

Abstract
Colorectal cancer is the third largest cancer in worldwide and has been proven to be closely related to the intestinal microbiota. Many reports and clinical studies have shown that intestinal microbial behavior may lead to pathological changes in the host intestines. The changes can be divided into epigenetic changes and carcinogenic changes at the gene level, which ultimately promote the production and development of colorectal cancer. This article reviews the pathways of microbial signaling in the intestinal epithelial barrier, the role of microbiota in inflammatory colorectal tumors, and typical microbial carcinogenesis. Finally, by gaining a deeper understanding of the intestinal microbiota, we hope to achieve the goal of treating colorectal cancer using current microbiota technologies, such as fecal microbiological transplantation.
Keywords: microbiota, inflammation, carcinogenesis, colorectal cancer
Abbreviations
- AP‐1
activating protein 1
- AMPs
antimicrobial peptides
- BFT
B. fragilis toxin
- CDI
C. difficile infection
- CDT
cell death toxin
- CIN
chromosome instability
- CLR
C‐lectin‐like receptor
- CECs
colonic epithelial cells
- CRC
colorectal cancer
- CIMP
CpG island methylator phenotype
- CD
Crohn's disease
- COX‐2
cyclooxygenase‐2
- CDT
cytolethal distending toxin
- CNF
cytotoxic necrotizing factor
- DAMPs
damage‐associated molecular patterns
- DCs
dendritic cells
- ETBF
enterotoxigenic Bacteroides fragilis
- FMT
fecal microorganism transplantation
- GC
gastric cancer
- IBD
inflammatory bowel disease
- IRF
interferon regulatory factor
- IRAK1
IL‐1R‐associated kinases 1
- IFN
interferon
- IL
interleukin
- IEC
intestinal epithelial cell
- LRRs
leucine‐rich repeats
- MAMPs
microbe‐associated molecular patterns
- MDP
muramyl dipeptide
- MSI
microsatellite instability
- MMR
mismatch repair
- MAPK
mitogen‐activated protein kinase
- MyD88
myeloid differentiation factor 88
- NF‐κB
NF‐kappaB
- NLR
nod‐like receptor
- NTBF
nonenterotoxigenic B. fragilis
- PAMPs
pathogen‐associated molecular patterns
- PRR
pattern recognition receptor
- PKS
polyketone acid synthetase
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- RELMβ
resistin‐like moleculesβ
- RLR
Rig‐I‐like receptor
- sIgA
secretory immunoglobulin A
- SMO
spermine oxidase
- TAK
transforming growth factor kinase
- Th17
T helper cell 17
- TJs
tight junctions
- TLR
toll‐like receptor
- TRAF
TNF receptor‐associated factor
- TFFs
trefoil factor peptides
- TNF
tumor necrosis factor
- IFN‐β
type I IFN
- UC
ulcerative colitis
Introduction
As the world's third‐largest cancer, colorectal cancer (CRC) retains a high morbidity and mortality in our country, which imposes a severe burden on the health system and on patients.1 There are hundreds of kinds of microorganisms in human intestines, forming a symbiotic system with intestinal cells to maintain the intestinal environment. The typical bacteria include Escherichia coli, Enterococcus faecalis, and Bacteroides fragilis.2 Microbes have their own pathogenicity and carcinogenicity. For example, a class of toxins released from E. coli is known as cell death toxins (CDTs). They act directly on the intestinal epithelial cells and cause a highly proliferative epithelium in normal intestinal epithelial cells. The proliferating intestinal epithelial cells form an adenoma and continue to invade the submucosa of the intestinal mucosa, eventually leading to cancerous changes. Enterococcus faecalis destroys DNA via free radicals, such as active oxygen and active nitrogen.3 Some carcinogenic mechanisms may involve a variety of different signaling pathways. Their interactions, promotions, substitutions, and combinations result in intestinal microorganism induced CRC. The role of these mechanisms is inseparable from the subsequent inflammatory response, which eventually leads to CRC.4 This review provides an overview of the intestinal epithelial barrier structure and microbial signal transduction pathways, the role of microbes in inflammation‐induced colorectal neoplasms, and a detailed review of typical microbial carcinogenesis. Finally, we expect colorectal cancer may be treated using certain current technologies for the typical microbiota, such as fecal microbiological transplantation.
The Importance of the Intestinal Epithelial Barrier and Microbial Signals in the Regulation of Intestinal Homeostasis
Intestinal epithelial cells (IECs) are continuous physical barriers formed by single cells that separate the intestinal flora from the deeper intestinal tissue. Epithelial cells are networked together by tight junctions (TJs) and provide a paracellular seal (Fig. 1). This not only blocks the paracellular space, indicating ion flux between tissues, but also maintains cell polarity.5 Intestinal mucus is the first barrier between the intestinal tract and mucous tissue. It is mainly comprises a large amount of modified glycoprotein mucus.6 Jakobsson et al.7 found that the filtration function of colonic mucus depends on the microbial community. The intestinal microbiota are also active participants in maintaining intestinal homeostasis. Lipopolysaccharide acts as an endotoxin, and microbiota containing lipopolysaccharide interfere with the function of the epithelial barrier, leading to chronic inflammation and CRC. However, the intestinal microbiota can regulate the renewal and reorganization of TJs of intestinal epithelial cells, thereby enhancing the barrier function.8 The mucus layer of the intestinal epithelium is a sterile environment containing some biomolecules, such as secretory immunoglobulin A (sIgA), antimicrobial peptides (AMPs),9 microbe‐associated molecular patterns (MAMPs), trefoil factor peptides (TFFs), resistin‐like molecules β (RELMβ), and Fc‐γ binding proteins.10 A study using aseptic mice showed that the thickness of the mucus layer was reduced compared to the rodents with an intact microbiota.11 On the basis of IECs, there are also plasma cells, macrophages, and dendritic cells (DCs). These cells have a simple nature and limited inflammatory cytokine expression in the healthy state.12 However, in inflammatory bowel disease (IBD), the number of these immune cells will increase.13 Meanwhile, the expression of endothelial cell adhesion molecules in IBD also increases.14
Figure 1.
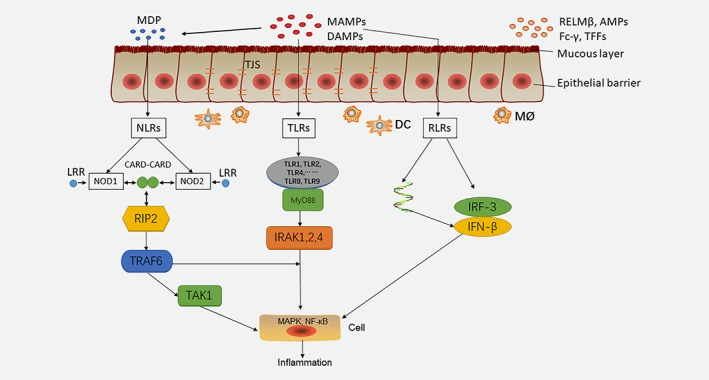
A schematic diagram of the signaling pathways of microorganisms in the intestinal epithelium. Among them, NLR, TLR and RLR family members provide significant microbial signaling pathways in the intestinal epithelium. DAMPs and MAMPs on the epithelial barrier activate signaling pathways through different receptors. MAMPs activate muramyl dipeptide (MDP) and recognize NLRs. NOD1 and NOD2 are active in intestinal cells and can recognize caspase recruitment domains (card‐card). NOD1 and NOD2 interact with RIP2, stimulates TRAF6 and recognizes TAK1, triggers MAPK and NF‐κB. TLRs include TLR1, TLR2, and TLR4, which bind to MyD88 and activate NF‐κB by binding to IRAK1, 2, and 4. RLRs are capable of recognizing viral RNA, releasing IFN‐β, and activating NF‐κB. If the signal pathways are disrupted, inflammation may occur, leading to cancerous lesions.
