guidelines (Nichols et al, 2008; Castaman et al, 2013; Laffan et al, 2014; Leebeek & Eikenboom, 2016).
Type 1 VWD is diagnosed when the VWF:RCo/VWF:Ag ratio is >0·6. Type 2 VWD is diagnosed based on an abnormal function of VWF (VWF:RCo/VWF:Ag ratio ≤0·6). Type 2 VWD is further subdivided in type 2A, 2B, 2M and 2N (Sadler et al, 2006). Type 2A and type 2B VWD are characterized by a loss of high molecular weight (HMW) multimers, which are the most active haemostatic multimers. Loss of HMW multimers lead to a moderate to severe bleeding phenotype. The typical gain of function mutations causing type 2B VWD lead to a higher affinity to bind the platelet glycoprotein (GP)1bα receptor, and subsequently leads to increased platelet binding and aggregation (Leebeek & Eikenboom, 2016). Therefore, type 2A and type 2B VWD patients can be distinguished from each other using the ristocetin‐induced platelet adhesion (RIPA) test, which is positive with low concentrations of ristocetin in type 2B VWD. Type 2M VWD is characterized by a decreased platelet adhesion or collagen binding, without loss of HMW VWF multimers. Type 2N VWD is caused by a missense mutation in the D’‐D3 domain of the VWF gene, leading to decreased VWF‐FVIII binding (Leebeek & Eikenboom, 2016). These patients have generally low FVIII compared to VWF levels. Based on the most recent International Society on Thrombosis and Haemostasis (ISTH) classification, type 3 VWD is defined as undetectable VWF levels (VWF:Ag ≤ 5 iu/dl) (Sadler et al, 2006). However, this definition will include also patients with severe type 1 VWD. Therefore, it has recently been proposed that type 3 VWD would be more precisely diagnosed by assessing VWF propeptide (VWFpp) levels and defining only those individuals with VWFpp levels ≤5 iu/dl as type 3 VWD (Sanders et al, 2015).
Several diagnostic tests have been developed to assess the functionality of VWF. The most widely used test is the VWF:RCo test. Due to the fact that the originally developed assay is elaborate and time consuming, has a poor sensitivity and high variation, several new activity tests have been developed in recent years (Bodo et al, 2015; Boender et al, 2018). In a recent study, the four currently most widely used VWF activity assays were compared in a cohort of 661 well‐defined VWD patients (Boender et al, 2018). The assays were found to be highly correlated with each other. However, in some patients, discrepancies in VWF activity assays resulted in a different classification of the type of VWD. The VWF:RCo assay, using ristocetin to activate VWF and whole platelets, was not sensitive enough to classify 18% of the total study population and misclassified half of the genotypically identified 2B patients. Also the VWF:GPIbM test, using gain‐of‐function GPIb fragments that bind spontaneously to VWF, misclassified some definite type 2 and type 3 VWD patients (Boender et al, 2018).
Our institution’s VWD diagnostic algorithm is based on recently published reviews on VWD (Ng et al, 2015; De Jong & Eikenboom, 2016; Leebeek & Eikenboom, 2016). In our laboratory we measure FVIII:C, VWF:Ag (in‐house assay), the automated VWF:GPIbM assay and VWF:CB assay to test the VWF activity, followed by multimer analysis and, in case of reduced HMW multimers, also the RIPA assay. In case of low FVIII we perform a FVIII: binding to VWF test to diagnose or exclude type 2N.
Genetic testing may be considered in patients with severe VWD. Genetic counselling may be required in families with type 3 disease: for instance, in cases where a couple already has a child with type 3 disease (Peake & Goodeve, 2010). Mutation analysis of both parents is necessary to identify the causative mutations and thereby enable an early diagnosis with chorionic villus sampling. In nearly all type 2 VWD patients a mutation in the VWF gene is found, but the clinical use of knowing the mutation in daily practice is debatable. Many patients with severe type 1 disease also have mutations, but this is strongly dependent upon the level of VWF (Leebeek & Eikenboom, 2016). The yield of genetic testing is over 90% in individuals with VWF levels <15 iu/dl, but in individuals with levels between 16 to 30 iu/dl it is only around 75% (Goodeve et al, 2007).
In our centre we do not routinely perform mutation analysis in VWD, unless it has clinical implications. Indications for performing mutation analysis are counselling for family planning in type 3, and diagnosing type 2B. We also perform mutation analysis in VWD type 2 patients, to enable the diagnosis of, or exclude, VWD in their unborn child by amniotic fluid sampling at 32–34 weeks of pregnancy and to assess whether an atraumatic delivery is necessary. We also perform genetic testing for scientific purposes, for instance in the Willebrand in the Netherlands (WiN) study (Boender et al, 2018).
Bleeding phenotype of severe VWD
von Willebrand disease is characterized mainly by mucocutaneous bleeding symptoms, including menorrhagia, nose bleeds, easy bruising and gastro‐intestinal bleeding (de Wee et al, 2012). In contrast to haemophilia patients, in whom factor levels are well associated with the severity of the bleeding phenotype, this is less clear in VWD. The bleeding phenotype of VWD is highly heterogeneous. Even patients with the same type of VWD and comparable levels of VWF activity can have a variable bleeding phenotype (de Wee et al, 2012; Flood et al, 2016).
In severe VWD the bleeding phenotype is clearly different from milder forms, mainly because of the co‐existing strongly reduced FVIII:C levels (Leebeek & Eikenboom, 2016). This was also observed in the WiN study, in which 40% of the included patients with VWD had severe VWD based on VWF levels (antigen or activity) <10 iu/dl (de Wee et al, 2012). Metjian et al (2009) reported the bleeding phenotype in 150 VWD patients with VWF levels <10 u/dl. Almost all patients experienced bleeding episodes and >90% of patients had been treated with factor concentrates. Joint and muscle bleeds had occurred in 45% and 28% of patients, respectively. Also in the WiN‐study, 58% of patients with type 3 disease reported joint bleeds, for which they received haemostatic treatment, whereas only 12% of patients with type 2 reported joint bleeds (van Galen et al, 2015). Joint bleeds in VWD patients may also result in arthropathy, as is observed in patients with haemophilia. In a recent study of 49 VWD patients with a verified history of joint bleeds, arthropathy was observed in 40% of the cases (van Galen et al, 2017). This was associated with more pain, functional limitations and less social participation compared to control VWD patients without joint bleeds. These patients had comparable outcomes to moderate severe haemophilia A patients (van Galen et al, 2018).
Type 2 VWD patients are a heterogeneous group and bleeding severity may differ between the various subtypes. Patients with type 2B have a more severe bleeding phenotype compared to other type 2 patients, due to the lack of HMW multimers and thrombocytopenia (Federici et al, 2009; de Wee et al, 2012). A prospective study showed that patients with type 2A had a more severe bleeding phenotype than patients with type 2M, which was mainly due to more gastro‐intestinal bleeding in the 2A patients. Because this was not related to FVIII or VWF levels, it is suggested that this was also due to the lack of HMW multimers in type 2A (Castaman et al, 2012).
In recent years, bleeding scores have been used to assess the severity of the bleeding phenotype. This was originally developed to improve the diagnosis of patients suffering from a bleeding tendency who were referred to specialized haematology clinics (Tosetto et al, 2006; Rodeghiero et al, 2007; Tosetto et al, 2008). By obtaining a well‐structured bleeding history on 13 items and scoring those items for severity of bleeding, a total bleeding score can be obtained. Several scores have been developed over the last years (Rydz & James, 2012). The ISTH Bleeding Assessment Tool (ISTH‐BAT) is currently the most widely used score (Rydz & James, 2012) and is helpful in diagnosing bleeding disorders, including VWD. Patients with a normal ISTH BAT score have a very low risk of having an inherited bleeding disorder. However, this may depend on age and on the haemostatic challenges that an individual has encountered. Bleeding scores have also been used to study the severity of the bleeding tendency in individuals previously diagnosed with VWD. Despite the fact that the bleeding symptoms may strongly vary between individuals, patients with severe VWD, including types 2 and 3, have clearly higher scores than type 1 patients (de Wee et al, 2012).
Federici et al (2014) investigated whether the bleeding score can be used to predict future bleeding and clinical outcomes in VWD patients (Federici et al, 2014). In a prospective study, VWF:RCo of <10 iu/dl and a high bleeding score (>10) were associated with an increased risk of developing a bleeding episode in the year to come. A bleeding score >10 remained highly associated with bleeding that needed haemostatic treatment after multivariate analysis. This may also be useful to identify severe VWD patients in need of intensive treatment before surgery or other interventions, and may also identify patients who may benefit from long term prophylaxis with FVIII/VWF concentrate (Federici et al, 2014).
An increased burden of bleeding symptoms and the bleeding score are strongly associated with a reduced health‐related quality of life (HRQoL) both in children and adults with VWD (de Wee et al, 2010; de Wee et al, 2011a). A more severe bleeding phenotype was associated with lower scores on the domains of physical functioning, bodily pain and general health (Atiq et al, 2019, de Wee et al, 2011a, 2011b). Type 3 VWD patients had a lower HRQoL compared to types 1 and 2. Bleeding scores were also strongly associated with HRQoL. Given that chronic pain, mainly related to joint bleeds, and arthropathy are strongly related to physical and mental health, efforts should be made to prevent and manage these bleeding episodes (McLaughlin et al, 2017).
Treatment of severe VWD patients
Severe VWD patients should be treated in specialised centres that have experience with the management of bleeding disorders, especially when patients are in need of treatment with factor concentrates. FVIII/VWF concentrates should be readily available and these centres should have a haemostasis laboratory with the ability to measure VWF and/or FVIII levels immediately (Castaman et al, 2013).
The goal of treatment in VWD patients is to primarily improve platelet adhesion and aggregation by increasing VWF and, secondly, to improve fibrin formation by increasing FVIII levels in the circulation (Heijdra et al, 2017). This can be achieved by stimulating endogenous VWF and FVIII release from Weibel‐Palade Bodies by desmopressin, or infusion of exogenous VWF with or without FVIII, (Castaman et al, 2013; Leebeek & Eikenboom, 2016). Based on these approaches, several treatment options are currently available.
