

Abstract
Polycyclic aromatic hydrocarbons (PAHs) that contain both five‐ and seven‐membered rings are rare, and those where these rings are annulated to each other and build azulene units have, to date, mainly been generated in minute amounts on surfaces. Herein, a rational approach to synthesize soluble contorted PAHs containing two embedded azulene units in the bulk is presented. By stepwise detachment of tert‐butyl groups, a series of three azulene embedded PAHs with different degrees of contortion has been made to study the impact of curvature on aromaticity and conjugation. Furthermore, the azulene PAHs showed high fluorescence quantum yields in the NIR regime.
Keywords: azulene, chlorination, helicenes, polycyclic aromatic hydrocarbons, UV/Vis spectroscopy
Deep Blue! Soluble contorted PAHs with two embedded azulene moieties with different degrees of curvature are synthesized and spectroscopically compared.
The bright blue color of azulene has attracted chemists since derivatives of it were first isolated from chamomile, yarrow, and wormwood.1 After diverse failed attempts to synthesize it, finally Plattner and Pfau were successful, starting from Δ9‐octalin.2 Later on, a more efficient route was described by Ziegler and Hafner, starting from cyclopentadiene and 2,4‐dinitrophenyl pyridinium chloride.3, 4 Other approaches followed5 and even nowadays new synthetic routes to azulene and their derivatives for example, by gold catalysis are being developed.6, 7 The origin of the blue color of azulene as non‐alternant isomer of the colorless naphthalene has fascinated theoretical and physical chemists dealing with it.8
A large number of kata‐condensed π‐extended azulene derivatives have been made and their chemical and photophysical properties have been studied.9 In contrast to the kata‐condensed π‐extended azulenes, those with additional benzene rings peri‐condensed between the five‐ and the seven‐membered rings are rarer.10 Although an azulene with one peri‐condensed benzene was realized already in 1958,11 efforts to synthesize azulene compounds with two peri‐condensed aromatic units have failed,12 because these structures are proposed to have biradical triplet ground state character,13 making them highly reactive.
It is worth mentioning that besides the natural occurrence of azulene derivatives in plants, its scaffold is found as a defect in graphenes.14 It was also proposed that azulene‐based polycyclic aromatic hydrocarbons (PAHs) of defined structure are found in interstellar space, since the interpretation of some measured IR/Raman spectra could not be fitted to a variety of PAHs containing six‐membered rings exclusively.15
There are a very few successful approaches known for synthesizing PAHs with azulene units embedded from precursor monolayers on surfaces.16 However, to the best of our knowledge, there are only a few examples of soluble azulene‐based PAHs that have been synthesized from solution on a larger scale.17, 18 One is the example recently published by Konishi and co‐workers,17 in which the PAH contains besides one azulene core, six more fused six‐membered rings. Very recently, the same group presented a small PAH that contains two fused azulene units and two additional peri‐fused six‐membered rings, giving overall four rings.18c This compound shows significant biradical character.
Herein we present the synthesis of large soluble PAHs containing two embedded azulene units and 15 fused aromatic rings overall. To the best of our knowledge, this represents the largest PAH containing at least two formal azulene units. Based on our research on diarenoperylenes,19 we recently found that Scholl‐type oxidations of 1 a and 1 b with both FeCl3 in nitromethane as well as with DDQ and trifluoromethanesulfonic acid gave five‐membered ring‐cyclized products 2 a and 2 b exclusively.20, 21 We envisioned combining this highly selective Scholl‐type reaction in a manner that would generate an additional seven‐membered ring (Scheme 1),22 preferably in the same step, resulting in azulene formation during Scholl‐cyclization, something that has previously only been observed on Au(111) surfaces.16a
Scheme 1.
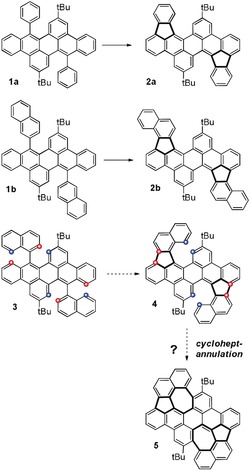
Conceptual approach to bis‐azulene embedded PAH 5 by concerted cyclopent‐ and cycloheptannulation.
