

Abstract
Objective
To investigate the multifactorial processes underlying cognitive aging based on the hypothesis that multiple causal pathways and mechanisms (amyloid, vascular, and resilience) influence longitudinal cognitive decline in each individual through worsening brain health.
Methods
We identified 1,230 elderly subjects (aged ≥50 years) with an average of 4.9 years of clinical follow‐up and with amyloid positron emission tomography, diffusion tensor imaging, and structural magnetic resonance imaging scans from the population‐based Mayo Clinic Study of Aging. We examined imaging markers of amyloid and brain health (white matter microstructural integrity and cortical thinning), systemic vascular health preceding the imaging markers, and early to midlife intellectual enrichment to predict longitudinal cognitive trajectories. We used latent growth curve models for modeling longitudinal cognitive decline.
Results
All the pathways (amyloid, vascular, resilience) converged through their effects on cortical thinning and worsening cognition and together explained patterns in cognitive decline. Resilience and vascular pathways (aging process, sex differences, education/occupation, and systemic vascular health) had significant impact on white matter microstructural integrity. Education/occupation levels contributed to white matter integrity through systemic vascular health. Worsening white matter integrity contributed to significant cortical thinning and subsequently longitudinal cognitive decline. Baseline amyloidosis contributed to a significant proportion of cognitive decline that accelerated with longer follow‐up times, and its primary impact was through cortical thinning.
Interpretation
We developed an integrated framework to help explain the dynamic and complex process of cognitive aging by considering key causal pathways. Such an approach is important for both better comprehension of cognitive aging processes and will aid in the development of successful intervention strategies. ANN NEUROL 2019;86:866–877
Successful interventions to prevent cognitive decline with age can only be designed through the development of integrated models that are able to successfully predict cognitive trajectories in the elderly by considering multiple pathways. The recent availability of longitudinal cognitive trajectories in population‐based cohorts with neuroimaging markers of disease and health provides an opportunity to further the mechanistic understanding of the cognitive aging process.
Alzheimer disease (AD) pathologies explain an important proportion of cognitive loss in aging and dementia.1 However, the extent of impact of higher amyloid burden on cognitive decline in the cognitively unimpaired (preclinical stages of AD clinical syndrome) is highly debated. Although it is now understood that the impact of amyloid on cognition is mediated through tau deposition and neurodegeneration, the assumption that amyloidosis is a single deterministic cause of cognitive aging in the population is not true.2, 3 To successfully predict cognitive outcomes, it is important to consider other key processes that substantially influence cognitive aging along with amyloidosis: vascular health, age‐related changes, and resilience mechanisms.
Microstructural integrity measured using diffusion tensor imaging (DTI) magnetic resonance imaging (MRI) is sensitive to brain changes due to small vessel disease, which is common in the elderly.4 We recently found that integrity of the small fibers in the genu of the corpus callosum measured using DTI‐based fractional anisotropy (FA) captured both variability in systemic vascular health and visible cerebrovascular injury in the form of white matter (WM) hyperintensities.5 There are also likely age‐related, noncerebrovascular contributions to genu FA. Aging‐related loss of WM tissue composition and integrity has been observed with an anterior–posterior gradient (last‐in‐first‐out theory) with greater anterior WM degradation.6, 7, 8 Due to observable age‐related changes seen in the frontal WM, genu FA measurement serves to capture both age‐related WM degradation and cerebrovascular health.
Resilience mechanisms have been studied extensively in the context of coping with AD pathologies,9 and education is the most commonly used proxy for studying resilience in pathology and imaging studies.10, 11 Here, we used a previously developed education/occupation score to capture intellectual enrichment in early life and midlife to understand the resilience pathway.12, 13
The overall goal of this study was to model cognitive aging in the population as a multifactorial process based on the hypothesis that longitudinal cognitive decline can be modeled by taking into account measurable entities that index multiple causal pathways and mechanisms. In this work, we used latent growth curve models (LGCMs) for modeling longitudinal cognitive decline. These models are useful for studying between‐person differences and within‐person change. LGCMs have been extensively used for mapping the courses and causes of repeated measures outcomes.14
Subjects and Methods
Participants
All participants were enrolled in the Mayo Clinic Study of Aging (MCSA), a longitudinal population‐based study of Olmsted County, Minnesota residents. The Olmsted County population was enumerated using the Rochester Epidemiology Project (REP) medical records linkage system.15, 16 A recent study from our group showed that the selected MCSA study participants were representative of the Olmsted County, Minnesota population.17 The complete details of the MCSA study design and diagnostic criteria have been previously discussed by Petersen et al18 and Roberts et al.19 Regarding standard protocol approvals, registrations, and patient consents, the study was approved by the Mayo Clinic and Olmsted Medical Center institutional review boards, and informed consent was obtained from all participants or their surrogates.
We identified all 1,230 individuals who were at least 50 years of age with amyloid positron emission tomography (PET) scans at baseline from MCSA and had at least 2 clinical follow‐ups (with complete neuropsychological evaluation at all visits), spanning the period from January 2006 to April 2018. At the time of the scans, 1,132 were cognitively unimpaired, 93 had mild cognitive impairment, 3 were diagnosed with AD clinical syndrome, and 2 had a missing clinical diagnosis due to incomplete data. Sex, years of education, and major occupation were ascertained at the baseline clinical visit. We used age at the time of the MRI scan as a covariate. Similar to our previous work, we calculated education and occupation composite score as a combination of education and the major occupation in each participant's life.20
Indicator of Systemic Vascular Health
As previously described by Rocca et al,21 as well as consolidated in our recent paper,22 we searched for and included the following 7 cardiovascular and metabolic conditions in a 5‐year capture frame from REP diagnostic indices for each individual before their MCSA visit: hypertension, hyperlipidemia, cardiac arrhythmias, coronary artery disease, congestive heart failure, diabetes mellitus, and stroke. We formed a composite score for these cardiovascular and metabolic conditions (CMC) as the summation of the presence or absence of these conditions. Specific International Classification of Diseases, 9th Revision (ICD‐9) and ICD‐10 codes pooled together have been published previously.22
Amyloid Assessment from Amyloid PET scans
The acquisition, processing, and summary measure details for amyloid PET scans acquired on the MCSA study participants have been discussed previously.23 A global amyloid load (standardized uptake value ratio [SUVR]) was computed for each subject by calculating median uptake in the prefrontal, orbitofrontal, parietal, temporal, anterior cingulate, and posterior cingulate/precuneus regions of interest (ROIs) divided by the median uptake in the cerebellar crus gray matter (GM) ROI.