Desmopressin
The most used treatment option to increase VWF and FVIII levels in patients with mild or moderate VWD is to administer desmopressin (1‐deamino‐8‐d‐arginine vasopressin, DDAVP) (Heijdra et al, 2017). Desmopressin stimulates endogenous VWF release from the endothelium into the circulation. Unfortunately type 3 patients are unresponsive due to the lack of endogenous VWF and only a limited number of severe type 1 and type 2 VWD patients can be treated with desmopressin (Leebeek & Eikenboom, 2016).
In a large study on severe VWD patients with VWF levels below 10 iu/dl at the time of desmopressin administration, in which response to desmopressin was defined as increase of plasma FVIII:C and VWF:RCo of at least 3‐fold over baseline, levels of at least 30 iu/dl, and a bleeding time of 12 min or less 2 h after the end of desmopressin infusion, most type 1 patients did not respond (Federici, 2004). Moreover, other factors, such as the type, pathophysiology and genotype of VWD, determine the response to desmopressin (Castaman et al, 2008; Sharma & Flood, 2017). Therefore, severe VWD type 1 and 2 patients without contra‐indications for desmopressin, should receive a test dose of desmopressin (0·3 μg/kg intravenously) to evaluate whether they respond sufficiently (Federici, 2008). In severe VWD patients who respond well, treatment with desmopressin is often sufficient to treat minor bleeding and to prevent bleeding during minor surgery or interventions. In case of a life‐threatening bleeding or major surgery, VWF/FVIII concentrates are needed. Moreover, repeated desmopressin use is limited by tachyphylaxis and side‐effects that may occur, including hypotension and hyponatraemia (Svensson et al, 2014).
Plasma‐derived factor VWF (/FVIII) concentrates
Plasma‐derived factor concentrates are the treatment of choice in patients with severe VWD, and in patients that do not respond to desmopressin or in whom desmopressin is contraindicated (Heijdra et al, 2017). VWF/FVIII concentrates are derived from plasma and are mostly purified by heparin ligand chromatography or ion‐exchange chromatography (Peyvandi et al 2019a). Viral inactivation is mostly performed with solvent detergent and dry heat to prevent virus transfer (Heijdra et al, 2017). There are several brands of plasma‐derived VWF/FVIII concentrates available, with a large variation in VWF:RCo/VWF:Ag ratio (from 0·29 to 2·4) and VWF:RCo/FVIII:C ratio (from 0·81 to >10) (Heijdra et al, 2017). Therefore, it is important to take into account the exact VWF and FVIII values of a concentrate (Castaman et al, 2013; Leebeek & Eikenboom, 2016).
Upon infusion of VWF and FVIII concentrates, 1 iu VWF:RCo/kg increases VWF to around 1·5 iu/dl, whereas 1 iu FVIII:C/kg increases plasma FVIII levels to around 2 iu/dl (Heijdra et al, 2017). Based on this presumed in vivo recovery, the VWF:RCo/FVIII:C ratio of the used product, and the targeted VWF and FVIII levels, the dose of concentrate that should be administered can be determined (Table 1).
Table 1.
Use of factor concentrates in severe VWD patients.
| Indication | Dose* (iu/kg) | Target levels of VWF:RCo and FVIII:C (iu/dl) | Treatment duration (days) | |
|---|---|---|---|---|
| Day 1 | After day 1 | |||
| Bleeding | ||||
| Mild to moderate | 20–40 | Peak >50–80 | Through >30 | 1–3 |
| Severe | 50 | Peak >100 | Through >50 | 3–10 |
| Intervention | ||||
| Dental extraction | 25 | Peak >50 | 1 | |
| Minor surgery | 30–60 | Peak >50–80 | Through >30 | 1–5 |
| Major surgery | 50–60 | Peak >100 | Through >50 | 7–10 |
| Delivery | 40–50 | Peak >100 | Through >50 | 3–7† |
The concentrate dose depends on the type and brand of concentrate.
Three to five days after vaginal labour, and 5–7 days after caesarean section, depending on bleeding and factor levels.
Factor concentrates can be administered by once or twice daily bolus infusions, or by continuous infusion. During surgery or repeated VWF/FVIII concentrate infusions, it is recommend to measure both VWF:RCo and FVIII:C daily to ensure that the target levels of VWF and FVIII are reached (Table 1). Despite the fact that it is still disputed whether normalisation of VWF, FVIII or both are needed to achieve adequate haemostasis in VWD patients, we strive for normalisation of both factors during surgery or other interventions, as is also suggested in most guidelines (Table 1) (Castaman et al, 2013; Laffan et al, 2014; Leebeek & Eikenboom, 2016; Mannucci & Franchini, 2017). It has recently been shown that current treatment strategies may lead to both under‐ and overdosing (Hazendonk et al, 2018a), and may be caused by pharmacokinetic and pharmacodynamic differences of VWF and FVIII (Pipe et al, 2016).
An important physiological mechanism that should be considered in treatment with VWF/FVIII concentrates is that VWD patients have normal FVIII synthesis, but increased FVIII proteolysis because of reduced FVIII binding to VWF, resulting in low FVIII levels. In patients with severe VWD, FVIII levels may be less than 10 iu/dl, thereby worsening the bleeding phenotype and increasing the bleeding risk during interventions (de Wee et al, 2012). Therefore, an increase of both VWF and FVIII by infusion of VWF/FVIII concentrates is needed. However, increase of VWF levels after administration of concentrates will also lead to an increment of endogenous FVIII, because of reduced FVIII proteolysis. This may lead to high FVIII levels after several days of treatment with VWF/FVIII concentrate (Mannucci, 2004). These high FVIII levels may be associated with an increased risk of thrombosis, especially after major surgery (Coppola et al, 2012). Therefore, it is important to measure VWF and FVIII:C regularly to assess the risk for developing thrombosis and to adjust dosing.
Purified VWF plasma concentrates without FVIII are available in some countries. Pure VWF concentrates increase VWF levels immediately, whereas FVIII levels increase gradually over time. By binding of endogenous FVIII to the exogenous VWF, FVIII is stabilised and FVIII degradation prevented, thereby gradually increasing circulating FVIII levels. Most patients reach FVIII levels of >60 iu/dl after a 6‐h infusion of pure VWF concentrate (Borel‐Derlon et al, 2007). In case of planned surgery, the first dose of pure VWF can be given the evening prior to surgery. Additional FVIII should be co‐administered if pure VWF concentrates are administered in emergency situations, such as severe bleeding or shortly before emergency surgery in patients with severe VWD, in order to increase FVIII levels immediately (Heijdra et al, 2017).
Recombinant VWF concentrate
Recombinant VWF (rVWF) concentrate (VEYVONDI®, Takeda Pharmaceutical Company Limited, Tokyo, Japan) was registered in 2015 for on demand treatment of bleeding in VWD patients (Turecek et al, 2009). Recombinant VWF is synthesized from a Chinese Hamster Ovary (CHO) cell line that co‐expresses VWF and F8 genes. Subsequently, this is purified by immune‐affinity chromatography, which results in a rVWF molecule with >99% purity (Peyvandi et al, 2019a). Thus, rVWF only contains VWF and does not contain FVIII. Therefore, patients that are treated with rVWF in acute situations may need additional FVIII infusion(s), as is done when pure VWF plasma concentrates are used. rVWF concentrate is not exposed to ADAMTS13 during the manufacturing process and therefore has more large, haemostatic highly active VWF multimers. Furthermore, during treatment with rVWF there is no risk of viral transmission and there may be a lower chance of developing an allergic reaction (Peyvandi et al, 2019a).
The first phase 3 trial in rVWF, in which it was combined with rFVIII concentrate, showed that on‐demand rVWF treatment had an excellent haemostatic efficacy for bleeding episodes in 96·9% of patients, whereas a good haemostatic efficacy was reached in the remaining patients (Gill et al, 2015). No thrombotic events or severe allergic reactions occurred, suggesting that rVWF is safe to use. A recent phase 3 trial investigating rVWF in 15 severe VWD patients indicated that rVWF was also effective in patients undergoing elective surgery (Peyvandi et al, ). One patient developed deep vein thrombosis, which could possibly be treatment related. Both previous trials showed that rVWF has good pharmacokinetic and pharmacodynamic properties. Due to stabilization of endogenous FVIII by rVWF binding, nearly all VWD patients with mean baseline FVIII:C levels of 16 iu/dl, achieved FVIII:C above 60 iu/dl at 6 h post‐rVWF‐infusion (Gill et al, 2015; Peyvandi et al, 2019b). In two cases, FVIII was administered because FVIII:C was <60 iu/dl. A recent review suggested that rVWF can be administered as monotherapy, without FVIII concentrates, if an immediate rise in FVIII:C is not necessary, FVIII:C levels are above 40 iu/dl or during elective surgeries (Peyvandi et al, 2019a).
Two phase 3 trials are currently ongoing which investigate the use of rVWF as long‐term prophylaxis (NCT02973087) to prevent bleeding episodes, and the use of rVWF concentrate in paediatric patients (NCT 02932618). In the coming years it will become clear in which patients and in which settings rVWF can be used for the prevention and treatment of bleeding in VWD.
Supportive treatment of severe VWD
Besides increasing VWF and FVIII levels, VWD patients may also benefit from supportive treatment. Given that the haemostatic system is a balance between coagulation and fibrinolysis, inhibition of fibrinolysis may have a beneficial effect in VWD patients, especially in the management of mucocutanous bleeding. Furthermore, women with VWD with menorrhagia may benefit from hormonal therapy.