Accordingly, we synthesized the bisnaphthyl diareno precursor 3 from the corresponding bromide 6 19b by Suzuki–Miyaura cross‐coupling in 67 % yield (Scheme 2). First, we tested FeCl3 in nitromethane/DCM for the oxidative cyclization. A regioselective formation of five‐membered rings is observed; however, it is accompanied by a selective twofold chlorination, giving the double‐helicene product 7 in 94 % yield. It is worth mentioning that additional chlorinations during oxidative Scholl‐type cyclizations have been observed, but they are still very rare and in most cases lead to the isolation of minor side‐products.23 Although the oxidant was added in large access (20 equiv) no additional formation of seven‐membered rings occurred. The double‐helical molecular structure of 7 and the positions of the chloride atoms were unambiguously identified by single‐crystal X‐ray diffraction of crystals grown from 1,2‐dichlorobenzene/methanol (Figure 1). Compound 7 crystallizes as racemic mixture of both double helicene enantiomers (M,M) and (P,P) in the space group P2/c. It is worth mentioning that two peaks with comparable integral have been observed by chiral HPLC under ambient conditions, suggesting that the enantiomers are not interconverting to each other at room temperature. Each enantiomer forms a dimer where the aromatic helical units of adjacent molecules are arranged in a coplanar fashion by weak π stacking (dπ‐π is between 3.6 and 3.9 Å). The two aromatic halves of the molecule are twisted by approx. 39.4°. In this case it was difficult to assign the positions of the chloride substituents by NMR methods, but single‐crystal X‐ray diffraction data gave a clear answer in this respect, showing that chlorination occurred in para‐positions of the prior existing C−C biaryl bond. This is in contrast to the position of triflyloxylation we observed for the bisnaphthyl dibenzoperylene derivative, we described before.20
Scheme 2.
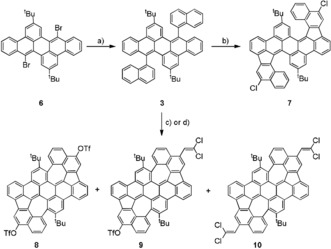
Synthesis of bisnaphthyldibenzoperylene 3 and reactions under oxidative conditions. a) 1‐Naphthylboronic acid, 10 mol % Pd2(dba)3, 31 mol % tBu3PHBF4, THF, 2 n K2CO3(aq), 80 °C, 16 h, 67 % yield. b) FeCl3 (20 equiv), CH3NO2/CH2Cl2, 0 °C to reflux, 1 h, 94 %. c) DDQ (8 equiv), TfOH/CH2Cl2, RT, 1 h, c (3)=1.67 mmol L−1 yields: 14 % (8), 30 % (9), 9 % (10). d) DDQ (10 equiv), TfOH/CH2Cl2, RT, 1 h, c (3)=3.33 mmol L−1 yields: 25 % (8), 11 % (9), 2 % (10).
Figure 1.
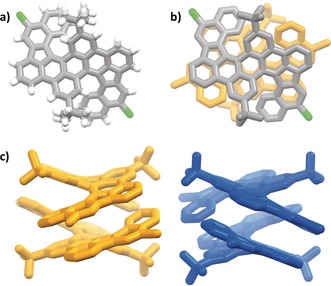
Single‐crystal X‐ray structure of PAH 7.29 a) Single enantiomer (P,P) in elemental colors (carbon: grey, hydrogen: white and chlorine green). b) Dimer of (P,P)pair. c) Side‐view of packing. (P,P)‐7 is shown in orange, (M,M)‐7 in blue. In (b) and (c) hydrogen atoms are omitted for clarity.
In contrast to the case of using FeCl3 as oxidant, with eight equivalents of DDQ in TfOH/DCM the desired cycloheptannulation in addition to the cyclopentannulation occurred, giving a mixture of azulene embedded PAHs 8 (14 %), 9 (30 %), and 10 (9 %), which were separated by column chromatography and identified and characterized by NMR and MS. For all those PAHs the cyclodehydrogenation reactions are accompanied by the selective addition of further moieties. As reported previously by our group,20 an additional bis(triflyloxylation) was observed for PAH 8. However, for the system described herein, the triflyloxylation is competing with the attachment of a dichlorovinylidene (DCV) unit giving PAHs 9 and 10 in 30 % and 9 % yield. To the best of our knowledge the introduction of DCV or similar groups at aromatic backbones under oxidative conditions is unprecedented to date during PAH‐cyclization. It is reasonable to assume that the DCV stems from the DCM solvent and given the high number of incorporated DCV groups it is excluded that these are from tetrachloroethane impurities. To date it is not clear by what mechanism the dichlorovinylidation is occurring and needs to be further investigated. To avoid the competing substitution by DCV rather than by OTf groups the amount of DCM was simply reduced by doubling the concentration of precursor 3 and slightly increasing the amount of DDQ from 8 to 10 equivalents, shifting the yields towards the desired bistriflate 8 from 14 % to 25 %, and significantly reducing the amount of the DCV‐containing PAHs 9 and 10 from overall 39 % to 13 %.
