

Abstract
The red‐spotted grouper Epinephelus akaara (E. akaara) is one of the most economically important marine fish in China, Japan and South‐East Asia and is a threatened species. The species is also considered a good model for studies of sex inversion, development, genetic diversity and immunity. Despite its importance, molecular resources for E. akaara remain limited and no reference genome has been published to date. In this study, we constructed a chromosome‐level reference genome of E. akaara by taking advantage of long‐read single‐molecule sequencing and de novo assembly by Oxford Nanopore Technology (ONT) and Hi‐C. A red‐spotted grouper genome of 1.135 Gb was assembled from a total of 106.29 Gb polished Nanopore sequence (GridION, ONT), equivalent to 96‐fold genome coverage. The assembled genome represents 96.8% completeness (BUSCO) with a contig N50 length of 5.25 Mb and a longest contig of 25.75 Mb. The contigs were clustered and ordered onto 24 pseudochromosomes covering approximately 95.55% of the genome assembly with Hi‐C data, with a scaffold N50 length of 46.03 Mb. The genome contained 43.02% repeat sequences and 5,480 noncoding RNAs. Furthermore, combined with several RNA‐seq data sets, 23,808 (99.5%) genes were functionally annotated from a total of 23,923 predicted protein‐coding sequences. The high‐quality chromosome‐level reference genome of E. akaara was assembled for the first time and will be a valuable resource for molecular breeding and functional genomics studies of red‐spotted grouper in the future.
Keywords: genome assembly, Hi‐C, Nanopore sequencing, red‐spotted grouper, RNA‐seq
1. INTRODUCTION
Groupers (Epinephelus spp.) are economically important marine fish species which are distributed worldwide in tropical and subtropical waters (Heemstra & Randall, 1993). The aquaculture of groupers is well developed in China, Japan and South‐East Asia due to their high nutrition value, high market value and a large consumer demand (Pierre et al., 2010). In China, the annual production of groupers exceeds 100,000 tons, generating gross revenue of $1.5 billion (The 8th National Grouper Breeding and Breeding Industrialization Forum in China). Among the Serranidae species, red‐spotted grouper (Epinephelus akaara), called Hong Kong grouper, is particularly preferred by consumers and is highly prized in China (Figure 1). Since the wild population of red‐spotted grouper has declined sharply in the past few decades (Morris, Roberts, & Hawkins, 2000), this species has been listed as endangered since 2003 (The 2004 IUCN red list of threatened species, http://www.iucnredlist.org). As a species of protogynous sequential hermaphrodite, most red‐spotted groupers start out as females and develop into males after exceeding one or several years, which increases the cost of obtaining mature male fish for breeding. Thus, it is important to investigate the mechanisms controlling the sex inversion of red‐spotted grouper (Huang, Zhou, Li, & Gui, 2009). Outbreaks of infectious diseases, such as the disease caused by nervous necrosis virus (NNV) infection, have become a serious threat to red‐spotted grouper culture industry (Tso & Lu, 2018). Investigation of molecular immunity and disease resistance mechanisms in red‐spotted grouper will benefit the sustainable development of red‐spotted grouper culture industry. Additionally, red‐spotted grouper is considered to be a good model for studies of sex inversion, development, genetic diversity and immunity (Huang et al., 2009; Mao, Lei, Alex, Hong, & Wang, 2012).
Figure 1.

The red‐spotted grouper (Epinephelus akaara) [Colour figure can be viewed at http://wileyonlinelibrary.com]
Currently, genomes from approximately 181 species of fish have been sequenced and are available at NCBI (https://www.ncbi.nlm.nih.gov/genome/browse#!/overview/fish). However, only limited genetic information is available for molecular breeding and functional genomics because the genome of E. akaara is absent. Genomic information, as an essential conservation and management tool (Nguyen et al., 2007; Timmins‐Schiffman, Friedman, Metzger, White, & Roberts, 2013), is needed for the protection and long‐term survival of the endangered the red‐spotted grouper. Genomic data and resources can aid us in fully understanding the diversity and population dynamics of the species and can provide a sound support for suitable conservation measures.
Compared to second‐generation sequencing technologies, which constructs a complete genome by assembling numerous small sequence reads, thus leaving many gaps, third‐generation sequencing technologies produce long reads and enable the production of a genome with a high level of completeness. The recent technological advances of the Nanopore sequencing technology (ONT, Oxford, UK) enable the production of long single reads with the longest 2,272,580 bases (https://nanoporetech.com/about-us/news/longer-and-longer-dna-sequence-more-two-million-bases-now-achieved-nanopore). The long reads assembly is possible to span tricky regions that have not been sequenced. For example, the ultra‐long reads produced by ONT enabled assembly and phasing of the 4‐Mb major histocompatibility complex (MHC) locus in human genome (Jain et al., 2018). Recently, many researchers have reported the value of long Nanopore reads in improving the contiguity and completeness of assembled genomes (Austin et al., 2017; Mondal, Rawal, Gaikwad, Sharma, & Singh, 2017; Tan et al., 2018). In the case of Murray cod (Maccullochella peelii), the inclusion of Nanopore reads for scaffolding followed by subsequent gap‐closing using Illumina data led to a 29% reduction in the number of scaffolds and a 55% and 54% increase in the scaffold and contig N50, respectively (Austin et al., 2017). With the potential of the minimal cost and reduced time requirements of Nanopore sequencing, Nanopore long read‐only assembly is beginning to be adopted in the genomics field (Jain et al., 2018; Michael et al., 2018; Schmidt et al.., 2017). However, its application has rarely been reported except in model animals, such as flies and nematode (Eccles et al., 2018; Miller, Staber, Zeitlinger, & Hawley,). In this study, we assembled a high‐quality genome of red‐spotted grouper using a combination of Nanopore long‐read sequencing and Hi‐C sequencing. Our reference genome should lay a strong foundation for genome science‐based molecular breeding of red‐spotted grouper with improved phenotypes of high yield, rapid growth and resistance to diseases and stress.
