

Abstract
Intracellular CuI is controlled by the transcriptional regulator CueR, which effectively discriminates between monovalent and divalent metal ions. It is intriguing that HgII does not activate transcription, as bis‐thiolate metal sites exhibit high affinity for HgII. Here the binding of HgII to CueR and a truncated variant, ΔC7‐CueR, without the last 7 amino acids at the C‐terminus including a conserved CCHH motif is explored. ESI‐MS demonstrates that up to two HgII bind to CueR, while ΔC7‐CueR accommodates only one HgII. 199mHg PAC and UV absorption spectroscopy indicate HgS2 structure at both the functional and the CCHH metal site. However, at sub‐equimolar concentrations of HgII at pH 8.0, the metal binding site displays an equilibrium between HgS2 and HgS3, involving cysteines from both sites. We hypothesize that the C‐terminal CCHH motif provides auxiliary ligands that coordinate to HgII and thereby prevents activation of transcription.
Keywords: coordination modes, CueR metalloregulatory protein, mercury, metal ion selectivity, perturbed angular correlation (PAC) spectroscopy
Intracellular CuI is controlled by the transcriptional regulator CueR, which effectively discriminates between monovalent and divalent metal ions. Here, the binding of HgII to CueR and a truncated variant, ΔC7‐CueR without the last seven amino acids at the C‐terminus including a conserved CCHH motif, is investigated.
The CueR metalloregulatory protein controls cellular copper homeostasis by activating the transcription of cueO and copA genes in prokaryotes and some eukaryotes.1 CueR responds to CuI, AgI and AuI, but not to the divalent ions HgII or ZnII.2 SC‐XRD studies on Escherichia coli CueR and EXAFS in solution revealed that the inducer metal ions are coordinated by C112 and C120 residues in a linear, bis‐cysteinate fashion.2, 3 These two cysteines are essential to the protein function, as shown by mutation studies (C112S and/or C120S) both in vitro3 and in vivo.4
CueR proteins from various bacteria contain two additional well conserved cysteines at the C‐terminal, disordered segment of the protein (Figure 1).2 Crystal structures of the activator and the repressor forms of the DNA‐bound CueR dimer suggest that a two‐turn helix between the metal binding loop and the CCHH motif may have a key role in the protein function.5 Upon AgI binding, the activator conformation is stabilized by the docking of the C‐terminal helix (via residues I122, I123, L126) into an opened, hydrophobic pocket, formed by residues of the dimerization helix and the DNA‐binding domain. This results in a small “scissoring” movement and bending of the DNA chain allowing the transcription to be carried out by the RNA polymerase. The allosteric role of the C‐terminal helix was confirmed by constructing the CuI‐independent constitutive activator (T84V/N125L/C112S/C120S) and the constitutive repressor (truncation from I122) mutants of CueR.5
Figure 1.
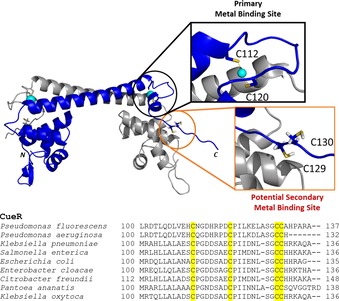
Structure of CueR (E.coli) (PDB id.: 1Q05‐modified) showing the potential metal binding sites (top). Sequence alignment of CueR proteins from various organisms (bottom). Conserved cysteine residues are highlighted in yellow.
Several representative examples can be found in the literature where non‐cognate metal ions bind to a metalloprotein with the same or even higher affinity than the inducer metal ion. However, despite the high affinity binding of non‐cognate metal ions, they cannot trigger the functional structural change of the protein, because the coordination number or geometry differ.6, 7, 8, 9 Thus, studying the interaction of metallo‐regulatory proteins with non‐cognate metal ions may provide a deeper insight into the mechanism of metal ion selection and the regulation of the transcription.8
Although CueR is one of the most thoroughly characterized proteins in the MerR family, the mechanism of discrimination between mono‐ and divalent metal ions is still not fully understood. Surprisingly, HgII does not trigger the activation of transcription by CueR,2 despite its well‐known preference for a bis‐thiolate coordination environment.10 O'Halloran et al. determined a CuI‐binding sensitivity of the CueR protein (1–2×10−21 m) based on an in vitro transcriptional assay.2 Our previous studies on model peptides of the metal binding loop of CueR also showed that these fragments bind CuI with a high affinity.11 However, according to model peptide studies12, 13 and QM/MM calculations,14 HgII ions may be coordinated even more efficiently. Moreover, HgII is also able to bind to a CC sequence,15 and therefore coordination of HgII ion by the CCHH motif is also highly probable.