To our delight, we were able to grow high‐quality single crystals of PAH 10 with two DCV groups to prove the formation of the two azulene units by slow vapor diffusion of chloroform/methanol. Azulene PAH 10 has a negative curvature and is also a racemic crystal containing both enantiomers (Figure 2). Although it is by definition not a double helicene, its absolute stereochemistry can best be described as such, or by assigning the two chiral biaryl axes formed during cycloheptannulation. The quality of the data allows the bond lengths and angles of the PAH backbone to be discussed in more detail. The longest C−C bonds (1.50 Å) are the biaryl axes formed during cycloheptannulation. With the exception of one bond to the five‐membered ring, which is between 1.37 and 1.39 Å, and the bond shared with the substituted six‐membered rings, the remaining five C−C bonds are significantly longer (between 1.42 and 1.50 Å), which is in contrast to pure azulene, where all bond lengths, with exception of the shared one with the cycloptadienyl ring (1.49 Å), are between 1.37 and 1.42 Å.24 This concludes that the embedded azulene units are of less aromatic character than azulene itself (see also discussion on nuclear independent chemical shifts below).
Figure 2.
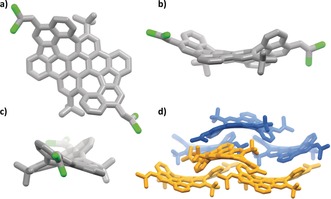
Single‐crystal X‐ray structure of PAH 10.29 a, b, d) Molecular structure of one enantiomer (M,M) from different perspectives. Grey: carbon; green: chlorine. d) Packing. The two enantiomers are highlighted in blue (P,P) and orange (M,M). Hydrogen atoms and solvate molecules have been omitted for clarity.
As demonstrated before, the triflate groups in PAH 8 are synthetically very valuable, because they can undergo palladium‐catalyzed cross‐coupling reactions for example, with boronic acids or esters. Palladium‐catalyzed cross‐coupling of PAH 8 with para‐tert‐butyl phenyl boronic acid or mesityl boronic acid gave the azulene PAHs 11 and 12 in 79 % and 93 % yield, respectively (Scheme 3). Both compounds are still well soluble so that they could be fully characterized in solution. Furthermore, the tert‐butyl groups in the bay‐positions of the mesityl‐substituted PAH 12 were removed by treating the sample with AlCl3 in toluene at 70 °C to give PAH 13 in 57 % yield along with some one‐fold de‐tert‐butylated PAH 14 (14 %). From doubly de‐tert‐butylated PAH 13 we were able to get single‐crystals for X‐ray diffraction analysis (Figure 3). The compound crystallizes in the space group Pc as a racemate of two stereoisomers forming π‐dimers (dπ‐π is between 3.7 and 3.9 Å). Unfortunately, the data quality was not sufficient to discuss the bond lengths and angles in more detail. Because the sterically bulky tert‐butyl groups are removed, the whole structure is somehow planarized in comparison to PAH 12 but still negatively curved.
Scheme 3.
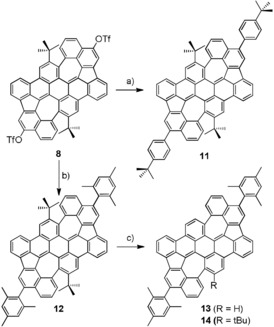
a) 4‐Tert‐Butylphenyl boronic acid, 10 mol % Pd(PPh3)4, toluene/EtOH, 2 N K2CO3(aq), 100 °C, 16 h, 79 % yield. b) Mesityl boronic acid, 10 mol % Pd(PPh3)4, toluene/EtOH, 2 n K2CO3(aq), 100 °C, 16 h, 93 %. c) AlCl3, toluene, 70 °C, 24 h, 57 % (13) and 14 % (14).
