

ABSTRACT
Ligamentum flavum hypertrophy (LFH) is the most important component of lumbar spinal canal stenosis. Although the pathophysiology of LFH has been extensively studied, no method has been proposed to prevent or treat it. Since the transforming growth factor‐β (TGF‐β) pathway is known to be critical in LFH pathology, we investigated whether LFH could be prevented by blocking or modulating the TGF‐β mechanism. Human LF cells were used for the experiments. First, we created TGF‐β receptor 1 (TGFBR1) knock out (KO) cells with CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 biotechnology and treated them with TGF‐β1 to determine the effects of blocking the TGF‐β pathway. Subsequently, we studied the effect of CCN5, which has recently been proposed to modulate the TGF‐β pathway. To assess the predisposition toward fibrosis, α‐smooth muscle actin (αSMA), fibronectin, collagen‐1, collagen‐3, and CCN2 were evaluated with quantitative real‐time polymerase chain reaction, western blotting, and immunocytochemistry. The TGFBR1 KO LF cells were successfully constructed with high KO efficiency. In wild‐type (WT) cells, treatment with TGF‐β1 resulted in the overexpression of the messenger RNA (mRNA) of fibrosis‐related factors. However, in KO cells, the responses to TGF‐β1 stimulation were significantly lower. In addition, CCN5 and TGF‐β1 co‐treatment caused a notable reduction in mRNA expression levels compared with TGF‐β1 stimulation only. The αSMA protein expression increased with TGF‐β1 but decreased with CCN5 treatment. TGF‐β1 induced LF cell transdifferentiation from fibroblasts to myofibroblasts. However, this cell transition dramatically decreased in the presence of CCN5. In conclusion, CCN5 could prevent LFH by modulating the TGF‐β pathway. © 2019 The Authors. Journal of Orthopaedic Research ® published by Wiley Periodicals, Inc. on behalf of Orthopaedic Research Society. J Orthop Res 37:2634–2644, 2019
Keywords: ligamentum flavum, CRISPR, CCN5, TGF‐β1, myofibroblast
More than 70% of the general population experiences chronic back pain at least once during their lifetime.1 Among the many possible diseases that could cause chronic back pain, lumbar spinal canal stenosis (LSCS) has shown increased occurrence in the elderly population, and it is expected to become more common with the increase in lifespan.2 LSCS is associated with chronic back pain and radiculopathy in the legs; it also commonly causes neurogenic claudication, which greatly affects the quality of daily life of elderly patients.
In addition to bulging discs and facet joint hypertrophy, ligamentum flavum hypertrophy (LFH) is a key contributing factor to LSCS. Research on the pathophysiology of LFH has been of great interest to spine specialists, and the majority of specialists have suggested that tissue inflammation, angiogenesis, and fibrosis lead to LFH.3, 4, 5 Although the development of LFH cannot be described by a single pathway or a single biomechanical change, the relationships between LFH and several key factors, including transforming growth factor‐β1 (TGF‐β1), angiopoietin‐like protein 2 (ANGPTL2), vascular endothelial growth factor (VEGF), and several other cytokines have been investigated.3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 It is widely known that among these cytokines, TGF‐β1 is the main component of LFH that leads to LSCS. Several studies have elucidated the key links of TGF‐β to inflammation,11, 12, 13 angiogenesis,4, 5 extracellular matrix (ECM) regulation,9, 10 and myofibroblast transdifferentiation3 leading to LFH. However, convincing evidence is still lacking regarding whether inhibition of the TGF‐β pathway can actually prevent LFH. More importantly, to date, no method has been reported to prevent or reverse the development of LFH.
The association of the TGF‐β pathway and inflammation is present beyond LFH, and recent studies have found that the CCN family proteins (CCN1–6), which are involved in ECM regulation and therefore called matricellular proteins, are important regulators of inflammation.14, 15, 16 Although some CCNs, such as CCN2, are pro‐hypertrophic and pro‐fibrotic, CCN5 has been shown to be anti‐fibrotic and anti‐hypertrophic in cardiac tissue.17, 18 CCN5 levels were recently found to be reduced in hypertrophied myocardial tissue, and treatment of myocardial tissue with CCN5 showed anti‐hypertrophic effects and inhibited the transition of fibroblasts to myofibroblasts.17 Although there are no previous studies on the effects of CCNs on LF, we considered the influence of CCNs on the myofibroblast transition in LFH as well as other previous findings on the effects of CCNs on the myocardium, and we hypothesized that CCN5 could potentially modulate the pathophysiology of LFH via the TGF‐β pathway.
In this study, we sought to investigate the relationship between LFH and TGF‐β using CRISPR (clustered regularly interspaced short palindromic repeats) gene editing technology, which is an innovative biotechnology but has not been effectively applied in spine research. To this end, we used CRISPR gene editing to block the TGF‐β pathway by knocking out transforming growth factor‐β receptor 1 (TGFBR1) and investigated the effects of the TGF‐β pathway blockade on the fibroblast transition and regulation of the LF ECM. Next, we sought to assess the potential anti‐fibrotic effect of CCN5 on LF by comparing the experimental outcomes after TGF‐β1 treatment. In conclusion, we believe that CCN5, by targeting the TGF‐β pathway, is a potential anti‐hypertrophic treatment for LFH.
METHODS
Human LF Tissue Harvest and Cell Culture
This study was conducted after approval from the Institutional Review Board of our institute. A total of four LF tissues were collected during lumbar spine surgery. As the assay was designed to verify the mechanism from the early stage of fibrosis, fresh and young LF cells were required. We used non‐hypertrophied LFs (<3 mm thickness) obtained from disc herniation patients (not LSCS patients), age under 40. After a phosphate‐buffered saline (PBS; Welgene, Gyeongsan‐si, Korea) wash, tissues were minced and incubated for 1 h at 37°C in Dulbecco's modified Eagle's medium (DMEM; Welgene, Gyeongsan‐si, Korea) with 0.2% collagenase type I (Gibco, Life Technologies, Grand Island, NY). We filtered the suspension with a 100‐μm mesh cell strainer (Falcon BD, Franklin Lakes, NJ) and centrifuged at 300 rcf for 5 min. The pellet was resuspended, seeded on to a cell culture plate (Costar, Corning, NY), and incubated with DMEM containing 10% fetal bovine serum (FBS; Welgene). Subsequent experiments were conducted using cells from the second passage. All experiments were conducted twice per single patient's cell and total two patients’ cells were tested (total four experiments per single assay) to verify the results. Two samples were used for CRISPR KO assay and two samples were used for CCN5 assay. In CRISPR KO assay, WT and KO cells were derived from identical tissues, and paired experiments were performed, and the same experiment was repeated with a different sample to verify the reproducibility.
CRISPR/Cas9‐Mediated TGFBR1 KO Cell Construction
SpCas9 and single guide RNA (sgRNA) vector lipofection
SpCas9 (Streptococcus pyogenes cas9), which is known to be the most efficient gene editing tool of CRISPR/Cas9, was used in this experiment. The CMV promotor‐driven SpCas9 expression vector was used for the experiment. A total of seven target sites were selected from TGFBR1 exon 3, which included the 5′‐NGG‐3′ protospacer adjacent motif (PAM), using an open source bioinformatics tool (http://www.rgenome.net/). The sites were cloned in to the pRG vector, in which sgRNA is transcribed under the U6 promoter (Figs. 1 and 2A and Table 1). Lipofection was conducted with Lipofectamine 2000 (Life Technologies, Carlsbad, CA) for 1 × 105 cells/well in 24‐well plates according to the manufacturer's instructions and incubated for 48 h.
Figure 1.
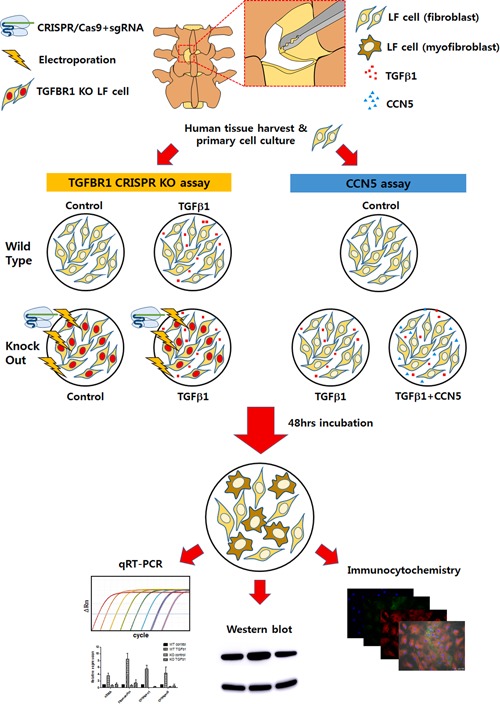
Overall scheme of the experiments. Ligamentum flavum (LF) tissue was harvested from patients and cultured to cells. Transforming growth factor‐β receptor 1 (TGFBR1) CRISPR knock out (KO) cells were created and the TGF‐β1 stimulation assay was conducted. CCN5 was added and the effect was validated with quantitative real‐time polymerase chain reaction (qRT‐PCR), western blotting, and immunocytochemistry. [Color figure can be viewed at wileyonlinelibrary.com].
Figure 2.

Sequence information of transforming growth factor‐β receptor 1 (TGFBR1) knock out (KO) target sites and polymerase chain reaction (PCR) primers are indicated (A). Target #5 showed the highest indel rate (88.2–89%) (B). The four major indel patterns (ID 1, 2, 4, and 5) and the wild‐type pattern (ID 3) are shown (C). [Color figure can be viewed at wileyonlinelibrary.com].
Table 1.
TGFBR1 Target Sequences With Protospacer Adjacent Motif (PAM) and Primer Sequences For Targeted Deep Sequencing
| Names | Sequence |
|---|---|
| TGFbR1 sgRNA target #5 | TAAAAGGGCGATCTAATGAAGGG |
| TGFbR1 sgRNA target #1 | GAACTGGCAGCTGTCATTGCTGG |
| TGFbR1 sgRNA target #2 | GAGATGCAGACGAAGCACACTGG |
| TGFbR1 sgRNA target #3 | CCATCGAGTGCCAAATGAAGAGG |
| TGFbR1 sgRNA target #4 | CCTCTTCATTTGGCACTCGATGG |
| TGFbR1 sgRNA target #6 | AGATCGCCCTTTTATTTCAGAGG |
| TGFbR1 sgRNA target #7 | GATCGCCCTTTTATTTCAGAGGG |
| First primer_forward | TTATTTCACTCGAGGCCCTTTTTCAGTAAA |
| First primer_reverse | GGGTTAGCTGCAGATCATGTGAATATC |
| Deep seq primer_forward | TTATTTCACTCGAGGCCCTTTTTCAGTAAA |
| Deep seq primer_reverse | CAGAACCTGACGTTGTCATATCATAAATTA |
Purification of the recombinant SpCas9 protein
The recombinant SpCas9 protein was purified from Escherichia coli BL21 (DE3) cells cultured in Luria‐Bertani (LB) medium at 18°C overnight after induction with 0.5 mM isopropyl β‐d‐1‐thiogalactopyranoside (IPTG). Harvested cells were resuspended in a lysis buffer (50 mM NaH2PO4 [pH 8], 300 mM NaCl, 10 mM imidazole) and lysed via ultrasonication. After centrifugation for 30 min at 15,000g, the supernatant was applied to Ni‐NTA agarose beads (Qiagen Inc., Valencia, CA) to bind SpCas9. The Ni NTA agarose beads were washed with wash buffer (50 mM NaH2PO4 [pH 8], 300 mM NaCl, 20 mM imidazole) and eluted with elution buffer (50 mM NaH2PO4 [pH 8], 300 mM NaCl, 250 mM imidazole). The purified SpCas9 was dialyzed against a buffer of 20 mM 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid (HEPES) (pH 7.5), 300 mM NaCl, 1 mM dithiothreitol (DTT), and 20% (vol/vol) glycerol.
Ribonucleoprotein (RNP) electroporation
Seven sgRNAs were synthesized by in vitro transcription. The sgRNAs were transcribed by T7 RNA polymerase in 40 mM Tris‐HCl (pH 7.9), 6 mM MgCl2, 10 mM DTT, 10 mM NaCl, 2 mM spermidine, NTPs, and an RNase inhibitor. The reaction mixture was incubated at 37°C for 8 h. The sgRNAs were purified using PCR purification kits (GeneAll, Seoul, Korea) and quantified using a NanoDrop spectrophotometer. An Amaxa Nucleofector machine (Amaxa/Lonza, Cologne, Germany) and a Neon Transfection System (Invitrogen, Basel, Switzerland) were used for electroporation. CMV promoter‐driven green fluorescent protein (GFP) (Amaxa/Lonza) was pre‐tested to optimize the electroporation conditions for human LF cells.
Optimization of human LF cell CRISPR/Cas9 KO
As there were no references on the subject of human LF cell CRISPR/Cas9 KO, we optimized the entire process. The conventional SpCas9 and sgRNA vector lipofection method produced extremely low indel rates for all seven targets (<1%); therefore, we decided to establish more effective methods. To this end, we tested 30 electroporation conditions with GFP: 6 conditions with the Amaxa and 24 conditions with the Neon. Of the conditions, IMR‐90 for the Amaxa and 1,100 voltage/30 width/1 pulse for the Neon was best suited to the human primary LF cells regarding both cell viability and transfection efficiency. Among serial RNP concentrations, 10 μg of SpCas9 protein with 6.5 μg of sgRNA per 1 × 105 cells was the most optimal condition.
Analysis of KO efficiency
After 48 h incubation after transfection, DNA was isolated from the cells with a DNeasy Blood & Tissue Kit (Qiagen) according to the manufacturer's instructions. Serial PCRs were performed for targeted deep sequencing (Figure 2A and Table 1). A Miniseq sequencing system (Illumina, San Diego, CA) was used for next generation sequencing (NGS), and fastq files were analyzed with an open source bioinformatics tool (http://www.rgenome.net/) to determine the insertion/deletion (indel) rate, which is a direct measure of the KO efficiency.
Wild type (WT) and TGFBR1 KO cell assay with exogeneous TGF‐β1‐conditioned medium
WT and KO cells were sub‐cultured in 24‐well plates at a density of 1 × 105 cells/well. After 24 h, the culture medium was replaced to DMEM with 1% FBS. For the both cell types, exogenous recombinant human TGF‐β1 (Peprotech, Rocky Hill, NJ) was added to a 10 ng/ml final concentration in the TGF‐β1 groups, and an equal amount of PBS was added in the control groups. The exogeneous TGF‐β1 concentration was determined based on previously published12 and our Supplementary Data (Supplementary Fig. S1). After 48 h of incubation, the cells were harvested for qRT‐PCR.
Exogeneous CCN5‐ and TGF‐β1‐conditioned medium assay
CCN5 was purchased from the company (Peprotech). WT cells were divided into three groups (control, TGF‐β1, and TGF‐β1 + CCN5) and sub‐cultured in 24‐well plates at a density of 1 × 105 cells/well. After 24 h, the culture medium was replaced with DMEM with 1% FBS; TGF‐β1 (10 ng/ml) and CCN5 (200 ng/ml) were added to the relevant groups. The exogeneous CCN5 concentration was determined based on previously published17 and our Supplementary Data (Supplementary Fig. S2). After 48 h of incubation, cells were prepared for qRT‐PCR, western blotting, and immunocytochemistry.
Gene Expression Analysis Using qRT‐PCR
Total RNA was extracted from the cells with an RNeasy mini kit (Qiagen) according to the manufacturer's instructions. Extracted RNA was reverse transcribed to complementary DNA (cDNA) with a Transcriptor First Strand cDNA Synthesis Kit (Roche, Basel, Switzerland). RT‐PCR was performed with a LightCycler 480 II and a LightCycler480 SYBR Green I Master (Roche). The relative expression levels of the target mRNAs, α‐smooth muscle actin (αSMA), fibronectin, collagen‐1, collagen‐3, and CCN2 were internally standardized using glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) expression and then compared with the control group. αSMA, which expresses at the cell membrane, was used as a representative marker of myofibroblasts. CCN2 is well a known pro‐fibrotic marker, and fibronectin/collagen‐1/‐3 are representative fibrosis products.17, 18 Target mRNA primer sequences are provided in the Supplementary Data (Supplementary Table S1). Results are shown as fold changes compared with the control group using the mean ± standard error of the mean (SEM).
αSMA western blot
LF cells were lysed with radioimmunoprecipitation assay buffer (Biosesang, Seongnam‐si, Korea). After centrifuging at 13,000 rpm for 15 min at 4°C, the supernatant was collected, and protein concentration was measured by the Bradford method using a Pierce BCA Protein Assay Kit (Thermo Scientific, Pierce Biotechnology, Rockford, IL). A total of 20 μg of protein per sample was separated by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis and transferred to a 0.2‐μm nitrocellulose membrane. The membrane was then blocked for 1 h with 5% blocking solution at room temperature (RT) and then incubated at 4°C overnight with the following primary antibodies: anti‐αSMA (Sigma‐Aldrich, St. Louis, MO) and anti‐GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA). The membranes were then exposed to the following secondary antibodies: anti‐mouse immunoglobulin G (IgG) and anti‐rabbit IgG (Jackson ImmunoResearch Labs, West Grove, PA). The signal was detected by adding SuperSignal West Pico Chemiluminescent Substrate and SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific, Pierce Biotechnology, Rockford, IL) to the membrane and exposing to X‐ray film.
Transdifferentiation Detection Using Immunocytochemistry
αSMA and vimentin were detected. Vimentin is a cytoskeletal component of mesenchymal cells and widely used as a marker to identify fibroblasts (including myofibroblasts). Cultured cells were fixed with 4% paraformaldehyde (PFA) for 15 min in a 35‐mm glass bottom culture dish (MakTek, Ashland, MA) and blocked with 5% normal goat serum for 1 h at RT. As primary antibodies, mouse anti‐αSMA and rabbit anti‐vimentin were incubated overnight at 4°C. Goat anti‐mouse Alexa Fluor 488 and Goat anti‐rabbit Alexa Fluor 594 conjugates were stained for 1 h at RT, and nuclei were counterstained with 4′,6‐diamidino‐2‐phenylindole (DAPI) for 10 min at RT, and then mounted. Samples were examined using a light microscope (DMI4000B, Leica, Wetzlar, Germany).
Statistical analyses
For statistical analyses, Mann–Whitney tests were used and p < 0.05 was taken to indicate statistical significance. Data are shown as mean ± SEM.
RESULTS
CRISPR KO cells of the LF
Construction of TGFBR1 KO cells with high indel frequency
We sought to investigate the function of TGF‐β pathway in the fibrosis of LF. To this end, we conducted CRISPR‐mediated KO of TGFBR1 gene in LF cells. In brief, we tested seven different CRISPR RNP complexes that target TGFBR1 genes with 30 electroporation conditions. After optimization of the type of CRISPR RNP and the electroporation condition, we first attempted to isolate a single‐cell colon. However, the attempt failed because of cell growth limitations of human primary LF cell. Under standard culture conditions, the LF fibroblasts naturally undergo transition to myofibroblasts at passage 5–6, and cell division stops due to senescence. Pure KO cell line is technically impossible to purify in such short passages with the traditional method. Therefore, we attempted to use the Cas9‐GFP vector for FACS sorting. However, the LF cells died after sorting because the primary cells were weak, and they grew weaker after transfection due to harsh conditions such as mechanical cell sorting. Therefore, we maximized the indel frequency so that the cell would mimic the KO cell line. After meticulous optimization for the complete process, we constructed two KO cell lines with high and consistent indel rates: 88.2% and 89% (Fig. 2B). Major indel patterns were one or two base pair deletions, indicating that successful frameshift mutations had occurred (Fig. 2C).
Fibrosis is Blocked With the TGFBR1 KO
TGF‐β1 upregulates fibrosis‐related mRNA expression in WT cells
In the WT cell experiment, the TGF‐β1‐conditioned medium group showed a 4.39 ± 2.70‐fold overexpression of αSMA mRNA levels compared with the control. As αSMA is a myofibroblast marker, this result indicates that the LF cells were forced to transition from fibroblasts to myofibroblasts upon TGF‐β1 stimulation. Furthermore, fibronectin (10.20 ± 6.76), collagen‐1 (6.72 ± 4.49), and collagen‐3 (6.16 ± 2.56) mRNA expression levels were also overexpressed in the TGF‐β1‐treated group (Fig. 3, Supplementary Fig. S1). These results indicate that TGF‐β1 stimulated the fibroblast to myofibroblast transition, and ultimately promoted fibrosis by over‐producing the ECM components listed above. These results are consistent with our previous study3.
Figure 3.
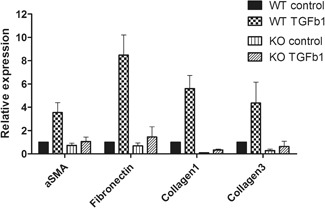
Transforming growth factor‐β receptor 1 (TGFBR1) knock out (KO) assay. TGF‐β1 stimulated α‐smooth muscle actin (αSMA), fibronectin, collagen‐1 and ‐3, and messenger RNA (mRNA) expression. TGFBR1 KO cell shows low αSMA, fibronectin, collagen‐1, and ‐3 expression levels with and without TGF‐β1.
Overexpression of mRNAs stimulated by TGF‐β1 is significantly interrupted in KO cells
Both WT and KO experiments were conducted with identical tissue‐derived cells in the same passage. Duplicate experiments were conducted with additional cells in the same manner. Therefore, KO cell mRNA expression levels were standardized with the WT control group. The αSMA (0.91 ± 0.55), fibronectin (0.94 ± 0.44), collagen‐1 (0.10 ± 0.10), and collagen‐3 (0.41 ± 0.16) mRNA expression changes in the KO control group were similar or even lower than those of the WT control group. This suggests that the TGFBR1 was not functional in KO cells, and not only the extrinsic but also intrinsic TGF‐β1 was blocked; therefore, the baseline expression levels of αSMA, fibronectin, collagen‐1, and collagen‐3, which are related to TGF‐β pathway, were reduced. Comparison of the KO TGF‐β1 group with the WT TGF‐β1 group indicates that αSMA (1.43 ± 0.68), fibronectin (2.33 ± 0.56), collagen‐1 (0.42 ± 0.26), and collagen‐3 (1.07 ± 0.21) mRNA expression levels were significantly lower in the KO TGF‐β1 group. This indicates that blocking TGFBR1 can significantly suppress the downstream mRNA expression of the TGF‐β pathway. Data are shown in Figure 3 and Supplementary Figure S2.
Fibrosis Initiated by TGF‐β1 is Inhibited With CCN5
CCN5 inhibits the overexpression of fibrosis‐related mRNA levels stimulated by TGF‐β1
In CCN5 experiments, the TGF‐β1‐stimulated group showed higher mRNA expression levels for αSMA (3.00 ± 0.29), fibronectin (3.77 ± 0.39), collagen‐1 (5.65 ± 0.75), collagen‐3 (2.52 ± 0.45), and CCN2 (5.84 ± 0.40) compared with the control. Interestingly, this fold change decreased for αSMA (1.45 ± 0.18), fibronectin (2.43 ± 0.04), collagen‐1 (2.54 ± 0.07), collagen‐3 (1.01 ± 0.01), and CCN2 (3.62 ± 0.13) after CCN5 co‐treatment with TGF‐β1 (Fig. 4). We found that the pro‐fibrotic and fibrotic mRNA expression levels were reduced consistently, although the scale of the reduction was less dramatic than in the TGFBR1 KO group. This result suggests that CCN5 has the potential to potently reduce fibrosis, which is the mechanism underlying LFH.
Figure 4.
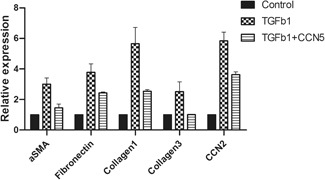
CCN5 assay. Transforming growth factor‐β1 (TGF‐β1) upregulated α‐smooth muscle actin (αSMA), fibronectin, collagen‐1, ‐3, and CCN2. CCN5 co‐treatment reduced the effect of TGF‐β1.
Protein levels of αSMA are reduced after CCN5 stimulation
Of the genes that were assessed by qRT‐PCR, αSMA was the only cell membrane protein that could be reliably detected from the cell at the protein level. Therefore, we assessed the αSMA protein levels from cell lysates. Consistent with the mRNA results, the TGF‐β1‐stimulated cells had higher αSMA protein levels. Interestingly, after CCN5 co‐treatment with TGF‐β1, αSMA levels were notably reduced. This result suggests, indirectly, that CCN5 could prevent the fibroblast to myofibroblast transdifferentiation (Fig. 5).
Figure 5.
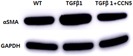
Intracellular protein analysis. Transforming growth factor‐β1 (TGF‐β1) stimulated α‐smooth muscle actin (αSMA) levels and CCN5 co‐treatment reduced αSMA levels. [Color figure can be viewed at wileyonlinelibrary.com].
CCN5 inhibits the fibroblast to myofibroblast transdifferentiation
Next, we assayed on the cell scale whether CCN5 could block the transition of fibroblasts to myofibroblasts. In the control group, LF cells appeared to be typically elongated and spindle‐shaped fibroblasts. We conducted vimentin staining to confirm that the cells were fibroblasts from LF tissue and not a result of contamination. αSMA staining was weak, which suggests that the cells had not transdifferentiated to myofibroblasts (Fig. 6A). After TGF‐β1 stimulation, this cell morphology changed dramatically to a plump stellate shape, and the cytoplasmic component expanded and showed a strong vimentin stain signal. Furthermore, the αSMA stain became prominent, indicating the transdifferentiation of fibroblasts to myofibroblasts (Fig. 6B). In the CCN5 + TGF‐β1 co‐treatment group, αSMA and vimentin stain signals were remarkably reduced, and the cell morphology was almost identical to the control group (Fig. 6C). We conclude that the results were consistent with the hypothesis that CCN5 could reduce fibrosis via downregulation of the TGF‐β signaling pathway.
Figure 6.

Immunocytochemistry: 4′,6‐diamidino‐2‐phenylindole (DAPI) (blue), vimentin (red), α‐smooth muscle actin (αSMA) (green). Ligamentum flavum shows fibroblast‐like morphology with vimentin stain (A). The transforming growth factor‐β1 (TGF‐β1)‐stimulated group shows myofibroblast‐like morphology with αSMA stain (B). The CCN5 + TGF‐β1 group shows similar cell morphology and stain characteristics to control rather than the TGF‐β1 group. [Color figure can be viewed at wileyonlinelibrary.com]
DISCUSSION
To understand the pathophysiologic process of LSCS and discover a possible therapeutic strategy for this degenerative disease, it is necessary to elucidate the biological mechanism of LFH. Accompanied with mechanical stress, inflammation/angiogenesis, and fibrosis of the LF have been considered the hallmark of LFH development in the literature. Sairyo et al.6 proposed a mechanical stress‐induced inflammation cascade leading to LFH via LF scar formation, and Hur et al.4 emphasized the role of angiogenesis as a key link coupling mechanical stress and LFH.
Although we still do not fully understand the pathophysiologic process of LFH, it is widely accepted that the TGF‐β signaling pathway is the core of the problem. TGF‐β1 triggers TGF‐β1/2 receptor activation and regulates the downstream molecular events, inducing the transitional change of fibroblasts to myofibroblasts.19, 20 The transition of fibroblasts to myofibroblasts is found in many cell types, and it is especially well documented in the field of cardiology as a key pathway leading to cardiac failure via myocardial hypertrophy and fibrosis.17, 18 Hur et al.3 for the first time, introduced the concept of fibrosis/hypertrophy pathomechanism into the field of LF and proposed that the myofibroblast transition induced by TGF‐β1 might be a key link between inflammation and LFH. The study showed the presence of myofibroblasts, which have contractile and ECM secretory functions, in the LF of LSCS patients by detecting αSMA. This study introduced the possibility that the transition of LF fibroblasts to myofibroblasts via TGF‐β pathway could be an initiating step of LFH development. Although there is growing evidence that the TGF‐β pathway plays a key role in the development of LFH, there are no published studies showing that KO of the TGF‐β pathway could actually inhibit the LFH process. In this current experimental study, by using the emerging biotechnology CRISPR gene editing tool, we successfully knocked out TGFBR1 and created a TGF‐β pathway KO LF cell model.
CRISPR is a hallmark of the bacterial immune system that actively destroys foreign viral or phage DNA. Modern biotechnology has made it possible to apply CRISPR to the genome editing of human cells. Various classes/types of CRISPRs are found in numerous bacteria. Of these CRISPRs, class 2/type II is most widely used because it contains a target binding module and a cleavage module in a single protein, Cas9, and the system is referred to as CRISPR/Cas9. SpCas9 is the most commonly used CRISPR/Cas9 because it is efficient. Accordingly, we conducted our experiments with SpCas9. Generally, the CRISPR/Cas9 experiments are very sensitive and extensive optimization of experimental conditions is required. Optimization conditions are well known for widely used cell lines such as HEK 293 T and Hela. However, to our knowledge, CRISPR experiments using primary cultures of LF cells have not been reported. Therefore, it was necessary to optimize all experimental conditions to obtain reliable results.
In this study, we successfully optimized the CRISPR/Cas9 KO conditions for human primary cultured LF cells. Therefore, it is of great significance that our study has brought this new technique into the field of spine research. By stimulating the TGFBR1 KO LF cells with TGF‐β, we discovered that blocking the TGF‐β pathway inhibited the expression of αSMA, which indicates the transdifferentiation of fibroblasts to myofibroblasts. End‐product proteins, especially ECM components, including fibronectin, collagen‐1, and ‐3, showed significantly decreased expression after TGF‐β1 stimulation when comparing the TGF‐β1 KO cells with wild‐type cells. This is important because it is the first experimental evidence that blocking TGFBR1 in LF cells can inhibit the TGF‐β pathway, indicating a potential target for LFH treatment via disruption of the fibroblast to myofibroblast transdifferentiation process.
Although we found that the progression of LFH represented by the fibroblast transition to myofibroblasts can be blocked by knocking out TGFBR1, there are still great obstacles to overcome in using these findings in a clinical setting, or even conducting an in vivo study. TGF‐β1 is a polypeptide that controls cell growth/proliferation and differentiation, and this role is not limited to certain cell types.21, 22 Knocking out TGFBR1 can be lethal to a host by affecting the host systemically. Therefore, we sought an alternative method to downregulate the LFH mechanism in a safer way by assessing CCN5 as a candidate molecule. As explained in the introduction, CCNs (sometimes called matricellular proteins) are involved in cell‐to‐cell signaling and play important roles in ECM regulation.14, 15, 16 CCN5 is unique because it lacks a C‐terminal repeat domain and acts as a negative regulator of other CCNs, especially CCN2.23, 24 Several recent studies have established the anti‐fibrotic and anti‐hypertrophic effects of CCN5, and the majority of them have shown that CCN5 may modulate the TGF‐β pathway when acting on fibroblasts.17, 18, 24
In our experiments, the presence of myofibroblasts in LF cells was confirmed by detecting the expression of αSMA. Treatment of LF cells with TGF‐β1 led to significant increases in mRNA expression of αSMA as well as end‐product ECM proteins. When TGF‐β1 treatment was accompanied with CCN5, not only αSMA levels but also fibronectin, collangen‐1, and ‐3 expression levels were significantly reduced. CCN2, which is a pro‐fibrotic and pro‐hypertrophic CCN protein, was also found to show the same significance, suggesting that CCN5 has a significant anti‐fibrotic and anti‐hypertrophic effect on LF cells. Not limited to the level of mRNA expression, we assayed whether intracellular protein level of αSMA was reduced after CCN5 treatment. Furthermore, to confirm whether the transdifferentiation of fibroblasts to myofibroblasts was inhibited by CCN5 on the cell phenotype, we performed immunocytochemical staining for αSMA and vimentin. Although the transdifferentiation of fibroblasts was significant in cells stimulated with TGF‐β1 (shown by significantly enhanced expression of αSMA), the expression of αSMA was almost equivalent to the control cells when CCN5 was added with TGF‐β1 stimulation. The bright field cell morphology changes also showed identical results. Taken together, the data suggest that CCN5 is capable of inhibiting the transdifferentiation of fibroblasts to myofibroblasts.
The application of CCN5 for anti‐hypertrophic purpose is still at its early phase of medical research as possible therapeutics for fibrosis. Previous studies, in the field of cardiology, suggested that CCN5 could be delivered by adeno associated virus via intravenous route for clinical application.17, 18 However, the assessment of safety and efficacy of the methods require extensive further studies. In case of LSCS, we anticipate that it will be important, in the subsequent studies, to construct a LSCS animal model to test diverse delivery methods such as direct protein delivery with local needle puncture and AAV‐mediated intravenous delivery. The efficacy and side effects of CCN5 due to delivery methods has to be investigated in conjunction to the toxicology.
In conclusion, we successfully generated an in vitro model of TGF‐β pathway blocking by constructing TGFBR1 KO cells with CRISPR genome editing tool. Using this model, we found that the TGF‐β pathway blockage led to the suppression of fibroblast to myofibroblast transdifferentiation. CCN5, a recently discovered member of the CCN family, showed significant anti‐fibrotic and anti‐hypertrophic effects on the LF cells stimulated by TGF‐β1 and inhibited LF myofibroblast transdifferentiation at the cell level (Fig. 7). We conclude that the results strongly suggest that CCN5 is a possible candidate for the clinical treatment of LFH and LSCS, and further in vivo experiments should provide more insight to harness the potential of CCN5 for medical applications.
Figure 7.
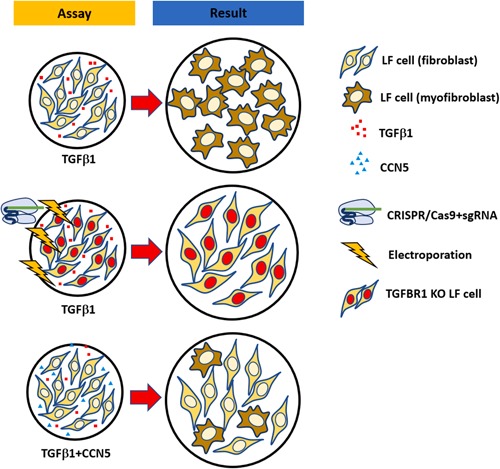
A model of how CCN5 may intervene fibrosis via transforming growth factor‐β (TGF‐β) pathway. Exogeneous TGF‐β1 induces fibroblast to myofibroblast transdifferentiation. Knock out (KO) of transforming growth factor‐β receptor 1 (TGFBR1), reduces the effect of TGF‐β1. Treatment of CCN5 decreases the transdifferentiation of fibroblast to myofibroblast by exogenous TGF‐β1. [Color figure can be viewed at wileyonlinelibrary.com]
ACKNOWLEDGMENTS
This study was approved by the Institutional Review Board of Clinical Trial Center of Korea University Anam Hospital (approval no. 2018AN0379). This study was supported by grants from the Basic Science Research Program through the National Research Foundation (NRF) funded by the Korean Ministry of Education, Science and Technology (KR) (NRF‐2017R1D1A1B03035760, NRF‐2019R1C1C1010602) to J.W.H. and (NRF‐2017R1D1A1B03035094, NRF‐2017R1E1A1A01074529, NRF‐2018M3A9H3021707) to J.K.H. This study was also supported by Korea University, Republic of Korea (K1722461, K1808641, K1809751) to J.W.H.
Supporting information
Supporting information.
AUTHORS’ CONTRIBUTION
J.W.H. and J.K.H. designed the experiments. J.W.H., S.Y., W.K.K., T.B., S.H.K., J.B.L., T.H.C., J.Y.P., K.K., and J.K.H. performed the experiments. J.W.H., W.K.K., J.K.H., S.Y., and K.K. wrote the manuscript. All authors have read and approved the final submitted manuscript.
Sunghyeok Ye, Woo‐Keun Kwon, and Taegeun Bae contributed equally to this work.
Conflicts of interest: None. Grant sponsor: Korean Ministry of Education, Science and Technology; Grant numbers: NRF‐2017R1D1A1B03035760, NRF‐2019R1C1C1010602 (J.W.H.); Grant sponsor: Korean Ministry of Education, Science and Technology; Grant numbers: NRF‐2017R1D1A1B03035094, NRF‐2017R1E1A1A01074529, NRF‐2018M3A9H3021707 (J.K.H.); Grant sponsor: Korea University, Republic of Korea; Grant numbers: K1722461, K1808641, K1809751 (J.W.H.).
Contributor Information
Junho K. Hur, Email: jhur@khu.ac.kr.
Junseok W. Hur, Email: hurjune@gmail.com.
References
REFERENCES
- 1. Andersson GB. 1999. Epidemiological features of chronic low‐back pain. Lancet 354:581–585. [DOI] [PubMed] [Google Scholar]
- 2. Kalichman L, Cole R, Kim DH, et al. 2009. Spinal stenosis prevalence and association with symptoms: the Framingham Study. Spine J 9:545–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hur JW, Bae T, Ye S, et al. 2017. Myofibroblast in the ligamentum flavum hypertrophic activity. Eur Spine J 26:2021–2030. [DOI] [PubMed] [Google Scholar]
- 4. Hur JW, Kim BJ, Park JH, et al. 2015. The mechanism of ligamentum flavum hypertrophy: introducing angiogenesis as a critical link that couples mechanical stress and hypertrophy. Neurosurgery 77:274–282. [DOI] [PubMed] [Google Scholar]
- 5. Moon HJ, Park YK, Ryu Y, et al. 2012. The angiogenic capacity from ligamentum flavum subsequent to inflammation: a critical component of the pathomechanism of hypertrophy. Spine (Phila Pa 1976) 37:E147–E155. [DOI] [PubMed] [Google Scholar]
- 6. Sairyo K, Biyani A, Goel V, et al. 2005. Pathomechanism of ligamentum flavum hypertrophy: a multidisciplinary investigation based on clinical, biomechanical, histologic, and biologic assessments. Spine (Phila Pa 1976) 30:2649–2656. [DOI] [PubMed] [Google Scholar]
- 7. Nakamura T, Okada T, Endo M, et al. 2015. Angiopoietin‐like protein 2 promotes inflammatory conditions in the ligamentum flavum in the pathogenesis of lumbar spinal canal stenosis by activating interleukin‐6 expression. Eur Spine J 24:2001–2009. [DOI] [PubMed] [Google Scholar]
- 8. Park JB, Lee JK, Park SJ, et al. 2005. Hypertrophy of ligamentum flavum in lumbar spinal stenosis associated with increased proteinase inhibitor concentration. J Bone Joint Surg Am 87:2750–2757. [DOI] [PubMed] [Google Scholar]
- 9. Amudong A, Muheremu A, Abudourexiti T. 2017. Hypertrophy of the ligamentum flavum and expression of transforming growth factor beta. J Int Med Res 45:2036–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cao YL, Duan Y, Zhu LX, et al. 2016. TGF‐β1, in association with the increased expression of connective tissue growth factor, induce the hypertrophy of the ligamentum flavum through the p38 MAPK pathway. Int J Mol Med 38:391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Löhr M, Hampl JA, Lee JY, et al. 2011. Hypertrophy of the lumbar ligamentum flavum is associated with inflammation‐related TGF‐β expression. Acta Neurochir (Wien) 153:134–141. [DOI] [PubMed] [Google Scholar]
- 12. Nakatani T, Marui T, Hitora T, et al. 2002. Mechanical stretching force promotes collagen synthesis by cultured cells from human ligamentum flavum via transforming growth factor‐β1. J Orthop Res 20:1380–1386. [DOI] [PubMed] [Google Scholar]
- 13. Park JB, Chang H, Lee JK. 2001. Quantitative analysis of transforming growth factor‐beta 1 in ligamentum flavum of lumbar spinal stenosis and disc herniation. Spine (Phila Pa 1976) 26:E492–E495.11679833 [Google Scholar]
- 14. Jun JI, Lau LF. 2011. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat Rev Drug Discov 10:945–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kular L, Pakradouni J, Kitabgi P, et al. 2011. The CCN family: a new class of inflammation modulators? Biochimie 93:377–388. [DOI] [PubMed] [Google Scholar]
- 16. Holbourn KP, Acharya KR, Perbal B. 2008. The CCN family of proteins: structure‐function relationships. Trends Biochem Sci 33:461–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jeong D, Lee MA, Li Y, et al. 2016. Matricellular protein CCN5 reverses established cardiac fibrosis. J Am Coll Cardiol 67:1556–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yoon PO, Lee MA, Cha H, et al. 2010. The opposing effects of CCN2 and CCN5 on the development of cardiac hypertrophy and fibrosis. J Mol Cell Cardiol 49:294–303. [DOI] [PubMed] [Google Scholar]
- 19. Meng X, Nikolic‐Paterson DJ, Lan HY. 2016. TGF‐β: the master regulator of fibrosis. Nat Rev Nephrol 12:325–338. [DOI] [PubMed] [Google Scholar]
- 20. Baum J, Duffy HS. 2011. Fibroblasts and myofibroblasts: what are we talking about? J Cardiovasc Pharmacol 57:376–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Matsuura I, Chiang KN, Lai CY, et al. 2010. Pin1 promotes transforming growth factor‐β‐induced migration and invasion. J Biol Chem 285:1754–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wrighton KH, Lin X, Feng XH. 2009. Phospho‐control of TGF‐β superfamily signaling. Cell Res 19:8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen CC, Lau LF. 2009. Functions and mechanisms of action of CCN matricellular proteins. Int J Biochem Cell Biol 41:771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu H, Li P, Liu M, et al. 2015. CCN2 and CCN5 exerts opposing effect on fibroblast proliferation and transdifferentiation induced by TGF‐β. Clin Exp Pharmacol Physiol 42:1207–1219. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.