With the present work we aim to explore the role of the C‐terminal CCHH motif with a particular focus on the binding of HgII to CueR. To achieve this, we studied the HgII‐interaction of E. coli CueR and its truncated variant, lacking seven C‐terminal residues (including the CCHH motif), ΔC7‐CueR. The integrity of this variant was confirmed by CD spectroscopy and electrophoretic mobility shift assay, see Figure S3.
A series of ESI‐MS spectra were recorded with the two protein variants, see Figures 2, S4 and S5. The disappearance of the signals of the apo‐form in the presence of 1.0 equivalent of HgII implies that HgII ions display high affinity to both proteins. The spectra obtained at twofold HgII‐excess per protein clearly demonstrate the availability of two binding sites for HgII ions in the Wild‐type (WT) CueR. These are most likely the metal ion binding loop formed by C112 and C120, and the C‐terminal CCHH motif. Participation of the latter CCHH sequence motif in HgII binding is supported by the lack of signals corresponding to a Hg2‐ΔC7‐CueR complex, even at twofold HgII‐excess over the truncated protein. Both the Hg‐CueR and Hg2‐CueR species are observed at 1.0 equivalent HgII, suggesting that there is no significant difference in the HgII‐binding affinities of the two sites.
Figure 2.
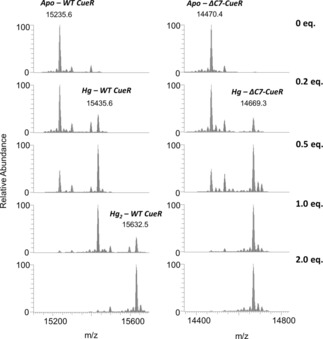
Deconvoluted native ESI‐MS spectra of the WT and truncated CueR in the absence and presence of 0.2, 0.5, 1.0 and 2.0 equivalents of HgII ions. Individual samples contained 20 μm protein in a 10 mm NH4HCO3 buffer, 0.5 mm TCEP, pH 7.5.
199mHg‐perturbed angular correlation (PAC) spectroscopy12, 13, 16, 17, 18, 19, 20 was used to elucidate the metal site structures and dynamics at the nanosecond timescale, see Figure 3 and Supporting Information Figure S6. At pH 6.0 and HgII:CueR of 0.2 and 1.0, the signals agree well with a HgS2 coordination geometry, that is, coordination of HgII by two cysteinates.18 This is also the case at HgII:CueR of 2.0, although a slightly larger linewidth is observed, in particular for the first peak at around 1.4 rad ns−1. This line broadening presumably reflects the occupation of two HgS2 sites, and it can originate either from minor differences in structure of the two sites, or from metal site dynamics at the nanosecond time scale becoming more pronounced upon binding of the second HgII (Figure 3).
Figure 3.
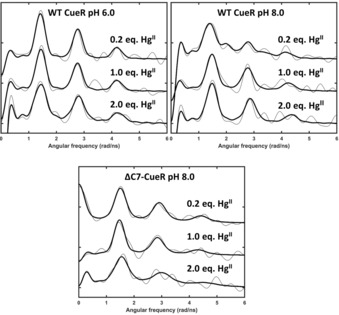
Experimental (grey) and fitted (black) 199mHg PAC spectra of WT and truncated CueR in the presence of DNA with 0.2, 1.0 and 2.0 equivalents of HgII. Top left: WT at pH 6.0; top right: WT at pH 8.0 c WT CueR=12 μm, 0.5 equiv. DNA, and bottom: ΔC7‐CueR at pH 8.0 c ΔC7‐CueR=8.4 μm, 0.5 equiv. DNA.