Innate receptors, such as pathogen‐associated molecular patterns (PAMPs) and damage‐associated molecular patterns (DAMPs), play an important role in the innate immune response and are able to identify molecular patterns. These molecular patterns include Nod‐like receptors (NLRs), C‐lectin‐like receptors (CLRs), Rig‐I‐like receptors (RLRs), and toll‐like receptors (TLRs).15 Although mice with pattern recognition receptor (PRR) and signal transduction defects have been modeled, skin‐specific PRR knockout mice are still needed to demonstrate the role of bacterially derived signals in intestinal homeostasis.16 Over the last few years, we have witnessed a significant expansion in the number of reports associated with the contribution of the NLR family members to IBD pathobiology.17
NLRs are cytoplasmic receptors and are highly conserved throughout evolution, attesting to their important role in host defense.18 MAMPs activate muramyl dipeptide (MDP) and recognize NLRs. Among them, nucleotide binding oligomerization domain containing (NOD)1 and NOD2 are active in intestinal cells and can recognize caspase recruitment domains (card‐card).19 Under the stimulation of leucine‐rich repeats (LRRs), which are involved in bacterial‐sensing during pathogenesis, NOD1 and NOD2 interact with the receptor‐interacting protein 2 (RIP2), which stimulates tumor necrosis factor (TNF) receptor‐associated factor 6 (TRAF6) and recognizes transforming growth factor kinase 1(TAK1), triggering mitogen‐activated protein kinase (MAPK) and NF‐kappa B (NF‐κB) signaling.20 By contrast, there has been little research on CLRs.
The RLR signaling pathway induces the phosphorylation and homodimerization of interferon regulatory factor 3 (IRF3) and upregulates the transcription of type I IFN (IFN‐β).21 Meanwhile, RLRs can be used for virus detection. RNA viral infection plays a key role in the early production and subsequent expression of IFN‐β, which induces an inflammatory response, thereby inhibiting viral replication.22 Therefore, IFN‐β identifies the virus RNA and induces the congenital antiviral response.23 Remarkably, like other innate immune pathways, upon stimulation, RLR signaling is markedly amplified by multiple feed‐forward loops in self‐regulatory or autocrine/paracrine ways.24 TLRs are usually sensitive to microbial components, DNA, and RNA fragments.25 TLRs can be located on the cell surface or in the intracellular compartment, with the specific ligand completing the feedback and associated with a specific adaptor that activates the cascade of downstream signals.26
TLRs include TLR1, TLR2, and TLR4, and bind to myeloid differentiation factor 88 (MyD88) and activate NF‐κB by binding to IL‐1R‐associated kinases 1, 2, and 4 (IRAK1, 2, and 4).27 TRAF6 mediates the activation of NF‐κB induced by MyD88 and IRAK.28 TLRs are strongly expressed in human rectal adenocarcinoma cells, especially TLR2 and TLR4.29 In addition to TLR3, MyD8830 signaling is usually triggered by adapters, which initiates a signal cascade, which eventually activates transcription factors, such as NF‐κB, IRF,31 and activating protein 1 (AP‐1).32
Inflammatory Cytokine‐Mediated Signaling Pathway Leading to Colorectal Cancer
The occurrence of malignant tumors is inseparable from chronic inflammation. Accumulating evidence confirms that the collapse of the symbiotic relationship is important in the pathogenesis of IBD.33 It is generally believed that chronic colon inflammation from ulcerative colitis (UC) or Crohn's disease (CD) may increase the risk of colon cancer.34 In the past few decades, the incidence of UC in western countries has been very high. However, recent studies have found that the incidence of IBD is increasing steadily in newly industrialized, non‐Western countries.35 In IBD, mucosal lesions are caused by disorders of the intestinal microbiota.36 Intestinal microbiota also drive IBD pathogenicity through pro‐inflammatory factors or restriction of protective compounds.37 This hints at the joint induction of intestinal inflammation by a variety of microbes and the possibility of causing CRC.38 The inflammatory cytokines associated with CRC are primarily intermediate mediators, such as microbial metabolites. A large amount of metabolites in the blood come from the intestines, which support the important role of metabolites in formation of microbial‐cytokines and construction of intestinal microenvironment.39 However, there has been little research in this area. The symbiotic and pathogenic microbiota also induce local inflammation by invading the normal colon tissue, and accelerate tumorigenesis by promoting the genotoxicity of colonic epithelial cells, thus promoting the development of CRC.40 The DNA in the IECs undergoes modification (including nitration, oxidation, methylation and deamination) by chronic inflammation, which leads to abnormal proliferation. This process may contribute to the activation or progression of CRC.41 Innate immune cells, such as macrophages, dendritic cells (DCs), and adaptive immune cells are recruited in response to inflammation.42 The mitotic immune system is further developed with the participation of symbiotic bacteria. Macrophages, DCs, and natural killer (NK) cells proliferate and release proinflammatory cytokines, such as interleukin (IL)‐12, IL‐23, tumor necrosis factor α (TNF‐α), and INF‐γ (Fig. 2). These factors activate cells of the adaptive immune system, including T lymphocytes, B lymphocytes, and various inflammatory mediators.43The inflamed colorectal epithelial cells do not form an effective surface barrier to reject the invasion of the symbiotic bacteria and their derivatives. As a result of this defect in the barrier function, symbiotic bacteria become the driving force for inducing and maintaining the tumor to promote inflammation.44 Genes or cells undergo mutations, proliferation, or apoptosis under the action of inflammation and gradually develop carcinogenic phenotypes. Nuclear factor kappa (NF‐κB) is the link between inflammation and cancer, which mainly regulates cell survival and immunity.45 IL‐6 is one of the major cytokines induced by NF‐κB. IL‐6 produced in the lamina propria activates the signal transduction and activator of transcription (STAT3) signaling pathway in IECs. This pathway promotes cell proliferation, inhibits apoptosis and other tumorigenic pathways to promote tumorigenesis.46 Two transcription factors, NF‐κB and STAT3, are essential for inflammation‐promoted cancer development and progression. NF‐κB signaling pathway is activated by TNF‐α and IL‐17, and is associated with cytokines. STAT3 is activated with the help of IL‐6, IL‐21, IL‐22, and IL‐23.40 In a mouse model, the development of colitis was shown to require the expression of IL‐23. At the same time, IL‐23 is crucial in the regulation of T helper cell 17 (Th17)47 function and IL‐17 production.48 The increase in IL‐17 expression appears in the colons of patients with UC and CD.49 The NF‐κB pathway also serves as an important regulator of the genes encoding TNF and Cyclooxygenase‐2 (COX‐2),50 which are often highly overexpressed in inflammatory bowel disease, as well as in colorectal adenomas and adenocarcinomas. Other key innate components of the inflammatory response that contribute to CRC progression include reactive nitrogen species (RNS) and reactive oxygen species (ROS), which serve as genotoxic compounds promoting the accumulation of mutations within proliferating epithelial cells.51
Figure 2.