MRI Acquisition and Processing
MRI was acquired on three 3T General Electric (Boston, MA) scanners. FreeSurfer version 5.3 was used to estimate average cortical thickness in AD signature regions (entorhinal cortex, inferior temporal, middle temporal, fusiform) as a single measure, which was used as a surrogate for aging‐ and AD‐related neurodegeneration.23 The DTI acquisition and processing protocol was previously published,5 and we used FA of the genu of the corpus callosum as our measure of cerebrovascular health.
Cognitive Performance
The MCSA neuropsychological battery consists of 9 tests covering 4 cognitive domains, as previously described.18, 19 In this work, we used a global cognitive z score that was estimated from the z transformation of the average of the 4 domain z scores (executive function, language, memory, and visuospatial performance) based on the entire MCSA population as a cognitive outcome variable.
Statistical Analyses
LGCMs for Cognitive Decline
Our primary model was the LGCM, a type of structural equation model (SEM), used to describe the complex pathways preceding and leading to cognitive decline. The LGCMs were run using all available data, with a maximum of 7 visits per subject. SEM path analyses require assumptions regarding the ordering of the variables. We used 3 methods to determine the ordering: inherent temporal ordering, theoretical reasoning, and results from 2‐panel longitudinal SEMs. Age, sex (male), APOE4, and cycle number (which represents the total number of clinical visits) are exogenous variables and were placed at level 1. We positioned education/occupation, an early/midlife variable, at the second level and systemic vascular health, measured in the 5 years preceding the baseline visit, at the third level. Amyloid, genu FA, and thickness were all acquired at the baseline visit of the individual. Because extensive literature has shown that upstream amyloid causes downstream neurodegeneration, which underlies cognitive impairment,24 we placed amyloid at level 4, thickness at level 5, and the outcome measure of cognition at level 6. We were able to determine the order of genu FA with respect to thickness by first building a 2‐panel SEM, described below, that provided the final temporal ordering to the growth curve model. This allowed us to understand the causal associations between the variables and cognition.
In LGCMs, all subjects are assumed to have individual cognitive curves or trajectories over time of the same functional form, although the parameters describing the curves can differ. We put time into the curves explicitly through 3 latent variables: “intercept” (describing how high or low is cognition), “linear” (describing the slope of cognitive change over time), and “quadratic” (describing curvature in cognitive change over time). These latent variables would manifest through up to 7 repeated values of cognition (Cog1–Cog7) for each subject. Each of the 3 aspects of the cognitive curves could have a unique set of predictors. For example, in our model, age directly affected intercept and linearity but not quadratic. We fit the models using Mplus version 8,25 and used full information maximum likelihood to handle missing data. Genu FA and log (amyloid) were standardized in the models. We also looked at potential interactions of age × genu FA, age × log (amyloid), and genu FA × log (amyloid), but did not find any of the interactions to be significant. Forcing these interactions into the models also produced substantial lack of fit. We pruned the SEM using backward elimination from left to right and from right to left (both methods produced the same result) to get a parsimonious model.
Genu FA and Neurodegeneration: Two‐Panel SEM
To answer the question of causality between genu FA and neurodegeneration, we used longitudinal genu FA and thickness data from the first set of contiguous values available for each subject. We repeated the modeling using the last set of contiguous values for each subject to check for consistency. These specific analyses were restricted to 2 time points to maintain an adequate sample size; including more time points reduced the available sample size substantially. We ran 2‐wave, 2‐variable cross‐lagged panel models.26 These models address the question, “Does x cause y or does y cause x?” while controlling for past characteristics of the individuals. Genu FA and neurodegeneration at the first time point (wave 1) were used to predict genu FA and neurodegeneration at the second time point (wave 2). If, for example, genu FA at time 1 predicted neurodegeneration at time 2 but neurodegeneration at time 1 did not predict genu FA at time 2, we would have evidence that genu FA precedes neurodegeneration. We again ran these models in the SEM framework using Mplus version 8.25
Results
The participant characteristics by decade (50–59, 60–69, 70–79, 80–90+) are shown in Table 1. There were approximately equal numbers of males and females. There were some differences in APOE4 carrier status across decades (p = 0.04), and higher education/occupation was seen in the midlife cohort (50–69 years) compared to elderly cohort (70–90+). Because our 50‐ to 69‐year‐old cohort started in 2012 and the amyloid scanning was started in the 70 to 90+ cohort in 2008, the 70 to 90+ cohort had a longer cognitive follow‐up. As expected, cognition worsened with age.
Table 1.
Characteristics Table by Decade with the Mean (SD) Listed for the Continuous Variables and Count (%) for the Categorical Variables
| Characteristic | 50–59, n = 181 | 60–69, n = 368 | 70–79, n = 447 | 80–90+, n = 234 | p a |
|---|---|---|---|---|---|
| Demographics and APOE4 genotype | |||||
| Age, yr | 55.0 (2.5) | 64.8 (2.8) | 74.4 (2.7) | 83.7 (3.2) | |
| Males | 92 (51%) | 196 (53%) | 236 (53%) | 140 (60%) | 0.23 |
| APOE4 carrier | 51 (28%) | 111 (30%) | 135 (30%) | 48 (21%) | 0.04 |
| Primary predictors | |||||
| Education and occupation [0,0,1,0] | 13.0 (2.0) | 13.1 (2.4) | 12.4 (2.6) | 12.4 (2.9) | <0.001 |
| CMC | 0.9 (1.1) | 1.6 (1.3) | 2.3 (1.4) | 2.6 (1.5) | <0.001 |
| Genu FA [1,1,84,71] | 0.63 (0.04) | 0.62 (0.04) | 0.59 (0.05) | 0.56 (0.04) | <0.001 |
| Amyloid burden, SUVR | 1.27 (0.08) | 1.38 (0.19) | 1.52 (0.35) | 1.67 (0.47) | <0.001 |
| Thickness, mm [0,2,6,4] | 2.97 (0.12) | 2.95 (0.12) | 2.84 (0.15) | 2.74 (0.16) | <0.001 |
| Cognition‐related variables | |||||
| Baseline global cognition | 0.99 (0.73) | 0.50 (0.85) | −0.09 (0.94) | −0.58 (1.12) | <0.001 |
| Total cognitive visits | 3.7 (0.7) | 4.1 (0.9) | 5.4 (1.5) | 5.3 (1.4) | <0.001 |
| Follow‐up, yr | 3.5 (0.9) | 3.9 (1.0) | 5.8 (1.9) | 5.7 (1.8) | <0.001 |
| Cognitively impaired [0,1,1,0] | 1 (1%) | 17 (5%) | 39 (9%) | 39 (17%) | <0.001 |
The analysis for amyloid burden was done on the log‐transformed values. Brackets indicate the number of missing observations for each age group.
Probability values for differences between decade groups come from an analysis of variance for the continuous variables or a chi‐squared test for the categorical variables.
CMC = cardiovascular and metabolic conditions; FA = fractional anisotropy; SD = standard deviation; SUVR = standardized uptake value ratio.