2. MATERIALS AND METHODS
2.1. DNA sampling and sequencing
A male adult E. akaara (NCBI taxonomy ID: 215347), bred at the Fisheries Research Institute of Fujian, Xiamen, China (XMRG2392), was used for DNA sequencing (Figure 1). The DNA of E. akaara was extracted from muscle tissue using a Blood & Cell Culture DNA Midi Kit (Q13343, Qiagen, CA, USA), and then, the genomic DNA was used to construct Illumina and Nanopore libraries.
An Illumina library with insert sizes of 400 bp was constructed using an Illumina TruSeq Nano DNA Library Prep Kit and then sequenced on an Illumina HiSeq X Ten instrument. The raw data were filtered using the following strategies (Luo et al., 2012): (a) filtered reads with adapters; (b) trimmed reads with 2 low‐quality bases at the 5’ end and 3 low‐quality bases at the 3’ end; (c) filtered reads with N bases more than 10%; (d) filtered duplicated reads due to polymerase chain reaction amplification; and (e) filtered reads with low‐quality bases (≤5) >50%. All the obtained clean reads were applied to estimate the genome size and heterozygosity by a k‐mer analysis. A 17‐mer frequency distribution analysis was performed on the quality‐filtered reads from the Illumina sequencing to estimate the genome size of the red‐spotted grouper using jellyfish (Marçais & Kingsford, 2011).
For each Nanopore library, approximately 8 µg of gDNA was size‐selected (10–50 kb) with a Blue Pippin (Sage Science, Beverly, MA) and processed using the Ligation sequencing 1D kit (SQK‐LSK108, ONT, UK) according to the manufacturer's instructions. Nineteen libraries were constructed and sequenced on nineteen different R9.4 FlowCells using the GridION X5 sequencer (ONT, UK) for 48 hr each at the Genome Center of Nextomics (Wuhan, China). Base calling was subsequently performed on fast5 files using the ONT Albacore software (v0.8.4), and the “passed filter” reads representing data of somewhat higher quality were used for further downstream analyses.
2.2. RNA sampling and sequencing
Nine RNA samples were extracted from a male adult red‐spotted grouper (brain tissue) and a female adult red‐spotted grouper (eight tissues including brain, gill, gonad, heart, kidney, liver, muscle and spleen) using TRNzol Universal Reagent (Cat# DP424, TIANGEN). The adult E. akaara were bred at the Fisheries Research Institute of Fujian, Xiamen, China. The concentration and integrity of RNA samples were evaluated using a NanoDrop™ One UV–Vis spectrophotometer (Thermo Fisher Scientific, USA), Qubit® 3.0 Fluorometer (Invitrogen, USA) and Agilent Bioanalyzer 2100 (Agilent Technologies, USA). RNA samples with an integrity value over 7.2 were used for the subsequent cDNA library construction and sequencing. The cDNA library was prepared using the TruSeq Sample Preparation Kit (Illumina, CA, USA), and paired‐end sequencing with 150 bp was conducted on a HiSeq X ten platform (Illumina). The clean data were obtained by removing reads containing adapters, reads containing poly‐N and low‐quality reads from the raw data.
2.3. Oxford Nanopore assembly, correction and validation
The Oxford Nanopore sequencing data were assembled using Canu (Koren et al., 2017), and Canu assembly was generated using the default parameters and the complete Canu pipeline (Koren et al., 2017).
To further improve the accuracy of the assembly, nanopolish (v0.8.4) was used based on bwa mem –x ont2d (Jain et al., 2018). As Illumina data are known to have a lower overall error rate, five rounds of consensus correction were performed using Illumina reads mapped with bwa v0.7.12 (Li & Durbin, 2009)(bwa, RRID:SCR_010910) and pilon (Walker et al., 2014) (pilon, RRID:SCR_014731).
To evaluate the completeness genome of the assembly, we used benchmarking universal single‐copy orthologs v3.0.1 (Simão, Waterhouse, Ioannidis, Kriventseva, & Zdobnov, 2015) (busco, RRID:SCR_015008) to search the annotated genes in the assembly. Additionally, we further aligned all the RNA‐seq reads against the E. akaara genome assembly using HISAT2 (Kim, Langmead, & Salzberg, 2015) with default parameters.
2.4. Repeat annotation
The repeat sequences in the genome consisted of simple sequence repeats (SSRs), moderately repetitive sequences and highly repetitive sequences. The SSRs in the genome of E. akaara were identified with the MIcroSAtellite identification tool (misa, RRID:SCR_010765) (Thiel, Michalek, Varshney, & Graner, 2003), which can identify and locate simple and composite microsatellites. Repetitive sequences including tandem repeats and transposable elements (TEs) were searched for the E. akaara genome. The tandem repeats finder (trf, v4.07b) was used to annotate the tandem repeats (Benson, 1999). The transposable elements (TEs) were identified by a combination of homology‐based and de novo approaches. Repeat regions were searched against the repbase (v.22.11) library by repeatmasker v4.0.6 (Tarailo‐Graovac & Chen, 2009)(repeatmasker, RRID:SCR_012954). In addition, repeatmodeler v1.0.4 (repeatmodeler, RRID:SCR_015027) was used to construct a de novo repeat library, which built a repeat consensus database with classification information (Tarailo‐Graovac & Chen, 2009), and repeatmasker v4.0.6 was adopted to search the repeat regions against the built repeat library. RepeatProteinMask was used to search against the TE protein database using a WU‐BLASTX engine.