Fibrinolysis inhibitors
Fibrinolysis inhibitors prevent fibrin degradation by inhibiting the interaction between plasminogen and fibrin. The use of these agents is recommended in case of mucocutaneous bleeding, because of their high effectiveness in controlling bleeds associated with mucosa, in which high fibrinolytic activity is found (Sindet‐Pedersen, 1991). In addition, they have low costs and low risk of major side‐effects. The effectiveness of fibrinolysis inhibitors, such as tranexamic acid and epsilon‐aminocaproic acid, in patients with inherited bleeding disorders undergoing oral or dental surgery has recently been reviewed (van Galen et al, 2019). It was found that only a limited number of studies have been performed in haemophilia patients, whereas no studies were performed in VWD. The performed studies revealed a beneficial effect of systemically administered tranexamic acid and reduction of post‐operative bleeding (van Galen et al, 2019).
We prescribe tranexamic acid for (severe) VWD patients with mucosal tissue‐associated surgery, including tonsillectomy, hysterectomy and gastro‐intestinal surgery or with mucocutaneous bleeding: 1000 mg four times a day (total 4000 mg daily) in adults and 25 mg/kg/day divided over 2–3 doses per day in children for 7 days. In patients undergoing dental surgery we additionally prescribe mouth wash 4 times daily with oral tranexamic solution.
Hormonal treatment
Menorrhagia is a major clinical problem in women with VWD and is associated with a reduced quality of life (Kadir et al, 1998; de Wee et al, 2012). About 80% of women with VWD experience menorrhagia and need some kind of treatment for their symptoms, in contrast to 5–10% of the general population (de Wee et al, 2011b). Moreover, hysterectomy is performed because of menorrhagia in 20% of women with VWD, which is nearly twice as high compared to the general population (de Wee et al, 2011b).
VWD women with menorrhagia should first be investigated by a gynaecologist to exclude anatomical and hormonal abnormalities. If no gynaecological abnormalities are found, oral contraceptives containing progesterone and oestrogens are recommended for menorrhagia treatment (Kadir et al, 1998). Women can choose to have a contraception‐free week or continuous treatment (without a week stop). Both methods have been shown to be effective in decreasing bleeding severity and bleeding days in healthy women (Kwiecien et al, 2003). Tranexamic acid can also be used during the menstrual period to decrease blood loss (Bonnar & Sheppard, 1996).
In case oral contraceptives are not effective or if women do not want to use them, a levonorgestrel‐containing intrauterine device (IUD; Mirena®) may be an alternative. Adequate VWF and FVIII levels are required at the time of IUD placement, by infusion of either desmopressin or VWF/FVIII concentrates. If IUDs are not successful, desmopressin or VWF concentrates may be necessary to prevent and treat menorrhagia. This can be given prophylactically during the first few days of menstruation, and results in reduced blood loss in most women (Holm et al, 2015). If all these treatment options are not successful, then endometrial ablation or hysterectomy may be considered (Rubin et al, 2004).
Platelet transfusion
VWD patients with intractable bleeding, despite infusion of VWF/FVIII concentrate, can benefit from platelet transfusion (Castillo et al, 1991). In contrast to platelets of VWD patients, donor platelets contain functional VWF and may therefore deliver VWF at the site of injury. Several case series have shown that platelet transfusions can improve haemostasis and may stop bleeding episodes in VWD patients (Fressinaud et al, 1994; Castillo et al, 1997). Recent in vitro studies indicated that platelets restore thrombin generation in VWD type 3 (Szanto et al, 2019). No clinical data are available yet to indicate when platelet transfusions should be initiated in severe VWD patients with intractable bleeding. One should take the severity and site of bleeding into account when considering initiation of platelet transfusions. In case of joint bleeds, muscle bleeds, central nervous system bleeds or continuous lowering of haemoglobin despite VWF/FVIII concentrates, one should consider initiating platelet infusions.
Management in specific situations
Pregnancy and labour
For women with severe VWD, pregnancy and delivery carry a potential risk for bleeding and should be carefully monitored and managed (Fig 1). In the general population VWF and FVIII levels increase during pregnancy, especially in the third trimester. A similar increase is seen in mild type 1 VWD patients. At the time of labour, FVIII and VWF levels can be two to three times higher compared to baseline levels, and may thereby normalise (Sood et al, 2016). Therefore, VWF and FVIII levels should be measured at least once in the third trimester to be able to determine a treatment plan for delivery (Fig 1). After delivery, VWF and FVIII levels decrease to baseline levels within 3 weeks postpartum (James et al, 2015). In type 2 VWD, VWF:Ag and FVIII:C levels also increase during pregnancy, but VWF:RCo remains low (Castaman & James, 2019). Therefore, the increase of dysfunctional VWF protein may not normalise haemostasis in these women. In women with VWD type 3 no VWF is detectable and FVIII levels remain low during pregnancy, making them most prone to post‐partum haemorrhage (PPH).
Figure 1.
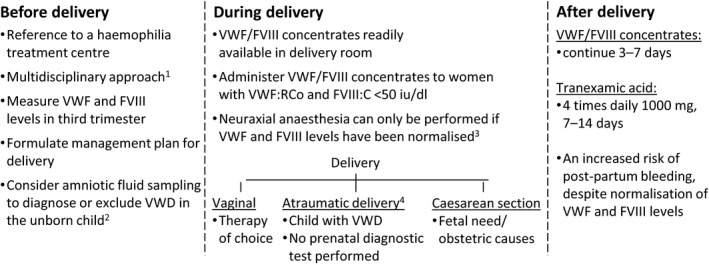
Management of delivery in severe VWD patients. 1Consists of a haematologist, clinical geneticist, gynaecologist, paediatric haematologist and anaesthesiologist. 2If the causative VWF gene mutation is known in the mother. 3Either physiologically or by factor infusion or desmopressin. 4No invasive management with vacuum or forceps.
During delivery, complications may not only concern the mother with VWD, but also the potentially affected child. Therefore, it is of utmost importance to follow a multidisciplinary approach (Fig 1). The monitoring and care of pregnant women with VWD should only be undertaken in hospitals that have experience with patients with bleeding disorders. Furthermore, a multidisciplinary team of (at least) a haematologist, paediatric haematologist, gynaecologist and anaesthesiologist with expertise in bleeding disorders should formulate a management plan for care during pregnancy and at the time of delivery (Fig 1).
Both the bleeding risk for the pregnant patient and the possibility of bleeding in an affected child have to be considered. In many patients with type 2, in whom the causative VWF mutation is known, we discuss with the patient and her partner the possibility of performing amniotic fluid aspiration at 32–24 weeks of pregnancy to establish or exclude the diagnosis of VWF in the child. The causative mutation should be known preferably before pregnancy. In case the child is affected by severe VWD, atraumatic labour should be advised. This is also advised in case no prenatal diagnostics have been performed or are inconclusive (Fig 1). In women with type 3 VWD caused by a double heterozygote mutation, this is generally not necessary, because the child will probably not be severely affected, although this may depend on the underlying mutations (Castaman et al, 2006; Peake & Goodeve, 2010).
Although several studies have been performed on postpartum haemorrhage in VWD patients, the quality of those observational studies is low (Kadir & Chi, 2006; James et al, 2015; Stoof et al, 2015). It is generally accepted that no treatment is necessary if the levels of VWF and FVIII are >50 iu/dl during third trimester. Desmopressin can be administered in women with type 1 VWD that are known to have a good desmopressin response (Nichols et al, 2008; Laffan et al, 2014). We only administer desmopressin after clamping the umbilical cord in order to prevent severe hypotension in the mother, which can harm the unborn child. There is still limited evidence for treatment recommendations using FVIII/VWF concentrates in pregnant VWD patients. In an observational study, James et al (2015) found no incidence of PPH in 17 type 1 VWD patients with VWF and FVIII levels >50 iu/dl during third trimester, who did not receive prophylactic treatment. In 15 women (7 with type 1 and 8 with type 2) with mean VWF:RCo of 34 (0–75) iu/dl during third trimester that were prophylactically treated with factor concentrate during labour, only one patient with type 2B VWD had excessive bleeding (James et al, 2015). VWD women who were prophylactically treated with factor concentrates had, despite treatment, more blood loss compared to women without VWD and untreated type 1 VWD women (James et al, 2015). Stoof et al (2015) observed PPH in over 50% of type 1 and type 2 VWD patients with VWF levels <50 iu/dl in the third trimester, despite VWF concentrate treatment aiming target levels of 100 iu/dl at labour. Based on these findings, they suggested that higher (more physiological) levels of 150–200 iu/dl, should be the target at the time of delivery. Current treatment guidelines may not be sufficient to prevent PPH in these women, and new treatment approaches aiming for higher VWF and FVIII levels may be needed to prevent PPH in this group (Ragni et al, 2017).
Thrombocytopenia may occur during pregnancy in women with type 2B VWD. It is recommended that platelet counts should be monitored regularly in the third trimester. In cases where platelet counts at delivery are <50 × 109/l, platelet transfusion is recommended with a target level >50 × 109/l (Kruse‐Jarres & Johnsen, 2018).
Neuraxial anaesthesia can only be performed if VWF and FVIII levels have been normalised. If levels have not been normalised at the time of labour, and neuraxial anaesthesia is preferred, prophylactic factor infusion or desmopressin should be given to ensure normal VWF and FVIII levels at the time of performing neuraxial anaesthesia (Nichols et al, 2008). However, it should be noted that some guidelines recommended against neuraxial anaesthesia in patients with type 2 or 3 VWD (Laffan et al, 2014).
For women with severe VWD, we infuse FVIII/VWF at a concentrate dosage of 40–60 VWF:RCo iu/kg at start of labour, continued twice daily 25 iu/kg for 3–5 days for vaginal labour, and 5–7 days after caesarean section, depending on bleeding and factor levels (Table 1). In addition, all women with VWD should, independent of their VWD severity and VWF levels, receive tranexamic acid peri‐ and at least for 7 days postpartum (Fig 1). Tranexamic acid lowers the risk of secondary post‐partum haemorrhage and is safe for pregnant women and for their child during lactation (Hawke et al, 2016).