Two‐Panel SEM
A subset of the individuals (n = 642) with 2 consecutive genu FA and thickness measurements at the same time points were selected to establish causality between genu FA and thickness for the 2‐panel SEM. The top panel of Figure 1 summarizes the 2‐panel SEM for genu FA and thickness. Thickness at time t = 0 predicted thickness at t = 1 (p < 0.001) and genu FA at t = 0 predicted genu FA at t = 1 (p < 0.001) after controlling for baseline characteristics of the individuals. Genu FA at t = 0 was predictive of thickness at t = 1 (p = 0.015), but thickness at t = 0 was not predictive of genu FA at t = 1 (p = 0.205). These results suggest that WM damage was upstream to thickness. This is also illustrated by the bottom panel of Figure 1. This independent model provided sufficient evidence for placing genu FA before thickness in our primary analysis.
Figure 1.
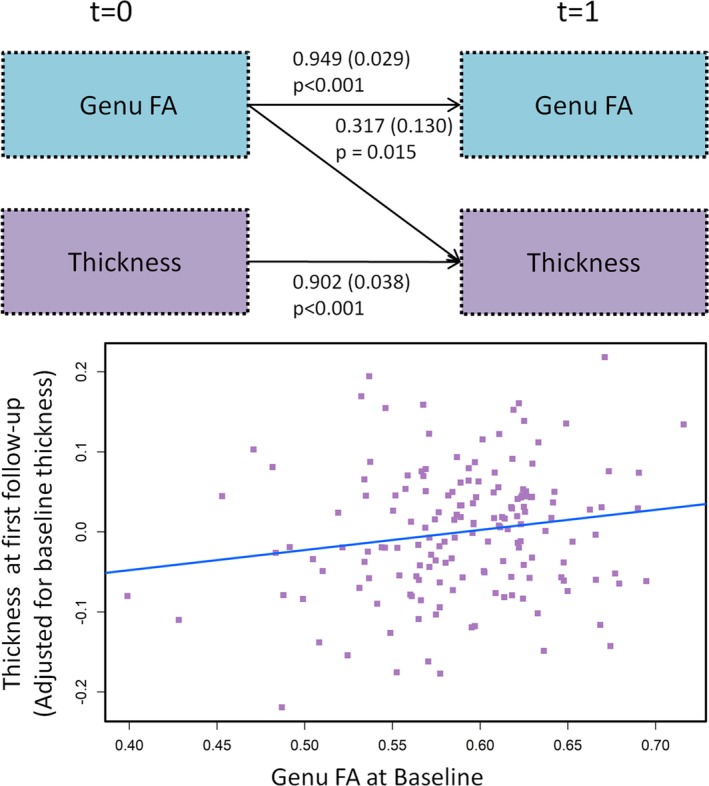
(Top panel) Two‐panel model showing the impact of worsening white matter microstructural changes as measured by genu fractional anisotropy (FA) on future neurodegeneration. The values shown by the arrows indicating significance refer to the estimate, standard error in parentheses, and p value. (Bottom panel) Plot of baseline genu FA versus thickness at first follow‐up after adjusting for baseline thickness.
Latent Growth Curve Model
The final parsimonious model for the latent growth curve SEM fit the data very well, with a root mean square error of approximation of 0.022 (95% confidence interval = 0.015–0.029), standardized root mean square residual of 0.022 (both should be <0.05 for a good fit model), confirmatory fit index of 0.996, and Tucker–Lewis index of 0.995 (both should be >0.95). The Supplementary Table describes all the paths in detail. Please note that the coefficients across all arrows cannot be compared but coefficients on arrows going to a particular outcome are comparable. The coefficients on arrows to a particular outcome are adjusted for each other, as in a multiple linear regression. Figure 2 summarizes the overall model, with arrows indicating the effects that are significant at p < 0.05. Whereas significant arrows originating from age, sex, APOE4 status, and cycle number are shown in gray, the solid black arrows show the impact of the primary predictors.
Figure 2.
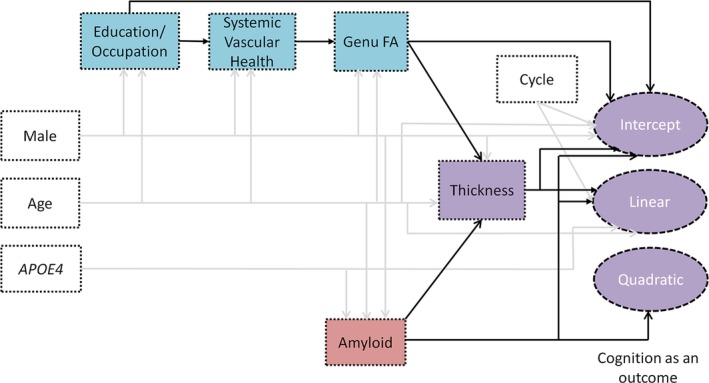
Final model with significant associations at p < 0.05 shown by the arrows. Whereas arrows originating from age, sex, APOE4 status, and cycle number are shown in gray, the solid black arrows show the impact of the primary predictors. The amyloid pathway is shown by the red box, the resilience and vascular pathway are shown by the blue boxes, and the purple boxes show the variables where the pathways converge. FA = fractional anisotropy.
Age, Sex, Education/Occupation, and Vascular Health Influence WM Microstructure
We first discuss the results pertaining to the resilience and vascular pathways that consist mainly of age, sex, education/occupation, and vascular health mechanisms in the growth curve SEM in Figure 3 (also summarized in Table 2). All these associations were significant at the p < 0.001 level. Older age was associated with lower education/occupation (improving education trends over decades), worse CMC, and lower genu FA. Male sex was associated with higher education/occupation, higher CMC, and higher genu FA. Lower education/occupation was a significant predictor of higher CMC. Higher CMC was a significant predictor of lower genu FA. APOE4 status did not have significant effects on the resilience and vascular pathways in this data.
Figure 3.
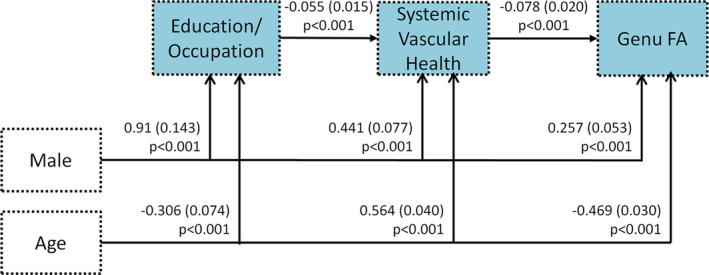
Resilience and vascular health pathways showing the impact of age, sex, education/occupation, and systemic vascular health on white matter microstructure as measured by genu fractional anisotropy (FA). The values shown by the arrows indicating significance refer to the estimate, standard error in parentheses, and p value.