The spectrum recorded with 0.2 equivalent of HgII per CueR at pH 8.0 is more complex than at pH 6.0. Qualitatively, the first peak is shifted to slightly lower frequency and exhibits considerable broadening, and the second peak (ca. 2.8 rad ns−1) is significantly attenuated, to the extent that it barely rises above the noise level. A reliable analysis of the data requires the inclusion of two nuclear quadrupole interactions (NQIs). One of these NQIs is very similar to that observed in the spectra at pH 6.0, most likely reflecting a HgS2 structure. The other NQI has a higher asymmetry parameter and a lower frequency, see Table S1, indicating a higher coordination number than 2. The lower frequency agrees well with an ideal trigonal planar HgS3 structure, but the relatively high asymmetry parameter rules out this possibility. However, in the simple angular overlap model (AOM),21 a T‐shaped HgS3 coordination geometry gives the same frequency as a trigonal planar structure, but an asymmetry parameter of 1. Thus, a HgS3 structure in between trigonal planar and T‐shaped, with the third ligand in a slightly longer Hg−S distance seems to be a plausible structural interpretation of the low frequency signal. It is also possible that the PAC data reflect a trigonal planar HgS2N structure, with a histidine coordinating, as this would give an asymmetry parameter different from zero. However, this seems less likely, given the thiophilicity of HgII, and the UV absorption data, vide infra. Finally, it is conceivable that the spectrum reflects intermediate (nanosecond) exchange between HgS2 and HgS3 structures. Notice that this entails a flip of principal axis of the electric field gradient tensor, which has V zz along the axis of HgS2 but perpendicular to the HgS3 plane, and therefore the asymmetry parameter will depend on the dynamics in a non‐trivial manner. It cannot be excluded that the data recorded at 1.0 equivalent of HgII also contain signals reflecting both of these species, but the reduced chi‐square does not improve significantly upon including a second NQI. Consequently, we have only included the high frequency NQI (HgS2) in the analysis. For the experiment with 2.0 equivalents of HgII the signal may be satisfactorily fitted with just one (high frequency) NQI, presumably reflecting HgS2 structure for both HgII bound to CueR (Figure 3).
Most interestingly, the 199mHg PAC spectrum recorded at pH 8.0 with 0.2 equivalents HgII for ΔC7‐CueR exhibits a signal reflecting only HgS2 structure (Figure 3). The fact that the ΔC7‐CueR HgII site exhibits a HgS2 structure strongly supports the interpretation presented above for the WT CueR: if HgS3 is formed by occupation of the functional site, a third thiolate is recruited from the CCHH motif, or vice versa, HgII binds to the CCHH motif and recruits one of the cysteines from the functional binding site. With 2.0 equivalents of HgII per ΔC7‐CueR at pH 8.0, the signal changes as compared to experiments with ≤1 equivalent HgII, presumably because the functional metal site is filled, and the additional HgII accommodates a coordination geometry other than linear HgS2 due to weak or non‐specific HgII adducts. This agrees well with the ESI‐MS data, where no Hg2‐ΔC7‐CueR was observed. Thus it is likely that the signal includes more than one NQI. Surprisingly, the signal shifts to slightly higher frequency, which is difficult to account for, except if a positive charge appears in the equatorial plane of HgS2, vide infra.
To further characterize the metal site coordination geometries, we applied UV absorption spectroscopy (Figure 4). HgII‐thiolate complexes possess characteristic charge transfer (CT) bands in the region of 230–300 nm. Moreover, features of the absorption spectrum reflect the coordination geometry of the complexes. Using Hg(SEt)2 and [Et4N][Hg(SBut)3] model compounds, the UV‐absorption spectra of linearly and trigonal planar coordinated HgII, respectively, were characterized.24 Linearly coordinated HgII‐thiolate species display a transition at around 230 nm.22 The increase of the coordination number shifts the absorption bands towards longer wavelengths.23, 25 The spectrum of a trigonal HgII‐thiolate complex has a characteristic absorption maximum at 245 nm with a distinct shoulder at around 290 nm.22 Qualitatively, the absorption difference spectra at sub‐equimolar HgII:WT CueR ratios exhibit a characteristic absorption at around 290 nm reflecting the presence of HgS3 structure (Figure 4), in agreement with the PAC data, vide supra. The PAC data indicate 40 % HgS3 and 60 % HgS2 at 0.2 equivalents HgII. We used the recorded spectrum with 2.0 equivalents HgII per WT CueR (Figure 4 A) to determine the molar absorption of the HgS2 species (green curve in Figure 4 C). Next, we predicted the pure HgS3 molar absorption spectrum (Figure 4 C, purple curve) by assuming that the experimentally determined spectrum is given by 0.6 HgS2+0.4 HgS3. The UV absorption spectra derived in this manner for HgS2 and HgS3 agree well with those reported in the literature,23 strongly supporting the interpretation of the PAC data presented above. We present molar absorption data at selected wavelength values in Table 1. The UV absorption spectra recorded for ΔC7‐CueR exclusively exhibit the signature of HgS2 structure, corroborating the interpretation of other experimental data. Surprisingly, the absorbance for ΔC7‐CueR continues to increase beyond 1.0 equivalent HgII and saturates at ca. 2:1 HgII:ΔC7‐CueR, indicating that the truncated protein can accommodate two HgII ions in a HgS2 coordination environment. This may be realized if a dinuclear Hg2S2 site is formed with the two thiolates as bridging ligands. Interestingly, this agrees with the unexpectedly high frequency observed by PAC spectroscopy, which can be explained by the presence of a positive charge (the second HgII) in the Hg2S2 structure, vide supra. The fact that the species with two HgII bound per CueR monomer is not observed in ESI‐MS implies that the binding of the second HgII is relatively weak.