Surgery and dental interventions
In patients undergoing surgery, the goal is to increase VWF and FVIII levels sufficiently to prevent bleeding. For major surgery, peak VWF and FVIII levels should be at least 100 iu/dl on day 1, to establish normal haemostasis (Table 1). In severe VWD patients, this can be achieved by infusion of plasma‐derived FVIII/VWF concentrate, pure VWF concentrate or rVWF. VWF concentrates should be administered at a loading dose of 50–60 VWF:RCo iu/kg in major surgery, followed by half of the dosage for maintenance every 8–24 h for major surgery and every 12–48 h for minor surgery (Castaman et al, 2013; Laffan et al, 2014; Leebeek & Eikenboom, 2016). After major surgery, trough levels should be at least 50 iu/dl for 7–10 days (Table 1). For minor surgery one can aim for peak target FVIII and VWF levels of 50–80 iu/dl on day 1 followed by trough levels of at least 30 iu/dl for 1–5 days, depending on the type of surgery. In patients undergoing dental extraction, peak target levels of at least 50 iu/dl on day 1 are sufficient to prevent bleeding. Moreover, local measures, including secure suturing, wound compression and good wound sealing are, in addition to fibrinolysis inhibitors, important to prevent re‐bleeding.
Treatment of gastrointestinal bleeding
Gastrointestinal bleeding is quite common and can be life threatening in severe VWD patients. In general, the prevalence of gastrointestinal bleeding is about 15% in VWD patients, but in patients with type 2 and type 3 VWD the prevalence is up to 27% (de Wee et al, 2012). Previous studies have shown that gastrointestinal bleeding almost exclusively occurs in patients that have a reduction of HMW VWF multimers, which is the case in type 2A, type 2B and type 3 VWD (Leebeek & Eikenboom, 2016). Gastrointestinal bleeding is more common in young, black or male VWD patients (Tsagianni et al, 2019). The most significant predictors of bleeding include angiodysplasia, diverticulitis, hepatitis C and smoking (Tsagianni et al, 2019).
VWD patients with gastrointestinal bleeding should be treated immediately with VWF/FVIII concentrates. In addition, endoscopic management is needed to be able to treat local sites of bleeding. Many VWD patients have recurrent gastrointestinal bleeding, despite treatment with factor concentrates. This may be caused by angiodysplasia or vascular malformations in the intestines, which give a higher chance for continuation of bleeding or re‐bleeding (Makris et al, 2015; Tsagianni et al, 2019). Many drugs have been used to treat VWD patients with persistent gastrointestinal bleeding. The effectiveness of octreotide and thalidomide has been shown in the treatment of angiodysplasia‐related gastrointestinal bleeding in the general population (Iannone et al, 2016). For VWD patients, this has only been reported in small case series (Bowers et al, 2000; Engelen et al, 2015). Statins, which have anti‐angiogenetic properties, have also been reported to reduce bleeding in intractable gastrointestinal bleeding in VWD patients (Sohal & Laffan, 2008; Alikhan & Keeling, 2010). The use of lenalidomide as treatment for angiodysplasia‐related gastro intestinal bleeding in VWD was recently reported to be successful in increasing bleed‐free duration (Khatri et al, 2018). It was also recently shown in one case with type 2A VWD that rVWF, containing high content of HMW multimers, was effective in the treatment of severe gastrointestinal bleeding that was unresponsive to other standard treatments for VWD (Brown, 2017). It is noteworthy that the previously mentioned case reports may be subjected to publication bias, and therefore well‐designed studies are needed to assess the efficacy and side‐effects of these drugs for the treatment of gastrointestinal bleeding in VWD patients.
In our centre, we only use thalidomide and statins in patient with recurrent gastrointestinal bleeding despite local measures and on‐demand infusion of FVIII/VWF concentrates. If bleeding persists we initiate long‐term prophylaxis with FVIII/VWF concentrate (Saccullo & Makris, 2016).
Patients with alloantibodies to VWF
A rare but severe side effect of treatment with plasma‐derived factor concentrates is development of alloantibodies against VWF. Alloantibodies against VWF form in response to treatment with plasma‐derived VWF factor ‐containing concentrates, and are mostly polyclonal IgG antibodies (James et al, 2013). The first results of the 3WINTER‐IPS project, which included a large cohort of 260 type 3 VWD patients, showed that a 6% prevalence of alloantibodies in type 3 VWD patients (Stufano et al, 2019). Recently the first type 2B VWD patient with alloantibodies was reported (Baaij et al, 2015). Patients with alloantibodies against VWF generally have a limited recovery and a rapid clearance of VWF after infusion of VWF concentrates (James et al, 2013; Franchini & Mannucci, 2018). Alloantibodies may also lead to immune complex formation and, subsequently, complement activation, causing life‐threatening anaphylactic allergic reactions (Bergamaschini et al, 1995; Franchini et al, 2009; James et al, 2013; Franchini & Mannucci, 2018). Therefore, VWF‐containing concentrates are contraindicated in patients with alloantibodies against VWF.
Several treatment options have been reported for the treatment of severe VWD patients with alloantibodies (Scott et al, 2018). Continuous infusion of high dose FVIII concentrates can be a treatment option, however high dosages of FVIII are needed because FVIII has a very short half‐life in these patients (Mannucci & Federici, 1995). Moreover, this may be contraindicated in patients with high titre VWF alloantibodies, because it was shown that it can precipitate anaphylactic reactions (Mannucci et al, 1981). In patients that do not have high‐titre alloantibodies and that do not have an allergic reaction to VWF, a high‐dose of VWF/FVIII concentrate may also be successful (Martin‐Salces et al, 2012). Furthermore, there is limited evidence to treat patients with alloantibodies to VWF with recombinant activated FVII (rFVIIa; Novoseven®) or FVIII inhibitor bypassing activity (FEIBA®) (Grossmann et al, 2000; Boyer‐Neumann et al, 2003), although one of the rare cases that was treated with rFVIIa developed venous thromboembolism (Boyer‐Neumann et al, 2003).
Long‐term prophylactic treatment
Prophylactic treatment with regular infusions of factor concentrate is currently the standard of care for patients with severe haemophilia A and B, but this is not the case in patients with severe VWD. Despite the fact that patients with severe VWD may have recurrent bleeds, including joint bleeds, gastrointestinal bleeds, severe nose bleeds and menorrhagia, only a few studies on the use of prophylaxis have been reported (Berntorp & Petrini, 2005; Abshire et al, 2015). In Sweden, prophylaxis has been given to children and adults with severe VWD for many decades, mainly in children with severe nose bleeds and adults with joints bleeds (Berntorp & Petrini, 2005). In an international survey by the VWD Prophylaxis Network covering 5300 VWD patients, 99 patients had received prophylaxis, predominantly for joint bleeding (41%), oral/nasal bleeding (24%) and gastro‐intestinal bleeding (12%) (Berntorp & Abshire 2006). Prophylaxis was given to 24% of patients with type 3, 1·8% with type 2 and only 0·2% with type 1.
The first prospective study on prophylaxis with VWF/FVIII concentrate in 12 severe VWD patients showed that a dose of 50 VWF:RCo iu/kg two or three times per week substantially reduced the number of mucosal and joint bleeding episodes (Abshire et al, 2015). The data from this prospective study were analysed together with a retrospective study and revealed significant reduction of bleeding rates for epistaxis, gastrointestinal bleeding, joint bleeding and menorrhagia. Patients with gastrointestinal bleeding needed three or more times per week prophylaxis, as well as higher dosages (Holm et al, 2015). Recently a prospective randomised Italian study was reported on the use of prophylaxis versus on‐demand treatment in 19 patients with severe VWD. This phase 3 study also showed a reduction of the number of bleeding events, although it had a small sample size and strong heterogeneity in the study population (Peyvandi et al, 2019c). A prospective phase 3 trial is ongoing to investigate the efficacy and safety of prophylactic use of recombinant VWF concentrate in patients with severe VWD (NCT02973087).
Currently, we initiate prophylactic treatment with FVIII/ VWF concentrates only in severe VWD patients that have repeated bleeding episodes, mostly concerning joint bleeding and gastro‐intestinal bleeding. Also, in patients with severe menorrhagia that do not respond to other treatment, such as oral contraceptives, intrauterine device and tranexamic acid, we initiate prophylactic VWF/FVIII concentrate during the menstrual period. Before starting prophylaxis, patients receive instructions for home treatment and self‐infusion of the coagulation factor concentrate. Prophylaxis is initiated with twice a week 50 VWF:RCo iu/kg intravenously and the dose is increased to three times a week if bleeding persists. The benefit and burden of prophylaxis is evaluated with the patients at least twice‐yearly to discuss continuing or stopping prophylaxis. Most patients are reluctant to discontinue treatment, especially in those with gastrointestinal bleeding, because of the observed benefit of prophylaxis (Saccullo & Makris, 2016).
Future developments in the treatment of severe VWD patients
Individualized treatment based on pharmacokinetic modelling
Recently we have shown that the current dosing of FVIII/VWF concentrate for surgical procedures based on body weight results in an overdosing in most patients with VWD (Hazendonk et al, 2018a). This suggest that individualised dosing based on a pharmacokinetic study of each patient planned for surgery may result in better outcome and may be more cost‐effective (Berntorp, 2018, Hazendonk et al, 2018a). Only limited studies have been performed so far on the use of pharmacokinetic‐based dosing in VWD (Lethagen et al, 2007; Di Paola et al, 2011). These studies revealed that pharmacokinetic analysis pre‐surgery was of limited value. Therefore better models should be developed based on both FVIII:C and VWF:RCo levels to improve accuracy of prediction, as is now also employed in haemophilia (Hazendonk et al, 2018b). We are currently performing a trial investigating the individualized treatment of VWD patients based on population pharmacokinetic models. In this trial, we calculate the optimum required dosages of desmopressin and VWF/FVIII concentrates based on VWF and FVIII measurements at baseline, 4, 24 and 48 h after administration of desmopressin or VWF/FVIII concentrates. Using population models, one can predict the dose needed by a patient to reach the required target levels, reducing sub‐ and supratherapeutical VWF and FVIII levels.
Gene therapy
Gene therapy has only been investigated in a preclinical setting for VWD (De Meyer et al, 2008; Wang et al, 2012; Portier et al, 2018). In patients with haemophilia, gene therapy has shown very good results in clinical trials (Perrin et al, 2019a). Compared to haemophilia, gene therapy for VWD seems more challenging, mainly because of the large size of the VWF gene, which makes it difficult to insert VWF cDNA in viral gene transfer vectors. In addition, VWF is synthesized mainly in the endothelium, which is difficult to target with gene therapy. So far, VWF liver‐specific gene transfer has been used in mice models leading to higher plasma VWF levels (Wang et al, 2012; Portier et al, 2018). More preclinical studies are needed before gene therapy can be used in clinical trials in patients suffering from severe VWD.
Conclusion
Management of severe VWD patients is more demanding than management of mild VWD/ type 1 patients. Severe VWD patients carry a high risk of bleeding in some specific situations, such as during delivery or surgery and after development of alloantibodies to VWF. Patients with severe VWD who lack HMW multimers also have a high risk of recurrent gastrointestinal bleeding due to angiodysplasia. Patients suffering from recurrent bleedings may benefit from prophylaxis, which is only offered to a small number of patients. Future prospective studies are needed to investigate the benefit of newer treatment options, to improve current management and update guidelines in these patients.
Author contributions
F. Leebeek and F. Atiq wrote the manuscript and critically revised the whole manuscript.
Conflict of Interest
F.W.G. Leebeek received research support from CSL Behring and Shire/Takeda for performing the Willebrand in the Netherlands (WiN) study, and is consultant for uniQure, Novo Nordisk and Shire/Takeda, of which the fees go to the institution. He also received travel support from Sobi and is DSMB member of a study sponsored by Roche. F. Atiq received the CSL‐Behring‐Heimburger Award 2018, and a travel grant from Sobi.
Supporting information
Data S1 . Literature search for severe VWD treatment.
Acknowledgements
We would like to thank Wichor M. Bramer for assistance with the literature search (see Data S1).
References
- Abshire, T. , Cox‐Gill, J. , Kempton, C.L. , Leebeek, F.W. , Carcao, M. , Kouides, P. , Donfield, S. & Berntorp, E. (2015) Prophylaxis escalation in severe von Willebrand disease: a prospective study from the von Willebrand Disease Prophylaxis Network. Journal of Thrombosis and Haemostasis, 13, 1585–1589. [DOI] [PubMed] [Google Scholar]
- Alikhan, R. & Keeling, D. (2010) Von Willebrand disease, angiodysplasia and atorvastatin. British Journal of Haematology, 149, 159–160. [DOI] [PubMed] [Google Scholar]
- Atiq, F. , Mauser‐Bunschoten, E.P. , Eikenboom, J. , van Galen, K.P.M. , Meijer, K. , de Meris, J. , Cnossen, M.H. , Beckers, E.A.M. , Laros‐van Gorkom, B.A.P. , Nieuwenhuizen, L. , van der Bom, J.G. , Fijnvandraat, K. & Leebeek, F.W.G. ; WiN Study Group . (2019) Sports participation and physical activity in patients with von Willebrand disease. Haemophilia, 25, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baaij, M. , van Galen, K.P. , Urbanus, R.T. , Nigten, J. , Eikenboom, J.H. & Schutgens, R.E. (2015) First report of inhibitory von Willebrand factor alloantibodies in type 2B von Willebrand disease. British Journal of Haematology, 171, 424–427. [DOI] [PubMed] [Google Scholar]
- Bergamaschini, L. , Mannucci, P.M. , Federici, A.B. , Coppola, R. , Guzzoni, S. & Agostoni, A. (1995) Posttransfusion anaphylactic reactions in a patient with severe von Willebrand disease: role of complement and alloantibodies to von Willebrand factor. Journal of Laboratory and Clinical Medicine, 125, 348–355. [PubMed] [Google Scholar]
- Berntorp, E. (2018) Replacement therapy during surgery in von Willebrand disease needs personalization. Haemophilia, 24, 338–340. [DOI] [PubMed] [Google Scholar]
- Berntorp, E. & Abshire, T. ; vWD PN Steering Committee . (2006) The von Willebrand disease prophylaxis network (vWD PN): exploring a treatment concept. Thrombosis Research, 118, S19–22. [DOI] [PubMed] [Google Scholar]
- Berntorp, E. & Petrini, P. (2005) Long‐term prophylaxis in von Willebrand disease. Blood Coagulation & Fibrinolysis, 16 (Suppl 1), S23–S26. [DOI] [PubMed] [Google Scholar]
- Bodo, I. , Eikenboom, J. , Montgomery, R. , Patzke, J. , Schneppenheim, R. & Di Paola, J. ; von Willebrand factor Subcommittee of the Standardization and Scientific Committee of the International Society for Thrombosis and Haemostasis . (2015) Platelet‐dependent von Willebrand factor activity. Nomenclature and methodology: communication from the SSC of the ISTH. Journal of Thrombosis and Haemostasis, 13, 1345–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boender, J. , Eikenboom, J. , van der Bom, J.G. , Meijer, K. , de Meris, J. , Fijnvandraat, K. , Cnossen, M.H. , Laros‐van Gorkom, B.A.P. , van Heerde, W.L. , Mauser‐Bunschoten, E.P. , de Maat, M.P.M. & Leebeek, F.W.G. ; WiN Study Group . (2018) Clinically relevant differences between assays for von Willebrand factor activity. Journal of Thrombosis and Haemostasis, 16, 2413–2424. [DOI] [PubMed] [Google Scholar]
- Bonnar, J. & Sheppard, B.L. (1996) Treatment of menorrhagia during menstruation: randomised controlled trial of ethamsylate, mefenamic acid, and tranexamic acid. BMJ, 313, 579–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borel‐Derlon, A. , Federici, A.B. , Roussel‐Robert, V. , Goudemand, J. , Lee, C.A. , Scharrer, I. , Rothschild, C. , Berntorp, E. , Henriet, C. , Tellier, Z. , Bridey, F. & Mannucci, P.M. (2007) Treatment of severe von Willebrand disease with a high‐purity von Willebrand factor concentrate (Wilfactin): a prospective study of 50 patients. Journal of Thrombosis and Haemostasis, 5, 1115–1124. [DOI] [PubMed] [Google Scholar]
- Bowers, M. , McNulty, O. & Mayne, E. (2000) Octreotide in the treatment of gastrointestinal bleeding caused by angiodysplasia in two patients with von Willebrand's disease. British Journal of Haematology, 108, 524–527. [DOI] [PubMed] [Google Scholar]
- Boyer‐Neumann, C. , Dreyfus, M. , Wolf, M. , Veyradier, A. & Meyer, D. (2003) Multi‐therapeutic approach to manage delivery in an alloimmunized patient with type 3 von Willebrand disease. Journal of Thrombosis and Haemostasis, 1, 190–192. [DOI] [PubMed] [Google Scholar]
- Brown, R. (2017) Recombinant von Willebrand factor for severe gastrointestinal bleeding unresponsive to other treatments in a patient with type 2A von Willebrand disease: a case report. Blood Coagulation & Fibrinolysis, 28, 570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaman, G. & James, P.D. (2019) Pregnancy and delivery in women with von Willebrand disease. European Journal of Haematology, 103, 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaman, G. , Rodeghiero, F. , Tosetto, A. , Cappelletti, A. , Baudo, F. , Eikenboom, J.C. , Federici, A.B. , Lethagen, S. , Linari, S. , Lusher, J. , Nishino, M. , Petrini, P. , Srivastava, A. & Ungerstedt, J.S. (2006) Hemorrhagic symptoms and bleeding risk in obligatory carriers of type 3 von Willebrand disease: an international, multicenter study. Journal of Thrombosis and Haemostasis, 4, 2164–2169. [DOI] [PubMed] [Google Scholar]
- Castaman, G. , Lethagen, S. , Federici, A.B. , Tosetto, A. , Goodeve, A. , Budde, U. , Batlle, J. , Meyer, D. , Mazurier, C. , Fressinaud, E. , Goudemand, J. , Eikenboom, J. , Schneppenheim, R. , Ingerslev, J. , Vorlova, Z. , Habart, D. , Holmberg, L. , Pasi, J. , Hill, F. , Peake, I. & Rodeghiero, F. (2008) Response to desmopressin is influenced by the genotype and phenotype in type 1 von Willebrand disease (VWD): results from the European Study MCMDM‐1VWD. Blood, 111, 3531–3539. [DOI] [PubMed] [Google Scholar]
- Castaman, G. , Federici, A.B. , Tosetto, A. , La Marca, S. , Stufano, F. , Mannucci, P.M. & Rodeghiero, F. (2012) Different bleeding risk in type 2A and 2M von Willebrand disease: a 2‐year prospective study in 107 patients. Journal of Thrombosis and Haemostasis, 10, 632–638. [DOI] [PubMed] [Google Scholar]
- Castaman, G. , Goodeve, A. & Eikenboom, J. ; European Group on von Willebrand Disease . (2013) Principles of care for the diagnosis and treatment of von Willebrand disease. Haematologica, 98, 667–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo, R. , Monteagudo, J. , Escolar, G. , Ordinas, A. , Magallon, M. & Martin Villar, J. (1991) Hemostatic effect of normal platelet transfusion in severe von Willebrand disease patients. Blood, 77, 1901–1905. [PubMed] [Google Scholar]
- Castillo, R. , Escolar, G. , Monteagudo, J. , Aznar‐Salatti, J. , Reverter, J.C. & Ordinas, A. (1997) Hemostasis in patients with severe von Willebrand disease improves after normal platelet transfusion and normalizes with further correction of the plasma defect. Transfusion, 37, 785–790. [DOI] [PubMed] [Google Scholar]
- Coppola, A. , Franchini, M. , Makris, M. , Santagostino, E. , Di Minno, G. & Mannucci, P.M. (2012) Thrombotic adverse events to coagulation factor concentrates for treatment of patients with haemophilia and von Willebrand disease: a systematic review of prospective studies. Haemophilia, 18, e173–187. [DOI] [PubMed] [Google Scholar]
- De Jong, A. & Eikenboom, J. (2016) Developments in the diagnostic procedures for von Willebrand disease. Journal of Thrombosis and Haemostasis, 14, 449–460. [DOI] [PubMed] [Google Scholar]
- De Meyer, S.F. , Vandeputte, N. , Pareyn, I. , Petrus, I. , Lenting, P.J. , Chuah, M.K. , VandenDriessche, T. , Deckmyn, H. & Vanhoorelbeke, K. (2008) Restoration of plasma von Willebrand factor deficiency is sufficient to correct thrombus formation after gene therapy for severe von Willebrand disease. Arteriosclerosis, Thrombosis, and Vascular Biology, 28, 1621–1626. [DOI] [PubMed] [Google Scholar]
- Di Paola, J. , Lethagen, S. , Gill, J. , Mannucci, P. , Manco‐Johnson, M. , Bernstein, J. , Nichols, W.L. & Bergman, G.E. (2011) Presurgical pharmacokinetic analysis of a von Willebrand factor/factor VIII (VWF/FVIII) concentrate in patients with von Willebrand's disease (VWD) has limited value in dosing for surgery. Haemophilia, 17, 752–758. [DOI] [PubMed] [Google Scholar]
- Engelen, E.T. , van Galen, K.P. & Schutgens, R.E. (2015) Thalidomide for treatment of gastrointestinal bleedings due to angiodysplasia: a case report in acquired von Willebrand syndrome and review of the literature. Haemophilia, 21, 419–429. [DOI] [PubMed] [Google Scholar]
- Federici, A.B. (2004) Clinical diagnosis of von Willebrand disease. Haemophilia, 10 (Suppl 4), 169–176. [DOI] [PubMed] [Google Scholar]
- Federici, A.B. (2008) The use of desmopressin in von Willebrand disease: the experience of the first 30 years (1977–2007). Haemophilia, 14 (Suppl 1), 5–14. [DOI] [PubMed] [Google Scholar]
- Federici, A.B. , Mannucci, P.M. , Castaman, G. , Baronciani, L. , Bucciarelli, P. , Canciani, M.T. , Pecci, A. , Lenting, P.J. & De Groot, P.G. (2009) Clinical and molecular predictors of thrombocytopenia and risk of bleeding in patients with von Willebrand disease type 2B: a cohort study of 67 patients. Blood, 113, 526–534. [DOI] [PubMed] [Google Scholar]
- Federici, A.B. , Bucciarelli, P. , Castaman, G. , Mazzucconi, M.G. , Morfini, M. , Rocino, A. , Schiavoni, M. , Peyvandi, F. , Rodeghiero, F. & Mannucci, P.M. (2014) The bleeding score predicts clinical outcomes and replacement therapy in adults with von Willebrand disease. Blood, 123, 4037–4044. [DOI] [PubMed] [Google Scholar]
- Flood, V.H. , Christopherson, P.A. , Gill, J.C. , Friedman, K.D. , Haberichter, S.L. , Bellissimo, D.B. , Udani, R.A. , Dasgupta, M. , Hoffmann, R.G. , Ragni, M.V. , Shapiro, A.D. , Lusher, J.M. , Lentz, S.R. , Abshire, T.C. , Leissinger, C. , Hoots, W.K. , Manco‐Johnson, M.J. , Gruppo, R.A. , Boggio, L.N. , Montgomery, K.T. , Goodeve, A.C. , James, P.D. , Lillicrap, D. , Peake, I.R. & Montgomery, R.R. (2016) Clinical and laboratory variability in a cohort of patients diagnosed with type 1 VWD in the United States. Blood, 127, 2481–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchini, M. & Mannucci, P.M. (2018) Alloantibodies in von Willebrand disease. Seminars in Thrombosis and Hemostasis, 44, 590–594. [DOI] [PubMed] [Google Scholar]
- Franchini, M. , Lippi, G. , Montagnana, M. , Targher, G. , Zaffanello, M. , Salvagno, G.L. , Rivolta, G.F. , Di Perna, C. & Tagliaferri, A. (2009) Anaphylaxis in patients with congenital bleeding disorders and inhibitors. Blood Coagulation & Fibrinolysis, 20, 225–229. [DOI] [PubMed] [Google Scholar]
- Fressinaud, E. , Federici, A.B. , Castaman, G. , Rothschild, C. , Rodeghiero, F. , Baumgartner, H.R. , Mannucci, P.M. & Meyer, D. (1994) The role of platelet von Willebrand factor in platelet adhesion and thrombus formation: a study of 34 patients with various subtypes of type I von Willebrand disease. British Journal of Haematology, 86, 327–332. [DOI] [PubMed] [Google Scholar]
- van Galen, K.P. , Sanders, Y.V. , Vojinovic, U. , Eikenboom, J. , Cnossen, M.H. , Schutgens, R.E. , van der Bom, J.G. , Fijnvandraat, K. , Laros‐Van Gorkom, B.A. , Meijer, K. , Leebeek, F.W. & Mauser‐Bunschoten, E.P. ; for the Win Study Group . (2015) Joint bleeds in von Willebrand disease patients have significant impact on quality of life and joint integrity: a cross‐sectional study. Haemophilia, 21, e185–192. [DOI] [PubMed] [Google Scholar]
- van Galen, K.P.M. , de Kleijn, P. , Foppen, W. , Eikenboom, J. , Meijer, K. , Schutgens, R.E.G. , Fischer, K. , Cnossen, M.H. , de Meris, J. , Fijnvandraat, K. , van der Bom, J.G. , Laros‐van Gorkom, B.A.P. , Leebeek, F.W.G. & Mauser‐Bunschoten, E.P. ; WiN Study Group . (2017) Long‐term impact of joint bleeds in von Willebrand disease: a nested case‐control study. Haematologica, 102, 1486–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Galen, K.P.M. , Timmer, M. , de Kleijn, P. , Leebeek, F.W.G. , Foppen, W. , Schutgens, R.E.G. , Eikenboom, J. , Meijer, K. , Fijnvandraat, K. , Laros‐van Gorkom, B.A.P. , Twisk, J.W. , Mauser‐Bunschoten, E.P. & Fischer, K. ; WiN Study Group . (2018) Long‐term outcome after joint bleeds in Von Willebrand disease compared to haemophilia A: a post hoc analysis. Thrombosis and Haemostasis, 118, 1690–1700. [DOI] [PubMed] [Google Scholar]
- van Galen, K.P. , Engelen, E.T. , Mauser‐Bunschoten, E.P. , van Es, R.J. & Schutgens, R.E. (2019) Antifibrinolytic therapy for preventing oral bleeding in patients with haemophilia or Von Willebrand disease undergoing minor oral surgery or dental extractions. Cochrane Database Systematic Review, 4, CD011385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill, J.C. , Castaman, G. , Windyga, J. , Kouides, P. , Ragni, M. , Leebeek, F.W. , Obermann‐Slupetzky, O. , Chapman, M. , Fritsch, S. , Pavlova, B.G. , Presch, I. & Ewenstein, B. (2015) Hemostatic efficacy, safety, and pharmacokinetics of a recombinant von Willebrand factor in severe von Willebrand disease. Blood, 126, 2038–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodeve, A. , Eikenboom, J. , Castaman, G. , Rodeghiero, F. , Federici, A.B. , Batlle, J. , Meyer, D. , Mazurier, C. , Goudemand, J. , Schneppenheim, R. , Budde, U. , Ingerslev, J. , Habart, D. , Vorlova, Z. , Holmberg, L. , Lethagen, S. , Pasi, J. , Hill, F. , Hashemi Soteh, M. , Baronciani, L. , Hallden, C. , Guilliatt, A. , Lester, W. & Peake, I. (2007) Phenotype and genotype of a cohort of families historically diagnosed with type 1 von Willebrand disease in the European study, Molecular and Clinical Markers for the Diagnosis and Management of Type 1 von Willebrand Disease (MCMDM‐1VWD). Blood, 109, 112–121. [DOI] [PubMed] [Google Scholar]
- Grossmann, R.E. , Geisen, U. , Schwender, S. & Keller, F. (2000) Continuous infusion of recombinant factor VIIa (NovoSeven) in the treatment of a patient with type III von Willebrand's disease and alloantibodies against von Willebrand factor. Thrombosis and Haemostasis, 83, 633–634. [PubMed] [Google Scholar]
- Hawke, L. , Grabell, J. , Sim, W. , Thibeault, L. , Muir, E. , Hopman, W. , Smith, G. & James, P. (2016) Obstetric bleeding among women with inherited bleeding disorders: a retrospective study. Haemophilia, 22, 906–911. [DOI] [PubMed] [Google Scholar]
- Hazendonk, H. , Heijdra, J.M. , de Jager, N.C.B. , Veerman, H.C. , Boender, J. , van Moort, I. , Mathot, R.A.A. , Meijer, K. , Laros‐van Gorkom, B.A.P. , Eikenboom, J. , Fijnvandraat, K. , Leebeek, F.W.G. & Cnossen, M.H. ; “OPTI‐CLOT” and “WIN” Study Group . (2018a) Analysis of current perioperative management with Haemate® P/Humate P® in von Willebrand disease: identifying the need for personalized treatment. Haemophilia, 24, 460–470. [DOI] [PubMed] [Google Scholar]
- Hazendonk, H. , van Moort, I. , Mathot, R.A.A. , Fijnvandraat, K. , Leebeek, F.W.G. , Collins, P.W. , Cnossen, M.H. ; “OPTI‐CLOT” Study Group . (2018b) Setting the stage for individualized therapy in hemophilia: What role can pharmacokinetics play? Blood Reviews, 32, 265–271. [DOI] [PubMed] [Google Scholar]