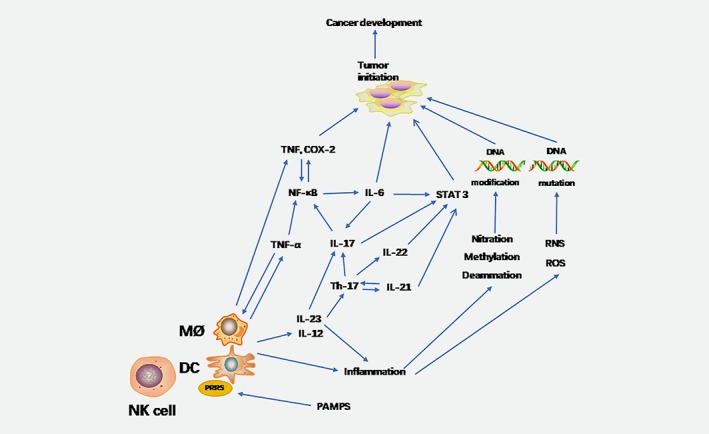
Inflammation oncogenic signaling pathways. PAMPs recognize surface PRRs such as macrophages and dendritic cells. The inflammatory cells release cytokines such as IL‐6, IL‐23, TNF‐α et al. IL‐6 activates the NF‐κB and signal transducer and STAT3 pathways, which in turn induce cancer. IL‐17 is an important cytokine that also activates the NF‐κB and STAT3 signaling pathways, but requires the help of IL‐23. At the same time, the release of ROS and RNS, and chemical modifications such as methylation and amination, can damage DNA and promote the development of cancer.
Typical Microbial Families Contributing to Colorectal Cancer
Enterococcus faecalis
E. faecalis is a gram‐positive facultative anaerobic bacterium. Recent research has linked E. faecalis with CRC, because the bacterium was found to aggregate at a higher level in fecal specimens from patients with CRC compared to that of healthy controls, and is more abundant in the adjacent tissues of cancer and CRC compared to healthy mucosa.52 In Il10 knockout mice, E. faecalis promoted colon inflammation, leading to dysplasia and CRC.53 One study also showed that E. faecalis, which can cause colitis after infection, can express TGF‐β in the intestinal epithelial cells of wild‐type mice, thereby activating the Smad signaling pathway.54 In this process, the dedifferentiation of TGF‐β signal transduction enhances the stem cell characteristics of CRC, thereby promoting its occurrence.55 This is associated with the deletion of the expression of the TLR2 protein and the inhibition of NF‐κB‐dependent pro‐inflammatory gene expression54 (Fig. 3). In contrast, Il10 gene‐deficient mice in IECs fail to suppress TLR2‐mediated pro‐inflammatory gene expression during colonization with E. faecalis.56 In addition, the Smad4 haploid environment not only affects colitis, but also has an impact on CRC.57 In addition to inducing chronic inflammation, E. faecalis can also generate extracellular superoxide and hydrogen peroxide. In vitro, DNA damage was induced by free radicals generated outside the cells.58 Infection by E. faecalis induced an intracellular ROS reaction that was independent of the oxphos system and impaired the mitochondrial genome in gastric cell culture. Finally, the microbiota can induce NF‐κB through DNA damage, leading to inflammation.59 ROS can cause chromosome instability (CIN), which may be related to the occurrence of CRC. In mammalian cells, E. faecalis can induce CIN via the production of superoxide. At the same time, E. faecalis seems to be involved in the bystander effect of COX‐2.60 These results validate a novel mechanism for CRC induction that involves endogenous CIN and cellular transformation arising through a microbiome‐driven bystander effect.61 At the same time, the extracellular superoxide formation of E. faecalis can enhance the expression of COX‐2 in macrophages and promote the CIN in IECs. The macrophages polarized by E. faecalis can induce malignant tumor aneuploidy or chromosome instability in primary colonic epithelial cells.62 These findings can explain the effect of E. faecalis on the incidence of CRC.
Figure 3.
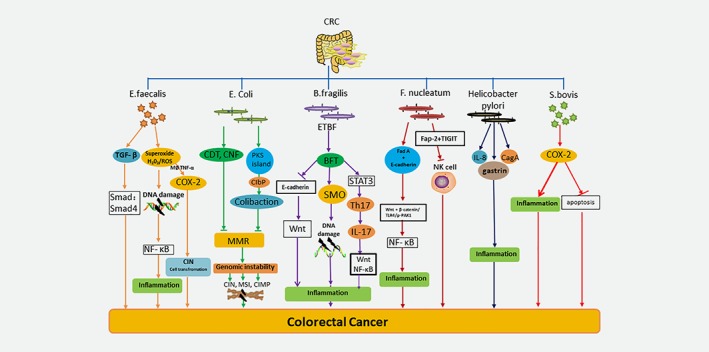
The carcinogenic mechanisms of six major microorganisms. E. faecalis causes colitis after infection and expresses TGF‐β in intestinal epithelial cells, thereby activating SMAD4 signaling pathway. E. faecalis also produces extracellular superoxide and hydrogen peroxide,inducing DNA damage. Meanwhile, it also appears to be involved in the bystander effect of COX‐2, the endogenous CIN and cell transformation caused by the release of TNF‐α from macrophages, results in cancer. Some strains of E. coli B2 produce CDT and CNF, which can directly lead to a DNA damage response and genomic instability. At the same time, the mismatch repair pathway is inhibited, leading to tumorigenesis. E. coli can also induce late bridges and chromosomal aberrations by genomic toxin‐containing pks islands and catalyze the synthesis of colibactin by clbP. Eventually it leads to CIN, MSI and CIMP, which leads to cancer. ETBF synthesizes BFT. BFT binds to CECs and stimulates the cleavage of E‐cadherin, thereby amplifying the Wnt and NF‐κB pathway and releasing pro‐inflammatory mediators to destroy DNA. At the same time, ETBF activates the signal transducer and activator of STAT3 signaling pathway, induces the production of Th17, releases interleukin IL‐17, and promotes colon tumor formation. BFT rapidly causes the expression of SMO and promotes SMO‐dependent ROS production and damages DNA in intestinal epithelial cell lines. These reactions cause tumor formation. By complexing with E‐cadherin on epithelial cells, F. nucleatum stimulates FadA, which activates wnt/β‐catenin/TLR4/p‐PAK1 signaling and upregulates oncogene expression. By releasing RNA into host cells and activating NF‐κB, F. nucleatum stimulates inflammation. F. nucleatum also inhibits natural killer (NK) cell activity in the tumor microenvironment, leading to colorectal tumorigenesis. Helicobacter pylori causes the metastasis of colorectal cancer caused by chronic gastritis. In addition, H. pylori infection may cause colorectal epithelial damage through inflammatory reactions such as IL‐8 mediated inflammatory responses. H. pylori virulent strains induce enhanced inflammatory responses, and expression of the cytotoxin‐associated gene A (CagA) gene may also lead to colorectal cancer. The S. bovis antigen induces the expression of COX‐2. With the help of prostaglandins, COX‐2 promotes cell proliferation and angiogenesis, and inhibits apoptosis, thus stimulating the cancer pathway.