Figure 3.
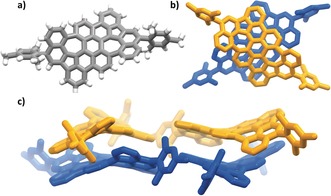
Single‐crystal X‐ray structure of PAH 14.29 a) Molecular structure. b) and c) Packing by weak π‐stacking. The two enantiomers are colored differently. (M,M): blue; (P,P): orange.
All compounds have been investigated by absorption and emission spectroscopy as well as by cyclovoltammetry (CV). Furthermore, frontier molecular orbitals and their corresponding energies have been calculated by DFT methods. All data are summarized in Table S1 in the Supporting Information. Herein, we discuss two series, namely the one with OTf and DCV substituents (PAHs 8–10) as well as mesityl‐substituted PAHs with various numbers of tert‐butyl groups (PAHs 12–14) (Figure 4, Table 1). The most bathochromically shifted peak (S0‐S1 transitions from HOMO to LUMO) in the UV/Vis spectrum of PAH 8 with two OTf groups is found at 628 nm and shifts with the degree of substitution with DCV groups to 636 nm (PAH 9) and finally to 644 nm (PAH 10), which can be simply explained by expansion of the π‐system by the olefinic double bond of each DCV unit. Most interestingly, the maxima of the peaks do not deviate that much from that of azulene itself.25 The emission is shifted as well from 648 through 662 to 671 nm, with fluorescence quantum efficiencies between ϕ=20 and 27 %.
Figure 4.
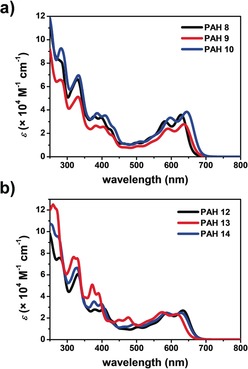
UV/Vis comparison of PAHs 8–10 and 12–14. All spectra were recorded in CHCl3 at room temperature (c(PAH=11–25 μmol L−1).
Table 1.
Summary of the photophysical and electrochemical characterization of PAHs 8–10 and 12–14.
|
Compound |
λ abs [nm][a,b] |
E g opt [eV][a,c] |
λ em (λ ex) [nm][a,d] |
ṽ [cm−1][e] |
Φ [%][f] |
E HOMO CV [eV][g] |
E LUMO CV [eV][g] |
E gap CV [eV] |
|---|---|---|---|---|---|---|---|---|
|
8 |
628 |
1.9 |
648 (280) |
491 |
20±1 |
−5.06 |
−3.29 |
1.77 |
|
9 |
636 |
1.8 |
662 (280) |
617 |
26±1 |
−4.95 |
−3.17 |
1.78 |
|
10 |
644 |
1.8 |
671 (282) |
625 |
24±1 |
−5.00 |
−3.27 |
1.73 |
|
12 |
633 |
1.8 |
658 (277) |
600 |
25±1 |
−4.95 |
−3.09 |
1.86 |
|
13 |
615 |
1.9 |
646 (268) |
780 |
18±1 |
−5.03 |
−3.22 |
1.81 |
|
14 |
628 |
1.8 |
658 (272) |
726 |
22±1 |
−4.95 |
−3.07 |
1.88 |
[a] Measured in CHCl3 at room temperature. [b] Absorption maximum at the longest wavelength. [c] Estimated from absorption onset. [d] Emission maximum. [e] Stoke's shift. [f] Fluorescence quantum yield. [g] Measured in 0.1 m nBu4NClO4 in CH2Cl2 at room temperature. The scan speed was 100 mV s−1, and ferrocene/ferrocenium (Fc/Fc+) was used as internal reference. E HOMO CV=−(E onset ox + 4.8 eV). E LUMO CV=−(E onset red + 4.8 eV).