- Heijdra, J.M. , Cnossen, M.H. & Leebeek, F.W.G. (2017) Current and emerging options for the management of inherited von Willebrand disease. Drugs, 77, 1531–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm, E. , Abshire, T.C. , Bowen, J. , Alvarez, M.T. , Bolton‐Maggs, P. , Carcao, M. , Federici, A.B. , Gill, J.C. , Halimeh, S. , Kempton, C. , Key, N.S. , Kouides, P. , Lail, A. , Landorph, A. , Leebeek, F. , Makris, M. , Mannucci, P. , Mauser‐Bunschoten, E.P. , Nugent, D. , Valentino, L.A. , Winikoff, R. & Berntorp, E. (2015) Changes in bleeding patterns in von Willebrand disease after institution of long‐term replacement therapy: results from the von Willebrand Disease Prophylaxis Network. Blood Coagulation & Fibrinolysis, 26, 383–388. [DOI] [PubMed] [Google Scholar]
- Iannone, A. , Principi, M. , Barone, M. , Losurdo, G. , Ierardi, E. & Di Leo, A. (2016) Gastrointestinal bleeding from vascular malformations: Is octreotide effective to rescue difficult‐to‐treat patients? Clinics and Research in Hepatology and Gastroenterology, 40, 373–377. [DOI] [PubMed] [Google Scholar]
- James, P.D. , Lillicrap, D. & Mannucci, P.M. (2013) Alloantibodies in von Willebrand disease. Blood, 122, 636–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James, A.H. , Konkle, B.A. , Kouides, P. , Ragni, M.V. , Thames, B. , Gupta, S. , Sood, S. , Fletcher, S.K. & Philipp, C.S. (2015) Postpartum von Willebrand factor levels in women with and without von Willebrand disease and implications for prophylaxis. Haemophilia, 21, 81–87. [DOI] [PubMed] [Google Scholar]
- de Jong, A. & Eikenboom, J. (2017) Von Willebrand disease mutation spectrum and associated mutation mechanisms. Thrombosis Research, 159, 65–75. [DOI] [PubMed] [Google Scholar]
- de Wee, E.M. , Mauser‐Bunschoten, E.P. , Van Der Bom, J.G. , Degenaar‐Dujardin, M.E. , Eikenboom, H.C. , Fijnvandraat, K. , de Goede‐Bolder, A. , Laros‐van Gorkom, B.A. , Meijer, K. , Raat, H. & Leebeek, F.W. ; Win Study Group . (2010) Health‐related quality of life among adult patients with moderate and severe von Willebrand disease. Journal of Thrombosis and Haemostasis, 8, 1492–1499. [DOI] [PubMed] [Google Scholar]
- de Wee, E.M. , Fijnvandraat, K. , de Goede‐Bolder, A. , Mauser‐Bunschoten, E.P. , Eikenboom, J.C. , Brons, P.P. , Smiers, F.J. , Tamminga, R. , Oostenbrink, R. , Raat, H. , van der Bom, J.G. & Leebeek, F.W. ; WiN Study Group . (2011a) Impact of von Willebrand disease on health‐related quality of life in a pediatric population. Journal of Thrombosis and Haemostasis, 9, 502–509. [DOI] [PubMed] [Google Scholar]
- de Wee, E.M. , Knol, H.M. , Mauser‐Bunschoten, E.P. , van der Bom, J.G. , Eikenboom, J.C. , Fijnvandraat, K. , De Goede‐Bolder, A. , Laros‐van Gorkom, B. , Ypma, P.F. , Zweegman, S. , Meijer, K. & Leebeek, F.W. ; for the Win Study Group . (2011b) Gynaecological and obstetric bleeding in moderate and severe von Willebrand disease. Thrombosis and Haemostasis, 106, 885–892. [DOI] [PubMed] [Google Scholar]
- de Wee, E.M. , Sanders, Y.V. , Mauser‐Bunschoten, E.P. , van der Bom, J.G. , Degenaar‐Dujardin, M.E. , Eikenboom, J. , de Goede‐Bolder, A. , Laros‐van Gorkom, B.A. , Meijer, K. , Hamulyak, K. , Nijziel, M.R. , Fijnvandraat, K. & Leebeek, F.W. ; WiN Study Group . (2012) Determinants of bleeding phenotype in adult patients with moderate or severe von Willebrand disease. Thrombosis and Haemostasis, 108, 683–692. [DOI] [PubMed] [Google Scholar]
- Kadir, R.A. & Chi, C. (2006) Women and von Willebrand disease: controversies in diagnosis and management. Seminars in Thrombosis and Hemostasis, 32, 605–615. [DOI] [PubMed] [Google Scholar]
- Kadir, R.A. , Sabin, C.A. , Pollard, D. , Lee, C.A. & Economides, D.L. (1998) Quality of life during menstruation in patients with inherited bleeding disorders. Haemophilia, 4, 836–841. [DOI] [PubMed] [Google Scholar]
- Khatri, N.V. , Patel, B. , Kohli, D.R. , Solomon, S.S. , Bull‐Henry, K. & Kessler, C.M. (2018) Lenalidomide as a novel therapy for gastrointestinal angiodysplasia in von Willebrand disease. Haemophilia, 24, 278–282. [DOI] [PubMed] [Google Scholar]
- Kruse‐Jarres, R. & Johnsen, J.M. (2018) How I treat type 2B von Willebrand disease. Blood, 131, 1292–1300. [DOI] [PubMed] [Google Scholar]
- Kwiecien, M. , Edelman, A. , Nichols, M.D. & Jensen, J.T. (2003) Bleeding patterns and patient acceptability of standard or continuous dosing regimens of a low‐dose oral contraceptive: a randomized trial. Contraception, 67, 9–13. [DOI] [PubMed] [Google Scholar]
- Laffan, M.A. , Lester, W. , O'Donnell, J.S. , Will, A. , Tait, R.C. , Goodeve, A. , Millar, C.M. & Keeling, D.M. (2014) The diagnosis and management of von Willebrand disease: a United Kingdom Haemophilia Centre Doctors Organization guideline approved by the British Committee for Standards in Haematology. British Journal of Haematology, 167, 453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin, M. , Aguila, S. , Schneppenheim, S. , Dalton, N. , Jones, K.L. , O'Sullivan, J.M. , O'Connell, N.M. , Ryan, K. , White, B. , Byrne, M. , Rafferty, M. , Doyle, M.M. , Nolan, M. , Preston, R.J.S. , Budde, U. , James, P. , Di Paola, J. & O'Donnell, J.S. (2017) Novel insights into the clinical phenotype and pathophysiology underlying low VWF levels. Blood, 130, 2344–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leebeek, F.W. & Eikenboom, J.C. (2016) Von Willebrand's Disease. New England Journal of Medicine, 375, 2067–2080. [DOI] [PubMed] [Google Scholar]
- Lethagen, S. , Kyrle, P.A. , Castaman, G. , Haertel, S. & Mannucci, P.M. ; HAEMATE P Surgical Study Group . (2007) von Willebrand factor/factor VIII concentrate (Haemate P) dosing based on pharmacokinetics: a prospective multicenter trial in elective surgery. Journal of Thrombosis and Haemostasis, 5, 1420–1430. [DOI] [PubMed] [Google Scholar]
- Makris, M. , Federici, A.B. , Mannucci, P.M. , Bolton‐Maggs, P.H. , Yee, T.T. , Abshire, T. & Berntorp, E. (2015) The natural history of occult or angiodysplastic gastrointestinal bleeding in von Willebrand disease. Haemophilia, 21, 338–342. [DOI] [PubMed] [Google Scholar]
- Mannucci, P.M. (2004) Treatment of von Willebrand's disease. New England Journal of Medicine, 351, 683–694. [DOI] [PubMed] [Google Scholar]
- Mannucci, P.M. & Federici, A.B. (1995) Antibodies to von Willebrand factor in von Willebrand disease. Advances in Experimental Medicine and Biology, 386, 87–92. [DOI] [PubMed] [Google Scholar]
- Mannucci, P.M. & Franchini, M. (2017) Von Willebrand's disease. New England Journal of Medicine, 376, 701. [DOI] [PubMed] [Google Scholar]
- Mannucci, P.M. , Ruggeri, Z.M. , Ciavarella, N. , Kazatchkine, M.D. & Mowbray, J.F. (1981) Precipitating antibodies to factor VIII/von Willebrand factor in von Willebrand's disease: effects on replacement therapy. Blood, 57, 25–31. [PubMed] [Google Scholar]
- Martin‐Salces, M. , Jimenez‐Yuste, V. , Alvarez‐Roman, M.T. , Rivas‐Pollmar, I. & Rodriguez de la Rua, A. (2012) Management of delivery with FVIII/VWF concentrates in a pregnant woman with type 3 von Willebrand disease and alloantibodies. Thrombosis and Haemostasis, 108, 796–798. [DOI] [PubMed] [Google Scholar]
- McLaughlin, J.M. , Munn, J.E. , Anderson, T.L. , Lambing, A. , Tortella, B. & Witkop, M.L. (2017) Predictors of quality of life among adolescents and young adults with a bleeding disorder. Health and Quality of Life Outcomes, 15, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metjian, A.D. , Wang, C. , Sood, S.L. , Cuker, A. , Peterson, S.M. , Soucie, J.M. & Konkle, B.A. ; HTCN Study Investigators . (2009) Bleeding symptoms and laboratory correlation in patients with severe von Willebrand disease. Haemophilia, 15, 918–925. [DOI] [PubMed] [Google Scholar]
- Ng, C. , Motto, D.G. & Di Paola, J. (2015) Diagnostic approach to von Willebrand disease. Blood, 125, 2029–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols, W.L. , Hultin, M.B. , James, A.H. , Manco‐Johnson, M.J. , Montgomery, R.R. , Ortel, T.L. , Rick, M.E. , Sadler, J.E. , Weinstein, M. & Yawn, B.P. (2008) von Willebrand disease (VWD): evidence‐based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia, 14, 171–232. [DOI] [PubMed] [Google Scholar]
- Peake, I.R. & Goodeve, A.C. (2010) Genetic testing for von Willebrand disease: the case for. Journal of Thrombosis and Haemostasis, 8, 13–16. [DOI] [PubMed] [Google Scholar]
- Perrin, G.Q. , Herzog, R.W. & Markusic, D.M. (2019a) Update on clinical gene therapy for hemophilia. Blood, 133, 407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyvandi, F. , Kouides, P. , Turecek, P.L. , Dow, E. & Berntorp, E. (2019a) Evolution of replacement therapy for von Willebrand disease: from plasma fraction to recombinant von Willebrand factor. Blood Reviews, 10.1016/j.blre.2019.04.001. [DOI] [PubMed] [Google Scholar]
- Peyvandi, F. , Mamaev, A. , Wang, J.D. , Stasyshyn, O. , Timofeeva, M. , Curry, N. , Cid, A.R. , Yee, T.T. , Kavakli, K. , Castaman, G. & Sytkowski, A. (2019b) Phase 3 study of recombinant von Willebrand factor in patients with severe von Willebrand disease who are undergoing elective surgery. Journal of Thrombosis and Haemostasis, 17, 52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyvandi, F. , Castaman, G. , Gresele, P. , De Cristofaro, R. , Schinco, P. , Bertomoro, A. , Morfini, M. , Gamba, G. , Barillari, G. , Jimenez‐Yuste, V. , Konigs, C. , Iorio, A. & Federici, A.B. (2019c) A phase III study comparing secondary long‐term prophylaxis versus on‐demand treatment with vWF/FVIII concentrates in severe inherited von Willebrand disease. Blood Transfusion, 4, 1–8. 10.2450/2019.0183-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pipe, S.W. , Montgomery, R.R. , Pratt, K.P. , Lenting, P.J. & Lillicrap, D. (2016) Life in the shadow of a dominant partner: the FVIII‐VWF association and its clinical implications for hemophilia A. Blood, 128, 2007–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portier, I. , Vanhoorelbeke, K. , Verhenne, S. , Pareyn, I. , Vandeputte, N. , Deckmyn, H. , Goldenberg, D.S. , Samal, H.B. , Singh, M. , Ivics, Z. , Izsvak, Z. & De Meyer, S.F. (2018) High and long‐term von Willebrand factor expression after Sleeping Beauty transposon‐mediated gene therapy in a mouse model of severe von Willebrand disease. Journal of Thrombosis and Haemostasis, 16, 592–604. [DOI] [PubMed] [Google Scholar]
- Ragni, M.V. , Machin, N. , James, A.H. , Seaman, C.D. , Malec, L.M. , Kessler, C.M. , Konkle, B.A. , Kouides, P.A. , Neff, A.T. , Philipp, C.S. & Brooks, M.M. (2017) Feasibility of the Von Willebrand disease PREVENT trial. Thrombosis Research, 156, 8–13. [DOI] [PubMed] [Google Scholar]
- Rodeghiero, F. , Tosetto, A. & Castaman, G. (2007) How to estimate bleeding risk in mild bleeding disorders. Journal of Thrombosis and Haemostasis, 5 (Suppl 1), 157–166. [DOI] [PubMed] [Google Scholar]
- Rubin, G. , Wortman, M. & Kouides, P.A. (2004) Endometrial ablation for von Willebrand disease‐related menorrhagia–experience with seven cases. Haemophilia, 10, 477–482. [DOI] [PubMed] [Google Scholar]
- Rydz, N. & James, P.D. (2012) The evolution and value of bleeding assessment tools. Journal of Thrombosis and Haemostasis, 10, 2223–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccullo, G. & Makris, M. (2016) Prophylaxis in von Willebrand disease: coming of age? Seminars in Thrombosis and Hemostasis, 42, 498–506. [DOI] [PubMed] [Google Scholar]
- Sadler, J.E. , Mannucci, P.M. , Berntorp, E. , Bochkov, N. , Boulyjenkov, V. , Ginsburg, D. , Meyer, D. , Peake, I. , Rodeghiero, F. & Srivastava, A. (2000) Impact, diagnosis and treatment of von Willebrand disease. Thrombosis and Haemostasis, 84, 160–174. [PubMed] [Google Scholar]
- Sadler, J.E. , Budde, U. , Eikenboom, J.C. , Favaloro, E.J. , Hill, F.G. , Holmberg, L. , Ingerslev, J. , Lee, C.A. , Lillicrap, D. , Mannucci, P.M. , Mazurier, C. , Meyer, D. , Nichols, W.L. , Nishino, M. , Peake, I.R. , Rodeghiero, F. , Schneppenheim, R. , Ruggeri, Z.M. , Srivastava, A. , Montgomery, R.R. & Federici, A.B. ; Working Party on von Willebrand Disease Classification . (2006) Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. Journal of Thrombosis and Haemostasis, 4, 2103–2114. [DOI] [PubMed] [Google Scholar]
- Sanders, Y.V. , Groeneveld, D. , Meijer, K. , Fijnvandraat, K. , Cnossen, M.H. , van der Bom, J.G. , Coppens, M. , de Meris, J. , Laros‐van Gorkom, B.A. , Mauser‐Bunschoten, E.P. , Leebeek, F.W. & Eikenboom, J. ; WiN Study Group . (2015) von Willebrand factor propeptide and the phenotypic classification of von Willebrand disease. Blood, 125, 3006–3013. [DOI] [PubMed] [Google Scholar]
- Scott, M. , Hay, C.R.M. , Elkhalifa, S. , Tower, C. , Cocker, M. & Thachil, J. (2018) Management of pregnancy in type 3 von Willebrand disease with alloantibodies. British Journal of Haematology, 182, 440–442. [DOI] [PubMed] [Google Scholar]
- Sharma, R. & Flood, V.H. (2017) Advances in the diagnosis and treatment of Von Willebrand disease. Blood, 130, 2386–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindet‐Pedersen, S. (1991) Haemostasis in oral surgery–the possible pathogenetic implications of oral fibrinolysis on bleeding. Experimental and clinical studies of the haemostatic balance in the oral cavity, with particular reference to patients with acquired and congenital defects of the coagulation system. Danish Medical Bulletin, 38, 427–443. [PubMed] [Google Scholar]
- Sohal, M. & Laffan, M. (2008) Von Willebrand disease and angiodysplasia responding to atorvastatin. British Journal of Haematology, 142, 308–309. [DOI] [PubMed] [Google Scholar]
- Sood, S.L. , James, A.H. , Ragni, M.V. , Shapiro, A.D. , Witmer, C. , Vega, R. , Bolgiano, D. & Konkle, B.A. (2016) A prospective study of von Willebrand factor levels and bleeding in pregnant women with type 1 von Willebrand disease. Haemophilia, 22, e562–e564. [DOI] [PubMed] [Google Scholar]
- Stoof, S.C. , van Steenbergen, H.W. , Zwagemaker, A. , Sanders, Y.V. , Cannegieter, S.C. , Duvekot, J.J. , Leebeek, F.W. , Peters, M. , Kruip, M.J. & Eikenboom, J. (2015) Primary postpartum haemorrhage in women with von Willebrand disease or carriership of haemophilia despite specialised care: a retrospective survey. Haemophilia, 21, 505–512. [DOI] [PubMed] [Google Scholar]
- Stufano, F. , Budde, U. , Baronciani, L. , Peyvandi, F. , Goodeve, A. , Schneppenheim, R. , Badiee, Z. , Esghi, P. , Ettorre, C. , Keikhaei, B. , Lassila, R. , Leebeek, F. , Mazzucconi, M.G. , Toogeh, G. , Trossaert, M. , Peake, I. , Mannucci, P.M. , Eikenboom, J. & Federici, A. (2019) Evaluation of the von Willebrand factor (VWF) inhibitor in a large cohort of European and Iranian patients previously diagnosed with type 3 von Willebrand disease (VWD3) enrolled into the 3WINTER-IPS Project. In: ISTH Conference 2019, Melbourne.
- Svensson, P.J. , Bergqvist, P.B. , Juul, K.V. & Berntorp, E. (2014) Desmopressin in treatment of haematological disorders and in prevention of surgical bleeding. Blood Reviews, 28, 95–102. [DOI] [PubMed] [Google Scholar]
- Szanto, T. , Nummi, V. , Jouppila, A. , Brinkman, H.J.M. & Lassila, R. (2019) Platelets compensate for poor thrombin generation in type 3 von Willebrand disease. Platelets, 1–9. [DOI] [PubMed] [Google Scholar]
- Tosetto, A. , Rodeghiero, F. , Castaman, G. , Goodeve, A. , Federici, A.B. , Batlle, J. , Meyer, D. , Fressinaud, E. , Mazurier, C. , Goudemand, J. , Eikenboom, J. , Schneppenheim, R. , Budde, U. , Ingerslev, J. , Vorlova, Z. , Habart, D. , Holmberg, L. , Lethagen, S. , Pasi, J. , Hill, F. & Peake, I. (2006) A quantitative analysis of bleeding symptoms in type 1 von Willebrand disease: results from a multicenter European study (MCMDM‐1 VWD). Journal of Thrombosis and Haemostasis, 4, 766–773. [DOI] [PubMed] [Google Scholar]
- Tosetto, A. , Castaman, G. & Rodeghiero, F. (2008) Bleeding scores in inherited bleeding disorders: clinical or research tools? Haemophilia, 14, 415–422. [DOI] [PubMed] [Google Scholar]
- Tsagianni, A. , Comer, D.M. , Yabes, J.G. & Ragni, M.V. (2019) Von Willebrand disease and gastrointestinal bleeding: a national inpatient sample study. Thrombosis Research, 178, 119–123. [DOI] [PubMed] [Google Scholar]
- Turecek, P.L. , Mitterer, A. , Matthiessen, H.P. , Gritsch, H. , Varadi, K. , Siekmann, J. , Schnecker, K. , Plaimauer, B. , Kaliwoda, M. , Purtscher, M. , Woehrer, W. , Mundt, W. , Muchitsch, E.M. , Suiter, T. , Ewenstein, B. , Ehrlich, H.J. & Schwarz, H.P. (2009) Development of a plasma‐ and albumin‐free recombinant von Willebrand factor. Hamostaseologie, 29 (Suppl 1), S32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Rosenberg, J.B. , De, B.P. , Ferris, B. , Wang, R. , Rivella, S. , Kaminsky, S.M. & Crystal, R.G. (2012) In vivo gene transfer strategies to achieve partial correction of von Willebrand disease. Human Gene Therapy, 23, 576–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 . Literature search for severe VWD treatment.