2.5. Gene prediction
Gene models were constructed, which incorporates ab initio prediction, homology‐based prediction and RNA‐seq‐assisted prediction. For ab initio gene prediction, repeat regions of the E. akaara genome were first masked based on the result from the repeat annotation, and then snap (V2013‐02‐16) (Korf, 2004) and augustus v3.2.2 (augustus: Gene Prediction, RRID:SCR_008417) (Stanke et al., 2006), which are trained to model parameters from homologous genes of BUSCO, were employed to generate gene structures. For homology‐based prediction, protein sequences from 8 sequenced vertebrates, Ctenopharyngodon idellus (Wang et al., 2015), Cynoglossus semilaevis (Chen et al., 2014), Takifugu rubripes (Kai et al., 2011), Gasterosteus aculeatus (Jones et al., 2012), Lates calcarifer (Shubha et al., 2016), Oreochromis niloticus (Conte, Gammerdinger, Bartie, Penman, & Kocher, 2017), Oryzias latipes (Kasahara et al., 2007) and Danio rerio (Genome Reference Consortium, GRCz11; released in May 2017, https://www.ncbi.nlm.nih.gov/genome/?term=Danio+rerio), were initially mapped onto the E. akaara genome using tBlastn (Mount, 2007). Subsequently, exonerate (v2.2.0) (Slater & Birney, 2005) was used to polish BLAST hits to obtain exact intron/exon positions. Furthermore, E. akaara RNA‐seq data from nine tissues sequenced in this study and four tissues provided by Sun Yat‐sen University were used to identify candidate exon regions and donor and acceptor sites by TransDecoder (https://github.com/TransDecoder/TransDecoder) based on RNA‐seq alignments to the genome using Tophat. Finally, the three respective annotation files were integrated using evidencemodeler (EVM) v1.1.1 (Haas et al., 2008) to produce a consensus gene set. The gene set was aligned to the transposon database by transposonpsi (v08222010) with default parameters. Any gene homology to transposons was removed from the final gene set.
2.6. Gene function annotation
The functional annotation of the predicted genes of E. akaara was performed by alignment to the SwissProt, TrEMBL, KEGG and GO databases using blastall and kaas (v2.1) (Moriya, Itoh, Okuda, Yoshizawa, & Kanehisa, 2007). Motifs and domains were annotated using interproscan v5.24 (Jones et al., 2014)( interproscan, RRID:SCR_005829) by searching against publicly available databases including prodom (prodom, RRID:SCR_006969), prints (prints, RRID:SCR_003412), pfam (pfam, RRID:SCR_004726), smrt, panther (panther, RRID:SCR_004869) and prosite (prosite, RRID:SCR_003457).
2.7. Noncoding RNA annotation
Noncoding RNAs, including rRNAs, snRNAs, miRNAs and tRNAs, were identified by adopting infernal v1.1.2 (Nawrocki & Eddy, 2013) using the rfam database (release 13.0) (Kalvari et al., 2018) for the E. akaara genome using BLASTN (Camacho et al., 2009) (E‐value ≤ 1e−5). Transfer RNA was predicted using trnascan‐se v1.3.1 (trnascan‐se, RRID:SCR_010835) software with default parameters for eukaryotes (Lowe & Eddy, 1997). The rRNAs and their subunits were predicted using rnammer v1.2 (Lagesen et al., 2007).
2.8. Chromosome assembly by Hi‐C
An improved Hi‐C procedure (Rao et al., 2014) was adapted for red‐spotted grouper bred at the Fisheries Research Institute of Fujian, Xiamen, China (XMRG201305).. In brief, a 100 μl blood sample was treated with 1% formaldehyde for 10 min at room temperature to induce cross‐linking. The reaction was quenched by adding 2.5 M glycine to 0.2 M for 5 min. Nuclei were digested with 100 units of DpnII and marked by biotin‐14‐dCTP (Invitrogen), and then ligated by T4 DNA Ligase. After incubating overnight to reverse cross‐links, the ligated DNA was sheared into 300‐ to 600‐bp fragments. The DNA fragments were blunt‐end repaired and A‐tailed, followed by purification through biotin–streptavidin‐mediated pulldown. Finally, the Hi‐C libraries were quantified and sequenced in an Illumina NovaSeq 6,000 instrument. The Hi‐C library generated 112.83 Gb data (Table 1), of among which 384.80 million clean paired‐end reads were aligned using bowtie2 (v2.3.2) with the end‐to‐end model (‐very‐sensitive ‐L 30). The paired reads were mapped to the genome independently, but only the reads for which both pairs could be uniquely mapped to the assembled genome were used in further analysis. In total, 262.94 million paired reads could be uniquely mapped to the flank sequences of DpnII‐cleaved sites in the assembled genome. Reads with low mapping quality (maq < 20), multiple hits, duplications and singletons were discarded. Subsequently, the contact count among each contig was calculated and normalized by standardizing the digestion sites of DpnII on the genome sketch.
Table 1.
Genome assembly statistics and postprocessing of E. akaara
| Genome assembly of E. akaara (2n = 24) | Canu | Canu + Nanopolish | Canu + Nanopolish+Pilon 5X | Canu + Nanopolish+Pilon 5X + Hi‐C |
|---|---|---|---|---|
| Total assembly size of contigs (bp) | 1,124,444,644 | 1,131,374,363 | 1,135,521,910 | |
| Number of contigs | 2,055 | 2,055 | 2,055 | |
| N50 contig length (bp) | 5,194,129 | 5,229,032 | 5,248,933 | |
| N90 contig length (bp) | 130,058 | 131,300 | 132,032 | |
| Longest contig (bp) | 25,455,544 | 25,638,949 | 25,751,680 | |
| Total assembly size of scaffolds (bp) | 1,135,726,210a | |||
| Number of scaffolds | 24 | |||
| N50 scaffold length (bp) | 46,028,906 | |||
| N90 scaffold length (bp) | 35,838,671 |
Unanchored contig base count is not included.
We then used Lachesis (https://github.com/shendurelab/LACHESIS) to cluster, order and orient the contigs. First, 2,055 contigs were clustered into 24 chromosome groups (Figure 2) according to the agglomerative hierarchical clustering algorithm with the following parameters:
CLUSTER MIN RE SITES = 100;
CLUSTER MAX LINK DENSITY = 2;
CLUSTER NONINFORMATIVE RATIO = 1.5;
ORDER MIN N RES IN TRUNK = 60;
ORDER MIN N RES IN SHREDS = 60).
Figure 2.