Comparing PAHs 12–14 of the second series is even more interesting, because these structures have all the same π‐system, but a different degree of curvature, giving further insight into the influence of the changes of curvature on the aromatic character. PAH 12 with the two tert‐butyl groups present has the longest wavelength absorption at 633 nm (S0‐S1 transition from HOMO to LUMO), which shifts to 628 nm for PAH 14 with one tert‐butyl and to 615 m for PAH 13 without tert‐butyl groups. Geometry optimizations (B3LYP/6–311G*) reveal that the π‐system is gradually planarized by removal of tert‐butyl groups and thus becomes more aromatic, which is in agreement with an observation we have made before for hexabenzoovalenes.26 To strengthen this hypothesis, NICS(0) values were calculated for geometry‐optimized structures of PAHs 12 and 13 (see Supporting Information) and the sums of the NICS values ΣNICS of the PAH backbone compared. As expected, the more contorted the system, the less aromatic it is. The most contorted PAH 12 with two tert‐butyl groups has ΣNICS=−42.5, whereas PAH 14 is slightly more aromatic (ΣNICS=−44.5). The most negative sum of NICS(0) values is found for de‐tert‐butylated PAH 13 with ΣNICS=−47.4, which is in agreement with the observation made by UV/Vis spectroscopy. PAHs 12–14 show emission in a similar range (647 to 658 nm) and quantum efficiencies are ϕ=25 % for the more rigid PAH 12 and 18 % for the more flexible PAH 13, suggesting thermal paths for loss of energy.
To summarize, larger contorted and soluble PAHs with negative curvature containing two azulene cores have been synthesized by cyclopent‐ and cycloheptannulation in one step. As observed before for another system,20 the cyclodehydrogenation reaction is accompanied by further selective functionalization of the PAH in the periphery. Besides triflyloxylation, the introduction of two dichlorovinylene groups has been found, which is, to the best of our knowledge, unprecedented for such systems. The OTf groups can be transformed to aryl units under transition metal catalyzed cross‐coupling conditions. Herein, we attached solubilizing mesityl groups to be able to remove the tert‐butyl groups in the bay regions to study the relationship between aromaticity and the degree of curvature. The flatter, the more aromatic the PAH is. All azulene PAHs show a distinct emission close to the near‐infrared (NIR) regime between 650 and 670 nm with relatively high quantum yields ϕ between 20 and 30 %. The emission is already in the regime of the NIR‐window of biological tissues, making the bis‐triflate 8 especially interesting, because it can be decorated with further functional groups or moieties to interact or react selectively with specific sites on biological probes for imaging purposes similar to graphene quantum dots,27 which is ongoing in our laboratory. Furthermore, as long as the tert‐butyl groups are present, analysis of the PAHs by chiral HPLC suggest that these are separable, which makes the azulene PAH 8 a potentially interesting building block for the synthesis of larger chiral curved PAHs.28
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
X.Y. is thankful for a PhD scholarship from the Chinese Scholarship Council (CSC). All authors are grateful for funding of this project by the Deutsche Forschungsgemeinschaft (DFG) within the collaborative research center SFB 1249 on “N‐heteropolycycles as functional materials”. Support by the state of Baden‐Württemberg through bwHPC and the DFG through grant no INST 40/467‐1 FUGG (JUSTUS cluster) is acknowledged.
X. Yang, F. Rominger, M. Mastalerz, Angew. Chem. Int. Ed. 2019, 58, 17577.
References
- 1.
- 1a. Hansen H.-J., Chimia 1996, 50, 489–496; [Google Scholar]
- 1b. Hansen H.-J., Chimia 1997, 51, 147–159; [Google Scholar]
- 1c. Gordon M., Chem. Rev. 1952, 50, 127–200; [Google Scholar]
- 1d. Hafner K., Angew. Chem. 1958, 70, 419–430. [Google Scholar]
- 2. Plattner P. A., St. Pfau A., Helv. Chim. Acta 1937, 20, 224–232. [Google Scholar]
- 3.
- 3a. Ziegler K., Hafner K., Angew. Chem. 1955, 67, 301–301; [Google Scholar]
- 3b. Hafner K., Justus Liebigs Ann. Chem. 1957, 606, 79–89. [Google Scholar]
- 4.
- 4a. Rösler H., König W., Naturwissenschaften 1955, 42, 211; [Google Scholar]
- 4b. Ziegler K., Angew. Chem. 1955, 67, 301–301. [Google Scholar]
- 5.