Table 2.
Total, Direct, and Indirect Effects Seen on Components of the Resilience, Vascular, and Amyloid Pathways (Shown in Fig 2)
| Path | Total Effect | Direct Effect | Total Indirect Effect | |||
|---|---|---|---|---|---|---|
| Estimate (SE) | p | Estimate (SE) | p | Estimate (SE) | p | |
| Education/occupation, systemic vascular health, and genu FA as outcomes | ||||||
| Age [decade] → education/occupation | −0.306 (0.074) | <0.001 | −0.306 (0.074) | <0.001 | — | — |
| Male → education/occupation | 0.910 (0.143) | <0.001 | 0.910 (0.143) | <0.001 | — | — |
| Age [decade] → CMC | 0.581 (0.040) | <0.001 | 0.564 (0.040) | <0.001 | 0.017 (0.006) | 0.007 |
| Male → CMC | 0.391 (0.076) | <0.001 | 0.441 (0.077) | <0.001 | −0.050 (0.016) | 0.002 |
| Education/occupation → CMC | −0.055 (0.015) | <0.001 | −0.055 (0.015) | <0.001 | — | — |
| Age [decade] → genu FA | −0.514 (0.028) | <0.001 | −0.469 (0.030) | <0.001 | −0.045 (0.012) | <0.001 |
| Male → genu FA | 0.227 (0.053) | <0.001 | 0.257 (0.053) | <0.001 | −0.030 (0.010) | 0.002 |
| Education/occupation → genu FA | 0.004 (0.002) | 0.008 | — | — | 0.004 (0.002) | 0.008 |
| CMC → genu FA | −0.078 (0.020) | <0.001 | −0.078 (0.020) | <0.001 | — | — |
| Amyloid as an outcome | ||||||
| Age [decade] → log [amyloid] | 0.443 (0.026) | <0.001 | 0.443 (0.026) | <0.001 | — | — |
| Male → log [amyloid] | −0.100 (0.050) | 0.045 | −0.100 (0.050) | 0.045 | — | — |
| APOE4 → log [amyloid] | 0.599 (0.055) | <0.001 | 0.599 (0.055) | <0.001 | — | — |
CMC = cardiovascular and metabolic conditions; FA = fractional anisotropy; SE = standard error.
Predictors of Amyloid Pathway
In the final model (see Fig 2), age and APOE4 status (p < 0.001) and female sex (p = 0.045) were significant predictors of amyloid deposition. Baseline amyloidosis did not have significant associations with education/occupation, CMC, or genu FA (resilience and vascular pathways discussed above).
Convergence of All Pathways on Neurodegeneration and Cognitive Decline
The resilience, vascular, and amyloid pathways converged through their effects on cortical thickness and cognitive decline (see Fig 2) as described here.
With thickness as an outcome, the main predictors were as follows. Older age (p < 0.001) and male sex (p = 0.033) were significant direct predictors of lower thickness (see Fig 2 and Table 3). Genu FA and amyloidosis directly impacted thickness, with the impact of 1 standard deviation (SD) change in genu FA (estimate = 0.041, standard error [SE] = 0.005; p < 0.001) being greater than the impact of 1 SD change in log (amyloid) (estimate = −0.022, SE = 0.004; p < 0.001) on thickness. APOE4 status indirectly impacted thickness through its impact on amyloidosis (p < 0.001). Education/occupation (p = 0.011) and CMC (p < 0.001) indirectly impacted thickness through genu FA.
Table 3.
Total, Direct, and Indirect Effects Seen on Thickness and Cognition
| Path | Total Effect | Direct Effect | Total Indirect Effect | |||
|---|---|---|---|---|---|---|
| Estimate (SE) | p | Estimate (SE) | p | Estimate (SE) | p | |
| Thickness as an outcome | ||||||
| Age [decade] → thickness | −0.089 (0.004) | <0.001 | −0.058 (0.005) | <0.001 | −0.031 (0.003) | <0.001 |
| Male → thickness | −0.017 (0.008) | 0.033 | −0.029 (0.008) | <0.001 | 0.011 (0.003) | <0.001 |
| APOE4 → thickness | −0.013 (0.003) | <0.001 | — | — | −0.013 (0.003) | <0.001 |
| Log [amyloid] → thickness | −0.022 (0.004) | <0.001 | −0.022 (0.004) | <0.001 | — | — |
| Education/occupation → thickness | <0.001 (<0.001) | 0.011 | — | — | <0.001 (<0.001) | 0.011 |
| Genu FA → thickness | 0.041 (0.005) | <0.001 | 0.041 (0.005) | <0.001 | — | — |
| CMC → thickness | −0.003 (0.001) | <0.001 | — | — | −0.003 (0.001) | <0.001 |
| Cognition as an outcome | ||||||
| Age [decade] → intercept | −0.630 (0.032) | <0.001 | −0.400 (0.034) | <0.001 | −0.230 (0.024) | <0.001 |
| Male → intercept | −0.191 (0.051) | <0.001 | −0.347 (0.047) | <0.001 | 0.155 (0.026) | <0.001 |
| APOE4 → intercept | −0.086 (0.017) | <0.001 | — | — | −0.086 (0.017) | <0.001 |
| Log [amyloid] → intercept | −0.143 (0.025) | <0.001 | −0.127 (0.025) | <0.001 | −0.016 (0.005) | 0.001 |
| Cycle number → intercept | 0.075 (0.015) | <0.001 | 0.075 (0.015) | <0.001 | — | — |
| Education/occupation → intercept | 0.139 (0.009) | <0.001 | 0.138 (0.009) | <0.001 | 0.001 (<0.001) | 0.016 |
| Genu FA → intercept | 0.160 (0.028) | <0.001 | 0.130 (0.029) | <0.001 | 0.030 (0.008) | <0.001 |
| CMC → intercept | −0.012 (0.004) | 0.001 | — | — | −0.012 (0.004) | 0.001 |
| Thickness → intercept | 0.726 (0.171) | <0.001 | 0.726 (0.171) | <0.001 | — | — |
| Age [decade] → linear | −0.064 (0.006) | <0.001 | −0.030 (0.007) | <0.001 | −0.033 (0.005) | <0.001 |
| Male → linear | −0.001 (0.003) | 0.640 | — | — | −0.001 (0.003) | 0.640 |
| APOE4 → linear | −0.039 (0.010) | <0.001 | −0.019 (0.010) | 0.049 | −0.020 (0.005) | <0.001 |
| Log [amyloid] → linear | −0.033 (0.009) | <0.001 | −0.028 (0.009) | 0.001 | −0.005 (0.001) | <0.001 |
| Cycle number → linear | −0.015 (0.003) | <0.001 | −0.015 (0.003) | <0.001 | — | — |
| Education/occupation → linear | <0.001 (<0.001) | 0.016 | — | — | <0.001 (<0.001) | 0.016 |
| Genu FA → linear | 0.010 (0.002) | <0.001 | — | — | 0.010 (0.002) | <0.001 |
| CMC → linear | −0.001 (<0.001) | 0.001 | — | — | −0.001 (<0.001) | 0.001 |
| Thickness → linear | 0.237 (0.030) | <0.001 | 0.237 (0.030) | <0.001 | — | — |
| Age [decade] → quadratic | −0.002 (0.001) | 0.018 | — | — | −0.002 (0.001) | 0.018 |
| Male → quadratic | <0.001 (<0.001) | 0.125 | — | — | <0.001 (<0.001) | 0.125 |
| APOE4 → quadratic | −0.003 (0.001) | 0.020 | — | — | −0.003 (0.001) | 0.020 |
| Log [amyloid] → quadratic | −0.004 (0.002) | 0.017 | −0.004 (0.002) | 0.017 | — | — |
CMC = cardiovascular and metabolic conditions; FA = fractional anisotropy; SE = standard error.
With cognition as an outcome, the main predictors were as follows. For intercept, older age, male sex, higher amyloid, lower cycle number (which reflects practice effects), lower education/occupation, lower genu FA, and lower thickness were direct predictors of lower baseline cognition (p < 0.001). Age (in decades; total effect estimate = −0.63, SE = 0.032) and thickness (total effect estimate = 0.726, SE = 0.171) had the largest effects given the scales used. For the linear component, older age (direct estimate = −0.030, SE = 0.007; p < 0.001), APOE4 status (direct estimate = −0.019, SE = 0.010; p = 0.049), higher amyloid (direct estimate = −0.028, SE = 0.009; p = 0.001), higher cycle number (direct estimate = −0.015, SE = 0.003; p < 0.001), and lower thickness (direct estimate = −0.237, SE = 0.030; p < 0.001) were significant direct predictors of faster linear decline. Lower thickness (total estimate = −0.237, SE = 0.030; p < 0.001) had the largest effect given the scales used. For the quadratic component, higher amyloid was the only direct predictor of the quadratic component of cognition (p = 0.017), with age and APOE4 having significant indirect effects on the quadratic component of cognition through amyloid.
Baseline Amyloidosis Associated with Accelerated Decline in Cognition with Longer Follow‐up
The primary predictors from the resilience and vascular pathways were associated with baseline cognition directly and also associated indirectly with the linear component of cognitive decline through cortical thickness. In contrast, amyloidosis had both direct associations with all components of cognitive decline (intercept, linear, and quadratic) and an indirect impact on cognitive decline through thickness (intercept and linear components). This stronger overall influence of amyloidosis can be observed in Figure 4, which illustrates the impact of baseline amyloidosis in a 75‐year‐old male with APOE4 carrier status in our study, and with median education/occupation score of 12.53. The low and high levels of amyloid burden (low = 1.30 SUVR and high = 1.71 SUVR) and genu FA (low = 0.55 and high = 0.63) were defined by the 20th and 80th percentiles. With shorter follow‐up times, the effects of genu FA and amyloidosis were similar and linear in nature, but the quadratic effect of amyloidosis causes nonlinearity (acceleration) of the trajectories with longer follow‐up times.
Figure 4.
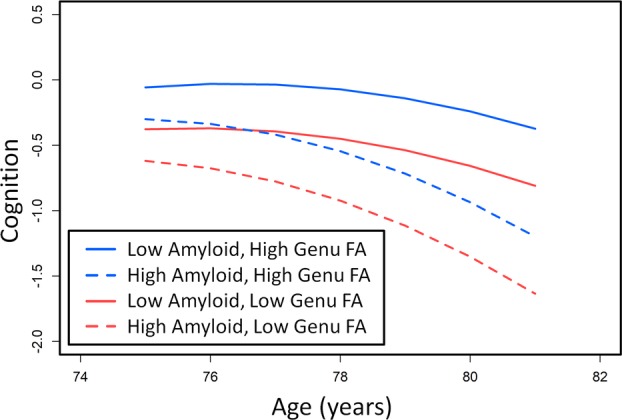
The cognition trajectories for a 75‐year‐old male APOE4 carrier with mean education/occupation at high and low levels of genu fractional anisotropy (FA) and amyloid deposition defined by 20th and 80th percentiles.
Complex and Dynamic Process of Cognitive Aging
As individuals age, we would expect the values for amyloid, genu FA, and cognition to change. A cross‐sectional sample of the MCSA should reveal different values for these variables in different age decades reflecting the complex processes. At ages 55, 65, 75, and 85 years, we selected male or female participants who were an APOE4 carrier or noncarrier at the 20th (low) versus 80th (high) percentiles of their age group depending on their characteristics, as shown in the table in Figure 5. Then we compared the predicted cognitive trajectories of the best profile (high education/occupation, low amyloid, high genu FA), shown by the blue curves, with the worst profile (low education/occupation, high amyloid, low genu FA), shown by the red curves, for that specific age decade. These comparisons as shown in Figure 5 illustrate the complex process of cognitive aging. There is also noted nonlinearity due to practice effects in the 50–60 individuals. This effect is attenuated in older age individuals, because they have significantly greater neurodegeneration, which contributes to lesser extent of practice effects.27
Figure 5.
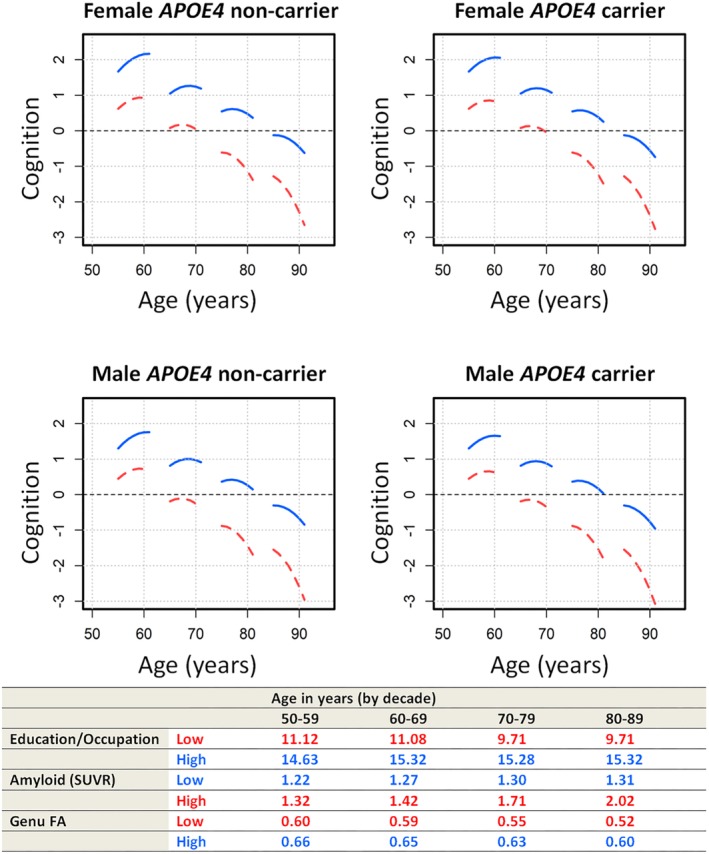
Complex process of cognitive aging. Red curves show worse values for all predictors (low education/occupation, high amyloid burden, low genu fractional anisotropy [FA]), and blue curves show the better values for all predictors (high education/occupation, low amyloid burden, high genu FA). Plots are shown for males versus females as well as APOE4 carriers versus APOE4 noncarriers. SUVR = standardized uptake value ratio. [Correction added on 25 October 2019, after first online publication: There was an error in the table portion at the bottom of Figure 5. The age range in the 3rd column has been changed from 60‐60 to 60‐69.]
Discussion
Cognitive aging is a multifactorial process. Individual variability in longitudinal cognitive trajectories can be approached through development of integrated models that consider key mechanisms and pathways that influence cognitive aging. The question of how these key pathways in the population—amyloid, resilience, and vascular—influence cognitive aging is critical for comprehending this complex process. Using latent growth curve SEMs, we observed some causal pathways and mapped the course of the cognitive trajectories with age as a function of these pathways. Laying down the theoretical framework for the dynamic and complex process of cognitive aging is important for the development of successful interventions.
Resilience and Vascular Pathways: Age, Sex, Intellectual Enrichment, Systemic Vascular Health, and WM Degradation
A primary mechanism through which resilience can help delay the onset of cognitive impairment is higher brain reserve or greater neurobiological capital,11, 28 which could be due to developmental differences, less age‐related damage, sex differences, higher education, or better cerebrovascular health. Here, we found that better WM microstructural integrity (greater brain reserve) was associated with being younger, male sex, higher education/occupation, and better systemic vascular health. Thus, both elements of the construct of resilience and of cerebrovascular disease were relevant to WM structural integrity.
As expected, age was associated with systemic brain health decline, all brain changes, and cognitive decline. Residual age effects in our model probably reflect mechanisms that are not captured by the ones we included. We also observed a significant impact of sex on the primary predictors. Sex differences in education/occupation are attributable to distinct gender roles in the past.29 Sex differences in systemic vascular health have been well documented, with greater vascular risk factors in males, especially before 80 years of age.30 WM integrity using FA is greater in males.31, 32 The significant sexual dimorphism in brain structure and organization has been suggested to be related to certain tasks33 and explains the sex differences in brain reserve and cognitive performance we observed between males and females. APOE4 status had minimal impact on the vascular and resilience pathways in these data. This is consistent with the finding that APOE4 impacts cognition primarily through an effect on AD pathophysiology and not cerebrovascular pathology.34, 35
An important result in the context of education/occupation was the association between higher education/occupation and greater genu FA that was explained through better systemic vascular health. Our results highlight that the resilience pathway from education/occupation to genu FA through systemic vascular health and the impact of genu FA on cognition directly and indirectly through cortical thickness contribute to greater “brain reserve” and thus explain reduced risk of dementia through higher education/occupation. In contrast to the brain reserve mechanism, the pathway from education/occupation that goes directly to cognition can be termed as the cognitive reserve or resilience mechanism,9, 28 because here education/occupation as a proxy explains why individuals at the different levels of amyloid pathology could have the same level of cognition due to differences in education/occupation scores (which is not explained by differences in brain structure). Surprisingly, there were no direct effects of education/occupation on the cortical thickness measure, because thickness may not be a good proxy for synaptic health, unlike fluorodeoxyglucose PET (where the literature has been more consistent).
This is the first study to our knowledge that establishes that resilience and vascular pathways drive degradation of WM microstructure, which then contributes to greater shrinkage of GM regions implicated in AD36 and aging,37 as demonstrated by the 2‐panel SEM. Figure 1 helps establish the crux of this argument using longitudinal data. This plays an important role in understanding the mechanisms through which resilience and vascular pathways influence cognitive decline. Although the disconnection of the brain due to WM degradation contributing to cognition has been discussed in the literature,38 the study of its impact through brain shrinkage clarifies the mechanisms through which brain changes influence cognitive trajectories.
Although there are suggested hypotheses regarding pathways39 (eg, blood–brain barrier dysfunction, cerebral blood flow changes) through which systemic vascular health may be causal to amyloid deposition, the pathology and imaging literature provides evidence for cerebrovascular injury in the absence of amyloid and tau accumulation and the impact of certain risk factors (eg, diabetes) on cerebrovascular injury but not on amyloid accumulation.40, 41 These ideas support the hypothesis that there exists a substantial amyloid independent effect of vascular health on brain and cognitive aging that we observed in the context of cognitive aging.
Amyloidosis and Cognitive Decline
Age and APOE4 have been repeatedly shown to be the chief risk factors for amyloidosis.42 Sex differences in amyloid deposition have been inconsistent in the literature. Here, we found that females had slightly greater amyloid (p = 0.045) in comparison to males, which is consistent with the borderline findings in a large postmortem study.43 Our group and others have shown that amyloid burden at baseline was associated with greater rate of cognitive decline and increased risk of dementia.2, 3, 44, 45, 46, 47
The growth curve modeling approach allowed us to observe 2 phenomena with respect to the amyloid pathway and cognitive decline. First, the impact of amyloid was significant on the quadratic component of decline, suggesting that baseline amyloid burden predicted accelerated decline (see Fig 4). A longer follow‐up allowed us to observe the accelerated worsening of cognition as a function of baseline amyloidosis. This emphasizes that amyloidosis is an early event in the cascade of AD events and that associations of amyloid with cognition become increasingly more pronounced with time. Second, in line with our previous work,48 we found that amyloid elevations are associated with cognitive decline and the association of amyloid with cognition was not completely mediated through the cortical thickness measure. Other work of ours49 suggests that even if tau PET were in our model, amyloidosis would still show an association with cognition. All these results suggest that (1) amyloid may be a proxy measure for other neurodegenerative processes not captured by thickness or by tau that are proximate to cognition; or (2) cortical thickness is an imperfect proxy measure for the neurodegenerative substrate of cognition, which is loss of function in synaptic connections. It is also possible that amyloid PET is likewise an imperfect measure of the toxic substrate of brain amyloidosis. Furthermore, we observed that the impact of amyloidosis at a given age was significantly worse, especially on the rate of cognitive decline (see Fig 4).
Convergence of Pathways
AD and cerebrovascular disease, the two main pathological changes, have differential impact on brain health early in the disease process. AD (plaques and tangles) has primarily been shown to trigger tau‐mediated neuronal death, altering GM structure. In contrast, cerebrovascular disease, specifically small vessel disease, has a predilection for WM. In this work, we found that the resilience, cerebrovascular, and AD pathways all converged on cortical thickness and cognitive decline. The amyloid pathway primarily acts through neuronal loss or GM degradation, whereas the resilience and vascular pathways measured through age, education/occupation, and systemic vascular health primarily act through WM degradation, which then causally impacts GM structural changes.
The clarification of the mechanisms and their convergence on brain aging and longitudinal cognitive trajectories has broad implications. First, it will contribute to improved designs of prevention trials for cognitive impairment. Figure 5 illustrates that multidomain trials targeting multiple important factors that are causal to brain and cognitive aging are needed to be able to age well along the blue curves versus the red curves that represents poor disease and health predictors. Second, cognitive aging is driven by aging at one end, and at the other end we observe a net sum of the effects of all mechanistic pathways that converge on downstream neurodegeneration and cognition. Therefore, the associations we observe between age, cortical thickness, or cognition and the first pathway as well as age, cortical thickness, or cognition and the second pathway might manifest as artificial associations between the two pathways. Caution is warranted about interpreting interactions between pathways due to sample characteristics. Finally, having a direct biological measure of age‐specific brain health and cerebrovascular health through WM integrity is helpful.
Strengths and Limitations
A key strength is the availability of the rich data resource of MCSA, with key measures of health and disease for studying cognitive aging. We also integrated series of tools that our group has developed over the years. A major weakness is that we took a modeling approach that considered only a selected number of variables that were global in nature. However, the global approach helped us simplify and model a complex problem. Another weakness is that important brain pathologies affecting cognition exist in the aging population but for which no biomarkers exist at this time, including alpha synuclein, TDP43, and microinfarctions. Thus, the significant impact of age seen on both cognitive decline and neurodegeneration that was not mediated through the resilience, vascular, and amyloid pathways could be due to the unmeasured causes of brain and cognitive aging. Ideally, we would prefer 3 or more waves for the longitudinal panel data analysis. Unfortunately, the length of follow‐up available at this time restricted the analysis to 2 waves with an adequate sample size. The longitudinal panel analysis could also be subject to regression to the mean, although we did not see evidence of this in plots for genu FA or thickness.
Author Contributions
P.V. and T.G.L. conceived and designed the study. All authors participated in data collection and analysis. P.V., T.G.L., and S.A.P. drafted the manuscript and figures.
Potential Conflicts of Interest
Nothing to report.
Supporting information
Supplemental Table: Complete results of the regressions of the final SEM model.
Acknowledgment
This work was supported by National Institute of Neurological Disorders and Stroke (NINDS) and National Institute on Aging (NIA) grants U01 AG006786 (principal investigator [PI]: R.C.P.), R01 NS097495 (PI: P.V.), R01 AG056366 (PI: P.V.), P50 AG016574/P1 (PI: P.V.), P50 AG016574 (PI: R.C.P.), R37 AG011378 (PI: C.R.J.), R01 AG041851 (PIs: C.R.J. and D.S.K.), and R01 AG034676 (REP PI: W. A. Rocca), the Gerald and Henrietta Rauenhorst Foundation, the Millis Family, the Alexander Family Alzheimer's Disease Research Professorship of the Mayo Foundation, the Alzheimer's Association (Zenith Fellows Award), the Liston Award, the Elsie and Marvin Dekelboum Family Foundation, the Schuler Foundation, and Opus building NIH grant C06 RR018898.
We thank A. L. Reddy and D. A. Reyes for their help with the figures; and all the study participants and staff of the MCSA, Mayo Alzheimer's Disease Research Center, and Aging Dementia Imaging Research Laboratory at the Mayo Clinic for making this study possible.
References
- 1. Boyle PA, Yu L, Leurgans SE, et al. Attributable risk of Alzheimer's dementia attributed to age‐related neuropathologies. Ann Neurol 2019;85:114–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baker JE, Lim YY, Pietrzak RH, et al. Cognitive impairment and decline in cognitively normal older adults with high amyloid‐β: a meta‐analysis. Alzheimers Dement (Amst) 2017;6:108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hedden T, Oh H, Younger AP, Patel TA. Meta‐analysis of amyloid‐cognition relations in cognitively normal older adults. Neurology 2013;80:1341–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Croall ID, Lohner V, Moynihan B, et al. Using DTI to assess white matter microstructure in cerebral small vessel disease (SVD) in multicentre studies. Clin Sci (Lond) 2017;131:1361–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vemuri P, Lesnick TG, Przybelski SA, et al. Development of a cerebrovascular magnetic resonance imaging biomarker for cognitive aging. Ann Neurol 2018;84:705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gunning‐Dixon FM, Brickman AM, Cheng JC, Alexopoulos GS. Aging of cerebral white matter: a review of MRI findings. Int J Geriatr Psychiatry 2009;24:109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Salat DH, Tuch DS, Hevelone ND, et al. Age‐related changes in prefrontal white matter measured by diffusion tensor imaging. Ann N Y Acad Sci 2005;1064:37–49. [DOI] [PubMed] [Google Scholar]
- 8. Brickman AM, Meier IB, Korgaonkar MS, et al. Testing the white matter retrogenesis hypothesis of cognitive aging. Neurobiol Aging 2012;33:1699–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arenaza‐Urquijo EM, Vemuri P. Resistance vs resilience to Alzheimer disease: clarifying terminology for preclinical studies. Neurology 2018;90:695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roe CM, Xiong C, Miller JP, Morris JC. Education and Alzheimer disease without dementia: support for the cognitive reserve hypothesis. Neurology 2007;68:223–228. [DOI] [PubMed] [Google Scholar]
- 11. Katzman R, Terry R, DeTeresa R, et al. Clinical, pathological, and neurochemical changes in dementia: a subgroup with preserved mental status and numerous neocortical plaques. Ann Neurol 1988;23:138–144. [DOI] [PubMed] [Google Scholar]
- 12. Vemuri P, Lesnick TG, Przybelski SA, et al. Vascular and amyloid pathologies are independent predictors of cognitive decline in normal elderly. Brain 2015;138(pt 3):761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vemuri P, Lesnick TG, Przybelski SA, et al. Association of lifetime intellectual enrichment with cognitive decline in the older population. JAMA Neurol 2014;71:1017–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Curran PJ, Obeidat K, Losardo D. Twelve frequently asked questions about growth curve modeling. J Cogn Dev 2010;11:121–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rocca WA, Yawn BP, St Sauver JL, et al. History of the Rochester Epidemiology Project: half a century of medical records linkage in a US population. Mayo Clinic Proc 2012;87:1202–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. St Sauver JL, Grossardt BR, Yawn BP, et al. Data resource profile: the Rochester Epidemiology Project (REP) medical records‐linkage system. Int J Epidemiol 2012;41:1614–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roberts RO, Knopman DS, Syrjanen JA, et al. Weighting and standardization of frequencies to determine prevalence of AD imaging biomarkers. Neurology 2017;89:2039–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Petersen RC, Roberts RO, Knopman DS, et al. Prevalence of mild cognitive impairment is higher in men. The Mayo Clinic Study of Aging. Neurology 2010;75:889–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roberts RO, Geda YE, Knopman DS, et al. The Mayo Clinic Study of Aging: design and sampling, participation, baseline measures and sample characteristics. Neuroepidemiology 2008;30:58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vemuri P, Lesnick TG, Przybelski SA, et al. Effect of lifestyle activities on Alzheimer disease biomarkers and cognition. Ann Neurol 2012;72:730–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rocca WA, Gazzuola‐Rocca L, Smith CY, et al. Accelerated accumulation of multimorbidity after bilateral oophorectomy: a population‐based cohort study. Mayo Clin Proc 2016;91:1577–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vemuri P, Lesnick TG, Przybelski SA, et al. Age, vascular health, and Alzheimer disease biomarkers in an elderly sample. Ann Neurol 2017;82:706–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jack CR Jr, Wiste HJ, Weigand SD, et al. Defining imaging biomarker cut points for brain aging and Alzheimer's disease. Alzheimers Dement 2017;13:205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jack CR Jr, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 2013;12:207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Muthén LK, Muthén BO. Mplus user's guide. Los Angeles, CA: Muthén & Muthén, 1998. –2017. [Google Scholar]
- 26. Duncan OD. Some linear models for two‐wave, two‐variable panel analysis. Psychol Bull 1969;72:177–182. [Google Scholar]
- 27. Machulda MM, Hagen CE, Wiste HJ, et al. Practice effects and longitudinal cognitive change in clinically normal older adults differ by Alzheimer imaging biomarker status. Clin Neuropsychol 2017;31:99–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stern Y, Arenaza‐Urquijo EM, Bartres‐Faz D, et al. Whitepaper: Defining and investigating cognitive reserve, brain reserve, and brain maintenance. Alzheimers Dement 10.1016/j.jalz.2018.07.219. [Epub ahead of print] Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mielke MM, Vemuri P, Rocca WA. Clinical epidemiology of Alzheimer's disease: assessing sex and gender differences. Clin Epidemiol 2014;6:37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mosca L, Barrett‐Connor E, Wenger NK. Sex/gender differences in cardiovascular disease prevention: what a difference a decade makes. Circulation 2011;124:2145–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Westerhausen R, Walter C, Kreuder F, et al. The influence of handedness and gender on the microstructure of the human corpus callosum: a diffusion‐tensor magnetic resonance imaging study. Neurosci Lett 2003;351:99–102. [DOI] [PubMed] [Google Scholar]
- 32. Liu F, Vidarsson L, Winter JD, et al. Sex differences in the human corpus callosum microstructure: a combined T2 myelin‐water and diffusion tensor magnetic resonance imaging study. Brain Res 2010;1343:37–45. [DOI] [PubMed] [Google Scholar]
- 33. Burgaleta M, Head K, Álvarez‐Linera J, et al. Sex differences in brain volume are related to specific skills, not to general intelligence. Intelligence 2012;40:60–68. [Google Scholar]
- 34. Mortimer JA, Snowdon DA, Markesbery WR. The effect of APOE‐epsilon4 on dementia is mediated by Alzheimer neuropathology. Alzheimer Dis Assoc Disord 2009;23:152–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim HJ, Ye BS, Yoon CW, et al. Effects of APOE epsilon4 on brain amyloid, lacunar infarcts, and white matter lesions: a study among patients with subcortical vascular cognitive impairment. Neurobiol Aging 2013;34:2482–2487. [DOI] [PubMed] [Google Scholar]
- 36. Schwarz CG, Gunter JL, Wiste HJ, et al. A large‐scale comparison of cortical thickness and volume methods for measuring Alzheimer's disease severity. Neuroimage Clin 2016;11:802–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fjell AM, McEvoy L, Holland D, et al. What is normal in normal aging? Effects of aging, amyloid and Alzheimer's disease on the cerebral cortex and the hippocampus. Prog Neurobiol 2014;117:20–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bennett IJ, Madden DJ. Disconnected aging: cerebral white matter integrity and age‐related differences in cognition. Neuroscience 2014;276:187–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sweeney MD, Kisler K, Montagne A, et al. The role of brain vasculature in neurodegenerative disorders. Nat Neurosci 2018;21:1318–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chui HC, Zheng L, Reed BR, et al. Vascular risk factors and Alzheimer's disease: are these risk factors for plaques and tangles or for concomitant vascular pathology that increases the likelihood of dementia? An evidence‐based review. Alzheimers Res Ther 2012;4:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Abner EL, Nelson PT, Kryscio RJ, et al. Diabetes is associated with cerebrovascular but not Alzheimer's disease neuropathology. Alzheimers Dement 2016;12:882–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jansen WJ, Ossenkoppele R, Knol DL, et al. Prevalence of cerebral amyloid pathology in persons without dementia: a meta‐analysis. JAMA 2015;313:1924–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Oveisgharan S, Arvanitakis Z, Yu L, et al. Sex differences in Alzheimer's disease and common neuropathologies of aging. Acta Neuropathol 2018;136:887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Petersen RC, Wiste HJ, Weigand SD, et al. Association of elevated amyloid levels with cognition and biomarkers in cognitively normal people from the community. JAMA Neurol 2016;73:85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vos SJ, Xiong C, Visser PJ, et al. Preclinical Alzheimer's disease and its outcome: a longitudinal cohort study. Lancet Neurol 2013;12:957–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Villemagne VL, Pike KE, Chetelat G, et al. Longitudinal assessment of Aβ and cognition in aging and Alzheimer disease. Ann Neurol 2011;69:181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Petersen RC, Lundt ES, Therneau TM, et al. Predicting progression to mild cognitive impairment. Ann Neurol 2019;85:155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Knopman DS, Lundt ES, Therneau TM, et al. Joint associations of beta‐amyloidosis and cortical thickness with cognition. Neurobiol Aging 2018;65:121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Knopman DS, Lundt ES, Therneau TM, et al. Entorhinal cortex tau, amyloid‐beta, cortical thickness and memory performance in non‐demented subjects. Brain 2019;142:1148–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table: Complete results of the regressions of the final SEM model.