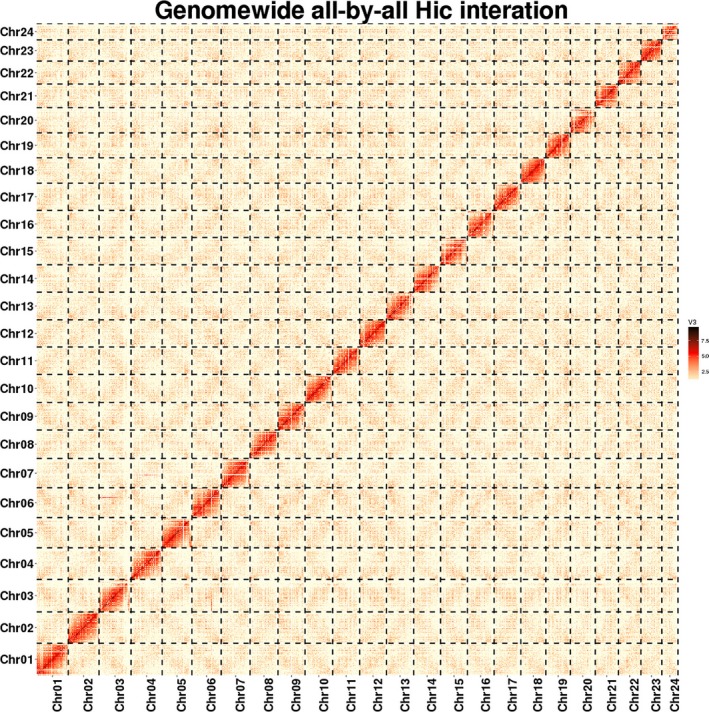
Red‐spotted grouper genome contig contact matrix using Hi‐C data. The colour bar illuminates the contact density from red (high) to white (low) [Colour figure can be viewed at http://wileyonlinelibrary.com]
The contigs within the chromosome groups were then constructed as branches based on the interaction strength among the contigs, and the most dependable branches were selected as roots to add the rest of the contigs into suitable positions and to produce a group with correct contigs order. Finally, the orientations of contigs within chromosomal groups were determined using WDAG based on interaction strength between two contigs directions.
3. RESULTS AND DISCUSSION
3.1. Initial characterization of the E. akaara genome
To estimate the genome size and heterozygosity of E. akaara, 54.7 Gb Illumina clean reads (Table S1) were used for k‐mer analysis. Based on the total number of 48,893,735,532 17‐mers and a peak 17‐mer depth of 44, the estimated genome size of E. akaara was calculated to be 1,111 Mb, and the estimated heterozygosity rate was approximately 0.375% (Table S2 and Figure S1).
3.2. De novo assembly of an E. akaara reference genome
We sequenced the genome of E. akaara with Oxford Nanopore sequencing technology. Nineteen Flow Cells yielded 111.05 Gb of data in total, of which 106.29 Gb was collected after filtering. The average read length and the N50 length were 18.35 kb and 26 kb, respectively (Table S3 and Figure S2). The average quality Q‐score was between 9.55 and 10.53 after filtering.
Canu, a state‐of‐the‐art genome assembler known to support Oxford Nanopore sequencing technology (Schmidt et al., 2017), was used with the E. akaara ONT sequenced data. The initial assembly of these ONT data resulted in an assembly size of 1,124 Mb (Table 1), which was close to the genome size estimated by the k‐mer distribution analysis (1,111 Mb), suggesting that the E. akaara genome size assembled by ONT reads is appropriate. To improve the Nanopore sequencing read‐level accuracy, two assembly polishing approaches were used: Nanopolish, which performs consensus‐based error correction of ONT reads, and Pilon, which used Illumina short reads to correct ONT reads (Loman, Quick, & Simpson, 2015). As shown in Table 1, by comparing the uncorrected, Nanopolish corrected, and Nanopolish followed by five rounds of Pilon assemblies, we found that Nanopolish and Pilon incrementally improved the continuity and completeness of the initial genome assembly, as judged by the marked increase in the contig N50 length (from 5.19 Mb to 5.25 Mb) and the longest contig length (from 25.46 Mb to 25.75 Mb). Finally, the length of the resulting assembled genome was 1,135 Mb with 2,055 contigs. To the best of our knowledge, this assembly is one of the most contiguous fish genome assemblies with the highest contig N50 when compared with the published genomes produced with other sequencing strategies (Table S4) (Austin et al., 2017; Conte et al., 2017; Kim et al.., 2018; Lehmann et al., 2018; Tan et al., 2018; Tørresen et al., 2017; Vij et al., 2016; Zhao et al., 2018), confirming that Nanopore long read‐only sequences can be effectively used to produce highly contiguous genome assemblies. The reference assembly of the E. akaara genome will be an invaluable molecular resource for supporting a range of genetic, genomic and phylogenetic studies specifically for grouper and more generally for other related fish species of the Perciformes.
Further, the genome completeness of the assembly was evaluated by BUSCO and RNA‐seq data. Approximately 96.8% of the complete BUSCO were found in the assembly (Table S5). When mapping the clean RNA‐seq data (149.1 Gb in total) to our assembly genome, the percentage of aligned reads ranged from 82.6% to 95.0% (Table S6). These results suggested that the genome assembly was complete and robust.
3.3. Chromosome assembly by Hi‐C data
95.55% (1,085,005,028 bp) of the total contig bases (1,135,521,910 bp) were reliably anchored to the 24 chromosomes, with a contig N50 of 5.20 Mb and a scaffold N50 of 46.03 Mb (Tables 2 and S4).
Table 2.
Summary statistics of annotated repeats
| Type | Repbase TEs | RepeatModeler | TE proteins | Combined TEs | ||||
|---|---|---|---|---|---|---|---|---|
| Length (bp) | Per cent of sequence (%) | Length (bp) | Per cent of sequence (%) | Length (bp) | Per cent of sequence (%) | Length (bp) | Per cent of sequence (%) | |
| DNA | 72,547,595 | 6.39 | 152,578,623 | 13.44 | 12,639,588 | 1.11 | 190,003,264 | 16.73 |
| LINE | 54,462,165 | 4.8 | 48,239,766 | 4.25 | 47,434,545 | 4.18 | 72,820,879 | 6.41 |
| LTR | 15,915,741 | 1.4 | 13,792,952 | 1.21 | 10,697,269 | 0.94 | 79,800,582 | 7.03 |
| SINE | 7,048,847 | 0.62 | 5,336,045 | 0.47 | 0 | 0 | 7,684,877 | 0.68 |
| Other | 2,402,087 | 0.21 | 14,324,885 | 1.26 | 620,554 | 0.06 | 47,787,296 | 4.21 |
| Unknown | 569,614 | 0.05 | 102,421,979 | 9.02 | 0 | 0 | 90,408,213 | 7.96 |
| Total | 152,946,049 | 13.47 | 336,694,250 | 29.65 | 71,391,956 | 6.29 | 488,505,111 | 43.02 |
3.4. Genome annotation
Repeat sequences were identified based on the assembled sequences of the E. akaara genome. A total of 545,286 SSRs were identified, including 190,558 mono‐, 261,169 di‐, 61,227 tri‐, 25,893 tetra‐, 5,335 penta‐ and 1,104 hexa‐nucleotide repeats (Table S8). Approximately 5.36% of the E. akaara genome was identified as tandem repeats (Table S9). Overall, the combined results of the homology‐based and de novo predictions indicated that repetitive sequences account for 43.02% of the genome, with DNA transposons representing the most abundant class of repeats accounting for 16.73% (Table 2).
For genome annotation, a total of 23,923 protein‐coding genes were identified in the E. akaara genome (Table 3). Compared with other existing published vertebrate genomes, the number of genes in E. akaara is similar to that in C. semilaevis (21,253 genes) and D. rerio (24,884 genes) (Table S10). In total, 23,808 genes were annotated in at least one of the databases, and up to 99.5% of E. akaara genes were functionally annotated (Table 4). Finally, 828 miRNAs, 792 rRNAs, 1,252 snRNAs and 2,608 tRNAs were also identified (Table 5). These results revealed that it is possible to obtain functional and highly contiguous genome assemblies covering most of the genes in gigabase‐sized fish genomes when using ONT in combination with Illumina data for polishing.
Table 3.
Summary statistics of predicted protein‐coding genes
| Gene set | Total number of gene | Average gene length (bp) | Average CDS length (bp) | Average exons number per gene | Average exon length (bp) | Average intron length (bp) |
|---|---|---|---|---|---|---|
| De novo | ||||||
| Augustus | 29,024 | 16,738 | 1,413.28 | 8.33 | 169.76 | 2,092.11 |
| SNAP | 59,925 | 26,835.73 | 1,417.17 | 10.27 | 137.93 | 2,740.60 |
| Homology | ||||||
| C. idellus | 53,989 | 5,519.60 | 901.7 | 4.39 | 205.53 | 1,363.34 |
| C. ynoglossus | 33,543 | 11,471.97 | 1,387.91 | 7.1 | 195.57 | 1,654.05 |
| D. rerio | 38,897 | 8,415.67 | 1,140.39 | 5.73 | 199.1 | 1,538.84 |
| G. aculeatus | 41,023 | 8,218.57 | 1,033.88 | 5.62 | 183.82 | 1,553.69 |
| L. calcarifer | 36,878 | 11,137.05 | 1,321.62 | 7.1 | 186.2 | 1,609.70 |
| O. niloticus | 41,660 | 9,565.48 | 1,305.17 | 6.56 | 198.92 | 1,485.36 |
| O. latipes | 46,255 | 6,769.88 | 987.84 | 5.1 | 193.58 | 1,409.27 |
| T. rubipes | 33,739 | 9,958.68 | 1,191.94 | 6.45 | 184.89 | 1,609.54 |
| Final set | ||||||
| Evm | 23,9,243 | 23,162.39 | 1,791.15 | 10.9 | 164.27 | 2,157.86 |
Table 4.
Summary statistics of functional annotated protein‐coding genes
| Type | Number | Per cent (%) |
|---|---|---|
| SwissProt | 22,223 | 92.9 |
| TrEMBL | 23,793 | 99.5 |
| KEGG | 13,828 | 57.8 |
| GO | 16,469 | 68.8 |
| InterProsScan | 21,892 | 91.5 |
| Total Annotated Genes | 23,808 | 99.5 |
| Predicted Genes | 23,923 | – |
Table 5.
Summary statistics of noncoding RNA
| Type | Copy number | Average length (bp) | Total length (bp) | Percentage (%) of genome |
|---|---|---|---|---|
| rRNA | ||||
| rRNA | 792 | 385.48 | 305,299 | 0.022804 |
| 18S | 47 | 1,742.36 | 81,891 | 0.006117 |
| 28S | 35 | 4,066.66 | 142,333 | 0.010632 |
| 5.8S | 47 | 148.04 | 6,958 | 0.00052 |
| 5S | 663 | 111.79 | 74,117 | 0.005536 |
| snRNA | ||||
| snRNA | 1,252 | 144.96 | 181,492 | 0.013557 |
| CD‐box | 230 | 122.29 | 28,127 | 0.002101 |
| HACA‐box | 107 | 146.43 | 15,668 | 0.00117 |
| splicing | 915 | 150.49 | 137,697 | 0.010285 |
| miRNA | 828 | 85.36 | 70,675 | 0.005279 |
| tRNA | 2,608 | 74.68 | 194,771 | 0.014548 |
4. CONCLUSION
In the present study, we assembled the most contiguous genome of E. akaara by de novo assembly of long sequence reads produced by Nanopore sequencing technology (GridION) and Hi‐C. The reference quality genome (1.135 Gb) of E. akaara has the assembled contig N50 of 5.25 Mb and a longest contig of 25.75 Mb. By utilizing Hi‐C technology, we assembled contigs into scaffolds that resulted in a chromosome‐level genome assembly with 24 chromosomes and a scaffold N50 length of 46.03 Mb. The genome was functionally annotated to produce a total of 23,808 (99.5%) protein‐coding genes. Our genome assembly results demonstrate that long sequence reads produced by Nanopore sequencing can be effectively used for genome assembly. Our well‐annotated genome allows us to identify genes and pathways related to sex determination and differentiation to reveal the mechanisms controlling the sex inversion of red‐spotted grouper, to screen for genetic variation conferring resistance to pathogen infection at the genome scale and to investigate genetic correlations with production traits of E. akaara in the future.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
AUTHOR CONTRIBUTIONS
H.G., J.H., Y.W., Z.H. and L.Z. designed research; H.G., K.L., M.S., S.W., Y.Z., Z.H., C.Z, Q.L., J.S., Z.W., D.C., L.L., D.W., J.H., Y.W., Z.H., L.Z. performed research and/or analysed data; H.G., K.L., M.S., Z.Z and S.W. wrote the study. All authors reviewed the manuscript.
Supporting information
ACKNOWLEDGEMENTS
We appreciate Nextomics Biosciences Institute for valuable comments and discussions.
Ge H, Lin K, Shen M, et al. De novo assembly of a chromosome‐level reference genome of red‐spotted grouper (Epinephelus akaara) using nanopore sequencing and Hi‐C. Mol Ecol Resour. 2019;19:1461–1469. 10.1111/1755-0998.13064
Ge, Lin , Shen and Wu are contributed equally.
Funding information
This research was funded by the Special Project for Basic Scientific Research of Public Welfare Institutes in Fujian Province (Development of molecular markers for antiviral necrosis of red‐spotted grouper No. 2019R1013‐10; Research on Visual Rapid Detection Technology of Grouper Viral Necrosis Virus No. 2019R1013‐2) and the Special funds for structural adjustment of the oceans and fisheries in Fujian Province (Development and application demonstration of rapid detection kit for main grouper of virus).
Data Availability Statement: The data sets supporting the results in this article are available in the NCBI. Raw sequencing reads have been deposited in the SRA (Sequence Read Archive) database with Bioproject ID PRJNA498398. The genome sequence data (Nanopore, Illumina and Hi‐C data) are available under the Accession nos SRX4953865, SRX4953864 and SRX5827484. And RNA‐seq data of 13 tested samples are available under the Accession nos SRX4953866‐SRX4953878. The assembly genome sequences (pseudochromosomes) and gene models are available via Dryad (https://datadryad.org/review?doi=doi:10.5061/dryad.4398b9f), and the repeat annotation is available via Dryad (https://datadryad.org/review?doi=doi:10.5061/dryad.s4g5m6f).
Contributor Information
Yilei Wang, Email: ylwang@jmu.edu.cn.
Jiang Hu, Email: huj@grandomics.com.
Zhongchi Huang, Email: hzc@fjta.com.
Leyun Zheng, Email: 981668817@qq.com.
DATA AVAILABILITY STATEMENT
The data sets supporting the results in this article are available in the NCBI. Raw sequencing reads have been deposited in the SRA (Sequence Read Archive) database with Bioproject ID PRJNA498398. The genome sequence data (Nanopore, Illumina and Hi‐C data) are available under the Accession nos SRX4953865, SRX4953864 and SRX5827484. And RNA‐seq data of 13 tested samples are available under the Accession nos SRX4953866‐SRX4953878. The assembly genome sequences (pseudochromosomes) and gene models are available via Dryad (https://datadryad.org/review?doi=doi:10.5061/dryad.4398b9f), and the repeat annotation is available via Dryad (https://datadryad.org/review?doi=doi:10.5061/dryad.s4g5m6f).
REFERENCES
- Austin, C. M. , Tan, M. H. , Harrisson, K. A. , Lee, Y. P. , Croft, L. J. , Sunnucks, P. , … Gan, H. M. (2017). De novo genome assembly and annotation of Australia's largest freshwater fish, the Murray cod (Maccullochella peelii), from Illumina and Nanopore sequencing read. Gigascience, 6(8), 1 10.1093/gigascience/gix063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson, G. (1999). Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Research, 27(2), 573–580. 10.1093/nar/27.2.573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho, C. , Coulouris, G. , Avagyan, V. , Ma, N. , Papadopoulos, J. , Bealer, K. , & Madden, T. L. (2009). BLAST+: Architecture and applications. BMC Bioinformatics, 10(1), 421 10.1186/1471-2105-10-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, S. , Zhang, G. , Shao, C. , Huang, Q. , Liu, G. , Zhang, P. , … Wang, J. (2014). Whole‐genome sequence of a flatfish provides insights into ZW sex chromosome evolution and adaptation to a benthic lifestyle. Nature Genetics, 46(3), 253–260. 10.1038/ng.2890 [DOI] [PubMed] [Google Scholar]
- Conte, M. A. , Gammerdinger, W. J. , Bartie, K. L. , Penman, D. J. , & Kocher, T. D. (2017). A high quality assembly of the Nile Tilapia (Oreochromis niloticus) genome reveals the structure of two sex determination regions. BMC Genomics, 18(1), 341 10.1186/s12864-017-3723-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles, D. , Chandler, J. , Camberis, M. , Henrissat, B. , Koren, S. , Le Gros, G. , & Ewbank, J. J. (2018). De novo assembly of the complex genome of Nippostrongylus brasiliensis using MinION long reads. BMC Biology, 16(1), 6 10.1186/s12915-017-0473-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, B. J. , Salzberg, S. L. , Zhu, W. , Pertea, M. , Allen, J. E. , Orvis, J. , … Wortman, J. R. (2008). Automated eukaryotic gene structure annotation using evidencemodeler and the program to assemble spliced alignments. Genome Biology, 9(1), R7 10.1186/gb-2008-9-1-r7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heemstra, P. C. , & Randall, J. E. (1993). FAO Species Catalogue. Vol. 16. Groupers of the World (Family Serranidae, Subfamily Epinephelinae). An Annotated and Illustrated Catalogue of the Grouper, Rockcod, Hind, Coral Grouper, and Lyretail Species Known to Date FAO Fisheries Synopsis. [Google Scholar]
- Huang, W. , Zhou, L. , Li, Z. , & Gui, J. F. (2009). Expression pattern, cellular localization and promoter activity analysis of ovarian aromatase (Cyp19a1a) in protogynous hermaphrodite red‐spotted grouper. Molecular & Cellular Endocrinology, 307(1), 224–236. 10.1016/j.mce.2009.04.003 [DOI] [PubMed] [Google Scholar]
- Jain, M. , Koren, S. , Miga, K. H. , Quick, J. , Rand, A. C. , Sasani, T. A. , Fiddes, I. T. (2018). Nanopore sequencing and assembly of a human genome with ultra‐long reads. Nature Biotechnology, 36(4), 338–345. 10.1038/nbt.4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, F. C. , Grabherr, M. G. , Chan, Y. F. , Russell, P. , Mauceli, E. , Johnson, J. , … Kingsley, D. M. (2012). The genomic basis of adaptive evolution in threespine sticklebacks. Nature, 484(7392), 55–61. 10.1038/nature10944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, P. , Binns, D. , Chang, H.‐Y. , Fraser, M. , Li, W. , McAnulla, C. , … Hunter, S. (2014). InterProScan 5: Genome‐scale protein function classification. Bioinformatics, 30(9), 1236–1240. 10.1093/bioinformatics/btu031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai, W. , Kikuchi, K. , Tohari, S. , Chew, A. K. , Tay, A. , Fujiwara, A. , Brenner, S. (2011). Integration of the genetic map and genome assembly of fugu facilitates insights into distinct features of genome evolution in teleosts and mammals. Genome Biology and Evolution, 3(3), 424–442. 10.1093/gbe/evr041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalvari, I. , Argasinska, J. , Quinonesolvera, N. , Nawrocki, E. P. , Rivas, E. , Eddy, S. R. , … Petrov, A. I. (2018). Rfam 13.0: shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Research, 46(D1), D335-D342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara, M. , Naruse, K. , Sasaki, S. , Nakatani, Y. , Qu, W. , Ahsan, B. , … Kasai, Y. (2007). The medaka draft genome and insights into vertebrate genome evolution. Nature, 447(7145), 714–719. [DOI] [PubMed] [Google Scholar]
- Kim, D. , Langmead, B. , & Salzberg, S. L. (2015). HISAT: A fast spliced aligner with low memory requirements. Nature Methods, 12(4), 357–360. 10.1038/nmeth.3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, L. , Bo‐Young, L. , Han, J. , Jeong, C. B. , Hwang, D. S. , Lee, M. C. , … Lee, J. S. (2018). The genome of the marine medaka Oryzias melastigma . Molecular Ecology Resources, 18(3), 656–665. 10.1111/1755-0998.12769 [DOI] [PubMed] [Google Scholar]
- Koren, S. , Walenz, B. P. , Berlin, K. , Miller, J. R. , Bergman, N. H. , & Phillippy, A. M. (2017). Canu: Scalable and accurate long‐read assembly via adaptive k‐mer weighting and repeat separation. Genome Research, 27(5), 722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf, I. (2004). Gene finding in novel genomes. BMC Bioinformatics, 5(1), 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagesen, K. , Hallin, P. , Rødland, E. A. , Stærfeldt, H. H. , Rognes, T. , & Ussery, D. W. (2007). RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Research, 35(9), 3100–3108. 10.1093/nar/gkm160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann, R. , Lightfoot, D. J. , Schunter, C. , Michell, C. T. , Ohyanagi, H. , Mineta, K. , Ravasi, T. (2018). Finding Nemo's Genes: A chromosome‐scale reference assembly of the genome of the orange clownfish Amphiprion percula . bioRxiv, 19, 570–585. 10.1101/278267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25(14), 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loman, N. J. , Quick, J. , & Simpson, J. T. (2015). A complete bacterial genome assembled de novo using only nanopore sequencing data. Nature Methods, 12(8), 733 10.1038/nmeth.3444 [DOI] [PubMed] [Google Scholar]
- Lowe, T. M. , & Eddy, S. R. (1997). trnascan‐se: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Research, 25(5), 955–964. 10.1093/nar/25.5.0955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, R. , Liu, B. , Xie, Y. , Li, Z. , Huang, W. , Yuan, J. , … Wang, J. (2012). SOAPdenovo2: An empirically improved memory‐efficient short‐read de novo assembler. Gigascience, 1(1), 18 10.1186/2047-217X-1-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao, M. G. , Lei, J. L. , Alex, P. M. , Hong, W. S. , & Wang, K. J. (2012). Characterization of RAG1 and IgM (mu chain) marking development of the immune system in red‐spotted grouper (Epinephelus akaara). Fish & Shellfish Immunology, 33(4), 725–735. 10.1016/j.fsi.2012.06.011 [DOI] [PubMed] [Google Scholar]
- Marçais, G. , & Kingsford, C. (2011). A fast, lock‐free approach for efficient parallel counting of occurrences of k‐mers. Bioinformatics, 27(6), 764–770. 10.1093/bioinformatics/btr011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael, T. P. , Jupe, F. , Bemm, F. , Motley, S. T. , Sandoval, J. P. , Lanz, C. , … Ecker, J. R. (2018). High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell. Nature Communications, 9(1), 541 10.1038/s41467-018-03016-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, D. E. , Staber, C. , Zeitlinger, J. , & Hawley, R. S. (2018). Genome report: highly contiguous genome assemblies of 15 drosophila species generated using nanopore sequencing. IEEE/ACM Transactions on Computational Biology & Bioinformatics, PP99, 1–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal, T. K. , Rawal, H. C. , Gaikwad, K. , Sharma, T. R. , & Singh, N. K. (2017). First de novo draft genome sequence of Oryza coarctata, the only halophytic species in the genus, Oryza. F1000research, 6, 1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya, Y. , Itoh, M. , Okuda, S. , Yoshizawa, A. C. , & Kanehisa, M. (2007). kaas: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Research, 35(Web Server), W182–W185. 10.1093/nar/gkm321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, A. V. , Roberts, C. M. , & Hawkins, J. P. (2000). The threatened status of groupers (Epinephelinae). Biodiversity & Conservation, 9(7), 919–942. [Google Scholar]
- Mount, D. W. (2007). Using the Basic Local Alignment Search Tool (BLAST). Csh Protocols, 2007(14), pdb.top17 10.1101/pdb.top17 [DOI] [PubMed] [Google Scholar]
- Nawrocki, E. P. , & Eddy, S. R. (2013). Infernal 1.1: 100‐fold faster RNA homology searches. Bioinformatics, 29(22), 2933–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, T. T. , Genini, S. , Bui, L. C. , Voegeli, P. , Stranzinger, G. , Renard, J.‐P. , … Nguyen, B. X. (2007). Genomic conservation of cattle microsatellite loci in wild gaur (Bos gaurus) and current genetic status of this species in Vietnam. BMC Genetics, 8, 77 10.1186/1471-2156-8-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre, S. , Gaillard, S. , Prévot‐D'Alvise, N. , Aubert, J. , Rostaing‐Capaillon, O. , Leung‐Tack, D. , & Grillasca, J. P. (2010). Grouper aquaculture: Asian success and Mediterranean trials. Aquatic Conservation Marine & Freshwater Ecosystems, 18(3), 297–308. 10.1002/aqc.840 [DOI] [Google Scholar]
- Rao, S. S. P. , Huntley, M. H. , Durand, N. C. , Stamenova, E. K. , Bochkov, I. D. , Robinson, J. T. , … Aiden, E. L. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell, 159(7), 1665–1680. 10.1016/j.cell.2014.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, V. A. , Denton, A. K. , Istace, B. , Wormit, A. , Geest, H. , … Usadel, B. (2017). De novo assembly of a new Solanum pennellii accession using nanopore sequencing. The Plant Cell, 29(10), 2336–2348. 10.1105/tpc.17.00521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão, F. A. , Waterhouse, R. M. , Ioannidis, P. , Kriventseva, E. V. , & Zdobnov, E. M. (2015). busco: Assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31(19), 3210–3212. 10.1093/bioinformatics/btv351 [DOI] [PubMed] [Google Scholar]
- Slater, G. S. C. , & Birney, E. (2005). Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics, 6(1), 31–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke, M. , Keller, O. , Gunduz, I. , Hayes, A. , Waack, S. , & Morgenstern, B. (2006). augustus: Ab initio prediction of alternative transcripts. Nucleic Acids Research, 34(Web Server), W435–W439. 10.1093/nar/gkl200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, M. H. , Austin, C. M. , Hammer, M. P. , Lee, Y. P. , Croft, L. J. , & Gan, H. M. (2018). Finding Nemo: Hybrid assembly with Oxford Nanopore and Illumina reads greatly improves the Clownfish (Amphiprion ocellaris) genome assembly. Gigascience, 7(3), 1 10.1093/gigascience/gix137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarailo‐Graovac, M. , & Chen, N. (2009). Using repeatmasker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics, 25(1), 4.10.11–14.10.14. 10.1002/0471250953.bi0410s25 [DOI] [PubMed] [Google Scholar]
- Thiel, T. , Michalek, W. , Varshney, R. K. , & Graner, A. (2003). Exploiting EST databases for the development and characterization of gene‐derived SSR‐markers in barley (Hordeum vulgare L.). Theoretical & Applied Genetics, 106(3), 411–422. 10.1007/s00122-002-1031-0 [DOI] [PubMed] [Google Scholar]
- Timmins‐Schiffman, E. B. , Friedman, C. S. , Metzger, D. C. , White, S. J. , & Roberts, S. B. (2013). Genomic resource development for shellfish of conservation concern. Molecular Ecology Resources, 13(2), 295–305. 10.1111/1755-0998.12052 [DOI] [PubMed] [Google Scholar]
- Tørresen, O. K. , Star, B. , Jentoft, S. , Reinar, W. B. , Grove, H. , Miller, J. R. , … Nederbragt, A. J. (2017). An improved genome assembly uncovers prolific tandem repeats in Atlantic cod. Bmc Genomics, 18(1), 95 10.1186/s12864-016-3448-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tso, C.‐H. , & Lu, M.‐W. (2018). Transcriptome profiling analysis of grouper during nervous necrosis virus persistent infection. Fish & Shellfish Immunology, 76, 224–232. 10.1016/j.fsi.2018.03.009 [DOI] [PubMed] [Google Scholar]
- Vij, S. , Kuhl, H. , Kuznetsova, I. S. , Komissarov, A. , Yurchenko, A. A. , Van Heusden, P. , … Orbán, L. (2016). Chromosomal‐level assembly of the asian seabass genome using long sequence reads and multi‐layered scaffolding. Plos Genetics, 12(4), e1005954 10.1371/journal.pgen.1005954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, B. J. , Abeel, T. , Shea, T. , Priest, M. , Abouelliel, A. , Sakthikumar, S. , … Earl, A. M. (2014). pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE, 9(11), e112963 10.1371/journal.pone.0112963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Lu, Y. , Zhang, Y. , Ning, Z. , Li, Y. , Zhao, Q. , … Zhu, Z. (2015). The draft genome of the grass carp (Ctenopharyngodon idellus) provides insights into its evolution and vegetarian adaptation. Nature Genetics, 47(6), 625 10.1038/ng.3280 [DOI] [PubMed] [Google Scholar]
- Zhao, X. , Luo, M. , Li, Z. , Zhong, P. , Cheng, Y. , Lai, F. , Yang, Y. (2018). Chromosome‐scale assembly of the Monopterus genome. GigaScience, 7(5), 10.1093/gigascience/giy046 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets supporting the results in this article are available in the NCBI. Raw sequencing reads have been deposited in the SRA (Sequence Read Archive) database with Bioproject ID PRJNA498398. The genome sequence data (Nanopore, Illumina and Hi‐C data) are available under the Accession nos SRX4953865, SRX4953864 and SRX5827484. And RNA‐seq data of 13 tested samples are available under the Accession nos SRX4953866‐SRX4953878. The assembly genome sequences (pseudochromosomes) and gene models are available via Dryad (https://datadryad.org/review?doi=doi:10.5061/dryad.4398b9f), and the repeat annotation is available via Dryad (https://datadryad.org/review?doi=doi:10.5061/dryad.s4g5m6f).