- 5a. Prinzbach H., Herr H.-J., Angew. Chem. Int. Ed. Engl. 1972, 11, 135–136; [Google Scholar]; Angew. Chem. 1972, 84, 117–118; [Google Scholar]
- 5b. Copland D., Leaver D., Menzies W. B., Tetrahedron Lett. 1977, 18, 639–640; [Google Scholar]
- 5c. Reiter S. E., Dunn L. C., Houk K. N., J. Am. Chem. Soc. 1977, 99, 4199–4201. [Google Scholar]
- 6. Carret S., Blanc A., Coquerel Y., Berthod M., Greene A. E., Deprés J.-P., Angew. Chem. Int. Ed. 2005, 44, 5130–5133; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 5260–5263. [Google Scholar]
- 7. Claus V., Schukin M., Harrer S., Rudolph M., Rominger F., Asiri A. M., Xie J., Hashmi A. S. K., Angew. Chem. Int. Ed. 2018, 57, 12966–12970; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 13148–13152. [Google Scholar]
- 8.
- 8a. Reid D. H., Stafford W. H., Ward J. P., J. Chem. Soc. (Res.) 1955, 1193–1201; [Google Scholar]
- 8b. Koutecký J., Hochman P., Michl J., J. Phys. Chem. 1964, 40, 2439–2456; [Google Scholar]
- 8c. Gründler W., Monatsh. Chem. 1983, 114, 155–176; [Google Scholar]
- 8d. Michl J., Thulstrup E. W., Tetrahedron 1976, 32, 205–209. [Google Scholar]
- 9.
- 9a. Nunn J. R., Rapson W. S., J. Chem. Soc. (Res.) 1949, 825–831; [Google Scholar]
- 9b. Nunn J. R., Rapson W. S., J. Chem. Soc. (Res.) 1949, 1051–1054; [Google Scholar]
- 9c. O'Leary M. A., Wege D., Tetrahedron Lett. 1978, 19, 2811–2814; [Google Scholar]
- 9d. Sperandio D., Hansen H.-J., Helv. Chim. Acta 1995, 78, 765–771; [Google Scholar]
- 9e. Kloster-Jensen E., Kováts E., Eschenmoser A., Heilbronner E., Helv. Chim. Acta 1956, 39, 1051–1067. [Google Scholar]
- 10.
- 10a. Todo E., Yamamoto K., Murata I., Chem. Lett. 1979, 8, 537–540; [Google Scholar]
- 10b. Tobe Y., Chem. Rec. 2015, 15, 86–96. [DOI] [PubMed] [Google Scholar]
- 11. Gardner P. D., Wulfman C. E., Osborn C. L., J. Am. Chem. Soc. 1958, 80, 143–148. [Google Scholar]
- 12. Das S., Wu J., Org. Lett. 2015, 17, 5854–5857. [DOI] [PubMed] [Google Scholar]
- 13. Baumgartner P., Weltin E., Wagnière G., Heilbronner E., Helv. Chim. Acta 1965, 48, 751–764. [Google Scholar]
- 14. Banhart F., Kotakoski J., Krasheninnikov A. V., ACS Nano 2011, 5, 26–41. [DOI] [PubMed] [Google Scholar]
- 15. Ricca A., Bauschlicher J. C. W., Allamandola L. J., Astrophys. J. 2011, 729, 94. [Google Scholar]
- 16.