Figure 4.
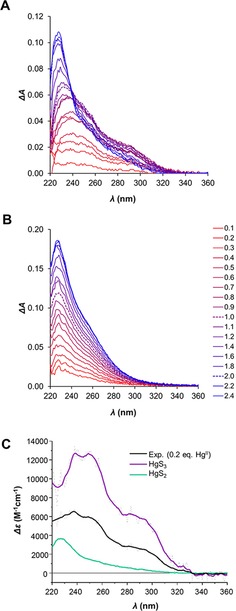
UV absorption difference spectra of WT CueR (A) and ΔC7‐CueR (B) titrated with HgII ions (0.1–2.4 equivalents). Spectra recorded in the presence of 1.0 and 2.0 equivalents of HgII are shown with dashed lines. pH 7.5, c WT CueR=14 μm, c ΔC7‐CueR=12 μm. (C) Estimated molar absorbance for the HgS2 and HgS3 species derived from the WT CueR UV absorption spectrum recorded with 2.0 equiv. HgII and 0.2 equiv. HgII combined with the relative population of the two species derived from 199mHg PAC data, see the text for details.
Table 1.
Spectroscopic properties of the HgS2 and HgS3 species compared to HgII/MerR and Hg/L16C complexes. The two entries for CueR are from this work, see Figure 4 C.
In Figure 5, we present model structures which agree with all the experimental data presented in this work. At pH 8.0 with 0.2 and 1.0 equivalent HgII, two species co‐exist, most likely the linear HgS2 and a HgS3 structure with the equatorial Hg‐S bond being longer than the other two. Such structures have also been observed in small, HgII containing inorganic compounds.26 The increased availability of deprotonated cysteines with increasing pH agrees well with this change in speciation observed from pH 6.0 to pH 8.0, that is, a change from HgS2 towards HgS3 coordination mode, and a similar trend has been observed for de novo designed proteins by Iranzo et al.18 The additional thiolate is most likely recruited from the CCHH motif, or vice versa, and may thus prevent the docking of the C‐terminal helix into the hydrophobic pocket, and consequently inhibit activation of transcription. The net negative charge of HgS3 may be stabilized due to the presence of lysine or arginine in the C‐terminal fragment of CueR in almost all the organisms listed in Figure 1. That is, we hypothesize that the CCHH motif is not involved in the function of CueR when sensing the monovalent coinage metals, but it does take part in binding of divalent metal ions, a mechanism that would account for the selectivity of CueR.
Figure 5.
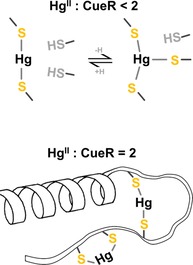
Model structures of HgII bound to WT CueR at pH 8.0. Binding of HgII to CueR gives rise to an equilibrium between HgS2 and HgS3 when HgII:CueR <2, and to pure HgS2 coordination upon addition of 2 HgII ions per protein monomer. This can only be accounted for if the CCHH C‐terminal motif participates in the coordination of HgII, see the text for details.
It may seem intriguing that with 1.0 equivalent HgII both the PAC and UV absorption spectra differ significantly from those recorded with 0.2 equivalent HgII. However, a simple probabilistic model qualitatively accounts for this change, assuming that the two sites are independent (i.e. distributing HgII randomly among the 4 metal sites of a protein dimer), and that population of two adjacent sites (the functional site and the C‐terminal site) leads to formation of HgS2, because there are no more cysteines locally available to form HgS3, see the Supporting Information for details. This very simple interpretation is to some extent supported by the ESI‐MS data, which display population of the Hg2‐CueR species when HgII and CueR are present in equimolar amounts. Obviously, the model is too simple because formation of HgS3 requires that cysteines from both metal binding sites are involved, but the alternative, that is, that one binding site (either the functional site or the C‐terminal site) binds HgII with significantly higher affinity than the other, does not agree with the spectroscopic data, because this would imply that the HgS2/HgS3 ratio should be the same at 0.2 and at 1.0 equivalent, nor with the ESI‐MS data, which indicate the presence of Hg2‐CueR already at 1.0 equivalent HgII. At 2.0 equivalents HgII, of course, there is no more possibility to form HgS3, because the protein is saturated with HgII in HgS2 structures. Similar geometrical rearrangement was observed in metallothioneins (by UV absorption) upon saturating the protein by the metal ion in a titration with HgII.27, 28 The function of the CCHH motif has also been studied by Stoyanov and Brown, using an in vivo assay to monitor the CueR controlled transcription.4 The double mutation of histidine (H131N/H132N) or cysteine residues (C129S/C130S) and truncation from G128 in E. coli CueR resulted in an only slightly altered induction of the transcription by cognate metal ions. Although experimental data were not presented, Stoyanov and Brown indicated that the selectivity of reaction with other, unspecified metal ions was not affected. To further explore this issue, a series of in vitro and in vivo transcriptional assays should be conducted.
In summary, we have demonstrated that up to two HgII ions bind with high affinity to WT CueR, one at the functional (C112 and C120) metal binding site, and the other at the C‐terminal CCHH motif. Moreover, under conditions where the protein is not saturated by HgII, a higher coordination number (presumably HgS3) is observed for WT CueR but not for ΔC7‐CueR, indicating that side chains from the CCHH motif may be recruited as auxiliary ligands at the functional metal site (or vice versa). This implies a mechanism where the specificity of CueR for monovalent coinage metal ions and against divalent metal ions is achieved by coordination to divalent metal ions by the CCHH motif, preventing the docking of the C‐terminal helix into the hydrophobic pocket,5 and consequently inhibiting activation of transcription. Indeed, the CCHH motif provides a selection of ligands that may participate in coordination of both soft and intermediate metal ions. As the findings presented here on HgII do represent a special case, the generalization to other divalent metal ions should be considered carefully.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge the financial support received from the Federal Ministry of Education and Research (BMBF) through grant 05K16PGA and from the European Union's Horizon 2020 Framework research and innovation program under grant agreement no. 654002 (ENSAR2). We further thank J. G. Correia (C2TN‐DECN‐IST‐UL) and project CERN‐FIS‐PAR‐0005–2017 FCT‐Portugal for technical assistance during the beam time and Jens‐Christian Navarro Poulsen and Morten J. Bjerrum (University of Copenhagen) for their support during expression and purification of the ΔC7‐CueR variant. We thank ISOLDE/CERN for beam time, EURONS and NICE for financial support. Financial support from the Hungarian National Research, Development and Innovation Office (GINOP‐2.3.2‐15‐2016‐00038, GINOP‐2.3.2‐15‐2016‐0001 and K 16/120130) is also acknowledged.
R. K. Balogh, B. Gyurcsik, É. Hunyadi-Gulyás, J. Schell, P. W. Thulstrup, L. Hemmingsen, A. Jancsó, Chem. Eur. J. 2019, 25, 15030.
Contributor Information
Prof. Lars Hemmingsen, Email: lhe@chem.ku.dk.
Prof. Attila Jancsó, Email: jancso@chem.u-szeged.hu.
References
- 1. Stoyanov J. V., Hobman J. L., Brown N. L., Mol. Microbiol. 2001, 39, 502–512. [DOI] [PubMed] [Google Scholar]
- 2. Changela A., Chen K., Xue Y., Holschen J., Outten C. E., Halloran T. V., Mondragón A., Science 2003, 301, 1383. [DOI] [PubMed] [Google Scholar]
- 3. Chen K., Yuldasheva S., Penner-Hahn J. E., O'Halloran T. V., J. Am. Chem. Soc. 2003, 125, 12088–12089. [DOI] [PubMed] [Google Scholar]
- 4. Stoyanov J. V., Brown N. L., J. Biol. Chem. 2003, 278, 1407–1410. [DOI] [PubMed] [Google Scholar]
- 5. Philips S. J., Canalizo-Hernandez M., Yildirim I., Schatz G. C., Mondragón A., O'Halloran T. V., Science 2015, 349, 877–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Golynskiy M. V., Gunderson W. A., Hendrich M. P., Cohen S. M., Biochemistry 2006, 45, 15359–15372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cavet J. S., Meng W., Pennella M. A., Appelhoff R. J., Giedroc D. P., Robinson N. J., J. Biol. Chem. 2002, 277, 38441–38448. [DOI] [PubMed] [Google Scholar]
- 8. Ma Z., Cowart D. M., Scott R. A., Giedroc D. P., Biochemistry 2009, 48, 3325–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Phillips C. M., Schreiter E. R., Guo Y., Wang S. C., Zamble D. B., Drennan C. L., Biochemistry 2008, 47, 1938–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.D. C. Bebout, Encyclopedia of Inorganic and Bioinorganic Chemistry 2011.