Escherichia coli
E. coli has a predominant place among aerobic‐anaerobic gram‐negative bacteria in the normal intestinal microbiota. Some virulent strains of E. coli that contain pathogenicity islands can infect the human gastrointestinal system and induce disease. E. coli is divided into four phylogenic groups (A, B1, B2, and D) according to the acquisition of virulence factors.63 Interestingly, some strains of E. coli from phylo group B2 are associated with CD, a chronic IBD that is a risk factor for CRC.64 At the same time, E. coli in group B2 can also contain the polyketone acid synthetase (pks) island containing the genotoxin. This hybrid peptide‐polyketone genotoxin is generated by a multi‐enzyme factory, which is encoded by the 54 kb pks genotoxicity island65 (Fig. 3). Infection with pks+ E. coli induces anaphase bridging, chromosome aberrations, aneuploidy, and tetraploidy.66 Mucus degradation enables an increase in pks+ E. coli adhesion, triggering increased DNA damage in colonic epithelial cells by colibactin.67 Bacteria‐host cell contact is required for the genotoxic effect of pks; therefore, an environment where bacteria can more readily take root in the epithelial layer could deliver more of the pks product colibactin to host cells in the epithelium.68 Deletion of the pks island decreased tumor number without altering inflammation, suggesting that colibactin contributes to tumor promotion independently of inflammation.69 Recent evidence shows that colibactin is synthesized via the peptidase activity of the editing enzyme, clbP, which is present in the pks island, indicating that the colibactin is synthesized as a prodrug.69 Vizcaino and others have used nuclear magnetic resonance spectroscopy and bioinformatics‐guided isotopic markers to describe the colibactin warhead. The warhead crosslinks duplex DNA in vitro, which is evidence for colibactin's DNA‐damaging activity.70 Some toxins can cause DNA damage, and then affect the cell cycle. E. coli harboring cytotoxic necrotizing factor (CNF) and cytolethal distending toxin (CDT) are particularly associated with CRC biopsies.71 In addition, DNA mismatch repair (MMR) plays a key role in sustaining genomic stability,72 which is a highly conserved biological pathway. MMR acts in the DNA damage response pathway, which degrades severely damaged cells and prevents both short‐term mutagenesis and long‐term tumorigenesis. Using a method involving depletion of DNA mismatch repair proteins,73 an E. coli effector protein stimulates host mutation. The pathogenic mechanisms that cause this condition included in three pathways: CIN, microsatellite instability (MSI), and the CpG island methylator phenotype (CIMP),74 which eventually lead to the occurrence of CRC.
Bacteroides fragilis
B. fragilis usually exists symbiotically and is believed to contribute to the host's nutritional status, together with mucosal and systemic immunity. By contrast, B. fragilis also induces human disease when colonic integrity is interfered with, which allows it to escape into the sterile peritoneum where it acts as an opportunistic pathogen.75 Among B. fragilis strains, only enterotoxigenic B. fragilis (ETBF) is associated with diarrheal disease. Alternatively, nonenterotoxigenic B. fragilis (NTBF) strains are believed to be a possible pro‐biotics but with the potential to induce colonic inflammation.76 The central part of the pathogenicity of ETBF is the synthesis of the B. fragilis toxin (BFT) (Fig. 3). BFT is a 20‐kDa zinc‐dependent metalloprotease toxin that binds to colonic epithelial cells (CECs) and stimulates cleavage of the tumor suppressor protein, E‐cadherin.77 E‐cadherin cleavage enhances procarcinogenic triggering, including Wnt signaling, CEC proliferation, and epithelial barrier disruption, which stimulate mucosal inflammation and the formation of colon tumors, as illustrated in murine models of colon carcinogenesis.78 At the molecular level, BFT binds to a specific colonic epithelial receptor, activating the Wnt and NF‐κB pathways. ETBF induces rapid activation of STAT3, both in the colonic epithelial cells, which are the targets of transformation in the colon, and in a subset of mucosal immune cells. STAT3 activation is required for Th17 cell development and, consistent with this, ETBF induces a rapid mucosal Th17 inflammatory response within 1 week of colonization.79 Colon tumors induced by ETBF also have a marked increase in STAT3 activation. IL‐17 is produced by a subpopulation of Th17 cells.80 The IL‐17‐dependent signaling pathway promotes NF‐κB and Wnt activation, and establishes an inflammatory tumor microenvironment in the gut.81, 82 Furthermore, administration of an IL‐17‐blocking antibody had an inhibitory effect on excess tumor formation, indicating that IL‐17 is necessary for tumorigenesis in this model.83 Spermine oxidase (SMO), a polyamine catabolic enzyme, is an epithelial source of inflammation‐induced ROS and DNA damage. BFT rapidly triggers SMO expression and contributes to SMO‐dependent ROS and DNA damage in intestinal epithelial cell lines. Mice that are infected with ETBF show elevated intestinal SMO expression.84 It has been suggested that SMO acts as a potential source of inflammation‐associated ROS, which is produced during polyamine catabolism. This causes apoptosis, DNA damage, and consequently, the formation of cancer.85
Streptococcus bovis/Streptococcus gallolyticus
S. bovis is implicitly associated with an expanded of a variety of cell and molecular modifications that may be linked with the appearance of CRC.86 The wall‐extracted S. bovis antigen induces the expression of COX‐2 (Fig. 3). With the help of prostaglandins, COX‐2 promotes cell angiogenesis and inhibits apoptosis. Thus, it can stimulate the cancer pathway.87 Using human CRC patient's feces and mucosa samples, Abdulamir and his colleagues showed enrichment of the bacteria compared to the healthy control group, without gastrointestinal lesions, which strengthened the connection between S. bovis and CRC.88 Thus, it appears that Streptococcus can provide a growth advantage in the tumor microenvironment and induce inflammation to promote carcinogenesis.
Fusobacterium nucleatum
F. nucleatum is an opportunistic commensal obligate anaerobic gram‐negative bacterium that is indigenous to the human oral cavity and has a role in periodontal disease. Recent data provide experimental support for tumor induction by F. nucleatum. F. nucleatum is more abundant in CRC tumor versus normal tissue.89 This bacterium induces the expansion of bone marrow‐derived immune cells, and promotes the expression of inflammatory genes in the small intestine and colon.90 Recent studies have uncovered oncogenic mechanisms that support a tumorigenic role of F. nucleatum. While Kostic's90 study unveiled an indirect method of interaction with tumor sites, Rubinstein's91 study demonstrated a direct interaction between F. nucleatum and host cells. F. nucleatum and other intestinal microbiota can also directly adhere to enterocytes, altering endothelial integrity, with possible oncogenic consequences. The best‐characterized associated factor is the external adhesin, FadA92 (Fig. 3). The unique FadA adhesin of F. nucleatum can bins to E‐cadherin, which activates β‐catenin signaling, inducing oncogenic responses in CRC cells.93 This is accompanied by increasing expression of transcription factors, oncogenes, Wnt, and inflammatory genes, together with growth promotion of CRC cells.94 By releasing RNA into the host cell and activating NF‐κB, F. nucleatum induced inflammation.95 NF‐κB is an internalized signal of the above process. Rubinstein et al. used FadA‐knock‐out mutants to demonstrate the effect of adhesion, and its effect on β‐catenin signaling cascades.65 A series of cytokines trigger the activation of NK cells, including IFN, IL‐2, IL‐12, IL‐15, and IL‐18. They recognize uncoordinated ligands to activate and/or suppress receptors. Alternatively, activation can be further accomplished by directly identifying pathogen‐associated molecular patterns. However, F. nucleatum inhibits NK cell activity in the tumor microenvironment, resulting in colorectal tumorigenesis.96 Fap‐2, which is a lactobacillus‐resistant nucleolus, is an inhibitor of lactose binding, and is involved in cell adhesion.97 Fap‐2 defends tumors from host immune attack. Abed et al. showed that F. nucleatum Fap‐2 attaches to beta‐D‐galactosyl(1–3)‐N‐acetyl‐D‐galactosamine (Gal‐GalNAc) as a polysaccharide receptor for CRC, reducing the ability of F. nucleatum to enhance CRC.98 T Cell immunoreceptor with Ig and ITIM domains (TIGIT) is an important inhibitory receptor on NK and T cells.99 Gur et al. proved that Fap‐2 binds to TIGIT on NK and T cells and interferes with the attack by the host immune system on F. nucleatum‐associated tumors.100 At the same time, under the control of TLR4/p‐PAK1/p‐β‐catenin S675 cascade, F. nucleatum enhances intestinal tumorigenesis in vivo.101 Invasive F. nucleatum activates catenin signaling through the TLR4/p‐PAK1/p‐β‐catenin S675 cascade, leading to CRC.102
Helicobacter pylori
H. pylori is a major factor in the development of stomach disorders and gastric cancer (GC).103 Some reports have indicated a potential relationship between H. pylori infection and colorectal neoplasms; however, this has been disputed by others. The pathophysiological mechanism of how H. pylori induces colorectal neoplasm remains unclear. Various mechanisms have been proposed to explain the correlation between H. pylori infection and CRC. First, H. pylori inhibits the gastric mucosal proton pump to reduce the secretion of gastric acid and increase the chances of the proliferation of other microbes.104 A meta‐analysis also showed that H. pylori may increase the risk of CRC; however, the evidence is insufficient.105 Second, H. pylori infection may cause damage to the colorectal epithelium through an inflammatory reaction, such as an inflammatory response mediated by IL‐8,49 which is a factor that corresponds with CRC (Fig. 3). In addition, virulent H. pylori strains expressing the CagA gene could also contribute to CRC by inducing enhanced inflammatory responses.106 To determine whether there is a correlation between colorectal cancer and CagA, Shmuely et al. detected the expression of serum IgG antibodies and CagA protein in 67 cases of colorectal adenocarcinomas. They found that CagA+ seropositivity was related to additional risk of gastric cancer and colonic cancer.107 Third, H. pylori infection promotes the secretion of gastrin, which may induce the proliferation of mucosal cells in the colon, leading to CRC.108 Increased plasma gastrin levels may induce higher mucosal cell proliferation in the colon and contribute to the development of CRC.109
Clinical Applications
We have shown that microbes are essential in the process of CRC development. Therefore, the protection of intestinal microbes can effectively reduce the occurrence of CRC. Although antibiotics and probiotics are frequently used, researchers have developed certain technologies to treat CRC associated with the above‐mentioned typical bacterial groups. Fecal microorganism transplantation (FMT) has recently been re‐evaluated as a promising method to treat diseases associated with microorganisms. Colonic enema or endoscopy is often used to introduce the distal gut flora from healthy donors into unhealthy recipients.110 FMT is most commonly used to treat recurrent Clostridium difficile infection (CDI) to replace the commensal microbiota that has been eliminated by antibiotic treatment.111 Reducing the adhesion of E. coli and targeting the host metabolism can affect the colonization of E. coli and prevent the occurrence of CRC.112 In the case of B. fragilis, researchers developed a biologically active recombinant BFT‐2 in vitro and found that a low dose of bioactive BFT‐2 could inhibit the formation of colorectal tumors when administered gastrically.113 In the case of F. nucleatum, xenografted mice with CRC were treated with antibiotics, which reduced the load of F. nucleatum and could reduce the overall growth of the tumor.114 CRC prediction based on fecal microbiota can provide accurate CRC prediction noninvasively with high accuracy. Although colonoscopy can visually observe the lesion, preoperative preparation is cumbersome. Moreover, the accuracy of the examination results can be affected by the skill and experience of the surgeon, and certain complications may occur. Painless colonoscopy reduces discomfort, but increases the risks associated with anesthesia. In addition, a limitation of short colonoscopy is that the working range is narrow and the examination is not comprehensive. Computed tomography (CT) colonoscopy may cause radiation damage and is expensive. Therefore, FMT, assisted by fecal DNA testing, could further improve the accuracy of CRC screening, and is a good choice for early screening and prognosis of CRC. We expect that these technologies will move out of the laboratory in the future and be applied in the treatment of colorectal cancer.
Conflict of interests
The authors declare that there is no conflict of interest.
Acknowledgements
We thank Hanjian Zhu and Jin Gao for their technical assistance. This work was supported by grants from Yangzhou Science and Technology Bureau Social Development Project (YZ2018087),training project of key talents of youth medicine in Jiangsu province, China (QNRC2016330) and Standardized diagnosis and treatment of key diseases in Jiangsu Province: standardized diagnosis and treatment of rectal cancer and rectal cancer liver metastasis and its clinical application (BE2015664).
Contributor Information
Dong Tang, Email: 83392785@qq.com.
Daorong Wang, Email: daorong666@sina.com.
References
- 1. Du LB, Li HZ, Wang YQ, et al. Report of colorectal cancer incidence and mortality in China, 2013. Zhonghua Zhong Liu Za Zhi 2017;39:701–6. [DOI] [PubMed] [Google Scholar]
- 2. Malys MK, Campbell L, Malys N. Symbiotic and antibiotic interactions between gut commensal microbiota and host immune system. Medicina 2015;51:69–75. [DOI] [PubMed] [Google Scholar]
- 3. Gagniere J, Raisch J, Veziant J, et al. Gut microbiota imbalance and colorectal cancer. World J Gastroenterol 2016;22:501–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Louis P, Hold GL, Flint HJ. The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol 2014;12:661–72. [DOI] [PubMed] [Google Scholar]
- 5. Capaldo CT, Powell DN, Kalman D. Layered defense: how mucus and tight junctions seal the intestinal barrier. J Mol Med 2017;95:927–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Swidsinski A, Loening‐Baucke V, Theissig F, et al. Comparative study of the intestinal mucus barrier in normal and inflamed colon. Gut 2007;56:343–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jakobsson HE, Rodriguez‐Pineiro AM, Schutte A, et al. The composition of the gut microbiota shapes the colon mucus barrier. EMBO Rep 2015;16:164–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Takiishi T, Fenero CIM, Camara NOS. Intestinal barrier and gut microbiota: shaping our immune responses throughout life. Tissue Barriers 2017;5:e1373208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol 2011;9:356–68. [DOI] [PubMed] [Google Scholar]
- 10. Coleman OI, Haller D. Bacterial signaling at the intestinal epithelial Interface in inflammation and Cancer. Front Immunol 2017;8:1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Swidsinski A, Sydora BC, Doerffel Y, et al. Viscosity gradient within the mucus layer determines the mucosal barrier function and the spatial organization of the intestinal microbiota. Inflamm Bowel Dis 2007;13:963–70. [DOI] [PubMed] [Google Scholar]
- 12. Tlaskalova‐Hogenova H, Farre‐Castany MA, Stepankova R, et al. The gut as a lymphoepithelial organ: the role of intestinal epithelial cells in mucosal immunity. Folia Microbiol 1995;40:385–91. [DOI] [PubMed] [Google Scholar]
- 13. Zanello G, Kevans D, Goethel A, et al. Genetics and innate and adaptive immunity in IBD. Nestle Nutr Inst Workshop Ser 2014;79:41–55. [DOI] [PubMed] [Google Scholar]
- 14. Cromer WE, Mathis JM, Granger DN, et al. Role of the endothelium in inflammatory bowel diseases. World J Gastroenterol 2011;17:578–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Elinav E, Henao‐Mejia J, Flavell RA. Integrative inflammasome activity in the regulation of intestinal mucosal immune responses. Mucosal Immunol 2013;6:4–13. [DOI] [PubMed] [Google Scholar]
- 16. Mu C, Yang Y, Zhu W. Crosstalk between the immune receptors and gut microbiota. Curr Protein Pept Sci 2015;16:622–31. [DOI] [PubMed] [Google Scholar]
- 17. Ringel‐Scaia VM, McDaniel DK, Allen IC. The goldilocks conundrum: NLR Inflammasome modulation of gastrointestinal inflammation during inflammatory bowel disease. Crit Rev Immunol 2016;36:283–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lei‐Leston AC, Murphy AG, Maloy KJ. Epithelial cell Inflammasomes in intestinal immunity and inflammation. Front Immunol 2017;8:1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ver Heul AM, Fowler CA, Ramaswamy S, et al. Ubiquitin regulates caspase recruitment domain‐mediated signaling by nucleotide‐binding oligomerization domain‐containing proteins NOD1 and NOD2. J Biol Chem 2013;288:6890–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maharana J, Dehury B, Sahoo JR, et al. Structural and functional insights into CARDs of zebrafish (Danio rerio) NOD1 and NOD2, and their interaction with adaptor protein RIP2. Mol Biosyst 2015;11:2324–36. [DOI] [PubMed] [Google Scholar]
- 21. Lavelle EC, Murphy C, O'Neill LA, et al. The role of TLRs, NLRs, and RLRs in mucosal innate immunity and homeostasis. Mucosal Immunol 2010;3:17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang J, Basagoudanavar SH, Wang X, et al. NF‐kappa B RelA subunit is crucial for early IFN‐beta expression and resistance to RNA virus replication. J Immunol 2010;185:1720–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Creagh EM, O'Neill LA. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co‐operate in innate immunity. Trends Immunol 2006;27:352–7. [DOI] [PubMed] [Google Scholar]
- 24. Shu XS, Zhao Y, Sun Y, et al. The epigenetic modifier PBRM1 restricts the basal activity of the innate immune system by repressing retinoic acid‐inducible gene‐I‐like receptor signalling and is a potential prognostic biomarker for colon cancer. J Pathol 2018;244:36–48. [DOI] [PubMed] [Google Scholar]
- 25. Sipos F, Furi I, Constantinovits M, et al. Contribution of TLR signaling to the pathogenesis of colitis‐associated cancer in inflammatory bowel disease. World J Gastroenterol 2014;20:12713–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reuven EM, Fink A, Shai Y. Regulation of innate immune responses by transmembrane interactions: lessons from the TLR family. Biochim Biophys Acta 1838;2014:1586–93. [DOI] [PubMed] [Google Scholar]
- 27. Della Mina E, Borghesi A, Zhou H, et al. Inherited human Irak‐1 deficiency selectively impairs TLR signaling in fibroblasts. Proc Natl Acad Sci U S A 2017;114:E514–E23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Muroi M, Tanamoto KI. TRAF6 distinctively mediates MyD88‐ and Irak‐1‐induced activation of NF‐kappaB. J Leukoc Biol 2008;83:702–7. [DOI] [PubMed] [Google Scholar]
- 29. Tchorzewski M, Lewkowicz P, Dziki A, et al. Expression of toll‐like receptors on human rectal adenocarcinoma cells. Arch Immunol Ther Exp (Warsz) 2014;62:247–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Warner N, Nunez G. MyD88: a critical adaptor protein in innate immunity signal transduction. J Immunol 2013;190:3–4. [DOI] [PubMed] [Google Scholar]
- 31. Ikushima H, Negishi H, Taniguchi T. The IRF family transcription factors at the interface of innate and adaptive immune responses. Cold Spring Harb Symp Quant Biol 2013;78:105–16. [DOI] [PubMed] [Google Scholar]
- 32. Ashida R, Tominaga K, Sasaki E, et al. AP‐1 and colorectal cancer. Inflammopharmacology 2005;13:113–25. [DOI] [PubMed] [Google Scholar]
- 33. Stecher B. The roles of inflammation, nutrient availability and the commensal microbiota in enteric pathogen infection. Microbiol Spectr 2015;3:297–320. [DOI] [PubMed] [Google Scholar]
- 34. Ullman TA, Itzkowitz SH. Intestinal inflammation and cancer. Gastroenterology 2011;140:1807–16. [DOI] [PubMed] [Google Scholar]
- 35. Ng SC, Shi HY, Hamidi N, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population‐based studies. Lancet 2018;390:2769–78. [DOI] [PubMed] [Google Scholar]
- 36. Manichanh C, Borruel N, Casellas F, et al. The gut microbiota in IBD. Nat Rev Gastroenterol Hepatol 2012;9:599–608. [DOI] [PubMed] [Google Scholar]
- 37. Seksik P. Gut microbiota and IBD. Gastroenterol Clin Biol 2010;34(Suppl 1):S44–51. [DOI] [PubMed] [Google Scholar]
- 38. Liu Z, Cao AT, Cong Y. Microbiota regulation of inflammatory bowel disease and colorectal cancer. Semin Cancer Biol 2013;23:543–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schirmer M, Smeekens SP, Vlamakis H, et al. Linking the human gut microbiome to inflammatory cytokine production capacity. Cell 2016;167:1125–36. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen J, Pitmon E, Wang K. Microbiome, inflammation and colorectal cancer. Semin Immunol 2017;32:43–53. [DOI] [PubMed] [Google Scholar]
- 41. Bayarsaihan D. Epigenetic mechanisms in inflammation. J Dent Res 2011;90:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lucas C, Barnich N, Nguyen HTT. Microbiota, inflammation and colorectal Cancer. Int J Mol Sci 2017;18:1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mandal P. Molecular mechanistic pathway of colorectal carcinogenesis associated with intestinal microbiota. Anaerobe 2018;49:63–70. [DOI] [PubMed] [Google Scholar]
- 44. Gil‐Cardoso K, Gines I, Pinent M, et al. Effects of flavonoids on intestinal inflammation, barrier integrity and changes in gut microbiota during diet‐induced obesity. Nutr Res Rev 2016;29:234–48. [DOI] [PubMed] [Google Scholar]
- 45. Candela M, Turroni S, Biagi E, et al. Inflammation and colorectal cancer, when microbiota‐host mutualism breaks. World J Gastroenterol 2014;20:908–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lasry A, Zinger A, Ben‐Neriah Y. Inflammatory networks underlying colorectal cancer. Nat Immunol 2016;17:230–40. [DOI] [PubMed] [Google Scholar]
- 47. Kempski J, Brockmann L, Gagliani N, et al. TH17 cell and epithelial cell crosstalk during inflammatory bowel disease and carcinogenesis. Front Immunol 2017;8:1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang R, Hasnain SZ, Tong H, et al. Neutralizing IL‐23 is superior to blocking IL‐17 in suppressing intestinal inflammation in a spontaneous murine colitis model. Inflamm Bowel Dis 2015;21:973–84. [DOI] [PubMed] [Google Scholar]
- 49. Rubin DC, Shaker A, Levin MS. Chronic intestinal inflammation: inflammatory bowel disease and colitis‐associated colon cancer. Front Immunol 2012;3:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ke J, Long X, Liu Y, et al. Role of NF‐kappaB in TNF‐alpha‐induced COX‐2 expression in synovial fibroblasts from human TMJ. J Dent Res 2007;86:363–7. [DOI] [PubMed] [Google Scholar]
- 51. Brennan CA, Garrett WS. Gut microbiota, inflammation, and colorectal Cancer. Annu Rev Microbiol 2016;70:395–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhou Y, He H, Xu H, et al. Association of oncogenic bacteria with colorectal cancer in South China. Oncotarget 2016;7:80794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Steck N, Hoffmann M, Sava IG, et al. Enterococcus faecalis metalloprotease compromises epithelial barrier and contributes to intestinal inflammation. Gastroenterology 2011;141:959–71. [DOI] [PubMed] [Google Scholar]
- 54. Ruiz PA, Shkoda A, Kim SC, et al. IL‐10 gene‐deficient mice lack TGF‐beta/Smad‐mediated TLR2 degradation and fail to inhibit proinflammatory gene expression in intestinal epithelial cells under conditions of chronic inflammation. Ann N Y Acad Sci 2006;1072:389–94. [DOI] [PubMed] [Google Scholar]
- 55. Nakano M, Kikushige Y, Miyawaki K, et al. Dedifferentiation process driven by TGF‐beta signaling enhances stem cell properties in human colorectal cancer. Oncogene 2018. [DOI] [PubMed] [Google Scholar]
- 56. Ruiz PA, Shkoda A, Kim SC, et al. IL‐10 gene‐deficient mice lack TGF‐beta/Smad signaling and fail to inhibit proinflammatory gene expression in intestinal epithelial cells after the colonization with colitogenic enterococcus faecalis. J Immunol 2005;174:2990–9. [DOI] [PubMed] [Google Scholar]
- 57. Szigeti R, Pangas SA, Nagy‐Szakal D, et al. SMAD4 haploinsufficiency associates with augmented colonic inflammation in select humans and mice. Ann Clin Lab Sci 2012;42:401–8. [PMC free article] [PubMed] [Google Scholar]
- 58. Huycke MM, Abrams V, Moore DR. Enterococcus faecalis produces extracellular superoxide and hydrogen peroxide that damages colonic epithelial cell DNA. Carcinogenesis 2002;23:529–36. [DOI] [PubMed] [Google Scholar]
- 59. McCool KW, Miyamoto S. DNA damage‐dependent NF‐kappaB activation: NEMO turns nuclear signaling inside out. Immunol Rev 2012;246:311–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang X, Allen TD, May RJ, et al. Enterococcus faecalis induces aneuploidy and tetraploidy in colonic epithelial cells through a bystander effect. Cancer Res 2008;68:9909–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang X, Yang Y, Huycke MM. Commensal bacteria drive endogenous transformation and tumour stem cell marker expression through a bystander effect. Gut 2015;64:459–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang X, Huycke MM. Extracellular superoxide production by enterococcus faecalis promotes chromosomal instability in mammalian cells. Gastroenterology 2007;132:551–61. [DOI] [PubMed] [Google Scholar]
- 63. Olesen B. Characterization of four Escherichia coli clonal groups. Acta pathol Microbiol Immunol Scand 2017;125(Suppl 139):1–28. [DOI] [PubMed] [Google Scholar]
- 64. Bonnet M, Buc E, Sauvanet P, et al. Colonization of the human gut by E. coli and colorectal cancer risk. Clin Cancer Res 2014;20:859–67. [DOI] [PubMed] [Google Scholar]
- 65. Allen‐Vercoe E, Jobin C. Fusobacterium and Enterobacteriaceae: important players for CRC? Immunol Lett 2014;162:54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cuevas‐Ramos G, Petit CR, Marcq I, et al. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci U S A 2010;107:11537–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dejea CM, Fathi P, Craig JM, et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 2018;359:592–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Arthur JC, Perez‐Chanona E, Muhlbauer M, et al. Intestinal inflammation targets cancer‐inducing activity of the microbiota. Science 2012;338:120–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Leung A, Tsoi H, Yu J. Fusobacterium and Escherichia: models of colorectal cancer driven by microbiota and the utility of microbiota in colorectal cancer screening. Expert Rev Gastroenterol Hepatol 2015;9:651–7. [DOI] [PubMed] [Google Scholar]
- 70. Vizcaino MI, Crawford JM. The colibactin warhead crosslinks DNA. Nat Chem 2015;7:411–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fais T, Delmas J, Cougnoux A, et al. Targeting colorectal cancer‐associated bacteria: a new area of research for personalized treatments. Gut Microbes 2016;7:329–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res 2008;18:85–98. [DOI] [PubMed] [Google Scholar]
- 73. Maddocks OD, Scanlon KM, Donnenberg MS. An Escherichia coli effector protein promotes host mutation via depletion of DNA mismatch repair proteins. mBio 2013;4:e00152–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Marmol I, Sanchez‐De‐Diego C, Pradilla Dieste A, et al. Colorectal carcinoma: a general overview and future perspectives in colorectal Cancer. Int J Mol Sci 2017;18:197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. May AK, Gleason TG, Sawyer RG, et al. Contribution of Escherichia coli alpha‐hemolysin to bacterial virulence and to intraperitoneal alterations in peritonitis. Infect Immun 2000;68:176–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wick EC, Sears CL. Bacteroides spp. and diarrhea. Curr Opin Infect Dis 2010;23:470–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Housseau F, Sears CL. Enterotoxigenic Bacteroides fragilis (ETBF)‐mediated colitis in min (Apc+/−) mice: a human commensal‐based murine model of colon carcinogenesis. Cell Cycle 2010;9:3–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sears CL, Geis AL, Housseau F. Bacteroides fragilis subverts mucosal biology: from symbiont to colon carcinogenesis. J Clin Invest 2014;124:4166–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Chung L, Thiele Orberg E, Geis AL, et al. Bacteroides fragilis toxin coordinates a pro‐carcinogenic inflammatory Cascade via targeting of colonic epithelial cells. Cell Host Microbe 2018;23:203–14. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Daoussis D, Andonopoulos AP, Liossis SN. Wnt pathway and IL‐17: novel regulators of joint remodeling in rheumatic diseases. Looking beyond the RANK‐RANKL‐OPG axis. Semin Arthritis Rheum 2010;39:369–83. [DOI] [PubMed] [Google Scholar]
- 81. Goktuna SI, Shostak K, Chau TL, et al. The Prosurvival IKK‐related kinase IKKepsilon integrates LPS and IL17A signaling cascades to promote Wnt‐dependent tumor development in the intestine. Cancer Res 2016;76:2587–99. [DOI] [PubMed] [Google Scholar]
- 82. Hata K, Andoh A, Shimada M, et al. IL‐17 stimulates inflammatory responses via NF‐kappaB and MAP kinase pathways in human colonic myofibroblasts. Am J Physiol Gastrointest Liver Physiol 2002;282:G1035–44. [DOI] [PubMed] [Google Scholar]
- 83. Dejea C, Wick E, Sears CL. Bacterial oncogenesis in the colon. Future Microbiol 2013;8:445–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Goodwin AC, Destefano Shields CE, Wu S, et al. Polyamine catabolism contributes to enterotoxigenic Bacteroides fragilis‐induced colon tumorigenesis. Proc Natl Acad Sci U S A 2011;108:15354–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Snezhkina AV, Krasnov GS, Lipatova AV, et al. The Dysregulation of polyamine metabolism in colorectal Cancer is associated with overexpression of c‐Myc and C/EBPbeta rather than Enterotoxigenic Bacteroides fragilis infection. Oxid Med Cell Longev 2016;2016:2353560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Fregoli L, Palmeri M, Palombo C, et al. Streptococcus bovis endocarditis as first clinical expression of an occult colorectal neoplasm. Int J Colorectal Dis 2015;30:145–6. [DOI] [PubMed] [Google Scholar]
- 87. Abdulamir AS, Hafidh RR, Abu Bakar F. The association of Streptococcus bovis/gallolyticus with colorectal tumors: the nature and the underlying mechanisms of its etiological role. J Exp Clin Cancer Res 2011;30:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Abdulamir AS, Hafidh RR, Bakar FA. Molecular detection, quantification, and isolation of Streptococcus gallolyticus bacteria colonizing colorectal tumors: inflammation‐driven potential of carcinogenesis via IL‐1, COX‐2, and IL‐8. Mol Cancer 2010;9:249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Flanagan L, Schmid J, Ebert M, et al. Fusobacterium nucleatum associates with stages of colorectal neoplasia development, colorectal cancer and disease outcome. Eur J Clin Microbiol Infect Dis 2014;33:1381–90. [DOI] [PubMed] [Google Scholar]
- 90. Kostic AD, Chun E, Robertson L, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor‐immune microenvironment. Cell Host Microbe 2013;14:207–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Rubinstein MR, Wang X, Liu W, et al. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E‐cadherin/beta‐catenin signaling via its FadA adhesin. Cell Host Microbe 2013;14:195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Fardini Y, Wang X, Temoin S, et al. Fusobacterium nucleatum adhesin FadA binds vascular endothelial cadherin and alters endothelial integrity. Mol Microbiol 2011;82:1468–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yu J, Chen Y, Fu X, et al. Invasive Fusobacterium nucleatum may play a role in the carcinogenesis of proximal colon cancer through the serrated neoplasia pathway. Int J Cancer 2016;139:1318–26. [DOI] [PubMed] [Google Scholar]
- 94. Amitay EL, Werner S, Vital M, et al. Fusobacterium and colorectal cancer: causal factor or passenger? Results from a large colorectal cancer screening study. Carcinogenesis 2017;38:781–8. [DOI] [PubMed] [Google Scholar]
- 95. Li YY, Ge QX, Cao J, et al. Association of Fusobacterium nucleatum infection with colorectal cancer in Chinese patients. World J Gastroenterol 2016;22:3227–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Bashir A, Miskeen AY, Bhat A, et al. Fusobacterium nucleatum: an emerging bug in colorectal tumorigenesis. Eur J Cancer Prev 2015;24:373–85. [DOI] [PubMed] [Google Scholar]
- 97. Coppenhagen‐Glazer S, Sol A, Abed J, et al. Fap2 of Fusobacterium nucleatum is a galactose‐inhibitable adhesin involved in coaggregation, cell adhesion, and preterm birth. Infect Immun 2015;83:1104–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Gholizadeh P, Eslami H, Kafil HS. Carcinogenesis mechanisms of Fusobacterium nucleatum. Biomed Pharmacother 2017;89:918–25. [DOI] [PubMed] [Google Scholar]
- 99. Stanietsky N, Simic H, Arapovic J, et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci U S A 2009;106:17858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Gur C, Ibrahim Y, Isaacson B, et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 2015;42:344–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Wu Y, Wu J, Chen T, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis in mice via a toll‐like receptor 4/p21‐activated kinase 1 Cascade. Dig Dis Sci 2018;63:1210–8. [DOI] [PubMed] [Google Scholar]
- 102. Chen Y, Peng Y, Yu J, et al. Invasive Fusobacterium nucleatum activates beta‐catenin signaling in colorectal cancer via a TLR4/P‐PAK1 cascade. Oncotarget 2017;8:31802–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Venerito M, Vasapolli R, Rokkas T, et al. Helicobacter pylori and gastrointestinal malignancies. Helicobacter 2015;20(Suppl 1):36–9. [DOI] [PubMed] [Google Scholar]
- 104. Saha A, Hammond CE, Beeson C, et al. Helicobacter pylori represses proton pump expression and inhibits acid secretion in human gastric mucosa. Gut 2010;59:874–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Zhang Y, Gao C, Zhai JH. meta‐analysis on the relationship between colorectal cancer and helicobacter pylori infection. Zhonghua liu xing bing xue za zhi = Zhonghua liuxingbingxue zazhi 2009;30:73–7. [PubMed] [Google Scholar]
- 106. Papastergiou V, Karatapanis S, Georgopoulos SD. Helicobacter pylori and colorectal neoplasia: is there a causal link? World J Gastroenterol 2016;22:649–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Shmuely H, Passaro D, Figer A, et al. Relationship between helicobacter pylori CagA status and colorectal cancer. Am J Gastroenterol 2001;96:3406–10. [DOI] [PubMed] [Google Scholar]
- 108. Inoue I, Kato J, Tamai H, et al. Helicobacter pylori‐related chronic gastritis as a risk factor for colonic neoplasms. World J Gastroenterol 2014;20:1485–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Hartwich A, Konturek SJ, Pierzchalski P, et al. Helicobacter pylori infection, gastrin, cyclooxygenase‐2, and apoptosis in colorectal cancer. Int J Colorectal Dis 2001;16:202–10. [DOI] [PubMed] [Google Scholar]
- 110. Peterson CT, Sharma V, Elmen L, et al. Immune homeostasis, dysbiosis and therapeutic modulation of the gut microbiota. Clin Exp Immunol 2015;179:363–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Walsh CJ, Guinane CM, O'Toole PW, et al. Beneficial modulation of the gut microbiota. FEBS Lett 2014;588:4120–30. [DOI] [PubMed] [Google Scholar]
- 112. Khan AA, Khan Z, Malik A, et al. Colorectal cancer‐inflammatory bowel disease nexus and felony of Escherichia coli . Life Sci 2017;180:60–7. [DOI] [PubMed] [Google Scholar]
- 113. Lv Y, Ye T, Wang HP, et al. Suppression of colorectal tumorigenesis by recombinant Bacteroides fragilis enterotoxin‐2 in vivo. World J Gastroenterol 2017;23:603–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Bullman S, Pedamallu CS, Sicinska E, et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 2017;358:1443–8. [DOI] [PMC free article] [PubMed] [Google Scholar]