- 16a. Hieulle J., Carbonell-Sanromà E., Vilas-Varela M., Garcia-Lekue A., Guitián E., Peña D., Pascual J. I., Nano Lett. 2018, 18, 418–423; [DOI] [PubMed] [Google Scholar]
- 16b. Mishra S., Lohr T. G., Pignedoli C. A., Liu J., Berger R., Urgel J. I., Müllen K., Feng X., Ruffieux P., Fasel R., ACS Nano 2018, 12, 11917–11927; [DOI] [PubMed] [Google Scholar]
- 16c. Skidin D., Eisenhut F., Richter M., Nikipar S., Krüger J., Ryndyk D. A., Berger R., Cuniberti G., Feng X., Moresco F., Chem. Commun. 2019, 55, 4731–4734; [DOI] [PubMed] [Google Scholar]
- 16d. Liu J., Mishra S., Pignedoli C. A., Passerone D., Urgel J. I., Fabrizio A., Lohr T. G., Ma J., Komber H., Baumgarten M., Corminboeuf C., Berger R., Ruffieux P., Müllen K., Fasel R., Feng X., J. Am. Chem. Soc. 2019, 141, 12011–12020. [DOI] [PubMed] [Google Scholar]
- 17. Konishi A., Morinaga A., Yasuda M., Chem. Eur. J. 2018, 24, 8548–8552. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Sasaki Y., Takase M., Okujima T., Mori S., Uno H., Org. Lett. 2019, 21, 1900–1903; [DOI] [PubMed] [Google Scholar]
- 18b. Kurotobi K., Kim K. S., Noh S. B., Kim D., Osuka A., Angew. Chem. Int. Ed. 2006, 45, 3944–3947; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 4048–4051; [Google Scholar]
- 18c. Konishi A., Horii K., Shiomi D., Sato K., Takui T., Yasuda M., J. Am. Chem. Soc. 2019, 141, 10165–10170; [DOI] [PubMed] [Google Scholar]
- 18d. Yamamoto K., Ie Y., Tohnai N., Kakiuchi F., Aso Y., Sci. Rep. 2018, 8, 17663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Zhang G., Rominger F., Zschieschang U., Klauk H., Mastalerz M., Chem. Eur. J. 2016, 22, 14840–14845; [DOI] [PubMed] [Google Scholar]
- 19b. Yang X., Rominger F., Mastalerz M., Org. Lett. 2018, 20, 7270–7273. [DOI] [PubMed] [Google Scholar]
- 20. Yang X., Hoffmann M., Rominger F., Kirschbaum T., Dreuw A., Mastalerz M., Angew. Chem. Int. Ed. 2019, 58, 10650–10654; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 10760–10764. [Google Scholar]
- 21.For oxylations during Scholl reactions, see also:
- 21a. Yang Y., Yuan L., Shan B., Liu Z., Miao Q., Chem. Eur. J. 2016, 22, 18620–18627; [DOI] [PubMed] [Google Scholar]
- 21b. Little M. S., Yeates S. G., Alwattar A. A., Heard K. W. J., Raftery J., Edwards A. C., Parry A. V. S., Quayle P., Eur. J. Org. Chem. 2017, 1694–1703. [Google Scholar]
- 22. Márquez I. R., Castro-Fernández S., Millán A., Campaña A. G., Chem. Commun. 2018, 54, 6705–6718. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Kawamura M., Tsurumaki E., Toyota S., Synthesis 2018, 50, 134–138; [Google Scholar]
- 23b. Bratz L. T., Von Niementowski S., Ber. Dtsch. Chem. Ges. B 1919, 52, 189–194; [Google Scholar]
- 23c. Grzybowski M., Skonieczny K., Butenschön H., Gryko D. T., Angew. Chem. Int. Ed. 2013, 52, 9900–9930; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 10084–10115; [Google Scholar]
- 23d. Grzybowski M., Sadowski B., Butenschön H., Gryko D., Angew. Chem. Int. Ed. 2019, https://doi.org/anie.201904934; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, https://doi.org/ange.201904934. [Google Scholar]
- 24. Dittrich B., Fabbiani F. P. A., Henn J., Schmidt M. U., Macchi P., Meindl K., Spackman M. A., Acta Crystallogr. Sect. B 2018, 74, 416–426. [DOI] [PubMed] [Google Scholar]
- 25. Plattner P. A., Heilbronner E., Helv. Chim. Acta 1947, 30, 910–920. [DOI] [PubMed] [Google Scholar]
- 26. Baumgärtner K., Chincha A. L. M., Dreuw A., Rominger F., Mastalerz M., Angew. Chem. Int. Ed. 2016, 55, 15594–15598; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15823–15827. [Google Scholar]
- 27.
- 27a. Nekoueian K., Amiri M., Sillanpää M., Marken F., Boukherroub R., Szunerits S., Chem. Soc. Rev. 2019, 48, 4281–4316; [DOI] [PubMed] [Google Scholar]
- 27b. Tian P., Tang L., Teng K. S., Lau S. P., Mater. Today 2018, 10, 221–258. [Google Scholar]
- 28. Rickhaus M., Mayor M., Juríček M., Chem. Soc. Rev. 2017, 46, 1643–1660. [DOI] [PubMed] [Google Scholar]
- 29.CCDC https://www.ccdc.cam.ac.uk/services/structures?id=doi:10.1002/anie.201908643 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from http://www.ccdc.cam.ac.uk/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary