

Abstract
Objectives
Many patients with Cushing's disease (CD) require chronic pharmacotherapy to control their hypercortisolism. We evaluated the efficacy and safety of long‐acting pasireotide during a long‐term extension study in patients with CD.
Design
Open‐label extension to a 12‐month Phase III study of long‐acting pasireotide in CD (N = 150; NCT01374906).
Patients
Patients with mean urinary free cortisol (mUFC) ≤ upper limit of normal (ULN) or receiving clinical benefit at core study end could continue long‐acting pasireotide during the extension.
Results
Eighty‐one of 150 (54.0%) enrolled patients entered the extension. Median overall exposure to pasireotide at study end was 23.9 months; 39/81 (48.1%) patients completed the extension (received ≥ 12 months’ treatment during the extension and could transit to a separate pasireotide safety study). mUFC was ≤ULN in 42/81 (51.9%), 13/81 (16.0%) and 43/81 (53.1%) patients at extension baseline, month (M) 36 and last assessment. Median mUFC remained within normal limits. Median late‐night salivary cortisol was 2.6 × ULN at core baseline and 1.3 × ULN at M36. Clinical improvements were sustained over time. Forty‐two (51.9%) patients discontinued during the extension: 25 (30.9%) before M24 and 17 (21.0%) after M24. Hyperglycaemia‐related AEs occurred in 39.5% of patients. Mean fasting glucose (FPG) and glycated haemoglobin (HbA1c) were stable during the extension, with antidiabetic medication initiated/escalated in some patients. Sixty‐six (81.5%) and 71 (88.9%) patients were classified as having diabetes (HbA1c ≥ 6.5%, FPG ≥ 7.0 mmol/L, antidiabetic medication use, or history of diabetes) at extension baseline and last assessment.
Conclusions
Long‐acting pasireotide provided sustained biochemical and clinical improvements, with no new safety signals emerging, supporting its use as an effective long‐term therapy for CD.
Keywords: Cushing syndrome, Cushing's disease, extension, hypercortisolism, pasireotide, Phase III, pituitary
1. INTRODUCTION
Cushing's disease (CD) is a rare endocrine disorder characterized by overproduction of cortisol by the adrenal glands, secondary to an adrenocorticotropic hormone (ACTH)–secreting pituitary adenoma (corticotropinoma).1 CD is associated with significant multisystem morbidity, impaired quality of life (QoL) and increased mortality if inadequately treated.2, 3
Selective resection of the corticotropinoma by transsphenoidal surgery is the first‐choice treatment for CD, but surgical success rates vary and depend on the expertise of the surgeon. CD persists in 10%‐35% of patients after surgery,4, 5 while recurrence rates range from 15% to 66% after a variable duration of remission.6, 7, 8 Medical therapy plays a prominent role in the management of persistent or recurrent CD, as well as in patients who are not candidates for surgery, such as those considered to be at high surgical risk and/or with acute complications of CD.2, 8, 9, 10
Given the chronic nature of CD, many patients require long‐term pharmacotherapy to achieve and maintain the goals of treatment: to control cortisol excess, reverse clinical features and complications of hypercortisolism, improve QoL, control tumour mass and increase life expectancy.8 A comprehensive understanding of the long‐term efficacy and safety of available medical treatment options is therefore central to achieving optimal outcomes for patients.
Based on the results of a 12‐month Phase III study,11 a long‐acting intramuscular formulation of pasireotide suitable for monthly administration has been approved in the EU, United States and other countries worldwide for the treatment of CD.12, 13 Here, we report the efficacy and safety of long‐acting pasireotide during a long‐term extension (median treatment duration: 23.9 months) to the Phase III study.
2. METHODS
2.1. Patients
This was an optional, open‐label extension to a 12‐month Phase III core study.11 Patients could enter the extension if they satisfied the following criteria at the end of the core study: mean urinary free cortisol (mUFC; mean of three 24‐hour samples collected within 2 weeks) not exceeding the upper limit of normal (ULN; 166.5 nmol/24 h) and/or considered by the investigator to be receiving significant clinical benefit from long‐acting pasireotide; demonstrated acceptable tolerability of pasireotide during the core study; and provided consent to participate in the extension.
2.2. Study design
During the core study, adults with confirmed CD and mUFC of 1.5‐5.0 × ULN were randomized to double‐blind pasireotide 10 or 30 mg every 28 days; dose titration was permitted based on efficacy/tolerability.11 Patients who entered the extension continued long‐acting pasireotide (without interruption) at the same dose that they received at month 12. Pasireotide dose could be increased (5 to 10 mg, 10 to 20 mg, 30 to 40 mg every 28 days) if mUFC > 1.0 × ULN, providing there was an interval of ≥3 months since last increase. Dose reductions were permitted at any time for tolerability (lowest permitted dose: 5 mg every 28 days).
During the extension, patients received long‐acting pasireotide for 12 months, at which point they continued in the study until an option was available to transit to a separate open‐label, long‐term safety study of long‐acting pasireotide (clinicaltrials.gov: NCT01794793; results not presented), which occurred once the last ongoing patient had received 12 months of treatment during the extension. Patients who had received an additional ≥12 months of treatment after the 12‐month core phase and had the option to enter the separate open‐label, long‐term safety study were considered to have completed the extension. The study ended once the last patient had received 12 months of treatment during the extension, and all patients had either transited to the long‐term safety study or discontinued treatment.
2.3. Assessments and outcomes during the extension
Prespecified outcomes of the extension included the following: changes in mUFC, plasma ACTH, and morning serum and late‐night salivary cortisol (LNSC); proportion of patients with controlled (≤ULN) or partially controlled (>ULN and ≥50% reduction from baseline) mUFC; change in pituitary tumour volume, clinical signs of hypercortisolism and health‐related QoL (Cushing's quality‐of‐life questionnaire [CushingQoL]); and safety and tolerability of long‐acting pasireotide.
During the extension, the following parameters were monitored at 3‐month intervals: clinical signs (systolic blood pressure [SBP] and diastolic blood pressure [DBP], body mass index [BMI] and waist circumference), mUFC, morning serum cortisol, LNSC and plasma ACTH. Bone mineral density (BMD; left total hip and lumbar spine [L1‐L4]) was measured at 6‐month intervals by Lunar or Hologic dual‐energy X‐ray absorptiometry (DXA) instruments. Patients were scanned on the same DXA instrument for the duration of the study. Pituitary tumour volume was measured at 6‐month intervals by magnetic resonance imaging (MRI) and assessed by a central reader.
Safety was assessed by monitoring adverse events (AEs). AEs were defined using the Medical Dictionary for Regulatory Activities version 18.1 and graded according to Common Terminology Criteria for Adverse Events version 3.0. Laboratory assessments, including haematological/blood biochemical measurements (fasting plasma glucose [FPG], glycated haemoglobin [HbA1c], thyroid and liver function tests), vital signs, gallbladder examinations and electrocardiograms, were assessed as described previously.11
2.4. Statistical methods
Efficacy and safety data are presented for patients who entered the extension. Analyses were performed once all patients had completed the extension or discontinued treatment. Morning serum cortisol, LNSC, morning plasma ACTH, mUFC and clinical signs were analysed descriptively. Actual/percentage changes were calculated from core study baseline (before pasireotide initiation), and from extension baseline (at end of core study [month 12]) for patients with evaluable assessments at core/extension baseline and the later time point. For the proportions of controlled and partially controlled responders, two‐sided 95% confidence intervals (CIs) were calculated. Efficacy data are presented up to month 36 and at each patient's last available assessment. Safety data are presented during the core study (up to month 12) and extension (after month 12 until study end). Definitions of diabetic status (diabetic, prediabetic and normoglycaemic) are included in the Appendix S1.
3. RESULTS
3.1. Study population
Eighty‐one of 150 patients (54.0%) who were enrolled in the core study entered the extension. Thirty‐nine (48.1%) patients completed the extension (ie received ≥ 12 months’ treatment during the extension and had opportunity to enter a separate long‐term pasireotide safety study; Figure 1); each of these patients opted to enrol in the separate long‐term safety study (results will be published separately). The remaining 42 (51.9%) patients discontinued long‐acting pasireotide during the extension: 25 (30.9%) discontinued prior to month 24 and 17 (21.0%) discontinued after month 24. The most common reasons for discontinuation during the extension phase were consent withdrawal (16.0%), unsatisfactory therapeutic effect (14.8%) and AEs (9.9%).
Figure 1.
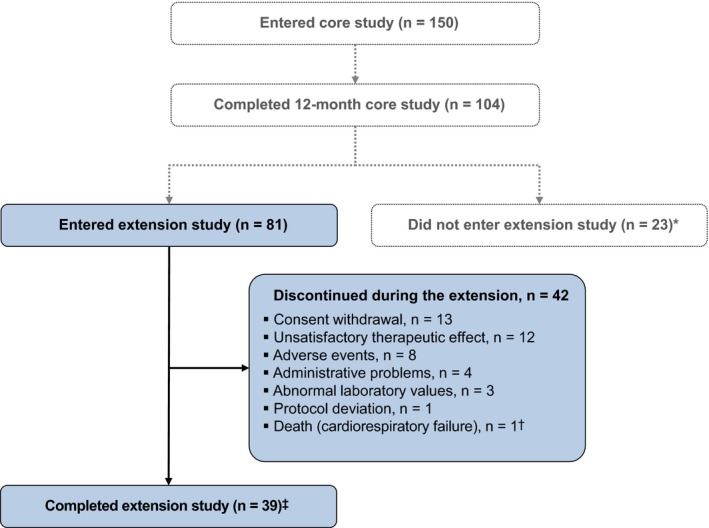
Patient disposition. *Reasons for not entering the extension were not recorded; †The death occurred 16 d after the 16th injection and was not suspected by the treating physician to be related to pasireotide11; ‡Patients were considered to have completed the extension if both of the following criteria were met: they had received an additional ≥12 mo of treatment during the extension; and the core study had been unblinded, which occurred once all 150 patients who were enrolled in the core study had reached month 12 or discontinued treatment. The AEs leading to discontinuation in 8 patients during the extension phase were as follows: diabetes mellitus, n = 2; hyperglycaemia, n = 3 (one of these patients also had increased gamma‐glutamyltransferase); hyperkalaemia, n = 1; endometrial cancer, n = 1; and acute cholecystitis, cholelithiasis, elevated liver enzymes, bilirubin increased, oedematous pancreatitis and ascites, n = 1 [Colour figure can be viewed at http://wileyonlinelibrary.com]
Median duration of pasireotide exposure from study start until end of extension was 23.9 months (range: 12.0‐55.3). At extension start, long‐acting pasireotide was administered at a dose of 5, 10, 30 and 40 mg every 28 days in 6, 15, 20 and 40 patients, respectively. During the extension, 12 (14.8%) patients had ≥1 dose level increase compared with the month 12 dose.
Table 1 shows the characteristics at core and extension baseline for the 81 patients who entered the extension.
Table 1.
Summary of baseline characteristics for patients who entered the extension phase (N = 81)
| Characteristic | Core baseline (month 0) | Extension baseline (month 12) |
|---|---|---|
| Mean age, years (SD) | 39.7 (12.8) | 40.7 (12.8) |
| Females, n (%) | 61 (75.3) | 61 (75.3) |
| Median time since diagnosis, months (IQR) | 26.9 (9.3‐65.9) | 37.9 (20.5‐77.0) |
| Pituitary adenoma, n (%)* | ||
| Nonvisible adenoma | 19 (23.5) | 17 (21.0) |
| Microadenoma | 34 (42.0) | 41 (50.6) |
| Macroadenoma | 26 (32.1) | 17 (21.0) |
| Missing | 2 (2.5) | 6 (7.4) |
| Baseline mUFC, × ULN | ||
| Mean (SD) | 2.6 (1.5) | 1.4 (1.3) |
| Median (IQR) | 2.3 (1.5‐3.4) | 1.0 (0.7‐1.8) |
| Previous treatment, n (%) | ||
| Previous pituitary surgery | 67 (82.7) | 67 (82.7) |
| Medical therapy | 32 (39.5) | 81 (100) |
| Diabetic status, n (%)† | ||
| Normal glucose tolerance | 35 (43.2) | 6 (7.4) |
| Prediabetic | 14 (17.3) | 9 (11.1) |
| Diabetic | 32 (39.5) | 66 (81.5) |
Abbreviations: IQR, interquartile range; SD, standard deviation.
Pituitary adenoma size defined according to maximum tumour diameter (microadenoma >0–<10 mm; macroadenoma ≥10 mm).
Diabetic status defined as follows: diabetic, patients with HbA1c ≥ 6.5%, FPG ≥ 126 mg/dL, prior history of diabetes mellitus, or receiving antidiabetic medication; prediabetic, patients not qualifying as diabetic and FPG >100–<126 mg/dL or HbA1c >5.7–<6.5%; and normal glucose tolerance, patients not qualifying as diabetic or prediabetic.
3.2. Long‐term efficacy
3.2.1. Change in mUFC and other biochemical markers of CD
At extension start, 42 (51.9%) patients had controlled mUFC (≤ULN). Eleven (13.6%) patients had partially controlled mUFC (>ULN but ≥50% decrease from core baseline), and 28 (34.6%) had uncontrolled mUFC (>ULN and <50% decrease from core baseline) but were considered to have been receiving clinical benefit from pasireotide treatment. Most patients (75.0%; n = 21/28) with uncontrolled mUFC at extension had mUFC ≤ ULN on ≥1 visit during the core study.
Overall, 38/81 (46.9%) patients had controlled mUFC at month 24 (after 12 months of treatment during the extension). The proportion of patients with controlled or partially controlled mUFC levels was stable from extension baseline to month 24 (Figure S1). Of the 42 patients with controlled mUFC at extension baseline, 27 (64.3%) had controlled mUFC at month 24, two (4.8%) had partially controlled mUFC, and three (7.1%) had uncontrolled mUFC; 10 (23.8%) had discontinued or did not have an mUFC assessment at month 24. Eleven of 39 (28.2%) patients with mUFC > ULN at extension baseline had controlled mUFC at month 24; each of these patients had mUFC ≤ ULN on ≥1 visit during the core study.
Of patients who received 36 months’ treatment, 13/18 (72.2%) had controlled mUFC at this time point. At the time of each patient's last available assessment, 43/81 (53.1%) patients were controlled, 16/81 (19.8%) were partially controlled and 22/81 (27.2%) were uncontrolled.
For patients with evaluable assessments, median mUFC levels remained within the normal range (15.9‐166.5 nmol/24 h) from extension baseline up to month 36 (Figure 2). For the subset of patients who received ≥12 and ≥24 months’ treatment during the extension, median mUFC levels remained within the normal range from extension baseline up to these time points. Median (range) change in mUFC from start of pasireotide treatment was −180.3 nmol/24 h (−1520.0, 667.7; n = 81 [−51.9% change]) at month 12, −236.4 nmol/24 h (−1541.8, 1109.3; n = 58 [−64.1%]) at month 24, and −267.2 nmol/24 h (−543.0, −21.1; n = 18 [−68.3%]) at month 36. Median (range) change in mUFC from extension baseline to last available assessment was −7.7 nmol/24 h (−651.2, 667.3; n = 78 [−5.9% change]).
Figure 2.
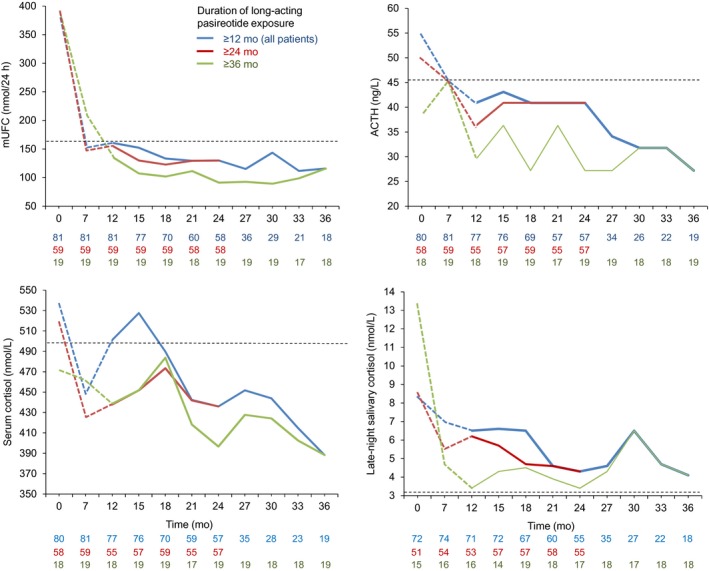
Median mUFC, ACTH, serum cortisol and LNSC over time, by duration of pasireotide treatment. Continuous lines represent data collected during the extension; dashed lines represent data collected during the core study for patients who were later enrolled in the extension. Horizontal reference lines represent the ULN range for mUFC (166.5 nmol/24 h), morning plasma ACTH (45.4 ng/L), morning serum cortisol (532.2 nmol/L) and LNSC (3.2 nmol/L). The number of patients contributing to the median is displayed under the X axis
Median levels of morning plasma ACTH, morning serum cortisol and LNSC were below core baseline levels at each 3‐monthly assessment during the extension (Figure 2 and Appendix S1), with levels within the normal range for plasma ACTH and morning serum cortisol at most time points. Median LNSC levels decreased from 2.6 × ULN (n = 72) at core baseline to 2.0 × ULN (n = 71) at extension baseline, 1.4 × ULN (n = 55) at month 24 and 1.3 × ULN (n = 18) at month 36.
3.2.2. Changes in pituitary tumour volume
Thirty‐four (42.0%) and 26 (32.1%) patients had a measurable microadenoma or macroadenoma, respectively, on MRI at start of pasireotide treatment; an adenoma was not visible in 19 (23.5%) patients. Of patients with a measurable adenoma at core baseline and month 24 (n = 35), 85.7% had a ≥20% reduction (n = 12; 34.3%) or <20% change (n = 18; 51.4%) in tumour volume between these time points (Table S1). No patient with a macroadenoma at study baseline had a ≥20% increase in tumour volume at month 24 or 36.
3.2.3. Change in clinical signs of hypercortisolism and health‐related QoL
Improvements in median SBP, DBP, BMI and waist circumference were sustained during the extension (Figure 3). For the 81 patients who entered the extension phase, median (range) changes from start of treatment to the end of the core study (month 12) were as follows: SBP, −4.0 mm Hg (−45.3, 27.3; n = 80); DBP, −2.0 mm Hg (−48.0, 19.3; n = 80); waist circumference, −5.0 cm (−38.0, 34.0; n = 75); and BMI, −1.9 kg/m2 (−11.2, 3.5; n = 80). Median (range) changes from extension baseline to last available assessment were as follows: SBP, 2.7 mm Hg (−41.7, 27.3; n = 79); DBP, 0.0 mm Hg (−22.0, 22.0; n = 79); waist circumference, 1.0 cm (−13.0, 21.0; n = 77); and BMI, 0.1 kg/m2 (−5.7, 4.1; n = 79).
Figure 3.
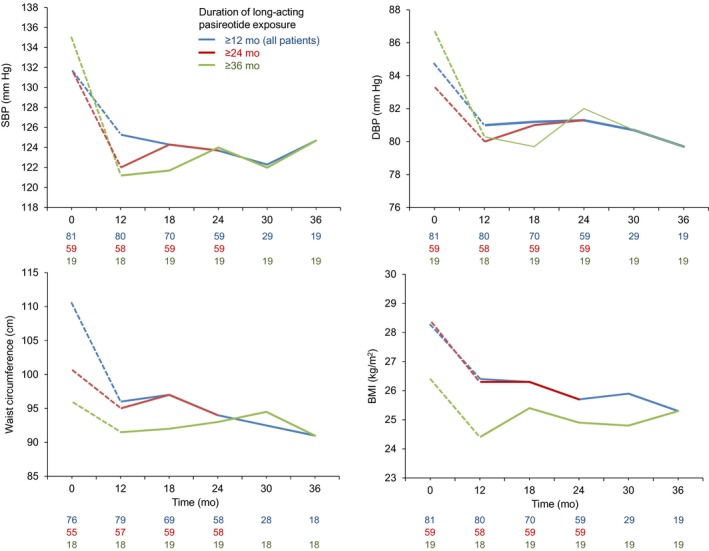
Median SBP, DBP, waist circumference and BMI, by duration of pasireotide treatment. Continuous lines represent data collected during the extension; dashed lines represent data collected during the core study for patients who were later enrolled in the extension
Forty‐five (55.6%) patients were receiving antihypertensive medication at extension baseline. Five patients received ≥1 additional agent, and two initiated antihypertensive medication during the extension. Conversely, five patients who were on antihypertensive medication at extension baseline were receiving ≥1 fewer agent at their last visit, one of whom was not receiving any blood pressure–lowering medication.
There were no clear changes in blood lipid levels or BMD (left total hip and lumbar spine) over 36 months’ treatment (Table S2). In total, 27 (33.3%) patients were receiving lipid‐lowering medication at extension baseline, none of whom had an increase in the number of prescribed medications during the extension. Four additional patients started lipid‐lowering drugs during the extension.
Median (range) CushingQoL score was 41.7 (6.3, 81.3; n = 79) prior to starting pasireotide. Median (range) CushingQoL score improved from core study baseline by 9.4 points (−31.3, 56.3; n = 78) at month 12, 8.3 points (−31.3, 29.2; n = 57) at month 24, and 10.4 points (−27.1, 31.3; n = 17) at month 36. Median (range) change in CushingQoL score from extension baseline to last observed value was −2.1 points (−52.1, 27.1; n = 76).
3.3. Long‐term safety
Most patients (74; 91.4%) experienced one or more AEs during the extension. Grade 3/4 events were reported in 31 (38.3%) patients (Table 2). Hyperglycaemia (23.5%) was the most commonly reported AE during the extension. When all AE terms related to hyperglycaemia were grouped, 32 (39.5%) patients experienced any hyperglycaemia‐related AE during the extension, which led to discontinuation in three patients. Fifty‐eight (71.6%) patients experienced a hyperglycaemia‐related AE during the 12‐month core study. Other common AEs (≥15% of patients) during the extension were nasopharyngitis (19.8%), cholelithiasis (18.5%) and diarrhoea (17.3%). Except for nasopharyngitis, these AEs occurred in fewer patients during the extension than the core study (Table 2). Gastrointestinal AEs were also less common during the extension than the core study: diarrhoea (17.3% vs 34.6%), nausea (9.9% vs 22.2%) and abdominal pain (2.5% vs 14.8%).
Table 2.
AEs (≥10% of patients during the core or extension phase), regardless of drug relationship, in the 81 patients who entered the extension
| AE, n (%) | Months 0‐12 | After month 12 | ||
|---|---|---|---|---|
| All grades | Grade 3/4 | All grades | Grade 3/4 | |
| Total | 80 (98.8) | 33 (40.7) | 74 (91.4) | 31 (38.3) |
| Hyperglycaemia | 32 (39.5) | 4 (4.9) | 19 (23.5) | 3 (3.7) |
| Nasopharyngitis | 14 (17.3) | 0 | 16 (19.8) | 0 |
| Cholelithiasis | 23 (28.4) | 1 (1.2) | 15 (18.5) | 2 (2.5) |
| Diarrhoea | 28 (34.6) | 0 | 14 (17.3) | 0 |
| Urinary tract infection | 6 (7.4) | 0 | 10 (12.3) | 0 |
| Headache | 11 (13.6) | 0 | 9 (11.1) | 0 |
| Diabetes mellitus | 15 (18.5) | 7 (8.6) | 8 (9.9) | 4 (4.9) |
| Nausea | 18 (22.2) | 0 | 8 (9.9) | 0 |
| Hypoglycaemia | 10 (12.3) | 2 (2.5) | 8 (9.9) | 2 (2.5) |
| Fatigue | 11 (13.6) | 0 | 7 (8.6) | 0 |
| Dizziness | 11 (13.6) | 1 (1.2) | 6 (7.4) | 0 |
| Abdominal pain | 12 (14.8) | 0 | 2 (2.5) | 1 (1.2) |
| Influenza | 11 (13.6) | 0 | 5 (6.2) | 0 |
| Oedema peripheral | 10 (12.3) | 0 | 5 (6.2) | 0 |
AEs are sorted in descending order of events during the extension. Patients who experienced an AE during the core study (months 0‐12) and the extension (after month 12) are counted in both columns.
Sixteen (19.8%) patients experienced a gallbladder/biliary tract–related AE during the extension, most commonly cholelithiasis (n = 15). One patient discontinued treatment because of cholelithiasis, increased blood bilirubin, and acute cholecystitis. Three (3.7%) patients experienced bradycardia‐related AEs during the extension, none of which led to discontinuation. Liver safety–related AEs occurred in nine (11.1%) patients, with four discontinuing treatment: increased hepatic enzymes with concomitant AEs of acute pancreatitis and acute cholecystitis, n = 1; increased hepatic enzymes, n = 1 (drug‐induced liver injury not objectified in clinical or subsequent laboratory findings); increased gamma‐glutamyltransferase with a concomitant AE of hyperglycaemia, n = 1; and increased alanine aminotransferase, n = 1.
One patient died from cardiac failure during the extension. This death was previously reported as part of the core study analysis,11 as the patient died before unblinding of the core study. The death was not suspected to be study drug related.
For patients who entered the extension, mean HbA1c and FPG levels increased from core baseline to month 12 (extension baseline). Mean HbA1c and FPG levels were stable during the extension (Figure 4). Mean (95% CI) changes in HbA1c and FPG from extension baseline to last observed value were 0.0% (−0.2, 0.2; n = 77) and −6.6 mg/dL (−14.5, 1.3; n = 78), respectively.
Figure 4.
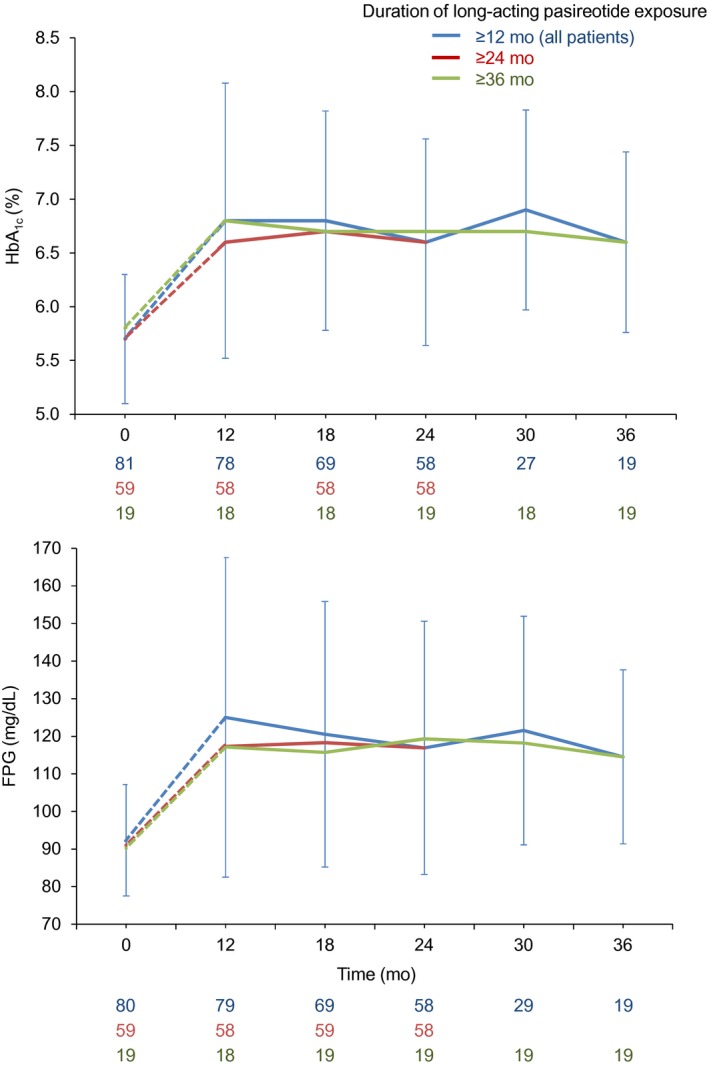
Mean (SD) FPG and HbA1c levels during the extension phase. Continuous lines represent data collected during the extension; dashed lines represent data collected during the core study for patients who were later enrolled in the extension
At extension baseline, 66 (81.5%), nine (11.1%) and six (7.4%) patients were classified as diabetic, prediabetic and normoglycaemic. At last available assessment, no patients had shifted from normoglycaemic to prediabetic/diabetic, while six (66.7%) had shifted from prediabetic to diabetic. Twelve patients (14.8%) had HbA1c ≥ 8% at their last assessment; nine of these patients had HbA1c ≥ 8% at extension baseline. One patient had shifted from prediabetic to normoglycaemic at their last assessment. Overall, 50/81 (61.7%) patients were receiving antidiabetic medication at extension baseline (one medication, n = 21; two medications, n = 14; three or more medications, n = 15). At last assessment, an increase in antidiabetic medications was reported in 10/35 (28.6%) patients. Eight of 31 (25.8%) patients who were not receiving antidiabetic drugs at month 12 were being treated with antidiabetic medication at their last assessment. Seventeen (21.0%) patients were receiving insulin at extension baseline; a further 10 (12.3%) patients started insulin therapy during the extension.
4. DISCUSSION
In this large, prospective, extension study, patients were treated with long‐acting pasireotide for a median of 2 years, allowing robust interpretation of its long‐term efficacy and safety. Long‐acting pasireotide provided sustained biochemical improvements and clinical benefit in a significant proportion of patients with CD and was generally well tolerated, with no new safety signals emerging over long‐term treatment.
Achieving and maintaining normal cortisol levels is a key treatment goal for patients with CD in order to prevent or alleviate the clinical signs and complications of hypercortisolism and improve life expectancy.8 Almost half (47%) of patients who entered this extension study had controlled mUFC after 2 years of long‐acting pasireotide treatment; 25% (n = 38/150) of patients who were enrolled in the core study had controlled mUFC at month 24. Baseline characteristics for patients who entered this extension study were similar to those reported previously for all patients who participated in the initial core phase of the study (N = 150).11 Patients could continue receiving pasireotide beyond month 24 until there was an option to enter a separate long‐term study of pasireotide. Over two‐thirds (72.2%; n = 13/18) of patients who were still participating in the extension at month 36 had controlled mUFC at this time point. Furthermore, over half (53%) of patients who entered the extension had a normal mUFC level at their last assessment, which was marginally higher than the proportion of responders at extension start. These results support the long‐term efficacy of pasireotide and are consistent with findings from a Phase III study, in which 69% (n = 11/16) of ongoing patients had controlled mUFC after 5 years of twice‐daily, subcutaneous pasireotide therapy.14, 15 Improvements in median mUFC, morning serum cortisol and LNSC levels were reported overall and for patients who received ≥2 or ≥3 years of treatment. Unlike mUFC and morning serum cortisol, median LNSC remained >ULN during the extension. Additional studies are needed to examine the effect of cortisol‐lowering therapies on salivary cortisol and the role of LNSC in monitoring medical treatment response.
Cardiovascular disease is the leading cause of death in patients with CD.16 Consequences of cortisol excess that can contribute to cardiovascular disease include hypertension, truncal obesity, impaired glucose tolerance, insulin resistance, dyslipidaemia and hypercoagulability.17 In our study, improvements in blood pressure, BMI and waist circumference were maintained for up to 3 years of pasireotide treatment. Such improvements may confer a reduction in mortality risk, given that small reductions in blood pressure can significantly reduce the risk of cardiovascular events in patients with hypertension.18 Contrary to previous reports, we did not observe any consistent changes in cholesterol or triglycerides during our study.18, 19
It is important to note that concomitant medications were allowed during this study, which could have contributed to the observed changes in clinical signs over time. Persistent hypercortisolism is associated with progressive bone degradation resulting in increased fracture risk for patients with CD.20 Encouragingly, BMD was stable throughout 36 months of pasireotide treatment, although additional studies are warranted to confirm the effects of pasireotide on the prevention/reversal of bone deterioration in patients with CD.
Hypercortisolism and its clinical effects detrimentally impact on QoL; the effects often persist after the resolution of hypercortisolism.21 Patients who participated in the current extension had significantly impaired QoL (CushingQoL score, 41.7 points) prior to starting treatment with long‐acting pasireotide. Importantly, CushingQoL score improved from baseline over 3 years of treatment, potentially reflecting the improvements in clinical signs of hypercortisolism during pasireotide treatment.
Pituitary tumour volume decreased or remained stable in most patients during long‐term treatment with pasireotide, consistent with previous reports.22, 23, 24, 25 Given its tumour‐shrinkage effects, it has been proposed that pasireotide may be particularly beneficial in patients with a clinically relevant tumour mass and/or tumour progression.9, 10, 26 This assertion is supported by our findings that almost half (46%; n = 6/13) of patients with a macroadenoma before initiation of pasireotide treatment had a significant (≥20% change) decrease in tumour volume by month 24, with the remainder exhibiting stable tumour size.
The safety profile of long‐acting pasireotide during this extension was similar to that during the core study, with no new safety signals emerging. AEs were generally mild/moderate and occurred less frequently in the extension than in the first 12 months of treatment. The lower rate of AEs in the extension phase than in the core study may be explained by the early onset and effective management of side effects during pasireotide treatment, while patients who tolerated pasireotide may have been more likely to continue into the extension phase. These findings are similar to those of a previous prospective study of subcutaneous twice‐daily pasireotide, in which AEs related to hyperglycaemia, liver safety, the gallbladder and bradycardia tended to emerge during the first 6 months of treatment and did not usually worsen over 5 years’ treatment.14
According to the American Diabetes Association and European Association for the Study of Diabetes, glycaemic targets should be individualized based on key patient characteristics. As such, while an HbA1c level of <7% represents a reasonable goal for many adults, less stringent targets (such as <8%) are appropriate in some patients, such as those with extensive comorbid conditions.27, 28 In our study, mean HbA1c and FPG levels were stable; mean HbA1c remained <7% over the course of the extension, and most (85%) patients had HbA1c < 8.0% at their last assessment.
Initiation and escalation of antidiabetic therapy was at the discretion of the investigator; 26% of patients who were not receiving antidiabetic therapy at extension baseline were receiving at least one agent at their last assessment, while 29% of patients who were already receiving 1‐2 antidiabetic drugs were receiving at least one additional agent. Taken together, these findings demonstrate that if hyperglycaemia occurs, it tends to emerge soon after initiation of pasireotide and can be effectively managed in some patients with antidiabetic therapy.
Few studies have examined the efficacy and safety of long‐term treatment with other medical therapies for CD. In a retrospective analysis of 51 patients who had received long‐term (≥2 years) ketoconazole treatment, a normal mUFC level was seen in 65% of patients at their last available assessment, with clinical improvements seen in hypertension, diabetes and hypokalaemia.29 One‐fifth (20.5%) of patients discontinued treatment because of intolerance.29 In a retrospective analysis of metyrapone treatment, 64% (n = 9/14) of patients who had an available mUFC assessment after ≥6 months of treatment had a normal mUFC level at their last available assessment, while there was a low incidence (11%; n = 4/38) of AEs in these patients.30 In another retrospective study, 40% (n = 21/53) of patients receiving cabergoline therapy initially achieved normal UFC levels, with treatment escape seen in 39% (n = 7/18) of patients who remained on treatment for ≥12 months.31 Published data on mifepristone and osilodrostat are limited to shorter durations of treatment.32, 33 The choice of medical treatment options should be tailored to the individual clinical situation, taking into consideration the patient, disease and tumour characteristics.8
5. CONCLUSIONS
Patients who entered this extension study exhibited sustained biochemical and clinical benefit over 3 years of treatment with long‐acting pasireotide, which was accompanied by improvements in QoL and tumour size control. The long‐term safety profile of pasireotide was favourable and consistent with that reported during the first 12 months of treatment. These data support the use of long‐acting pasireotide as an effective long‐term treatment option for some patients with CD.
CONFLICT OF INTEREST
Maria Fleseriu has received research support (paid to Oregon Health & Science University) from Novartis, Millendo Therapeutics and Strongbridge Biopharma, and scientific consultancy fees from Novartis and Strongbridge Biopharma. Stephan Petersenn had served as lecturer for Ipsen, Novartis and Pfizer, and as an advisory board member for Ipsen and Novartis. Beverly MK Biller has been the PI of research grants (paid to Massachusetts General Hospital) from Novartis, Millendo and Strongbridge Biopharma, and has received occasional consulting honoraria from Novartis and Strongbridge Biopharma. Pinar Kadioglu has no conflicts of interest to disclose. Christophe De Block has served as a lecturer and as an advisory board member for Ipsen and Novartis. Guy T'Sjoen has received scientific grants as principal investigator for Ipsen, Bayer Schering and Sandoz; consulting fees as an advisory board member for Ipsen and Novartis; and lecture fees as a speaker for Ferring and Novartis. Marie‐Christine Vantyghem has received funding as an investigator (paid to Lille University Hospital) from Novartis and Ipsen. Libuse Tauchmanova, Judi Wojna and Michael Roughton are employees of Novartis. John Newell‐Price has received research and consultancy fees (paid to the University of Sheffield) from Novartis, Diurnal, Ipsen, HRA Pharma and ONO Pharma. André Lacroix has received funding as an investigator or consultant from Novartis, Cortendo, Strongbridge Biopharma and GLWL Research, Inc.
Supporting information
ACKNOWLEDGEMENTS
This study was sponsored by Novartis Pharma AG. Financial support for medical editorial assistance was provided by Novartis Pharmaceuticals Corporation. We thank Robert Jenn PhD, Mudskipper Business Ltd, for medical editorial assistance with this manuscript, as well as the site investigators, study co‐ordinators and patients who participated in the trial.
Fleseriu M, Petersenn S, Biller BMK, et al. Long‐term efficacy and safety of once‐monthly pasireotide in Cushing's disease: A Phase III extension study. Clin Endocrinol (Oxf). 2019;91:776–785. 10.1111/cen.14081
Clinical Trial Registration Number: NCT01374906
DATA AVAILABILITY STATEMENT
Novartis is committed to sharing with qualified external researchers' access to patient‐level data and supporting clinical documents from eligible studies. These requests are reviewed and approved by an independent review panel on the basis of scientific merit. All data provided are anonymized to respect the privacy of patients who have participated in the trial, in line with applicable laws and regulations. This trial data availability is in accordance with the criteria and process described on http://www.clinicalstudydatarequest.com.
REFERENCES
- 1. Lacroix A, Feelders RA, Stratakis CA, et al. Cushing's syndrome. Lancet. 2015;386:913‐927. [DOI] [PubMed] [Google Scholar]
- 2. Pivonello R, De Leo M, Cozzolino A, et al. The treatment of Cushing's disease. Endocr Rev. 2015;36:385‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pivonello R, Isidori AM, De Martino MC, et al. Complications of Cushing's syndrome: state of the art. Lancet Diabetes Endocrinol. 2016;4:611‐629. [DOI] [PubMed] [Google Scholar]
- 4. Barker FG, Klibanski A, Swearingen B. Transsphenoidal surgery for pituitary tumors in the United States, 1996‐2000: mortality, morbidity, and the effects of hospital and surgeon volume. J Clin Endocrinol Metab. 2003;88:4709‐4719. [DOI] [PubMed] [Google Scholar]
- 5. Biller B, Grossman AB, Stewart PM, et al. Treatment of adrenocorticotropin‐dependent Cushing's syndrome: a consensus statement. J Clin Endocrinol Metab. 2008;93:2454‐2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aranda G, Ensenat J, Mora M, et al. Long‐term remission and recurrence rate in a cohort of Cushing's disease: the need for long‐term follow‐up. Pituitary. 2015;18:142‐149. [DOI] [PubMed] [Google Scholar]
- 7. Atkinson AB, Kennedy A, Wiggam MI, et al. Long‐term remission rates after pituitary surgery for Cushing's disease: the need for long‐term surveillance. Clin Endocrinol. 2005;63:549‐559. [DOI] [PubMed] [Google Scholar]
- 8. Nieman LK, Biller BM, Findling JW, et al. Treatment of Cushing's syndrome: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2015;100:2807‐2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cuevas‐Ramos D, Fleseriu M. Treatment of Cushing's disease: a mechanistic update. J Endocrinol. 2014;223:R19‐R39. [DOI] [PubMed] [Google Scholar]
- 10. Tritos NA, Biller B. Medical therapy for Cushing's syndrome in the twenty‐first century. Endocrinol Metab Clin North Am. 2018;47:427‐440. [DOI] [PubMed] [Google Scholar]
- 11. Lacroix A, Gu F, Gallardo W, et al. Efficacy and safety of once‐monthly pasireotide in Cushing's disease: a 12 month clinical trial. Lancet Diabetes Endocrinol. 2018;6:17‐26. [DOI] [PubMed] [Google Scholar]
- 12. Novartis Pharma AG. Signifor summary of product characteristics. 2013. http://www.signifor.com/european-product-characteristics.jsp. Accessed March 2019.
- 13. Novartis Pharmaceuticals Corporation . Signifor LAR prescribing information. 2019. http://www.pharma.us.novartis.com/product/pi/pdf/signifor_lar.pdf. Accessed March 2019.
- 14. Petersenn S, Salgado LR, Schopohl J, et al. Long‐term treatment of Cushing's disease with pasireotide: 5‐year results from an open‐label extension study of a Phase III trial. Endocrine. 2017;57:156‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schopohl J, Gu F, Rubens R, et al. Pasireotide can induce sustained decreases in urinary cortisol and provide clinical benefit in patients with Cushing's disease: results from an open‐ended, open‐label extension trial. Pituitary. 2015;18:604‐612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hardy ST, Loehr LR, Butler KR, et al. Reducing the blood pressure‐related burden of cardiovascular disease: impact of achievable improvements in blood pressure prevention and control. J Am Heart Assoc. 2015;4:e002276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ferrau F, Korbonits M. Metabolic comorbidities in Cushing's syndrome. Eur J Endocrinol. 2015;173:M133‐M157. [DOI] [PubMed] [Google Scholar]
- 18. Albani A, Ferraù F, Ciresi A, et al. Pasireotide treatment reduces cardiometabolic risk in Cushing's disease patients: an Italian, multicenter study. Endocrine. 2018;61:118‐124. [DOI] [PubMed] [Google Scholar]
- 19. Pivonello R, Petersenn S, Newell‐Price J, et al. Pasireotide treatment significantly improves clinical signs and symptoms in patients with Cushing's disease: results from a Phase III study. Clin Endocrinol. 2014;81:408‐417. [DOI] [PubMed] [Google Scholar]
- 20. Webb SM, Valassi E. Morbidity of Cushing's syndrome and impact of treatment. Endocrinol Metab Clin North Am. 2018;47:299‐311. [DOI] [PubMed] [Google Scholar]
- 21. Webb SM, Santos A, Resmini E, et al. Quality of life in Cushing's disease: a long term issue? Ann Endocrinol. 2018;79:132‐137. [DOI] [PubMed] [Google Scholar]
- 22. Simeoli C, Auriemma RS, Tortora F, et al. The treatment with pasireotide in Cushing's disease: effects of long‐term treatment on tumor mass in the experience of a single center. Endocrine. 2015;50:725‐740. [DOI] [PubMed] [Google Scholar]
- 23. Colao A, Petersenn S, Newell‐Price J, et al. A 12‐month Phase 3 study of pasireotide in Cushing's disease. N Engl J Med. 2012;366:914‐924. [DOI] [PubMed] [Google Scholar]
- 24. Katznelson L. Sustained improvements in plasma ACTH and clinical status in a patient with Nelson's syndrome treated with pasireotide LAR, a multireceptor somatostatin analog. J Clin Endocrinol Metab. 2013;98:1803‐1807. [DOI] [PubMed] [Google Scholar]
- 25. Lu L, Duan L, Jin Z, et al. Effective long‐term treatment of Cushing's disease with pasireotide ‐ a case report. Endocr Pract. 2013;19:e92‐e96. [DOI] [PubMed] [Google Scholar]
- 26. Petersenn S. How to manage pasireotide, when using as medical treatment for Cushing's disease. Endocrine. 2015;50:526‐528. [DOI] [PubMed] [Google Scholar]
- 27. American Diabetes Association . Standards of medical care in diabetes ‐ 2014. Diabetes Care. 2014;37(Suppl 1):S14‐S80. [DOI] [PubMed] [Google Scholar]
- 28. Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes: a patient‐centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2012;35:1364‐1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Castinetti F, Guignat L, Giraud P, et al. Ketoconazole in Cushing's disease: is it worth a try? J Clin Endocrinol Metab. 2014;99:1623‐1630. [DOI] [PubMed] [Google Scholar]
- 30. Daniel E, Aylwin S, Mustafa O, et al. Effectiveness of metyrapone in treating Cushing's syndrome: a retrospective multicenter study in 195 patients. J Clin Endocrinol Metab. 2015;100:4146‐4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ferriere A, Cortet C, Chanson P, et al. Cabergoline for Cushing's disease: a large retrospective multicenter study. Eur J Endocrinol. 2017;176:305‐314. [DOI] [PubMed] [Google Scholar]
- 32. Fleseriu M, Biller BM, Findling JW, et al. Mifepristone, a glucocorticoid receptor antagonist, produces clinical and metabolic benefits in patients with Cushing's syndrome. J Clin Endocrinol Metab. 2012;97:2039‐2049. [DOI] [PubMed] [Google Scholar]
- 33. Fleseriu M, Pivonello R, Young J, et al. Osilodrostat, a potent oral 11β‐hydroxylase inhibitor: 22‐week, prospective, Phase II study in Cushing's disease. Pituitary. 2016;19:138‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Novartis is committed to sharing with qualified external researchers' access to patient‐level data and supporting clinical documents from eligible studies. These requests are reviewed and approved by an independent review panel on the basis of scientific merit. All data provided are anonymized to respect the privacy of patients who have participated in the trial, in line with applicable laws and regulations. This trial data availability is in accordance with the criteria and process described on http://www.clinicalstudydatarequest.com.