- 11. Mesterházy E., Boff B., Lebrun C., Delangle P., Jancsó A., Inorg. Chim. Acta 2018, 472, 192–198. [Google Scholar]
- 12. Szunyogh D., Szokolai H., Thulstrup P. W., Larsen F. H., Gyurcsik B., Christensen N. J., Stachura M., Hemmingsen L., Jancsó A., Angew. Chem. Int. Ed. 2015, 54, 15756–15761; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15982–15987. [Google Scholar]
- 13. Szunyogh D., Gyurcsik B., Larsen F. H., Stachura M., Thulstrup P. W., Hemmingsen L., Jancsó A., Dalton Trans. 2015, 44, 12576–12588. [DOI] [PubMed] [Google Scholar]
- 14. Rao L., Cui Q., Xu X., J. Am. Chem. Soc. 2010, 132, 18092–18102. [DOI] [PubMed] [Google Scholar]
- 15. DeSilva T. M., Veglia G., Porcelli F., Prantner A. M., Opella S. J., Biopolymers 2002, 64, 189–197. [DOI] [PubMed] [Google Scholar]
- 16. Chakraborty S., Pallada S., Pedersen J. T., Jancso A., Correia J. G., Hemmingsen L., Acc. Chem. Res. 2017, 50, 2225–2232. [DOI] [PubMed] [Google Scholar]
- 17. Jancso A., Correia J. G., Gottberg A., Schell J., Stachura M., Szunyogh D., Pallada S., Lupascu D. C., Kowalska M., Hemmingsen L., J. Phys. G 2017, 44, 064003. [Google Scholar]
- 18. Iranzo O., Thulstrup P. W., Ryu S.-b., Hemmingsen L., Pecoraro V. L., Chem. Eur. J. 2007, 13, 9178–9190. [DOI] [PubMed] [Google Scholar]
- 19. Hemmingsen L., Sas K. N., Danielsen E., Chem. Rev. 2004, 104, 4027–4062. [DOI] [PubMed] [Google Scholar]
- 20. Butz T., Saibene S., Fraenzke T., Weber M., Nuclear Instruments and Methods in Physics Research Section A: Accelerators, Spectrometers, Detectors and Associated Equipment 1989, 284, 417–421. [Google Scholar]
- 21. Bauer R., Jensen S. J., Schmidt-Nielsen B., Hyperfine Interact. 1988, 39, 203–234. [Google Scholar]
- 22. Watton S. P., Wright J. G., MacDonnell F. M., Bryson J. W., Sabat M., O'Halloran T. V., J. Am. Chem. Soc. 1990, 112, 2824–2826. [Google Scholar]
- 23. Dieckmann G. R., McRorie D. K., Tierney D. L., Utschig L. M., Singer C. P., O'Halloran T. V., Penner-Hahn J. E., DeGrado W. F., Pecoraro V. L., J. Am. Chem. Soc. 1997, 119, 6195–6196. [Google Scholar]
- 24. Wright J. G., Natan M. J., MacDonnel F. M., Ralston D. M., O'Halloran T. V., Progress in Inorg. Chem. 1990, 38, 323–412. [Google Scholar]
- 25. Jalilehvand F., Leung B. O., Izadifard M., Damian E., Inorg. Chem. 2006, 45, 66–73. [DOI] [PubMed] [Google Scholar]
- 26. Manceau A., Nagy K. L., Dalton Trans. 2008, 1421–1425. [DOI] [PubMed] [Google Scholar]
- 27. Freisinger E., Inorg. Chim. Acta 2007, 360, 369–380. [Google Scholar]
- 28. Schicht O., Freisinger E., Inorg. Chim. Acta 2009, 362, 714–724. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary