

Abstract
Nutritional research is currently entering the field of personalized nutrition, to a large extent driven by major technological breakthroughs in analytical sciences and biocomputing. An efficient launching of the personalized approach depends on the ability of researchers to comprehensively monitor and characterize interindividual variability in the activity of the human gastrointestinal tract. This information is currently not available in such a form. This review therefore aims at identifying and discussing published data, providing evidence on interindividual variability in the processing of the major nutrients, i.e., protein, fat, carbohydrates, vitamins, and minerals, along the gastrointestinal tract, including oral processing, intestinal digestion, and absorption. Although interindividual variability is not a primary endpoint of most studies identified, a significant number of publications provides a wealth of information on this topic for each category of nutrients. This knowledge remains fragmented, however, and understanding the clinical relevance of most of the interindividual responses to food ingestion described in this review remains unclear. In that regard, this review has identified a gap and sets the base for future research addressing the issue of the interindividual variability in the response of the human organism to the ingestion of foods.
Keywords: digestion, food, gastrointestinal tract, gut microbiome, polymorphism
Despite numerous examples of interindividual variability in the processing of food by the human gastrointestinal tract, the knowledge on this topic, as illustrated for polymorphisms in mineral absorption, remains fragmented. The GutSelf review sets the base for motivating future research specifically addressing the issue of the interindividual variability in the response of the human organism to the ingestion of foods.
1. Introduction
The “food domain” and the “human domain” are tightly connected, and their reciprocal interactions are modulated by different factors, inter alia, ethnicity, culture (including gastronomy), economy, or politics. All such factors are drivers of different food choices and dietary patterns, and in turn modulate human physiology and health, via nutrient intake and absorption. A famous aphorism, which represents the connection between the food and human domains, was first used in a 1825 gastronomy book by Jean Anthelme Brillat‐Savarin, “Dis‐moi ce que tu manges, je te dirai ce que tu es.” [Tell me what you eat, and I will tell you what you are].1
Processing has long been used to ameliorate sensory, safety, and nutritional characteristics of food. The impact of processing is still an important research topic for both academia and the food industry. In regards to health, it is recognized that even minor changes in processing can lead to significant changes in the foods effect. However, the impact of changes in the most significant processing procedure applied by man on food, digestion in the gastrointestinal tract (GIT), is seldom considered.
As with any other human characteristic, physical or mental, events in the GIT are subject to variability between individuals. This variability can affect the final output, for example, leading to different digestion or absorption capacity of specific components, in turn leading to a different effect on physiology and health. In few words, “Tell me what you digest and absorb, and I will tell you what you are” would be more correct than Brillat–Savarin's aphorism. However to date, knowledge on human variability in the GIT is limited and fragmented, and has not been compiled in a comprehensive form encompassing all nutrients.
This review (GutSelf) provides an overview of the knowledge and published data on human interindividual variability in the processing of nutrients by the GIT. This task requests a multifactorial analysis specifically considering the class of nutrients (protein, fat, carbohydrates, vitamins, and minerals) as well as the functionality of the GIT (oral processing, intestinal digestion, and absorption). In addition, a range of interrelated variables modulating the processing of food by the GIT needs to be considered. In particular, published evidence for interindividual variability was identified for the following variables: chewing, nutrient sensing, saliva composition, nutrient digestibility, composition of the intestinal peptidome, enzymatic activity, genetic polymorphisms, body mass index (BMI), diet, circadian rhythm, and the gut microbiota. These variables can be categorized as intrinsic (e.g., genetic polymorphisms) or extrinsic (e.g., diet),2, 3 molecular (e.g., pepsin activity), or morphological (e.g., BMI), and of genetic (e.g., amylase polymorphism) or non‐genetic origin (although a non‐genetic origin is often difficult to demonstrate). In light of this complexity, this review is organized in three hierarchical levels: the first level corresponds to the classes of nutrient; the second level corresponds to the GIT functions; the third level corresponds to the variables associated with the interindividual variability.
Interindividual variability is rarely addressed by researchers as a primary endpoint. A systematic search of the literature based on a well‐defined selection of keywords was therefore unsuccessful, delivering either thousands of unspecific hits with a broad search strategy, or very few hits with narrow search strategies. Being the first of its kind, this review does not intend to be complete but, instead, to present the main evidence for such variability. Of note, the post‐absorptive fate of the nutrients, e.g., liver metabolism, is not part of this review. We also exclude the impact of aging on GIT processing as this topic has already been reviewed elsewhere.4 Finally, the European Cooperation in Science and Technology (COST) Action FA1403 (POSITIVe) has reviewed interindividual variation in response to consumption of plant food bioactives (https://www6.inra.fr/cost-positive) and the reader is referred to ongoing work and publications by the COST action on this topic.5, 6 Of note, although the concept of nutrikinetics covers absorption, distribution, metabolism, and excretion, our review focuses on the first part of this process (absorption) mediated by the GIT.
2. Proteins
2.1. Overview of Protein Processing by the GIT
The assimilation of proteins involves gastric and pancreatic enzymatic hydrolysis to luminal oligopeptides and free amino acids.7 The low pH in the stomach activates pepsinogen to pepsin. Pepsin cleaves peptide bonds at the aromatic amino acids, and results in a mixture of intermediate protein moieties and peptides. These are then delivered to the duodenum, where pancreatic proenzymes (trypsinogen, chymotrypsinogen, proelastase, and procarboxypeptidase) become activated by mucosal enterokinase and further breakdown these products into amino acids, dipeptides, and tripeptides. Part of the latter are hydrolyzed to amino acids by brush border dipeptidases.8 Then the remaining dipeptides and tripeptides, together with the amino acids, are absorbed by the enterocytes of the small intestine, via a specific group of amino acid transporters, and specific membrane proteins, such as peptide transporter 1 (PEPT1), for peptides.7
Amino acid transport activities in the enterocyte are frequently referred to as “systems”; the term indicates a functionally identified transport activity that appears to be similar in a variety of cell types. Amino acid transport systems have been described on both apical and basolateral membranes. They vary in solute specificity, being dependent of Na+, Cl−, H+, or K+. The basolateral system carriers may work in either direction and may represent an electroneutral or electrogenic transport process, depending on the luminal amino acid concentration and cellular demand and on the electrogenic driving forces.8 The encoded gene of amino acid transporters has been identified and belongs to the solute carrier (SLC) superfamily.9
Dipeptides and tripeptides are very efficiently absorbed in the small intestine. The process is indirectly Na+‐dependent, in that Na+ is necessary for the activity of Na+/H+ exchanger (NHE) 3 to generate a proton gradient for H+/peptide co‐transport. Thus, the two transport processes are functionally coupled. Peptides of four or more amino acids in length are poorly absorbed in a non‐carrier dependent mechanism. The carrier responsible for the intestinal uptake of peptides is known as peptide transporter 1 (PEPT1). It is encoded for by the SLC15A1 gene, and is expressed in the intestinal, and to a lesser extent, renal epithelia.
Inside enterocytes, most peptides are hydrolyzed, and the resulting amino acids are released together with those absorbed by amino acid transporters. Through the blood, amino acids are delivered to all tissues, where they serve as building blocks for protein synthesis, as precursors for a wide variety of bioactive molecules, and as energy metabolites.10
Among the variables presented in the last paragraph of the Introduction, a search of the literature has identified convincing evidence for the interindividual of the oral processing, digestion, and intestinal absorption of proteins by the GIT for the following variables: chewing, protein digestibility, composition of the intestinal peptidome, pepsin and chymotrypsin activity, and genetic polymorphisms.
2.2. Interindividual Variability in Oral Processing of Proteins
2.2.1. Chewing
In previous years, a large amount of evidence has shown that the kinetics of macronutrient breakdown during digestion, and thus, the bioavailability of nutrients, are strongly modulated by the structure the food adopts in the different compartments of the GIT.11 For solid and semi‐solid foods, the first step of disintegration occurs in the mouth, where the chewing process and saliva‐soaking result in the formation of a bolus that will be further swallowed. Significant interindividual variation in oral processing is observed within humans,12 with some people chewing their food intensively leading to the formation of small particles, and others only partly, resulting in large particles. This has been particularly described in elderly populations, where dental health problems may lead to a bolus made of large particles. For instance, Rémond et al.13 studied the digestion of beef meat in elderly with intact or impaired dentition. Twenty elderly volunteers aged 60–75 years were involved in the study. Ten volunteers had healthy natural dentition, with the remaining ten being edentulous and wore complete dentures. A rapid increase in plasma aminoacidemia and plasma leucine entry rate was observed after meat intake in healthy subjects. In complete denture wearers, the increase in leucine entry rate was delayed, and the amount of leucine appearing in peripheral blood during the whole postprandial period was lower than that in healthy subjects. Postprandial whole‐body protein synthesis was lower in denture wearers than in healthy subjects (30% compared with 48% of leucine intake, respectively). As such, this study showed that meat protein utilization for protein synthesis can be impaired by a decrease in chewing efficiency.
Following this study, Pennings et al. observed similar trends by following another strategy, comparing the digestion and amino acid bioavailability of minced beef versus beef steak.14 Ten older men (mean ± SEM age 74 ± 2 years) were randomly assigned to a crossover experiment that involved two treatments, in which they consumed 135 g of l‐[1‐13C]phenylalanine–labeled beef, which was provided as beef steak or minced beef. Meat consumption was combined with continuous intravenous l‐[ring‐2H5] phenylalanine and l‐[ring‐2H2] tyrosine infusion to assess beef protein digestion and absorption kinetics as well as whole‐body protein balance and skeletal muscle protein synthesis rates. Meat protein–derived phenylalanine appeared more rapidly in peripheral circulation after minced beef than after beef steak consumption. Also, its availability in the circulation during the 6 h postprandial period was greater after minced beef than after beef steak consumption (61 ± 3% compared with 49 ± 3%, respectively). Whole‐body protein balance was more positive after minced beef than after beef steak consumption (29 ± 2 compared with 19 ± 3 mmol phenylalanine kg–1, respectively). However, skeletal muscle protein synthesis rates did not differ between treatments when assessed over a 6 h postprandial period. In conclusion, minced beef was more rapidly digested and absorbed than beef steak, which resulted in increased amino acid availability and greater postprandial protein retention.
These two original studies clearly demonstrate that variability in chewing efficiency, and thus post‐oral processing food structure, can directly affect the kinetics of protein hydrolysis and the bioavailability of amino acids. Although to our knowledge, there are no other similar studies within a more general adult population, one can hypothesize that differences in chewing behavior between adults, even with healthy dentition, may regulate amino acid bioavailability.
2.3. Interindividual Variability in Digestion of Proteins
2.3.1. Protein Digestibility
Protein quality evaluation aims to assess the contribution of dietary protein in satisfying the metabolic needs for nitrogen and essential amino acids.15 To this aim, the Food and Agriculture Organization (FAO) has proposed indexes for evaluating protein quality.15, 16, 17 Protein digestibility mirrors the extent of protein digestion and intestinal absorption and thus the amount of amino acids made available for metabolism. Protein digestibility has been shown to fluctuate significantly according to the diet, especially between those of developed and developing countries, where in developing countries the diet is richer in anti‐nutritional factors and in fibers that can limit protein digestibility.18
Table 1 summarizes the true ileal and fecal digestibility and the variability of the values obtained. The coefficients of variation (CV) shown in the table provide an estimation of the interindividual variability assuming that the analytical variability is lower. Interestingly, the two proteins with the lowest ileal digestibility (<85%), likely due to their compacted structure19, 20 and the presence of trypsin inhibitor in raw egg,19 present the highest CV, with 42.7% CV for a raw egg digestibility of 51.3%19 and 10.5% CV for a rapeseed isolate digestibility of 84.0%,20 thus indicating a high interindividual variability. This could be the result of a different response from the proteases toward anti‐nutritional factors and/or toward resistant protein, which could be a result of the probable dependence on the levels of enzyme secretion and/or activity among individuals. It should be noted that the high CV for rapeseed digestibility was due to a sole subject, who showed no reason to be excluded,20 but without whom the CV was decreased by 3 (CV of 3.6%) for a digestibility of 87.1%. For the other protein sources, all other CV are below 8% suggesting low interindividual variability. This is probably due to the fact that these CV relate to more easily digestible dietary proteins (>85%), and that the variation between individuals is low in relation to this high digestibility. It could be interesting to estimate the interindividual variability in other proteins showing lower values of digestibility, such as plant proteins within their vegetal matrix containing anti‐nutritional factors. Endogenous losses should also be estimated for each subject as this may vary across individuals. The true fecal digestibility presented in Table 1 has been estimated after correction for a constant value of obligatory endogenous losses, which likely alters real variability.
Table 1.
True ileal and fecal digestibility in humans. Interindividual variability is represented by the coefficient of variation (CV)
| Protein source | Reference | True digestibility (%) | |||
|---|---|---|---|---|---|
| n | Mean | SD | CV | ||
| Ileal digestibility | |||||
| Animal protein | |||||
| Skimmed milk pasteurised | [231] | 5 | 95.5 | 0.9 | 0.9 |
| Protein milk alone | [232] | 7 | 94.8 | 1.6 | 1.7 |
| Protein milk with fat | [232] | 9 | 94.5 | 3.0 | 3.2 |
| Protein milk with sucrose | [232] | 9 | 94.6 | 1.5 | 1.6 |
| Intact casein | [233] | 6 | 94.1 | 1.5 | 1.5 |
| Hydrolysed casein | [233] | 5 | 92.3 | 1.3 | 1.4 |
| Bovine meat, cooked at 55 °C, 5 min | [234] | 8 | 94.1 | 2.0 | 2.1 |
| Bovine meat, cooked at 90 °C, 30 min | [234] | 8 | 90.1 | 5.9 | 6.6 |
| Egg protein, raw | [19] | 5 | 51.3 | 21.9 | 42.7 |
| Egg protein, cooked | [19] | 5 | 90.9 | 1.8 | 2.0 |
| Plant protein | 0.0 | ||||
| Wheat toast (15N gluten) | [235] | 9 | 90.3 | 4.3 | 4.8 |
| Soy protein isolate | [236] | 11 | 91.8 | 1.9 | 2.1 |
| Soy protein isolate with sucrose | [236] | 10 | 90.9 | 2.2 | 2.4 |
| Pea flour (Pisum Sativum, Solara cultivar) | [237] | 7 | 89.4 | 1.1 | 1.2 |
| Pea globulins (Pisum Sativum, Baccara cultivar) | [238] | 9 | 94.0 | 2.5 | 2.7 |
| Pea globulins + albumins (Pisum Sativum, Baccara cultivar) | [238] | 8 | 89.9 | 4.0 | 4.4 |
| Lupin flour (Lupinus Albus, cultivar Arès) | [239] | 7 | 91.0 | 3.0 | 3.3 |
| Rapeseed isolate (Brassica Napus L., Goeland cultivar) | [20] | 7 | 84.0 | 8.8 | 10.5 |
| Fecal digestibility | |||||
| Animal protein | |||||
| Egg | [240] | – | 97.0 | 3.0 | 3.1 |
| Egg, spray‐dried | [241] | 5 | 92.2 | 1.6 | 1.7 |
| Milk, cottage cheese | [241] | 4 | 99.1 | 1.8 | 1.8 |
| Milk, cheese | [240] | – | 95.0 | 3.0 | 3.2 |
| Tuna, canned | [241] | 4 | 89.9 | 1.0 | 1.1 |
| Meat, fish | [240] | – | 94.0 | 6.0 | 6.4 |
| Plant protein | |||||
| Maize | [240] | – | 85.0 | 4.0 | 4.7 |
| Rice, polished | [240] | – | 88.0 | 5.0 | 5.7 |
| Wheat, whole | [240] | – | 86.0 | 4.0 | 4.7 |
| Wheat, refined | [240] | – | 96.0 | 7.0 | 7.3 |
| Wheat gluten | [241] | 4 | 93.8 | 3.0 | 3.2 |
| Oatmeal | [240] | – | 86.0 | 7.0 | 8.1 |
| Soy flour | [240] | – | 86.0 | 7.0 | 8.1 |
| Soy isolate | [241] | 4 | 94.8 | 4.4 | 4.6 |
| Peanut flour | [241] | 4 | 90.8 | 2.0 | 2.2 |
| Mixed protein source | |||||
| Low fiber control diet | [242] | 7 | 95.1 | 1.5 | 1.6 |
| High fiber diet (coarse whole meal rye bread) | [242] | 7 | 90.7 | 1.4 | 1.5 |
| High fiber diet (fine whole meal rye bread) | [242] | 7 | 90.8 | 2.2 | 2.4 |
All ileal studies used 15N‐labelled protein sources ingested by healthy adults equipped by a naso‐ileal tube, except when noted.
Ileal digesta were collected in ileostomy patients.
True fecal digestibility was estimated by correcting apparent digestibility with a constant value of obligatory endogenous losses (9–12 mg N kg–1 body weight d–1).
2.3.2. Composition of the Intestinal Peptidome
Boutrou and colleagues21 equipped human volunteers with a double‐lumen nasogastric tube that migrated to the proximal jejunum to characterize the composition of the postprandial intestinal peptidome. Sample collection was performed for 6 h after the ingestion of 30 g 15N‐labeled casein or whey proteins. Nitrogen flow rates were measured, and peptides were identified by using mass spectrometry (MS). After casein ingestion, medium‐size peptides (750–1050 kDa) were released during 6 h, whereas larger peptides (1050–1800 kDa) were released from whey proteins in the first 3 h. Peptides originating from caseins that coagulate in the stomach and are released slowly in the small intestine, were observed all over the 6 h postprandial period, whereas the number of those released from whey proteins, which are “fast proteins” that remain soluble in the stomach and are rapidly transferred, digested, and absorbed in the small intestine, strongly peaked after 1 h and were nondetectable after 5 h. β‐Casein was the most important precursor of peptides, including bioactive peptides with various activities. The amounts of β‐casomorphins (β‐casein 57–, 58–, 59–, and 60–66) and β‐casein 108–113 released in the postprandial window were found in the lumen in concentrations sufficient to elicit the biological action of these peptides (i.e., opioid and antihypertensive, respectively). Of note, the amounts of peptides derived from casein and whey protein digestion were characterized by high standard deviations (SDs) (i.e., mostly significantly above 50%) across all peptide sizes and digestion times, which suggests a large variability in protein digestion among the subjects. These results indicate that omics analytical strategies (including peptidomics) potentially allow a holistic visualization of interindividual variability in food processing by the GIT, which takes into account a wide range of molecules (or groups of molecules) with potentially different degrees of interindividual variability.
2.3.3. Protease Activity
When comparing pepsin activity in subjects with ulcers versus healthy subjects, Le Veen and Hallinger22 observed dramatic interindividual variability. In healthy subjects, pepsin activity ranged from 188 to 8600 U mL–1 of gastric juice, whereas for patients suffering from ulcers, activities ranging from 940 U to 11 200 U mL–1. Similarly, Janowitz and Hollander23 found pepsin output ranging from 0 to 8335 HgbU h–1 in healthy individuals with a mean value of 4119 HgbU mL–1. Interestingly, these authors reported a positive correlation between the amounts of pepsin and acid secreted per hour, proposing that the peptic and parietal cells respond to common influences of vagal origin. These conclusions provide an early indication of the powerful potential of variables demonstrating high interindividual variability in human studies to propose relevant mechanistic hypothesises.
Of note, pepsin activity was at its lowest before food ingestion then gradually increased over the day.23 The changes are partly attributed to differences in pH of the gastric aspirates, the lowest values being observed when the pH was at its highest. Therefore, among the factors that can explain interindividual variability among subjects for pepsin activity in the stomach, the evolution of the pepsin secretion over the day also needs to be taken into account within each individual.
Using N‐benzoyl‐l‐tyrosine ethyl ester as substrate, Rick24 found chymotrypsin activities ranging from 28.4 to 154 U min–1 in adults. Parallel evolution of chymotrypsin and trypsin was described by Norman et al.25 in infants, who determined a trypsin/chymotrypsin ratio ranging from 0.5 to 2.0, varying slightly during the test meal within‐subject, but greatly from one infant to the other.
2.4. Interindividual Variability in Intestinal Absorption of Amino Acids and Peptides
2.4.1. Genetic Polymorphisms
The lack of intestinal absorption of amino acids or peptides is associated with a range of disorders, including cystinuria, Hartnup disorder, inflammatory bowel diseases (IBDs), obesity, and abnormal dietary behavior. However, most of these disorders are multifactorial and a direct link between them and intestinal absorption is difficult to establish. The following paragraphs present some of these cases.
Cystinuria is an autosomal inherited metabolic disorder characterized by impaired transport of cystine and dibasic amino acids in the proximal renal tubule and the GIT. Mutations in the amino acid transporters SLC3A1 and SLC7A9T are responsible for cystinuria.26, 27
Mutations in the neutral amino acid transporter B0AT1 (SLC6A19) is known to harbor mutations in Hartnup disorder. Hartnup disorder, an autosomal recessive defect, named after a English family with this condition in 1956, and results from impaired transport of neutral amino acids across epithelial cells in renal proximal tubules and intestinal mucosa. Symptoms include transient manifestations of pellagra (rashes), cerebellar ataxia, and psychosis. Using homozygosis mapping in the original family in whom Hartnup disorder was discovered, the location of one causative gene on chromosome 5p15 was confirmed. This region is homologous to the area of mouse chromosome 13 that encodes the sodium‐dependent amino acid transporter B(0)AT1. The protein product of SLC6A19, the Hartnup transporter, is expressed primarily in the intestine and renal proximal tubule, and functions as a neutral amino acid transporter.28 The lack of intestinal tryptophan transport appears to be responsible for most, if not all, clinical phenotypes of Hartnup disorder. In agreement with this observation, clinical symptoms are mainly observed in individuals with lower than normal plasma amino acid concentrations.10
Polymorphisms in the intestinal transporter PEPT1 (encoded by SLC15A1) are associated with IBDs, such as Crohn's disease (CD) and ulcerative colitis (UC), as the polymorphisms affect the gut epithelial barrier and interactions with bacteria. PEPT1 mediates intracellular uptake of bacterial products that can induce inflammation and NF‐κB activation upon binding to NOD2, a protein sensing bacterial peptidoglycan and stimulating host immune response, which is often mutated in CD. Twelve SLC15A1 single nucleotide polymorphisms (SNPs) were genotyped in a study conducted in 1783 individuals from two cohorts of Swedish and Finnish IBD patients and controls. The common allele (C) of a coding polymorphism (rs2297322, Ser117Asn) was associated with CD susceptibility in both cohorts, but with genetic effects in opposite directions (risk and protection, respectively). The best evidence of association was found in both populations when the analysis was performed on individuals not carrying NOD2 common risk alleles (Swedish cohort: OR 1.97, 95% confidence interval [CI] 1.34–2.92; Finnish cohort: OR 0.63, 95% CI 0.44–0.90). Compared to PEPT1‐Asn117 the PEPT1 variant encoded by the C allele (PEPT1‐Ser117) was associated with reduced signaling downstream of NOD2. Therefore, a functional polymorphism in SLC15A1 might modulate the inflammatory and antibacterial response in IBDs. However, further studies are needed to identify if the polymorphism truly contributes to disease susceptibility.29
Polymorphisms in neutral and cationic amino acid transporter ATB0+ (SLC6A14) are associated with obesity. Single nucleotide polymorphism (SNP) 22 510 C>G (rs2071877), located in the three untranslated regions of SLC6A14, has been strongly associated with obesity in the Finnish population. Furthermore, a haplotype including this SNP, together with SNP 20 649 C>T (rs2011162, located in intron 12), showed a significantly different allele frequency in obese versus control subjects. A study by Durand et al.30 genotyped SNPs 20 649 C>T and 22 510 C>G in 1267 obese and in 649 unrelated non‐obese normo‐glycemic subjects, all of French‐Caucasian origin. Both SNPs were associated with obesity ([OR] 1.23, 95% CI 1.04–1.45 and 1.36, 1.16–1.59, respectively). These results confirmed the observation of Suviolahti et al.31 in Finnish men (117 obese and 182 control subjects) as the most frequent alleles for both SNPs were associated with increased obesity. Durand et al.30 analyzed men and women separately and concluded that only women appeared to contribute to the association of obesity with SNP 20 649 C>T. On the other hand, both sexes contributed to the association with SNP 22 510 C>G.
Interestingly, Durand et al.30 further explored the potential contribution of SLC6A14 variants in dietary behavior. This study was motivated by the fact that SLC6A14 encodes for an amino acid transporter that may affect tryptophan availability for serotonin synthesis and that serotonin is a neurotransmitter involved in a wide spectrum of behaviors, including dietary habits. Applying the Three‐Factor Eating Questionnaire (TFEQ)32 to an obese cohort to measure hunger and disinhibition scores, SNPs in SLC6A14 were associated with the dietary behavior of obese women (BMIs 30–40 kg m–2), but not of morbidly obese women (BMI > 40 kg m–2). Moderately obese women homozygous for the at‐risk allele 22 510 C>G had also higher hunger and disinhibition scores. A similar result was observed with both scores for SNP 20 649 C>T. However, no association was observed when all obese adults were pooled as well as in the subgroup of obese adult men.
The mechanism by which SLC6A14 may participate in obesity and possibly in the control of dietary behavior is still uncertain and further studies are required. Nevertheless, genes involved in the serotonin signaling pathway have been associated with several obesity‐related phenotypes.30 However, the association between 22 510 C>G and the disinhibition and hunger scores of the validated TEFQ questionnaire, although modest and only observed in moderately obese women, suggests a role of SLC6A14 on food intake,27 in particular since disinhibition determines the loss of control over food intake under a variety of situations and is well correlated to binge eating and overeating.33
3. Fat
3.1. Overview of Fat Processing by the GIT
Triacylglycerols (TAGs) (also named triglycerides) represent the major class of lipids in food. Two additional classes of lipids, phospholipids (PLs) (in particular glycerophospholipids‐GPLs), and cholesterol esters (CEs) are also important sources of dietary lipids. Fat digestion takes place in three phases, namely the oral, gastric, and intestinal phases,34, 35 starting in the mouth with mastication.
The free fatty acids (FAs) present in the masticated food bolus activate sensory receptors on the taste buds.36 Activation of these receptors leads to the calcium‐mediated release of neurotransmitters and hormones that signal the presence of dietary fat to the brain.37 Oral FA signaling also modulates a range of nutritional factors including fat intake, appetite regulation, absorption of fat‐soluble nutrients such as vitamin A, and the storage of lipids in the GIT.38
Processing of TAGs by the GIT requires that the ingested fats be hydrolyzed into FAs and monoacylglycerols (MAGs). Because of their hydrophobicity TAGs need to be emulsified to be made available for hydrolysis.35 Only 10–30% of dietary fat is hydrolyzed in the stomach, which helps the digestive process create an emulsion by mechanical action.34, 39, 40 Gastric lipase hydrolyzes TAGs mainly at position sn‐3 containing short‐ and medium‐chain FAs (< C12) that are directly transferred to the blood.41
Fats enter the duodenum and stimulate the secretion of the peptides cholecystokinin (CCK) and secretin by enteroendocrine cells, stimulating the contraction of the gallbladder and the release of bile and pancreatic juice (containing lipase and colipase). Bile acts as a surfactant producing an emulsion of fine oil droplets increasing surface area and thus rendering lipids more easily accessible to the pancreatic enzymes.34, 42, 43, 44
After digestion, the hydrolyzed lipid‐soluble components (long‐chain FAs, 2‐MAGs, lyso‐PLs, free cholesterol (FC)) integrate with bile salts into mixed micelles to diffuse between the intestinal microvilli to interact with the luminal surface of the enterocytes. The FAs produced by fat digestion actively modulate lipid processing by the GIT. The binding of FAs to receptors on enteroendocrine cells promotes the activation of satiety hormones (leptin, CCK, peptide YY (PYY), glucagon‐like peptide 1 (GLP‐1)), the inhibition of hunger hormones (ghrelin), and the intestinal uptake of FAs.45 The absorption of lipids is a complex, two‐step process including lipid transfer from the intestinal lumen into enterocytes and their subsequent secretion into lymph/portal vein. The transport is mostly an active process driven by protein transporters, particularly at lower concentrations and may also involve diffusion when free FA concentration in the lumen exceeds that within cells.46 Protein‐mediated FA intestinal uptake involves CD36, FA transport protein 4,47 intestinal FA‐binding protein (I‐FABP; FABP2), and liver‐type FABP (LFABP; FABP1).48 The absorption of FAs and MAGs is highly efficient, with more than 95% of ingested dietary lipid being absorbed in vivo.49
Although the entire length of the small intestine can absorb cholesterol from the lumen, the major sites of absorption are the duodenum and proximal jejunum. Niemann–Pick C1‐Like 1 (NPC1L1) and ATP‐binding cassette transporters G5 and G8 (ABCG5/ABCG8) serve as opposing gatekeepers in the liver and intestine to tightly regulate whole body sterol homeostasis.50, 51 The efficiency of lipids absorption is determined by the net effect between influx and efflux of intraluminal molecules (mainly FAs and cholesterol) crossing the brush border membrane of the enterocyte.52 Cholesterol secretion by enterocytes occurs by apoB‐dependent (chylomicrons (CMs)) and apoB‐independent (HDL) pathways.53 Most of the absorbed cholesterol molecules coming from the diet and from the bile, reach the endoplasmic reticulum, where acyl‐CoA:cholesterol acyltransferase 2 (ACAT2) esterifies them. CEs are then assembled into the core of CM particles. Once across the intestinal microvillus membrane, FAs and MAGs are resynthesized to TAGs by acyl‐coenzyme A:monoacylglycerol acyltransferases (MGAT1, 2, and 5) and acyl‐coenzyme A:diacylglycerol acyltransferases (DGAT1 and 2). CMs assembly and secretion is critically dependent on microsomal triglyceride transfer protein (MTP) and apoB‐48.54, 55 The transmembrane 6 super family member 2 protein (TM6SF2) is also involved in packaging and secretion of dietary lipids in TAG‐rich lipoproteins.
Among the variables presented in the last paragraph of the Introduction, a search of the literature has identified convincing evidence for interindividuality in oral processing, digestion, and intestinal absorption of fat by the GIT for the following variables: FA sensing, genetic polymorphisms, saliva composition, BMI, diet, and circadian rhythm. These variables are summarized in a schematic manner in Figure 1.
Figure 1.
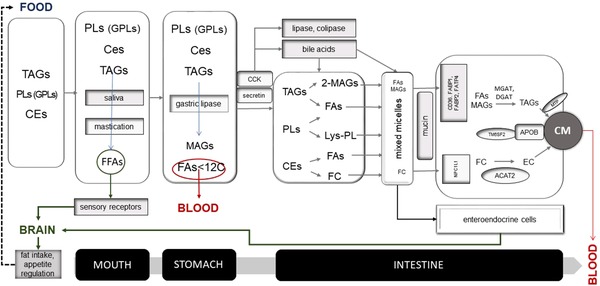
Overview of molecular variables contributing to inter‐individual variability in the processing of fat by the GIT.
3.2. Interindividual Variability in Oral Processing of Fat
3.2.1. FA Sensing
The first evidence for interindividual variability in fat tasting was published in 1997 by Tepper and Nurse.56 Their study revealed that, in contrast to non‐tasters, medium‐ and supertasters of the bitter compound 6‐n‐propylthiourail (PROP) were able to discriminate a salad dressing containing 40% fat from a dressing containing 10% fat. In a randomized, single blind, placebo‐controlled crossover design study, Kamphuis et al.57 later identified linoleic acid tasters and linoleic acid non‐tasters based on their ability to identify the presence of 10 µm linoleic acid. Mattes58 measured the oral detection thresholds for a range of FAs (linoleic acid, stearic acid, lauric acid, caproic acid) in healthy subjects. These thresholds covered 3–4 orders of magnitudes ranging from below 0.001% (w/v) to above 1% (w/v). Galindo et al.59 measured threshold concentrations for “fatty” orosensation between 0.1 and 0.4 mm for a range of FAs (C18:1–C20:4). The “scratchy” orosensation was detected at higher concentrations up to 2.2 mm and showed significantly higher interindividual variance relative to the “fatty” orosensation. Steward et al.60 determined the oral FA sensitivity of healthy subjects, which ranged from 0.02 to 6.4 mm and spanned two orders of magnitude for each of the FA tested (lauric acid, stearic acid, oleic acid, linoleic acid). Of note, within the group of hyposensitive subjects reported, significantly higher intake of total energy, fat, and carbohydrates (but not proteins), and monounsaturated and polyunsaturated fat was observed.
The results by Steward et al.60 were confirmed and complemented by a later study integrating dietary habits to the range of variables associated with oral FA sensing.61 In this study, the subjects were first characterized in their response to oleic acid and classified as hyposensitive and hypersensitive. Compared to the hypersensitive group, the hyposensitive group scored lower in the fat ranking test, consumed more energy, total fat (but not carbohydrates and proteins), saturated fat, and polyunsaturated fat. The dietary habits also differed, the hyposensitive group of subjects consuming greater amounts of full‐fat dairy, saturated fat from dairy, meat, eggs, and spreads as well as saturated fat from spreads. Hyposensitive subjects also consumed less low‐fat cheese. These differences in intake of foods and nutrients translated into differences into dietary habits, the hyposensitive subjects being less likely to adopt healthy habits such as substituting red meat with white meat and avoiding eating saturated fats.
3.2.2. Diet
Interindividual variability in fat sensing and intake might be confounded by the dietary patterns of the subjects. Mattes62 demonstrated that the acceptability of dietary fat during a fat reduction program is influenced by the composition of the diet, with the hedonic rating for high‐fat foods declining in subjects deprived of sensory exposure to fats. Later Stewart and Keast63 showed that subjects having consumed a low‐fat diet during 4‐weeks had an increased taste sensitivity to oleic acid.
Mattes64 has also investigated the impact of premeal fat stimuli on postprandial lipemia. Postprandial lipemia is a complex process that follows biphasic kinetics. The first phase, peaking within the first 30 min postprandially, results from hormonal signaling events preparing the organism for digestion of the meal. This phase releases TAGs in the blood that appear to be originating from intestinal lipid stores produced by the last previous meal. The second phase (peaking at 3–5 h) reflects the appearance of lipid in the blood resulting from the morning meal and de novo synthesis. Brief oral stimulation of humans with fat not only has a significant effect on postprandial lipemia, but is also characterized by significant interindividual variability. In particular, Mattes64 showed that a subgroup of subjects responded to a 10 s oral stimulation with cream cheese, by producing a significant postprandial TAGs response after the ingestion of safflower oil.
This last work illustrates the complex interactions taking place between food and the organism, which leads to individual responses. Variability does indeed not only result from differences in the genetic (see below) or health status of the subjects, but also depends on metabolic and hormonal stimulations that act within the short‐term time scale and may be significantly influenced by the dietary behavior of each subject.
3.2.3. Saliva Composition
Variation in the response of humans to the ingestion of FAs may be due to difference in salivary composition. Neyraud et al.65 investigated the variability of human saliva composition in human subjects for a range of parameters by comparing intrasubject variability with intersubject variability, as well as unstimulated saliva with saliva stimulated by chewing a piece of Parafilm. For all parameters investigated the interindividual variability was larger than the intrasubject variability. These parameters included salivary flow, protein content, lipolysis, amylolysis, proteolysis, lysozyme, anti‐oxidant activity, lipocalin, and uric acid. Of note, salivary flow, as well as lipolysis, lipocalin, proteolysis, and anti‐oxidant activity were stable over an 8‐month period. Similarly, fattiness perception and fat liking evidenced large interindividual variability. Lipolysis and fat perception as well as saliva flow rate and fat liking correlated positively. In conclusion, salivary characteristics and fat perception not only demonstrate significant interindividual variability, but their association suggests the composition of saliva may affect fat perception and liking.
3.2.4. Body–Mass Index
Evidence that BMI might contribute to the oral sensitivity of humans to FAs was provided in 2010 by Steward et al.60 A cohort of healthy subjects was evaluated for sensitivity to FAs for oleic, linoleic, and lauric acids. The hypersensitive subjects had significantly higher BMIs than the hyposensitive subjects. The same research group later confirmed these conclusions regarding the sensitivity to oleic acid of subjects with a narrower BMI range.61
Stewart and colleagues66 subsequently conducted a study in overweight/obese and lean men to identify difference in oleic acid sensing threshold. The detection threshold for oleic acid was significantly higher in the overweight/obese test group (7.9 mmol L–1) compared to the lean group (4.1 mmol L–1). Of note, the observed difference of 3.8 mmol L–1 in the mean threshold for oleic acid sensing between the two BMI groups, should be compared with the larger range of thresholds characterizing each of the subjects in this study (1 and 12 mmol L–1). Also, this small difference should be compared with the orders of magnitude of differences reported between individuals in Section 3.2.2. This shows that BMI contributes only to a minor extent to interindividual variability in fat sensing. In support to this conclusion, Stewart and Keast63 could not identify differences in the threshold to oleic acid sensing between one group of obese/overweight subjects and a group of lean subjects. These negative results were later confirmed by Tucker et al.67 However, Stewart and Keast63 also demonstrated that a high‐fat diet decreased taste sensitivity to oleic acid in the lean group of subjects but not in the overweight/obese group.
Taken together, these studies suggest that the contribution of the BMI to the interindividual variability of the oral sensibility to FAs plays a minimal role and that this contribution is modulated by the diet.
3.2.5. Genetic Polymorphisms
Family and twins studies, reviewed by Reed et al.,68 revealed that food preferences are largely determined by cultural transmission and individual experience. Interestingly, however, fat intake is more similar among monozygotic twins than among dizygotic twins, suggesting that heritability does contribute to interindividual variability in fat intake.
The first study showing an association between a gene and fat ingestive behavior in humans was published by Keller et al.69 The CD36 FA translocase is a ubiquitous membrane‐bound protein involved in a range of physiological immunological and metabolic processes, including the oral detection of FAs. The authors addressed association between five common polymorphisms of CD36 and the oral detection of fat in African‐American subjects using Italian dressings prepared with varying amounts of canola oil. A G>A variation in exon 1A (rs1761667) that reduces CD36 expression and is present in 45% of the subjects was associated with an increased ability to perceive creaminess. Also, a C>T variation in intron 11 (rs1527483) present in 12% of the subjects was associated with an increased ability to perceive fat content. The consideration that the ingestive behavior associated with variants of CD36 might impact on metabolism was indicated in the same study, with individuals with two (D/D) deletions at rs3840546 (12% of the subjects) having significant higher BMIs than I/I homozygotes and I/D heterozygotes. Taken together, this study provided evidence that polymorphisms in CD36 contribute to interindividual differences in the perception of fat and, further, that these differences impact on body weight via regulation of fat intake. Of note, however, the authors commented on work by other groups reporting opposite behavior associated with CD36 polymorphisms in other populations, suggesting the existence of additional mechanisms modulating the interindividual variability to fat sensing associated with CD3.
The findings of Keller et al.69 were later confirmed by Pepino et al.70 for the rs1761667 CD36 polymorphism in obese subjects. In this study, the detection threshold for oleic acid and triolein was significantly (eightfold) lower for subjects homozygous for the G allele than subjects homozygous for the A allele. Total energy, fat consumption, fat preference scores, and food cravings were, however, similar among the different alleles.
Genome‐wide association studies (GWAS) allow for a screening and identification of genes associated with particular phenotypes. Tanaka et al.71 made use of this approach, using a meta‐analysis of 12 observational studies, to identify genetic variants associated with macronutrient intake. A variant in the chromosome 19 locus (rs838145) was associated with lower fat consumption. A candidate gene in this region, fibroblast growth factor 21 (FGF21), encodes for a hormone produced by the liver and involved in lipid metabolism. Interestingly, FGF21 acts as a negative regulator of bile acid (BA) synthesis72 and polymorphisms in this protein might therefore contribute to interindividual variation in uptake of lipids such as cholesterol.
Further details on the genetic aspects of FA perception can be read in the reviews by Reed and Xia73 and Running et al.74
3.3. Interindividual Variability in Digestion of Fat
3.3.1. Diet
The quantity of gastric lipase excreted is influenced by the type of meal (a high‐fat diet elicits a higher output of gastric lipase than a low‐fat diet)75 and the given period (postprandial values are higher than fasting ones), whether in adults, children, or infants.39
3.3.2. Genetic Polymorphisms
The literature does not provide strong evidence for a role of genetic polymorphisms in fat digestion. At present, four common SNPs in CLPS (rs2766597, rs41270082, rs3748050, and rs3748051) have been investigated but evidence for their association with fat intake or obesity, not fat digestion, has so far only been provided by rodent models.76
3.4. Interindividual Variability in Intestinal Absorption of Fat
Usually, the entire fat digestion and absorption process lasts for 16–24 h if no more food is consumed after the initial meal.35, 77 The gastrointestinal transit of a standardized meal, containing 33% fat (mainly from dairy products), was analyzed by Madsen78 in 33 healthy volunteers of both genders aged from 23 to 65 years. Gastric emptying and intestinal transit times were not influenced by age, sex, or BMI. The interindividual variability in gastric emptying and small intestinal transit times was, however, substantial; by contrast, no difference was observed between subjects in colonic transit time. This data indicates that the factors governing interindividual variability in fat digestion are not understood and might be of genetic origin.
3.4.1. Diet
The composition of the duodenal fluids varies according to the nutritional state (fasted or fed) and the characteristics of ingested food. Intestinal lipid absorption has been shown to exhibit diurnal variations in humans, being maximized at mealtime.79 Notwithstanding, several parameters such as pH, lipolytic products, bile salts, phospholipids, osmolality, and surface tension display fluctuating patterns with high intersubject variability in response to the same meal.80
Different food components are known to reduce plasma cholesterol levels by interfering with diffusion of luminal cholesterol to the gut epithelium and/or inhibiting molecular mechanisms responsible for cholesterol uptake by the enterocyte. The capacity of soluble fibers to delay cholesterol absorption, probably via the reduction of biliary emulsification of cholesterol and the consequent delay of its diffusion from the lumen to gut epithelial cells,81 has been demonstrated in man.82 A likely mechanism of action of plant sterols is their competition with cholesterol for solubilization in micelles within the intestinal lumen, thus reducing the amount of cholesterol available for absorption, without any alteration of ABCG5 or ABCG8 expression in mice.83 A reduced saturated fat diet and a reduced saturated fat/high‐fiber diet have been shown to decrease cholesterol absorption by 7% and 10%, respectively, in ileostomy subjects.84 In cultured human enterocytes, eicosapentaenoic (EPA), docosahexaenoic (DHA), and PUFAs in general reduce cholesterol absorption by downregulating NPC1L1 protein expression.85, 86
The presence of dietary fat upregulates the expression of CD36,87 which plays a key role in the uptake of FAs.88 McKimmie et al.89 evidenced a considerable variability in the efficiency of absorption of individual FAs by the healthy adult gut. Absorption efficiency of saturated FAs is lower than MUFAs, or PUFAs, and is, as well, inversely correlated to the number of carbons in the fatty acyl chains. Different solubilization into BA mixed micelles, micellar diffusion through the unstirred water layer, and uptake by the enterocyte brush‐border membrane could be determinant of the observed differential absorption of FA, which is not related to gender or BMI.
During fasting, the enterocytes produce mostly VLDL. In fasting state, deprivation of lipid substrate causes degradation of ApoB‐48 and inhibition of nascent CM assembly. In postprandial state, the pre‐CM is transported via a pre‐CM transport vesicle to the Golgi apparatus90 and a mature CM is formed through acquisition of more neutral lipids. CM production is a highly regulated process, and postprandial plasma CM response displays a high interindividual variability. The amount and type of ingested fat is the major driver but other factors as non‐lipid nutritional factors (carbohydrates, proteins, fibers), hormones (insulin, GLP1, GLP2), nutraceuticals, and therapeutic interventions can significantly contribute.49
Overall, although food components, diet, and fasting state are not components of the interindividual variability, they could be confounders in nutritional studies aimed to evaluate variation in lipid digestion, absorption, and transport.
3.4.2. CD36 Expression
Diseases that impair secretion of bile, such as biliary obstruction or liver diseases, as well as diseases affecting the secretion of pancreatic enzymes, such as cystic fibrosis, lead to severe malabsorption of fats.91 In patients with liver cirrhosis, upregulation of intestinal glycosylated CD36 causes rapid absorption of LCFAs.92
3.4.3. Circadian Rhythm
Diurnal variation93 linked to fasting and postprandial states94 of BA synthesis rate also contributes to variability of absorption of dietary lipids. CM production undergoes diurnal variation, with concentration highest at night after feeding. This is coupled to changes in the gene expression of key proteins including MTP and ApoB, and disruptions in circadian rhythms have been proposed to contribute to dyslipidemia and insulin resistance.95
3.4.4. Body–Mass Index
Secretion of CCK and other gastrointestinal hormones, which is induced by cell surface receptor signaling stimulated by FAs,96, 97 is affected by BMI. Little et al.98, 99 found an inverse correlation between the number of CCK‐immunoreactive cells and BMI, which confirms that the gastrointestinal hormone responses to fats are attenuated in obesity, leading to impaired energy intake regulation with subsequent weight gain.
3.4.5. Genetic Polymorphisms Affecting Cholesterol Absorption and Serum TAGs
Interindividual variability is thought to also be due to genetic factors likely involving numerous genes implicated in lipid absorption, secretion, and clearance.
Given the fact that dozens of proteins are involved in BA synthesis and handling in the mammalian system, it can be anticipated that there are also numerous genetic factors underlying variability in BA profile and physiological processes sensitive to BA, including cholesterol absorption. Cholesterol absorption may also be modulated by the mucous coating the intestinal mucosa. Mucous is a diffusion‐limiting barrier, especially for cholesterol that may be extensively bound to surface mucins prior to transfer into the enterocyte. Physiological quantities of epithelial mucin are necessary for normal intestinal uptake and absorption of cholesterol in mice.100 Several polymorphisms in genes encoding for intestinal mucin have been identified, but their impact on intestinal absorption is still unknown.
Intestinal permeability and colon transit are affected by intraluminal BA. In particular, the G protein‐coupled bile acid receptor 1 (GPBAR1), which is expressed in the enteric nervous system and the enteroendocrine cells, has been demonstrated to mediate the prokinetic actions of BAs in a murine model.101, 102 In humans, there is evidence that genetic variation in GPBAR1 affects colon transit and total excretion of fecal BAs, predisposing affected subjects to irritable bowel syndrome (IBS).103
ACAT2 expression is restricted to the small intestine and liver,104 and its deficiency reduces cholesterol absorption rendering mice resistant to diet‐induced hypercholesterolemia, gallstone formation, and atherosclerosis.105, 106 He et al.107 showed that, in humans, three ACAT2 polymorphisms, 41A>G (Glu>Gly), 734C>T (Thr>Ile), and IVS4‐57_58 ins48 bp (D/I), associate with plasma lipid levels and coronary artery disease (CAD) susceptibility, but that their effects were not consistent across genders and ethnic groups. In a further study, the enzymatic activity of mutant Glu(14)Gly was found approximately two times higher than the wild‐type activity and was associated to plasma lipid levels and CAD risk.108 ACAT2 expression is restricted to the small intestine and liver,104 and its deficiency reduces cholesterol absorption rendering mice resistant to diet‐induced hypercholesterolemia, gallstone formation, and atherosclerosis.105, 106
An allele of the FABP‐2 promoter consisting of three polymorphisms located within 110 bp 5′ of the transcription initiation site has been described by Geschonke et al.,109 who evidenced an influence of the promoter polymorphism on the kinetics of the postprandial increase of TAGs. Another human genetic variant in FABP‐2, in which an alanine at codon 54 is substituted by a threonine, is associated with increased postprandial plasma TAGs.110 Increased excretion of TAGs in the feces of pPLA2‐KO mice on a high‐fat diet indicates that pPLA2 deficiency has a greater effect on the absorption of TAGs than PL hydrolysis,111 which is probably compensated for by other phospholipases.112 Polymorphisms in CD36 are associated with plasma vitamin E113 and carotenoid concentrations in humans.114 At present, the effect of CD36 variation on FA absorption has not been investigated. It is worth noting that in CD36 null mice there is a defect in the free FA uptake by the proximal intestine, which is compensated for by uptake in the distal intestine.115
As observed in MGAT2 or DGAT1 knockout animals, variation in MGAT and DGAT seems to have no effect on overall fat absorption but it delays transport of CMs into circulation.116 Intestine‐specific MTP ablation significantly reduces cholesterol absorption in mice.117 Mutations in the coding region of MTP have been reported to abolish the production of apoB‐containing lipoproteins and to lead to abetalipoproteinemia.118 Among the SNPs in MTP, the −493G/T in the promoter region has been mostly studied due to its plausible role in the modulation of lipid/lipoprotein profiles.119 Most but not all investigators reported an association between the MTP‐493T allele and low levels of serum total cholesterol (TC), LDL‐C, and apoB. The underlying reasons for the discrepancy in these findings might relate to differences in the populations studied, including differences in the diet.120 In MTP I128T SNP, the T128 variant confers reduced structural stability and decreased binding capacity to LDL particles. In Northern Europeans, the T128 allele is associated with lower plasma levels of TC and LDL‐C.121 This allele has been found to be protective against hyperlipidemia also in Chinese people.122 Another MTP polymorphism, Q297H, is associated with lipid homeostasis. Carriers of homozygous minor allele (297H) have significantly lower LDL‐C and non‐HDL‐C but higher risk for nonalcoholic fatty liver disease (NAFLD).123 Also carriers of a loss of function variant, (rs58542926, resulting in a glutamate to lysine substitution at residue 167) in TM6SF2 have an increased susceptibility to NAFLD with low circulating lipids.124 O'Hare et al.125 evidenced in two human cohorts that carriers of this TM6SF2 variant had improved fasting lipid profiles. In one cohort, variant carriers also exhibited significantly lower postprandial serum TAGs, suggestive of a role for TM6SF2 in the small intestine.
Familial hypobetalipoproteinemia, an autosomal codominant disorder, is characterized by molecular defects in APOB. Numerous polymorphisms of APOB have been described with an effect on lipid levels and cardiovascular risk. Nevertheless, not one of these associations has been consistently observed in a large number of studies, probably due to differences of ethnic groups and environmental factors.126
Finally, Desmarchelier et al.127 evaluated a combination of SNPs associated with the postprandial CM response in 33 healthy male volunteers. In this population, most of the interindividual variability in the postprandial CM response to dietary fat could be explained by a combination of 42 SNPs in 23 genes. Although it is likely that other genes as well as epigenetic factors are involved in the interindividual variation of postprandial CM response, this study clearly highlights the complexity of the problem.
3.4.6. Genetic Polymorphisms Affecting Phytosterol Absorption
Phytosterols (PSs) are plant sterols structurally similar to cholesterol. As such the genetic polymorphisms impacting on the absorption of fat, may also modulate processing of PSs by the GIT. As reported above, variability in plasma PS concentration among individuals is a reflection of either dietary intake and/or PS handling.128, 129, 130 As demonstrated in large cohorts, plasma PS concentration varies within a five‐ to tenfold range among individuals,131, 132 with the differences in variability remaining stable over time for a given individual.133 In studies where dietary intake of PSs has been controlled, wide interindividuality in plasma PS concentrations has been established and repeatedly demonstrated,134 indicating interindividual variability in PS handling.135
The genetic profile has been demonstrated to account for 80% of the variability.133 To date the majority of research attempting to identify genetic variability as justification for interindividuality in plasma PS concentration has focused on identifying SNP in genes encoding for ABCG5, ABCG8, and NPC1L1.
Functionally severe mutations in genes encoding for either ABCG5 or ABCG8 transporters, results in the rare lipid metabolic disorder, phytosterolemia. As ABCG5 or ABCG8 regulate intestinal sterol via efflux, this condition is characterized by hyperabsorption and decreased biliary excretion of dietary sterols. ABCG5 and ABCG8 favor PS transport, as such 30–100‐fold increases in non‐cholesterol sterols within plasma can be observed in people with phytosterolemia.136, 137 Although phytosterolemia is representative of an extreme case, more common sequence variants in ABCG5 or ABCG8 may have more subtle effects on sterol metabolism and contribute to interindividual variation in the plasma concentrations of PSs. Although research within this area is currently limited due to a preferential focus on cholesterol handling with these transporters, several polymorphisms for both ABCG5 (Q604E) and ABCG8 (T400K, D19H, M429V, and A632V) have been investigated in relation to PS handling.
Limited work has investigated the ABCG5 polymorphism, Q604E. Gylling and colleagues,138 in a cohort of 263 (114 male, 119 female; 53.1 ± 0.5 years) mild to moderate hypercholesterolemic subjects (serum cholesterol < 7.5 mmol L–1), identified that the Q604E polymorphism was significantly associated with low serum sitosterol in men. However, interestingly this was not seen in women. Q604E has additionally been investigated in relation to identifying an association with both cholesterol‐standardized serum campesterol and sitosterol concentrations, and changes in serum PS concentration after consumption of phytostanols. However, the ABCG8 polymorphism T400K demonstrated the most significant relationship with these two variables in comparison with the ABCG5 Q605E and ABCG8 A632V polymorphisms.139
In further regard to the ABCG8 polymorphism T400K, within the same study, additional analysis by Plat et al.139 evaluated the association between the three different alleles of the ABCG8 T400K polymorphism (TT, TK, and KK). The researchers found that the TT genotype had the largest reduction in serum PS concentration during consumption of phytostanols esters. This is indicative that the functionality of the ABCG5/G8 heterodimer is reduced in subjects with the TT genotype compared to TK and KK genotypes. In addition, interestingly this relationship was observed with no relationship with serum lipid and lipoprotein concentrations, suggesting that changes in the reduced functionality of the ABCG5/G8 heterodimer in the TT genotype, mainly affects plasma sterol concentrations, but not cholesterol.
The ABCG8 polymorphism T400K, in exon 8, as well as the ABCG8 D19H polymorphism in exon 1 have been shown to have the most pronounced effects in interindividuality, with these two sequence variations (D19H and T400K) being associated with lower serum phytosterol concentrations in a normolipidemic populations, indicating lower sterol excretion.133
The D19H polymorphism has been associated with lower serum campesterol and sitosterol than subjects without the allele in both normal and hypercholesterolemic subjects (serum cholesterol < 7.5 mmol L–1).133, 138, 140 This finding has also been reflected within a systematic review and meta‐analysis of ABCG5/G8 polymorphisms and markers of cholesterol metabolism.141 Within a pooled dataset of four studies, Jakulj and colleagues141 reported that the ABCG8 D19H polymorphism results in decreased total plasma PS concentration. Interestingly, the prevalence of the D19H polymorphism has been found to be rare in Asian and particularly Japanese populations, in addition to the ABCG8 SNP A632V. This was highlighted by Miwa and colleagues,142 when drawing comparisons between Japanese and Caucasian populations.
Within the same study by Miwa and colleagues,142 a novel ABCG8 polymorphism, M429V, was identified, within a population of Japanese hypercholesterolemic patients (n = 100, 48 men and 52 women, aged 30–87 years). The researchers found that individuals with the M429V polymorphism had significantly higher sitosterol serum levels, than non‐carriers. To our knowledge, this is the only study to have identified and investigated the M429V polymorphism in relation to PS metabolism to date.
Significantly less has been investigated in NPC1L1 related polymorphisms than ABCG5/8, with the majority of work investigating NPC1L1 polymorphisms in relation to cholesterol. However, non‐synonymous sequence variations in NPC1L1 have been shown to be five times more common in low absorbers of sterols, including PS, than in high absorbers.143 Which of the sequence variations were associated with lower absorption, however, was not identified. Further work to identify specific NPC1L1 related SNP, and their relation to PS absorption and metabolism is thus clearly required.
4. Carbohydrates
4.1. Overview of Carbohydrate Processing by the GIT
Based on their chemical structure carbohydrates can be grouped in monosaccharides and disaccharides (both commonly referred to as sugars), oligosaccharides (composed of malto‐oligosaccharides and nondigestible oligosaccharides), polysaccharides (composed of starch and non‐starch polysaccharides polyols) and sugar alcohols.144 The most common carbohydrates in foods are glucose and fructose (among the monosaccharides), sucrose and lactose (disaccharides), raffinose, stachyose, and fructo‐oligosaccharides (oligosaccharides), starch, cellulose, hemicellulose, pectin, β‐glucans, fructans, mucilages, and algal polysaccharides (polysaccharides), and sorbitol, mannitol, xylitol, lactotol, and maltotol (sugar alcohols).145
After carbohydrates are ingested, chewed, and mixed with salivary amylase, among others salivary juices, the bolus is swallowed and passed to the stomach through the esophagus. Once in the stomach, the acid neutralizes the salivary amylase halting further digestion of the carbohydrates and the chyme is formed. After the stomach, the chyme enters the small intestine and the duodenum. The pancreas releases the enzyme pancreatic amylase, which breaks the polysaccharides down into disaccharides. Afterward, enzymes such as lactase, and maltase break down the disaccharides into monosaccharides, which are then absorbed in the small intestine.146
Carbohydrates can be classified based on their digestion and absorption in the small intestine.144 Digestible carbohydrates are digested and absorbed in the small intestine whereas nondigestible carbohydrates (referred to as dietary fibers) are not digested by the small intestine and reach the large intestine to be fermented by the colonic microbiota. Glucose and fructose are thus absorbed directly into the capillaries within the villi of the small intestine. Most of the blood glucose is not stored in the liver but rather, via the action of insulin, rapidly passes through to the muscle and adipose tissue. Fructose, independently of insulin, converts in the liver to glucose, lactate, and/or fatty acids before passing into the blood stream where it can be oxidized for energy in other tissues.147 Therefore, fructose is considered a less direct source of energy. Di‐, oligo‐, and poly‐saccharides must be hydrolyzed to monosaccharides to be absorbed. Fructose is, thus digested in the small intestine by sucrase to produce glucose and fructose. Similarly, lactose is digested by lactase in the small intestine to produce glucose and galactose. The digestion of oligosaccharides and polysaccharides is more complex, the contribution of the small and large intestine depending on the physico‐chemical property of each carbohydrate. For some of these carbohydrates (e.g., maltodextrin), the combination of amylases with enzyme cleaving disaccharides (maltase, sucrase, isomaltase, lactase) is sufficient to complete digestion and absorption of the resulting monosaccharides in the small intestine. Others carbohydrates cannot (e.g., carbohydrates containing glycosidic linkages) or only partially (e.g., the digestible fraction of starch) be digested by the small intestine. These molecules are therefore fermented to various degrees by the colonic microbiota.144 Of note, oligo‐ and polysaccharides are considered more satiating than sugars because of their effect on processes such as gastric emptying and their transformation into short chain fatty acids (SCFAs), which can increase the secretion of hormones and peptides from enteroendocrine cells that result in increased satiety.147 Finally, sugars alcohols are not processed by the intestinal digestive system and are absorbed directly, although to different extents, into the circulation. Although some sugar alcohols, such as erythrytol are absorbed unmodified, others, such as lactitol, are fermented by the colonic microbiota prior to absorption.148
Factors that are likely to have an impact on interindividual differences in the response of humans to carbohydrate ingestion, as illustrated by the variability in the postprandial glycemic response,149 are numerous, including genetics, lifestyle, insulin sensitivity, exocrine pancreatic, sugar transporters activity levels, oral processing, and human gut microbiome composition. However, clear evidence for this interindividual variability has only been documented for a small number of dietary carbohydrates.
Among the variables presented in the last paragraph of the Introduction, a search of the literature has identified convincing evidence for interindividual variability in oral processing, digestion, and intestinal absorption of carbohydrates by the GIT for the following variables: chewing, genetic polymorphisms, and gut microbiota.
4.2. Interindividual Variability in Oral Processing of Carbohydrates
4.2.1. Chewing
Chewing mechanically breaks down complex carbohydrates in the mouth and salivary amylase, secreted by the salivary glands, breaks down the starches into the disaccharide maltose. The degree of mastication differs significantly among individuals and may contribute to the significant interindividual variation observed in the glycemic response to a food, due to the impact it has on food structure, and specifically particle size (see also the section on proteins).
A suitable example of this is the work by Zhu et al.,150 who identified that the elderly require a greater number of chews to form a bolus before swallowing, and investigated the impact of this chewing behavior on glucose metabolism. In a randomized cross‐over trial, comparing 15 with 40 chewing movements before swallowing, plasma concentrations of glucose, insulin, and gastric inhibitory peptide (GIP) were significantly higher when 40 chews were made, with late postprandial period becoming significantly lower. Their results suggested that increased number of chews within the oral phase, controlled glucose metabolism post‐oral processing by modulating the release of glucose in the blood. Looking specifically at chewing on food structure, Ranawana et al.151 studied the impact of food particle size on gastric emptying and the glycemic response. Within this study, smaller particles caused a significantly greater glycemic response than the larger particles. The insulin response was also significantly greater for the small particles than the large particles. The gastric emptying latency phase, lag, and half‐time were significantly shorter for the small particles. As such, smaller particles were identified to produce faster gastric emptying and greater glycemic and insulin response. Furthermore, Ranawana et al.152 used rice as the model to establish if interindividual variances in mastication and resulting degree of particle breakdown had an impact on in vitro and in vivo glycemic potency. In a randomized crossover design, with 15 subjects, the particle size distribution and in vitro digestibility of individuals' chewed rice were established together with their in vivo blood glycemic response. The content in rapidly digested starch in the masticated boluses, were measured during in vitro digestion. The particle size distribution of masticated rice varied significantly between participants. In vitro digestion of rice diminished as particle size increased. The degree of particle size breakdown, as a result of mastication, correlated with the content in rapidly digested starch in the chewed food bolus and initial digestion rate in vitro. The amount of undigested food remaining at the end of 120‐min in vitro digestion associated significantly with the percentage of particles greater than 2000 µm in masticated rice. The percentage of particles smaller than 500 µm correlated significantly with the in vivo glycemic response at 30 min post‐ingestion, although not with the total incremental area under the blood glucose curve. The gradation of usual mastication could consequently potentially affect both the scale and pattern of the glycemic response and may partially explain their interindividual differences. However, despite such findings, when all the food related factors including ingested particle size (mastication) are controlled for, interindividual variations in the glycemic and insulin response have still been observed which suggests additional factors than just oral processing contribute to interindividual differences in the glycemic response.
4.3. Interindividual Variability in Digestion of Carbohydrates
4.3.1. Genetic Polymorphisms and Amylase Copy Numbers
Low serum amylase (hypoamylasemia) has been described in certain common cardiometabolic diseases such as diabetes and metabolic syndrome. All of these diseases appear to have a common etiology of inadequate insulin activity due to insulin resistance and/or reduced insulin secretion. Some clinical studies have indicated that salivary amylase may be preferentially diminished in obese individuals, while others have shown that pancreatic amylase may be preferentially reduced in diabetic subjects with insulin dependence. Obesity, as a condition that elicits cardiometabolic diseases relating to insulin resistance (major contribution), may actually be a common determinant for low serum amylase in a general population.153 However, regardless of the accumulated evidence, the clinical relevance of serum, salivary, and pancreatic amylase and the underlying mechanisms have not been fully clarified.153
The genes encoding for α‐amylase are positioned in a cluster on the chromosome that comprises of the salivary amylase gene (AMY1), two pancreatic α‐amylase genes (AMY2A and AMY2B), and an associated pseudo gene. AMY1 displays a copy number variation (CNV), which is directly proportional to the salivary α‐amylase amount in saliva.154 Populations that evolved under high‐starch diets versus low‐starch diets possess a higher average copy number of AMY1, which reveals an intense positive selection imposed by diet on amylase copy number during evolution. The impact of CNV of AMY1 on human metabolism is broad involving evolutionary aspects of diet‐gene interaction, the glycemic response after starch consumption, the modulatory action of α‐amylase inhibitors on starch digestion, effects on taste perception and satiety, and influence on psychosocial stress. Also, other factors, such as hydration status, psychosocial stress level, and short‐term dietary habits also influence the α‐amylase content in saliva, all contributing to intraindividuality.154
Haplotypes differ in their structure based on the odd or even content of AMY1, which in turn is coupled to the copy number of pancreatic AMY2A and AMY2B.155 Most haplotypes have one copy each of AMY2A and AMY2B and have an odd number of copies of AMY1. In contrast, haplotypes carrying an even number of AMY1 have rearrangements causing CNV of AMY2A/AMY2B. Human populations possess different proportions of the basic haplotype classes.155 CNVs in AMY1, which varies more largely than the pancreatic amylase gene (AMY2A and AMY2B), correlated well with salivary and serum amylase levels. Additionally, low CNV of AMY1, indicating low salivary amylase, was linked with low taste perception/satiety insulin resistance, postprandial hyperglycemia, and obesity.
4.4. Interindividual Variability in Intestinal Absorption of Carbohydrates
4.4.1. Genetic Polymorphisms and Lactose Intolerance
Lactose intolerance is a frequent digestive problem. If the lactase enzyme is absent (alactasia) or deficient (hypolactasia), undigested lactose reaches the colonic microbiota, resulting in the production of SCFAs, hydrogen, and methane.156
Lactose intolerance might be genetic (primary hypolactasia and primary congenital alactasia) or environmentally induced (secondary or acquired hypoalactasia). In both cases, symptoms are produced by deficient levels of lactase in the lining of the duodenum.157 In the situation of primary or secondary lactase shortage, lactose passes throughout the GIT undigested or partly digested by enzymes generated by the intestinal bacterial flora. As a result, the undigested lactose molecules and products of bacterial digestion cause symptoms of lactose intolerance, such as abdominal pain, flatulence, gas, bloating, and diarrhea.158 The biological mechanisms leading to lactose malabsorption have been well described within numerous investigations, and several genetic, endoscopic, and physiological diagnostic tests are available. Lactose intolerance depends not only on insufficient lactase enzyme, but also on the amount of lactose, gastrointestinal motility, intestinal flora, small intestinal bacterial overgrowth, and sensitivity of the GIT to the formation of gas and other fermentation products of lactose digestion. The intestinal lactase activity normally decays in children, although approximately one‐third of humans now retain this activity during their life time.158
Lactase persistence (LP) is now common mainly in people of northern European descent and is due to the inheritance of autosomal‐dominant polymorphisms that prevent the epigenetic decline in lactase expression during childhood. The identification of genetic variants linked with LP or lactase non‐persistence (LNP) permits molecular recognition of the genetic predisposition of adult‐onset hypolactasia by DNA sequencing or restriction fragment length polymorphism analysis.158 Lukito et al.159 established that many individuals of European ancestry with the hereditary LP possess the C/T‐13 910 SNP in allele rs49882359 of MCM6, a gene that regulates the expression of the lactase LCT gene. Other lactase persistent populations are found in Africa and the Middle East with diverse genetic variants. These SNPs mirror the nutrient‐dependent ecological adaptation that took place as an evolutionary consequence of the agricultural revolution, which included dairying practices. This said, the gastrointestinal symptoms due to lactose malabsorption in the small intestine correlate, although not always clearly, with LNP. The lactose‐hydrolyzing activity of the colonic microbiome allows lactose fermentation to take place, so that the disaccharide is, consequently, not found in feces. The production of SCFAs and gases (carbon dioxide, hydrogen, methane), which can lead to the onset of symptoms, are also dose dependent. Up to 25 g of lactose can typically be consumed by individuals with LNP. However, the food matrix, dietary pattern, characteristics of the microbiome, age, as well as other factors are likely to alter tolerance. Hence, the idea that lactose intolerance is a disorder or disease of individuals with LNP is flawed and contains a cultural perspective.159
Interindividual variability in the metabolism of lactose has recently been demonstrated by Vionnet et al.156 who measured metabolites derived from lactose in the blood and urine of human adults subjects having ingested milk. These authors showed that not only lactose, but also galactose and two liver metabolites derived from galactose, galactitol, and galactonate demonstrated significant interindividual variability. In particular, a bimodal distribution of the postprandial kinetics of galactonate and galactitol in serum was observed in serum after milk ingestion (i.e., high responders and low responders; Figure 2). In 92% of the cases, the galactonate and galactitol responses matched the LCT genotype at rs4988235 for lactase persistence. The remaining 8% were proposed to reflect subjects whose lactose metabolism was misclassified based on genetic testing. The authors therefore proposed that the measurement of these metabolites after a lactose overload could be a better marker of lactase activity.
Figure 2.
Postprandial kinetics of the lactose‐derived metabolites galactonate (left) and galactitol (right) in the serum of subjects having ingested acidified milk. The relative intensities of galactonate and galactitol were measured by GC–MS. The red lines represent subjects with the CC genotype at the MCM6 rs49882359 allele. The orange lines represent subjects with the CT genotype. The green lines represent subjects with the TT genotype. Adapted from Vionnet et al.
The management of lactose intolerance comprises of a lactose‐reduced diet and enzyme substitutes. This approach is effective if symptoms are independently associated to dairy products. However, lactose intolerance can be part of a bigger intolerance to variably absorbed, fermentable oligo‐, di‐, monosaccharides, and polyols (FODMAPs). This phenomenon is observed in at least half of the patients with irritable bowel syndrome (IBS), who then need not only to control their lactose intake but also to decrease their dietary FODMAPs to recover from gastrointestinal symptoms. Of note, the long‐term effects of a dairy‐free, low FODMAPs diet on nutritional health and the fecal microbiome of humans are unclear.157
4.4.2. Genetic Polymorphisms and Fructose Malabsorption
Fructose is absorbed in the small intestine via the glucose transporter (GLUT) 5, which possesses a limited capacity of absorption, and transported into the blood by GLUT2. Fructose can be metabolized in the small intestine and liver by enzymes such as fructokinase, aldolase B, and triokinase.160 Carbohydrate response element‐binding protein (ChREBP) is a transcription factor involved in glycolytic and lipogenic gene expression linked to carbohydrate consumption. ChREBP target genes are implicated in fructolysis (Glut5, fructokinase), glycolysis (Glut2, liver pyruvate kinase), and lipogenesis (acetyl CoA carboxylase, fatty acid synthase).160
A deficiency in ChREBP may be responsible for fructose intolerance, as it impairs fructose transport and metabolism, when there is excessive consumption of fructose.161 Additionally, defects in fructose transporters like GLUT5 and GLUT2 and reduced expression of the genes involved in fructose metabolism, are likely to be other causes of fructose malabsorption.
4.4.3. Genetic Polymorphisms and Glucose Transport
Vega Lopez et al.162 compared the glucose response of healthy adults elicited on three different occasions after the consumption of 50 g of available carbohydrate, bread, and glucose, and concluded that the intraindividual responses between participants differed by 43%. These authors also mentioned that understanding all the sources of variability would be helpful in better defining the utility of glycemic index values.
One source of interindividual variability could be genetics. The sodium/glucose co‐transporter (SGLT)‐1 protein is a rate‐limiting factor for absorption of glucose and galactose in the small intestine, and it uses transmembrane sodium gradients to drive the cellular uptake of these molecules. Mutations on SGLT1 (missense, nonsense, and frameshift) can lead to wakened cellular glucose transport and cause glucose–galactose malabsorption (GGM). In the general population, the functional consequences of variants related to intestinal glucose absorption are uncharacterized. Seidelmann et al.163 therefore attempted to identify SGLT1 variants and to describe their clinical consequences. Seidelman et al.163 concluded that missense variants in SGLT1 protect from diet‐induced hyperglycemia in numerous populations. Reduced intestinal glucose uptake may protect from long‐term cardiometabolic outcomes, providing support for therapies that target SGLT1 function to prevent and treat metabolic conditions.
4.4.4. Gut Microbiota
Human digestive enzymes are not able to break down most complex carbohydrates and plant polysaccharides. As an alternative, these polysaccharides are metabolized by microbes, which generate SCFAs, as a by‐product of fermentation of these complex carbohydrates including acetate, propionate, and butyrate, which can be utilized as energy source by the body.164 It is well recognized that dietary intake of non‐digestible carbohydrates affects microbial fermentation and total bacterial numbers in the colon. New evidence from molecular ecology has also revealed that the total and type of non‐digestible carbohydrates (e.g., resistant starch, non‐starch polysaccharides, and prebiotics) influences the species composition of the intestinal microbiota both in short‐term dietary interventions and in answer to usual long‐term dietary intake. Interindividual variation in gut microbiota might, partially, indicate differences in dietary intake, but the response of the gut microbiota to dietary change can also vary between individuals.165
Among the numerous genes that have been identified in the human gut microbiome, those that encrypt carbohydrate‐active enzymes are of specific interest, as these enzymes are mandatory to digest most of our complex collection of dietary polysaccharides.166
Zeevi et al.149 showed that an algorithm combining personal clinical and microbiome features, could predict the postprandial glucose response of human subjects to the ingestion of food. Further, this algorithm was successful to propose a personalized dietary plan able to lower the postprandial glycemic response in an independent cohort of 26 subjects. To develop the algorithm, 46 898 postprandial glycemic responses were measured. The responses were highly variable among the individuals even when they consumed the same standardized meals. Of note, the glycemic response is also characterized by a significant intraindividual variability.
The presence of Proteobacteria and Enterobacteriaceae in the GIT is linked to poor glycemic control, and with diseases related to metabolic syndrome such as obesity, insulin resistance, and impaired lipid profile.167 Actinobacteria associations differ as Zeevi et al.149 found a positive association with the postprandial glycemic response to both glucose and bread, while Wu et al.168 reported that this phylum is associated with a high‐fat, low‐fiber diet.
The diversity of the gut microbial composition is reduced in approximately all modern chronic conditions studied.169 The “Western diet,” rich in animal protein, fats, and artificial additives, and deficient in fiber, beneficial microbes, plant phytochemicals, vitamins, and minerals, is considered to drive this decrease in gut microbial diversity by promoting gut dysbiosis, a situation in which one or more potentially harmful microorganisms are predominant, therefore, creating a disease‐prone situation. Recent dietary intervention studies indicate that diets rich in plant‐based, simply processed fiber could quickly reverse the effects of meat‐based diets on the gut microbiome.169 However, despite the clear evidence for the impact of diet on the gut microbiome, host genetics as well as the diversity and complexity of the gut microbiome might impair its dietary modulation on a personalized base.169
As detailed above, a large number of human intervention studies have confirmed that the nutritional behavior can lead to statistically significant changes in the composition of the gut microbiota. Roberfroid et al.170 showed that changes in the microbiota composition, in particular increases in Bifidobacteria, are indicators of intestinal health. Food products that cause a selective change in the gut microbiota composition and activity strengthen normobiosis and produce important physiological effects in the colon and in extra‐intestinal compartments leading to reduced risk of intestinal and systemic pathologies.170 Despite the fact that numerous factors shape the microbial composition of the colon, the diet remains an important factor because most microorganisms in the colon obtain energy for their growth by degrading and absorbing complex dietary compounds, particularly dietary fibers. Non‐digestible carbohydrates that bypass digestion in the upper GIT have a broad range of structures whose diversity has not yet been completely appreciated, in particular with regards to their metabolism by the gut bacteria. In that regard, Hamaker and Tuncil171 introduced the concept that individual chemical structures, often contained in fiber molecules, are specifically recognized by metabolic gene clusters in bacterial genomes. The large number of structural variations in dietary fibers is due to multiple genetic and environmental factors including the anatomical structure of the grain or plant from which the fibers are derived, the macro‐organization of the fiber polymers, or the size distribution of the carbohydrates in oligosaccharides and small polysaccharides. The structural complexity of dietary fibers could allow bacteria to survive in the aggressive colonic environment.171
5. Vitamins
5.1. Overview of Vitamin Processing by the GIT
Although only required in small amounts, vitamins are essential nutrients whose main task is to participate in controlling catalytic functions involved in metabolism. Based on their solubility, they have been categorized as water (B and C vitamins) and fat‐soluble (vitamins A, E, D, K). Only in some individual cases (retinol, calciferol and niacin) is the human body able to synthesize the vitamin from corresponding precursors, the provitamins. For all other vitamins, their intake through diet is essential.8, 172 Vitamins are mainly absorbed in the upper two parts of the small intestine, the duodenum and the jejunum. The duodenum is the main absorption site for vitamins A, B1 (thiamin), B2 (riboflavin), B3 (niacin), B7 (biotin), B9 (folate), and the lipid soluble vitamins D, E, and K. To a lesser extent, these vitamins are also absorbed in the jejunum, which is the main absorption site for B5 (pantothenic acid) and B6 (pyridoxine). In the distal part of the small intestine, the ileum, vitamin B12 and vitamin C are absorbed, as well as to a lesser extent folate and vitamins D and K.173 Some vitamins like B1 (thiamine), B2 (riboflavin), B7 (Biotin), B9 (folate), and vitamin K are also produced by the large intestinal bacteria, and therefore the colon is another possible site of absorption of this vitamins.174, 175, 176
The GIT is separated from the circulatory system by a multilayer of epithelial and other cells, which impede the vitamins to pass this epithelial wall. In humans and animals, the transport of vitamins across the small intestine is biphasic: mainly by active transport via specific carriers at low concentrations, and predominantly by passive diffusion down the osmotic gradient at higher concentrations.8, 177 Intestinal transport occurs either via carrier‐mediated Na+ dependent mechanisms (e.g., biotin, pantothenic acid) or via an acidic pH‐dependent, Na+‐independent carrier‐mediated mechanism (e.g., vitamin B6, riboflavin).8, 174 The lipid‐soluble vitamins A, D, E, and K are incorporated into CMs with other lipids and absorbed by the mucosa cells of the small intestine.8, 172 It was long assumed that fat‐soluble vitamins are absorbed by passive diffusion across the plasma membrane of enterocytes. However, new findings indicate that several membrane transporters, which play a key role in intestinal absorption of cholesterol, can also transport vitamin A and E.178
Water‐soluble vitamins are not stored, except for cobalamin. If the intake exceeds the requirements, most of the vitamin excess is excreted by urine and feces. In contrast, lipid‐soluble vitamins may be stored in significant amounts in the liver and fat tissue.172
Among the variables presented in the last paragraph of the Introduction, a search of the literature has identified convincing evidence for the interindividual variability of the intestinal absorption of vitamins by the GIT for the following variables: genetic polymorphisms.
5.2. Interindividual Variability in Intestinal Absorption of Vitamins
Literature reports an interrelationship between genetic polymorphism and vitamins. On the one hand, mutations may result in a modified vitamin bioavailability and intake; on the other hand, many vitamins influence genome expression, stability, and viability179 e.g., deficiencies impair nucleotide biosynthesis and thereby enhance polymerase error rates like single point mutations or DNA strand breaks. However, few gene variants are sufficiently penetrant to affect the average requirements to a greater degree than environmental factors179 and ancestry.180 The polymorphism most likely to influence the bioavailability of vitamins are presented below.
5.2.1. Genetic Polymorphisms and Vitamin C Absorption
In literature, substantial variability in the serum ascorbic acid response to a given amount of dietary vitamin is reported.181 Endogenous and exogenous factors may be responsible for this variation, including the two sodium‐dependent vitamin C transport proteins, SVCT1 and SVCT2. They are specific for the cotransport of sodium ions and ascorbic acid across cell membranes and encoded by SLC23A1 and SLC23A2, respectively. The transcriptional regulation of these genes controls the distribution of SVCTs in tissues and is therefore responsible for the maintenance of vitamin C levels in a wide range of cells, tissues, and extracellular fluids. Therefore, it can be assumed that genetic alterations in SLC23A1 and SLC23A2 will have a substantial effect on human vitamin C status. However, SLC23A1 knockout mice are still capable of absorbing ascorbic acid from the diet,182 but pharmacokinetics and viability of the offspring is severely affected.183 Although 1440 and 8165 variations of SVCT1 and SVCT2, respectively, are listed in the SNP database, not many of them have been verified in different populations, and most of them have neither been reported in the literature nor functionally characterized (Table 2).183 Whereas nearly all SNPs in SLC23A2 are shared between African Americans and Caucasians, a substantial number of SNPs in SLC23A1 are population specific in either Caucasians or African Americans. Apparently, SLC23A1 tolerates variations better than SLC23A2, indicating a higher physiological importance for the latter. However, it could not be concluded, from a human study by Cahill and El‐Sohemy, that the SVCT1 and SVCT2 genotypes modify the serum ascorbic acid response to dietary vitamin C.184 On the other hand, a collection of risk associations of several SNPs in SLC23A1 and SLC23A2 by Shaghaghi et al.183 indicated that both vitamin C uptake and storage might be involved in the pathogenesis of different diseases. Because these findings were reported on an individual basis in only small studies, a validation in larger cohorts is necessary.183 Of note, fasting serum ascorbic acid concentration is affected by one common polymorphism in SLC23A1 184 and the loss of SVCT1 results in reduced plasma levels of vitamin C due to lack of renal reabsorption. Therefore, the polymorphisms in SLC23A1 would increase the excretion of ascorbic acid from the body and alter the dose‐response relationship between plasma vitamin C levels and dietary vitamin C.182
Table 2.
Overview of polymorphism in genes involved in the processing of vitamins and β‐carotene by the GIT
| Protein | Gene | SNP (rs) | Reference |
|---|---|---|---|
| Vitamin C | |||
| Sodium‐dependent vitamin C transporter 1 | SLC23A1 | rs11950646 | [183, 243] |
| rs33972313 | [243] | ||
| rs10063949 | [182] | ||
| rs4257763 | [182] | ||
| rs6596473 | [182] | ||
| Sodium‐dependent vitamin C transporter 2 | SLC23A2 | rs6053005 | [183, 243] |
| rs6133175 | [183, 243] | ||
| rs6116569 | [183, 243] | ||
| rs1279683 | [244] | ||
| Glucose transporter type 2 | SLC2A2 | n.d. | [183] |
| Glucose transporter type 3 | SLC2A3 | n.d. | [183] |
| Vitamin B12 | |||
| Galactoside 2‐alpha‐L‐fucosyltransferase 2 | FUT2 | rs602662 | [188] |
| rs492602 | [188] | ||
| rs602662 | [188] | ||
| rs492602 | [188] | ||
| Transcobalamin‐1 | TCN1 | rs526934 | [188] |
| Transcobalamin‐2 | TCN2 | rs757874 | [188] |
| Cubillin | CUBN | rs1801222 | [188] |
| rs4748353 | [188] | ||
| Serum paraoxonase/arylesterase 1 | PON1 | rs3917577 | [188] |
| Gastric intrinsic factor | GIF | rs121434322 | [245] |
| rs35211634 | [245] | ||
| Protein amnionless | AMN | rs386834170 | [246] |
| rs386834169 | [246] | ||
| rs386834175 | [246] | ||
| rs386834165 | [246] | ||
| Folate | |||
| Folate transporter 1, FOLT | SLC19A1 | rs1051266 | [8] |
| rs2297291 | [247] | ||
| rs12659 | [247] | ||
| rs61510559 | [248] | ||
| Proton‐coupled folate transporter | SLC46A1 | rs2239907 | [8, 247] |
| rs5819844 | [247] | ||
| Folate receptor alpha | FOLR1 | rs35179028 | [8] |
| rs7928531 | [247] | ||
| Cubilin | CUBN | rs780635 | [188] |
| Folate Hydrolase 1 | FOLH1 | rs202676 | [187] |
| Vitamin A | |||
| Retinol binding protein 4 | RBP4 | (Ile59Asn) (rs121918584) | [196] |
| Gly75Asp (rs1218585) | [196] | ||
| c.248 + 1G>A | [195] | ||
| rs10882272 | [195] | ||
| Transthyretin | TTR | rs1667255 | [195] |
| Patatin‐like phospholipase domain‐containing 3 | PNPLA3 | rs738409 | [195] |
| β‐carotene oxygenase 1 | BCO1 | rs6564851 | [195] |
| BCO1 | rs12926540 | [195] | |
| BCO1 | rs7501331 | [195] | |
| BCO1 | rs12934922 | [195] | |
| Hepatic lipase | HL | rs1800588 | [195] |
| Lipoprotein lipase | LPL | S447X | [195] |
| Scavenger receptor class B member 1 | SCARB1 | SR‐BI intron 5 | [195] |
| rs61932577 | [249] | ||
| Cluster determinant 36 | CD36 | rs1984112 | [249] |
| CD36 | rs1761667 | [249] | |
| CD36 | rs7755 | [249] | |
| ATP‐binding cassette sub‐family A member 1 | ABCA1 | rs10991408* | [250] |
| ABCA1 | rs2791952 * | [250] | |
| ABCA1 | rs3887137 * | [250] | |
| ABCG5 | rs2278357 | [250] | |
| Apolipoprotein B‐100 | APOB | rs1042031 * | [250] |
| rs35364714 * | [250] | ||
| rs4643493 * | [250] | ||
| β‐carotene oxygenase 1 | BCO1 | rs7196470 | [250] |
| C‐X‐C Motif Chemokine Ligand 8 | CXCL8 | rs1247620 | [250] |
| rs1358594 | [250] | ||
| rs6834586 | [250] | ||
| Elongation Of Very Long Chain Fatty Acids Protein 2 | ELOVL2 | rs3798709 | [250] |
| rs911196 | [250] | ||
| rs9468304 | [250] | ||
| Intestine‐specific homeobox | ISX | rs16994824 | [250] |
| rs202313 | [250] | ||
| rs5755368 | [250] | ||
| Lipase C | LIPC | rs11857380 * | [250] |
| rs12185072 * | [250] | ||
| rs1869138 * | [250] | ||
| Polycystin 1 Like 2 | PKD1L2 | rs8043708 | [250] |
| Retinoid Isomerohydrolase | RPE65 | rs12139131 | [250] |
| rs4926340 | [250] | ||
| Superoxide Dismutase 2 | SOD2 | rs2501175 | [250] |
| Transcription Factor 7 Like 2 | TCF7L2 | rs946199 * | [250] |
| Vitamin D | |||
| Lipoprotein lipase | LPL | rs6586874 | [201] |
| rs10096561 | [201] | ||
| Intestine‐specific homeobox | ISX | rs5754862 | [201] |
| Ileal sodium/bile acid cotransporter | SLC10A2 | rs9558203 | [201] |
| rs9555166 | [201] | ||
| Vitamin D‐binding protein | GC | rs6845026 | [201] |
| Scavenger receptor class B member 1 | SCARB1 | rs12580803 | [201] |
| Pancreatic triacylglycerol lipase | PNLIP | rs2915775 | [201] |
| rs3010494 | [201] | ||
| 7‐dehydrocholesterol reductase | DHCR7 | rs11604724 | [201] |
| ATP‐binding cassette sub‐family A member 1 | ABCA1 | rs7043894 | [201] |
| Multidrug resistance protein 1 | ABCB1 | rs2235023 | [201] |
| rs10260862 | [201] | ||
| Apolipoprotein B‐100 | APOB | rs2854725 | [201] |
| Microtubule‐associated protein RP/EB family member 2 | MAPRE2 | rs1125425 | [201] |
| BET1 homolog | BET1 | rs10464587 | [201] |
| Arylamine N‐acetyltransferase 2 | NAT2 | rs4921920 | [201] |
| 25‐hydroxyvitamin D‐1 alpha hydroxylase | CYP27B1 | rs28934604 | [202] |
| 1,25‐dihydroxyvitamin D(3) 24‐hydroxylase | CYP24A1 | rs6068812 | [202] |
| Vitamin E | |||
| Ileal sodium/bile acid cotransporter | SLC10A2 | rs1571513 | [206] |
| rs9558203 | [206] | ||
| rs16961116 | [206] | ||
| rs12874168 | [206] | ||
| rs2065550 | [206] | ||
| Pancreatic triacylglycerol lipase | PNLIP | rs2915775 | [206] |
| rs3010494 | [206] | ||
| Sterol regulatory element‐binding protein 2 | SREBF2 | rs2839715 | [206] |
| rs4822062 | [206] | ||
| ATP‐binding cassette sub‐family G member 1 | ABCG1 | rs468320 | [206] |
| Niemann‐Pick disease type C1 | NPC1 | rs62001882 | [208] |
| rs141973731 | [208] | ||
| rs139659653 | [208] | ||
| rs114375162 | [208] | ||
n.d., not documented.
The star (*) indicates SNP that were associated with the variability of β‐carotene bioavailability due to their involvement in the postprandial metabolism of CMs [carry newly absorbed β‐carotene from the intestine to liver].
5.2.2. Genetic Polymorphisms and Vitamin B12 and Folate Absorption
The uptake of folate in the small, as well as in the large intestine, is a specific pH‐dependent, Na+ independent carrier‐mediated mechanism. Three specific carriers are known; the reduced folate carrier (RFC, the product of SLC19A1), the proton‐coupled folate transporter (PCFT/HCP1, the product of SLC46A1) and the GPI (glycosyl phosphatidyl‐inositol)‐anchored folate receptor 1.8, 185 However, only RFC and PCFT are known to be expressed and functional in intestinal tract.185 The RFC and PCFT are expressed at the apical membrane domain of the intestinal cells. While RFC is predominantly expressed in the lower part of the small intestine and in the large intestine, where pH is rather neutral/slightly alkaline, PCFT is mainly expressed at acidic pH, which is typical for the proximal part of the small intestine.174, 185 The process of folate absorption in the intestine is regulated by extracellular folate level. Thus, in folate deficiency the levels of expression of RFC and PCFT are increased in the jejunum, the ileum, and the colon.185, 186 Additionally, folate hydrolase 1 (FOLH1) an enzyme located on the intestinal brush border membrane is responsible for the cleavage of the polyglutamated tail from naturally occurring food folate before it can be absorbed. Cummings and colleagues showed, that carriers of the FOLH1 484 C variant could absorb naturally occurring food folate less well than T carriers.187
Vitamin B12 exists in two coenzyme forms, the methyl‐B12 and the 5′‐deoxyadenosyl‐B12. Blood and milk mainly contain methyl‐B12, whereas in most tissue the deoxyadenosil form accounts for most of vitamin B12.185 Dietary vitamin B12 can be ingested either in its free or in a protein bound form.
As widely known, vitamin B12 and folate metabolism are clearly linked and therefore genetic variants that modify vitamin B12 availability may also influence folate status.188 In a cross sectional study to identify associations between common SNPs in genes related to folate and vitamin B12 metabolism, or associated with B vitamin‐related chronic diseases and biomarkers of nutrient status in a population exposed to folic acid fortification, Zinck and co‐authors found 14 SNPs in 11 genes associated with red blood cell folate and serum vitamin B12 status. Only 6 of the 14 SNPs are related to protein location and function in the stomach or intestine (see Table 2). As already described elsewhere, common variants in exon 2 of fucosyltransferase 2 (FUT2) were also identified in this study and were associated with higher vitamin B12 status. The authors hypothesize that these variants reduce the risk for Helicobacter pylori infection mostly related to gastritis induced vitamin B12 malabsorption. A second hypothesis proposed that these FUT2 variants increase the secretion of a gastric intrinsic factor, a fucosylated glycoprotein required for the absorption of vitamin B12. In contrast to the improvement of vitamin B12 status by the above mentioned variations, two SNPs on the vitamin B12 binding proteins transcobalamin I (rs526934, intron 8) and II (rs757874, intron4) are associated with lower vitamin B12 status.188 Transcobalamin I is produced by salivary glands in the mouth and the gastric mucosal cells in the stomach, and protects vitamin B12 from acid degradation during the transport in the stomach. Transcobalamin II binds vitamin B12 after uptake by the enterocytes of the terminal ileum.189
The vitamin B12‐intrinsic factor complex receptor in the distal ileum is called cubilin (CUBN). The CUBN rs1801222 polymorphism results in a reduced functionality of CUBN, thereby lowering vitamin B12 absorption. A genetic variant that therefore may modify vitamin B12 availability and status indirectly influences the folate status. Because the vitamin B12 dependent MTR enzyme is required to convert 5‐methyltetrahydrofolate, this folate form is then taken up into cells from circulation, and converted to tetrahydrofolate, which no longer diffuses out of the cell.188
Of note, there are also relatively rare inherited defects in intestinal absorption of vitamin B12 (e.g., due to mutations in genes such as cobalamin binding intrinsic factor (CBLIF), amnion associated transmembrane protein (AMN), and CUBN).8
5.2.3. Genetic Polymorphisms and Vitamin A Absorption
In general, fat‐soluble vitamin absorption follows the same mechanisms as fat absorption.190 The absorption of vitamin A depends on its dietary form. From the over 600 carotenoids isolated from natural sources, only 60 have been detected in the human diet and 20 of these in human blood and tissues, with the most prominent being β‐carotene, α‐carotene, lycopene, lutein, and β‐cryptoxanthin.191 Retinol, retinal, retinoic acid, and retinyl esters are the different forms of retinoids, detected in animal sources.
Vitamin A (as retinol and retinyl ester) uptake by intestinal mucosa is preferentially localized in the proximal intestine. Interestingly, vitamin E presence significantly improves vitamin A uptake, when at medium and high concentrations (up to 40%).192
Information is limited regarding the physical forms of retinyl esters and carotenoids in the intestinal lumen, however it is clear from human and animal studies, that the co‐ingestion of dietary fat and vitamin E enhances the intestinal absorption of dietary vitamin A and carotenoids. Lipids also facilitate the conversion of pro‐vitamin A to vitamin A. Intestinal carotenoid and retinol absorption is preferentially localized in the proximal intestine and beside passive diffusion, also facilitated by specific active transporters at concentrations below saturation. Pro‐vitamin A carotenoids such as α‐ or β‐carotene are partly converted to retinol by oxygenase and reductase enzymes and the produced retinol is then available for uptake by intestinal mucosa.193, 194 Information on plant derived carotenoids is therefore only focused on this converted part.
Data on genetic variations in vitamin A bioavailability is scarce and most existing data focuses on β‐carotene.5 Both absorption efficiency and vitamin A tissue uptake vary by genetic variations in blood binding proteins such as retinol binding protein 4 (RBP4) affecting blood retinol concentrations.195 A GWAS has confirmed that SNPs in the two genes encoding the proteins that transport retinol in the blood, i.e., RBP4 and transthyretin (TTR), which cause amino acid substitutions at position 84 of the TTR molecule (Ser84 and Asn84), can significantly affect blood retinol concentrations.196 Additionally, blood levels of retinol were associated in some cases with a SNP in phospholipase domain containing 3.
Interestingly, genetic variations in proteins/enzymes located in tissues can also affect fasting blood retinol concentration. For example, an association between SNPs in beta‐carotene oxygenase 1 (BCO1) and blood retinol was found,197 suggesting that carotenoids significantly participate in blood retinol concentrations. As work in this area is limited, more research is required to identify additional SNPs relevant to vitamin A status and bioavailability.
5.2.4. Genetic Polymorphisms and Vitamin D Absorption
Vitamin D exists in two major forms, vitamin D2 (ergocalciferol) of plant and fungal origin and vitamin D3 (cholecalciferol) derived from animal food and endogenous synthetization in the skin. The intestinal absorption of vitamin D is similar to that of vitamin A.8 There are three known transporters: SRBI (scavenge receptor class B type 1), CD 36 (cluster determinant 36) and NPC1L1 (Neimann–Pick C1‐like 1), but the differences between absorption rate in the duodenum and the jejunum indicates the presence of another transporter, particularly expressed in the jejunum. Several other factors influence the bioavailability of vitamin D like nutrient status in the host, form, food matrix, amount, and composition of ingested food, amount of lipids, type of fatty acids, dietary fibers, activities of digestive enzymes, efficiency of transport across enterocytes, interactions with micronutrients, age of host, obesity, genetic variations, and therefore, contribute to the interindividual variability of vitamin D absorption.198, 199 Beside the absorption of vitamin D, also the endogenous synthesis varies, depending on the type and nature of skin pigments and the age of the host, as the vitamin D production in the skin, as well as the conversion of 25OHD into 1,25(OH)2D (active form) in the kidneys, decreases with age.190, 200
Current understanding regarding genetic polymorphisms related to the absorption and metabolism of vitamin D2 and D3 is limited. Nevertheless, high variability has been observed among individuals in response to vitamin D supplementation. Desmarchelier and colleagues201 assessed the relation between genetic polymorphisms and variability in vitamin D3 bioavailability. In 39 healthy adult men, a 34‐fold difference in area under the curve (AUC) was observed in postprandial vitamin D3 response. 17 SNPs (rs6586874, rs10096561, rs5754862, rs9558203, rs6845026, rs12580803, rs2915775, rs11604724, rs7043894, rs2235023, rs10260862, rs3010494, rs2854725, rs1125425, rs10464587, rs4921920, and rs9555166) located in or near 13 genes, which encode for several active transporter mechanisms (LPL, ISX, SLC10A2, GC, SCARB1, PNLIP, DHCR7, ABCA1, ABCB1, APOB, MAPRE2, BET1, and NAT2; see Table 2), which further supports the notion that, at nutritional doses, uptake of vitamin D is at least in part an active, rather than the previously thought passive process. These 17 SNPs explained 63.5% of the observed interindividual variance. In regards to metabolism, the enzymes 1a‐hydroxylase and 24‐hydroxylase have been identified to contribute to interindividual variability. Encoded for by CYP27B1 and CYP24A1, respectively, these enzymes are of critical importance in governing calcitriol (synthetic version of vitamin D3 used in clinical applications) concentration. These two enzymes work jointly to regulate the concentration of calcitriol at the tissue level. Genetic polymorphisms in both CYP27B1 and CYP24A1 have been identified. SNP's rs28934604 and rs6068812, respectively, have both been shown to significantly reduce calcidiol to calcitriol conversion rate in 1a ‐hydroxylase and 24‐hydroxylase.202, 203, 204
5.2.5. Genetic Polymorphisms and Vitamin E Absorption
Vitamin E is the term for a group of four different tocopherols and four different tocotrienols. They are all fat‐soluble antioxidants and α‐tocopherol is the best‐known and studied component of vitamin E. Vitamin E is essentially absorbed in the distal part of the intestine with a significant uptake in the jejunum.205 It is absorbed together with dietary fats incorporated into mixed micelles mainly by passive diffusion. However, newer findings show that several proteins, like NPC1L1, sterol regulatory element binding protein 2 (SREBP2) or scavenger receptor class B type I (SR‐B1), as well as CD36 molecule and apical sodium‐bile acid transporter, facilitate the uptake of vitamin E. Also inside the enterocyte, different candidates (e.g., L‐FABP or tocopherol‐associated protein) for the intracellular transport have been described.205 The bioavailability varies greatly from 10% to 81% due to influencing factors like composition of food matrix, presence and type of fat, pathologies, or genetic factors of the host.205 Studies in mice and Caco‐2 cells showed an interaction between vitamin D and vitamin E, reducing the vitamin E uptake by 35% in cases of a co‐intake of pharmacological doses of vitamin D. The reason could be that these two vitamins share common transport proteins like NPC1‐L1 and SR‐BI.
Current understanding regarding genetic polymorphisms related to the absorption and metabolism of vitamin E is limited. However, genetic polymorphisms in genes coding for vitamin E and lipid intestinal metabolism have been associated with a modulation of vitamin E bioavailability in humans, due to the shared intestinal transport pathway between dietary lipids and vitamin E (incorporation into mixed micelles).206
Borel and colleagues207 assessed the relation between genetic polymorphisms and variability in α‐tocopherol bioavailability in an α‐tocopherol supplementation trial. In 40 healthy adult men, an 81% postprandial variation in α‐tocopherol was observed. A total of 28 SNPs in or near 11 candidate genes were associated with 82% of interindividual variability of α‐tocopherol. Four of these genes (SLC10A2, PNLIP, SREBF2, and ABCG1), with their related SNPs (SLC10A2 rs1571513, rs9558203, rs16961116, rs12874168, rs2065550; PNLIP rs2915775, rs3010494; SREBF2 rs2839715, rs4822062; ABCG1 rs468320; see Table 2) were identified as being specifically associated with α‐tocopherol response.206, 207
The membrane protein NPC1L1 plays a crucial role in the absorption and movement of lipophilic compounds like cholesterol and vitamin E. Yamanashi and colleagues found four SNPs (Ala395Val (rs62001882), Gly402Ser (rs141973731), Arg417Trp (rs139659653), and Gly434Arg (rs114375162)) diminishing transport activity by 21% to 63% compared to the wild type, concluding, that this will influence bioavailability of vitamin E.208
Apolipoprotein A5 (APOA5), which is encoded by APOA5, plays an important role in regulating plasma triglyceride levels. Polymorphism in APOA5 is significantly associated with TAG concentration in the plasma. The specific SNP rs662799 of this gene, has been associated with higher vitamin E plasma concentration, suggesting that vitamin E transportation may be regulated by APOA5 in vivo.202, 209
6. Minerals
6.1. Overview of Mineral Processing by the GIT
Minerals and electrolytes intestinal absorption and secretion processes are governed by active and passive mechanisms. Both mechanism may occur simultaneously but also may occur as associated transport with other substances.210 Mineral intestinal absorption depends on the small or large intestine and, even more, between the three small intestinal segments (duodenum, jejunum, ileum). In addition, some minerals are present in non‐absorbable forms, such as the insoluble ferric iron (Fe3+), which needs to be reduced to absorbable ferrous iron (Fe+2) by duodenal brush border cytochrome B reductase 1 (DCYTB; CYBRD1), as well as luminal Cu2+ to Cu+. Dietary ascorbic acid can also mediate Fe3+ reduction.8
6.1.1. Intestinal Absorption of Minerals
Three general intestinal absorption mechanisms have been identified for minerals: 1) non‐transporter mediated passive diffusion driven by a concentration gradient (paracellular pathway), 2) transport mediated by a membrane transporter (active transport), and 3) mineral co‐transport with other substances such as sugar or amino acids.
The paracellular pathway consists of substances diffusion through the enterocyte tight junctions driven by the concentration gradient, but is also governed by epithelial ionic electrical resistant selectivity. This mechanism does not shows directional discrimination.211 Although no protein channels or transporters are involved in paracellular transport, the lateral intercellular space can influence the paracellular electrical resistance, as occurs with the claudin proteins during Ca2+ absorption. Besides, a “solvent drag effect,” linked transport with oxalates of some minerals may occur too.8
Transcellular mineral transport may be mediated by a membrane transporter protein. Ca is absorbed by the saturable transcellular transporters TRPV6 and TRPV5 at low luminal concentrations, whereas active transport by the apical l‐type channel Cav1.3 occurs at high concentrations. The active transporters TRPM7 and TRPM6 are responsible for Mg2+ absorption. Divalent metal ions are mainly transported by the divalent metal transporter 1 (DMT1, also known as DCT1 or Nramp2), and by other transporters (SLC39A4, (Zip4); SLC31A1 (CTR1); SLC39A8 (Zip8), and Zip14A/Zip14B). Several proteins belonging to the SLC26 “sulphate permease” transporter family are responsible for SO4− and oxalate anion intake, in a Na+ independent manner (SLC26A1 (SAT1); SLC26A2 (DTDST), and SLC26A3 (DRA)), together with oxalate intestinal intake driven by SLC26A7 (SUT2). Others active transport mediated by SLC26 proteins may occur, such as anion electroneutral exchangers (DRA/CLD exchanger; SLC26A3) or the ion exchangers PAT1 (SLC26A6a), capable to exchange Cl−/oxalates, SO4−/oxalates, SO4−/Cl−, and Cl−/OH−. Electrogenic Na+ absorption happens mediated by the epithelial sodium channels (ENaC), however, a not yet completely defined Na+/HCO3− cotransporter (NBC) may act too.8
A mineral–mineral or mineral–nutrient intestinal cotransport may also occur. SLC9 Na+/H+ exchangers (as SLC9A3) drive electroneutral NaCl absorption, whereas a Na+/H+ antiporter (NHE2, NHE3, and NHE8 isoforms) has also been identified.8 Phosphate intake may occur through an apical Na+‐phosphate cotransporter driven by NaPi‐IIb (SLC34A4).212
In addition, other mineral transport may occur associated to other nutrients, such as SGLT1, which allows a Na+‐glucose cotransport (in a 2:1 ratio) mediated by a Na+ electrochemical gradient, or the diverse SLC6 and SLC1 transporters, which allow the transport of some amino acids in a Na+ manner.8
6.1.2. Intestinal Secretion of Minerals and Mineral Exit
When mineral secretion or efflux is considered, different mechanisms have been identified for the different minerals. Ca basolateral enterocyte exit occurs by two mechanisms enhanced by vitamin D3: mainly via Ca2+‐ATPase PMCA1b (80% contribution) and through the Na+/Ca2+ exchanger NCX1 (SLC8A1).
PAT1 (SLC26A6) is responsible for oxalate cellular efflux and intestinal secretion.
After Zn apical absorption across Zip4 transporter, vesicular transport driven by the Znt protein occurs, allowing Zip5 basolateral transport, whereas metallothionein (MT) acts as a intracellular Zn binding protein.
Fe basolateral exit, probably shared for Cr and Mg exit, is driven by the SLC40A1 (FPN1) coupled to an Fe2+ oxidation process (iron oxidase hephaestin, HEPH), allowing binding oxidized iron to transferrin.
Once Cu+ is absorbed, it is transferred to the basolateral membrane by a number of intracellular chaperones, such as ATOX1, which delivers Cu+ to ATP7A P‐type ATPase present in the trans‐Golgi network (for cuproenzyme synthesis) or at the basolateral membrane.8
Among the variables presented in the last paragraph of the Introduction, a search of the literature has identified convincing evidence for the interindividual variability of the intestinal absorption of minerals by the GIT for the following variables: genetic polymorphisms.
6.2. Interindividual Variability in Intestinal Absorption of Minerals
6.2.1. Genetic Polymorphisms Affecting the Intestinal Processing of Minerals
Diverse studies have been conducted to elucidate the existence and influence of polymorphism and/or mutations in the mineral absorption and metabolism (Figure 3). For example, congenital chloride diarrhea (CLD) is an autosomal recessive disorder of intestinal electrolyte absorption, characterized by persistent secretory diarrhea, causing dehydration and hypoelectrolytemia. CLD is caused by mutations in the solute carrier family 26, member 3 gene (SLC26A3, CLD or DRA), which encodes a Na+‐independent Cl−/HCO3− (or OH−) exchanger. A wide variety of different mutations in SLC26A3 have been associated with CLD with no apparent evidence of phenotype–genotype correlation. The clinical course of CLD, however, is variable and may rather depend on environmental factors and compensatory mechanisms than mutations.213
Figure 3.
Overview of polymorphisms involved in mineral absorption by the GI tract.
Various chronic diseases have been associated to mineral cellular transporters polymorphisms. However, most of the results do not show a clear link between the disorders and a lack or dysfunction of the intestinal absorption. Most studies focusing on the impact of these polymorphisms in other organs such as kidney or liver.214, 215 Thus, apart from iron for which significant evidences are available (see next section),215 only limited information exists on the impact of polymorphisms on the intestinal absorption of minerals. This evidence is summarized below.
Intestinal absorption of zinc happens through an active transport mediated by Zip4 and DMT1.8 Besides, numerous proteins having been shown to be involved in zinc metabolism and homeostasis through the influx, chelating, sequestrating, coordinating, releasing, and efflux of zinc, with metallothioneins, Zrt‐ and Irt‐like proteins (ZIP), and Zn transporters (ZnT) being among them.216 Metallothioneins are cysteine‐rich proteins allowing the binding of biologically essential metals and managing metal homeostatic regulation, as well as absorbing heavy metals. In the case of zinc, the metallotheonin is an intracellular binding protein, acting as a storing and reservoir protein. Research was conducted in a Turkish population, with the aim of identifying the correlation between a polymorphism in the core promoter region‐5 A/G of metallothionein 2A (MT2A) and Cd, Pb, Zn, and Cu levels in blood samples. The genotype AA (typical homozygote) was found at 86.6% frequency, the heterozygote AG at 12.8% frequency and the atypical homozygote GG at 0.6% frequency. The atypical heterozygote correlated significantly with Cd, Pb, and Zn levels in blood, but not with Cu levels. The authors suggested a genetic predisposition in the accumulation of the heavy metals Pb and Cd caused by different intestinal absorption mediated by 2A (MT2A) polymorphism. In addition, the GG population was characterized by lower Zn levels in blood but higher Cd and Pb levels.217 The lower Zn levels in the GG population can be explained by the fact that competitive intestinal transport occurs when different metal ions share the same protein transporter. However, more research is necessary to further understand these relationships.
DMT1 transporter is a protein especially involved in duodenal Fe transport, but also in Zn, Mn, Co, Cd, Cu, Ni, and Pb uptake.217 Low levels of Fe cause higher DMT1 intestinal expression, whereas overexpression of this transporter increases the intestinal Fe absorption as well as Pb and Cd absorption too. Four major mammalian DMT1 isoforms have been discover, named A/+IRE, 1A/–IRE, 2/+IRE, and 2/‐IRE. The +IRE forms are mainly located in the apical side of the epithelial cells, especially in the duodenum. A study conducted by Kayaalti et al.217 investigated the influence of DMT1 IVS4+44 C/A polymorphism (located in the intron 4 of DMT1) in Fe serum levels, as well as Pb and Cd blood levels. The genotype frequencies were 49.8% for the CC typical homozygotes, 38.3% for the heterozygote, and 11.9% for the AA atypical homozygote. The authors suggested that DMT1 IVS4+44 C/A polymorphism is associated with interindividual variations in blood Fe and Pb levels, but not Cd, since this protein transporter is mainly responsible for intestinal uptake of dietary divalent metals. The CC genotype group had higher Fe and Pb blood levels, indicating that the absence of the A allele leads to higher iron and Pb intestinal absorption. Thus, carriers of the CC genotype may be more susceptible to metal toxicity as compared to those with AC and AA genotypes.217
Abrams et al.218 carried out a study of calcium absorption and vitamin D receptor (VDR) Fok1 polymorphism. A significant relationship was found between this polymorphism and calcium absorption, whereas no relation was found for other VDR polymorphisms such as ApaI, BsmI, and Taq1 genotypes. The same results were obtained in estrogen receptor genotypes for XbaI and PvuII, as well as for calcitonin receptor genotypes. The homozygotes FF, to VDR Fok1 polymorphism, showed higher calcium fractional absorption at the end of the study, with FF being lower compared to Ff and ff genotypes. These results suggest that the F allele, and therefore the FF genotype, allows higher calcium absorption. In agreement with this result, calcium levels at the beginning of the study was significantly higher in the FF genotype group (315 ± 19 mg d–1) in comparison with Ff (258 ± 17 mg d–1) and ff (224 ± 27 mg d–1) genotypes.218
6.2.2. Genetic Polymorphisms Affecting the Intestinal Processing of Iron
Iron is an essential mineral with a dietary intake estimated as 20–30 mg d–1, however, only 1.5 mg is absorbed by adult males. Adult females require additional 1.5–2 mg d–1ay to compensate for losses through menstrual bleeding and fetal requirements during pregnancy. The human body contains 3–5 g of iron stores, mostly present in hemoglobin or myoglobin.8 Significant sources of variation in the indices of iron status include age, sex, body–mass index, and genetic factors.219
No iron excretion pathways exist in humans, excepting losses in epithelial cells and fluids such as blood, tears, and sweat.220 Therefore, iron absorption is the primary determinant of iron balance, where a homeostatic mechanism avoids the iron overload. Iron bioavailability is conditioned by the digestion process, modulated in turn by the presence of dietary substances with iron‐binding (e.g., phytate, tannins) or reducing capacities (e.g., ascorbic acid). In addition, since divalent metal ions share diverse intestinal absorption transporters, the intestinal transport of iron may compete with other divalent cations uptake.221
Since non‐heme iron is absorbed by DMT1, the insoluble Fe+3 reduction to Fe+2 must be mediated by the duodenal brush border cytochrome b reductase 1 (DCYTB; CYRD1) as well as by dietary ascorbic acid.220 Conversely, heme iron transport occurs via the low affinity heme transporter HCP1 (SLC46A1). Both iron transporters are overexpressed in response to hypoxia8 whereas DMT1 increases to anemia.220
The absorbed Fe+2 may either be bound to ferritin or be exported into the bloodstream.220 Fe2+ is transported into vesicles containing either ferroportin (FPN1) or hephaestin (HEPH), crossing the enterocyte cytoplasm to the basolateral membrane.222 The exit of Fe across the enterocyte basolateral membrane occurs through the transporter FPN1, coupled to the ferroxidase activity of the basolateral HEPH, allowing the iron binding to transferrin protein in the interstitial fluid and plasma for body distribution.222
The liver hepcidin antimicrobial peptide (HAMP) is secreted from the liver in response to the body iron stores and inflammation (via IL‐6).8 HAMP directly binds to FPN1 causing to be internalized and degraded, thereby blocking cellular iron export. Adequate or high iron stores in the liver result in increasing of HAMP secretion and a decreasing of FPN1 mRNA expression, lowering FPN1 levels. The result of this repositioning of the FPN1 from the basolateral membrane is to reduce iron efflux from across the basolateral membrane of the duodenocyte, and thereby a decrease in iron absorption.221, 222
Hereditary hemochromatosis (HH) is a heterogeneous disorder of iron metabolism related to the HFE gene.223 HFE protein is found in duodenal crypt cells associated with b‐2 microglobin and transferrin receptors. HFE mutations prevent Fe from being taken into the crypt cells, leading to Fe deficiency in the duodenal crypt cells. This, in turn, increases DMT1 expression and activity, resulting in abnormally elevated iron absorption. Progressive accumulation of iron may provoke serious clinical consequences including diabetes, heart disease, arthritis, and cirrhosis.221, 222
Most people are homozygous for the C282Y mutation of HFE, with the minority being heterozygotes (C282Y, H63D), but the clinical penetrance is low. Since hepcidin expression is inappropriately low in hemochromatosis disorder, it has been suggested that the HFE protein is required for normal regulation of hepcidin synthesis. However, clinical penetrance is low in C282Y homozygotes, so that the phenotype is not related to HFE alone.221, 222 The most common form of the disease is classic or type 1 HH, mainly caused by a bi‐allelic missense pCys282Tyr (c.845G N A) mutation in HFE. The role of rs10421768, rs235756, rs2230267, and rs1439816 polymorphisms, respectively in HAMP, BMP2, FTL, and SLC40A1 (these genes are involved in iron metabolism pathways, principally in the hepcidin expression cascade) in hereditary hemochromatosis expressivity, suggests a hypothetical multifactorial model, characterized by a principal gene (HFE in HH type 1) and minor genetic and environmental factors that still must be fully elucidated.223
Hepcidin expression is also low in juvenile hemochromatosis, a rare but severe iron loading disorder associated with mutations of HFE2 (hemojuvelin). As several other mutations are responsible for hereditary hemochromatosis, this condition is now separated into four categories: 1) Type 1 (HFE mutations), 2) Type 2 HFE2 (hemojuvelin) mutation, 3) Type 3 (TfR2 mutation), Type 4 (ferroportin mutation).221
The ferroportin (FPN1) Q248H polymorphism, which typically occurs in sub‐Saharan and African Natives and African Americans, has been associated with higher ferritin serum levels. In a study by Rivers et al.,224 10.4% of cases with high serum ferritin levels in an American population were heterozygotes for Q248H, compared with 6.7% of the heterozygotes who had normal ferritin level. In addition, serum ferritin levels were associate with CYBRD1 rs884409,225 a polymorphism localized to the proximal region of the CYBRD1 promoter. In this study, participants with one or two copies of the minor allele of rs884409 had lower levels of serum ferritins. This polymorphism decreases the basal activity of the promoter by 30%. Decreased expression of CYBRD1 in turn results in a lower reduction of Fe, an essential step in intestinal iron intake. Of note, a reduced intestinal iron intake can be a protective mechanism against iron overload in C282Y homozygotic populations.
Mutations in FPN1 (SLC40A1) lead to an autosomal dominant form of hemochromatosis (type 4), also known as ferroportin disease.226 Also, rare mutations in DMT1 (SLC11A2) are responsible for an absorptive defect leading to hypochromic microcytic anemia. Secondary interference with DMT1 function can occur in intestinal disorders like coeliac disease, which damages the duodenal villus structure rendering patients unable to absorb iron. Therefore, patients who are Fe‐deficient and nonresponsive to iron supplementation need to be investigated for coeliac disease.
Much less is known about the genetic basis of non‐diet related iron deficiency. A polymorphism in the transferrin protein (G277S) has been associated with iron deficiency, but no effect of the SNP on the iron‐binding capacity of transferrin was observed from in vitro studies. A human iron absorption study also failed to show a significant difference between heterozygote and wild‐type individuals, although there was a difference in the slope of the curve describing the relationship between body iron stores (serum ferritin) and percentage iron absorption.221
7. Discussion
From a research point of view, the realization that human metabolism has significant interindividual variability fundamentally leads to a personalization of human physiology. The GutSelf review therefore fills an important gap by providing, for the first time, a global overview of the inter‐individual variability in the processing of nutrients by the human GIT.
The first lesson learned from our analysis of the literature was that interindividual variability is yet to be investigated as a key “primary endpoint” with nutritional research. This said, the GutSelf review highlights multiple “regions” of the multidimensional space describing the interaction of foods with the human GIT. The bioavailability of amino acids derived from dietary proteins varies depending on the quality of the chewing process, the amounts and proteolytic activity of pepsin, pancreatic and intestinal proteases, modulating protein hydrolysis, and the polymorphisms in amino acid transporters, modulating amino acid absorption. Variability in fat processing by the GIT is characterized by significant differences in the ability of humans to sense fatty acids, in particular due to polymorphisms in CD36, by variability in the activity of gastric lipase, and by variability in the absorption and secretion into the circulation of fatty acids, cholesterols, or phytosterols, via a range of, as to yet, uncharacterized biochemical and genetic factors, including transporters involved in the intestinal processing of CMs. With regard to carbohydrates, the importance of the chewing process, the role of the copy number of the amylase genes, the presence of an active lactase, the functionality of glucose transporters, as well as the diversity of the microbiota are clear elements mediating interindividual variability in the digestion and absorption of this group of nutrients. Variability in the bioavailability of vitamins is mostly mediated by polymorphism in intestinal transporters such SVCT vitamin C transporters, FUT2 and CBN for vitamin B12, CYP27B1 and CYP24A1 for vitamin D, APOA5 for vitamin E. Finally, although scarce, studies on the genetic variability of the gastrointestinal absorption of minerals seem to indicate that the bioavailability of minerals is potentially influenced by polymorphisms in receptors, the most obvious polymorphism being reported in genes, such as HFE, FPN1, CYBRD1, FPN1, or DMT1, involved in iron transport.
That the microbiota has been most obviously reported to contribute to the interindividual variability in the GIT processing of carbohydrate is symptomatic of the fact that carbohydrates are the major source of energy and, consequently, growth, for intestinal microbes.166 Although variability in the gut microbiota is largely recognized as an important factor in personalized nutrition,227 only few examples directly associate a differential processing of nutrients by the GIT with variation in the gut microbiota, among them the metabolism of flavonoids228 and the production of trimethylamine‐N‐oxide.229 Finally, the metabolic interplay between nutrients, the gut microbiota, and the host is complex and multidirectional, each component feeding back molecular or cellular signals to the others following their own stimulation.227, 230 In this context, the current study designs do not allow to filter out how variability in the gut microbiota modulates the metabolism of nutrients by the GIT.
Despite numerous indications of interindividual variability in the processing of food by the human GIT, the knowledge on this topic remains fragmented to a large extend because of a lack of specific focus of researchers on these issues. Also, a quantitative understanding of the clinical relevance of most of the interindividual responses to food ingestion remains unknown. Fostering personalized nutrition will request a conceptual analysis of the identified variables triggering interindividual variability in the response of human to dietary intake. To more precisely identify these variables, human nutritional trials will need to systematically report data on individual subjects. In addition, as interindividual variability of a biological variable can only be claimed if the precision of the analytical methods measuring the variable is better than the biological variability, the performance of the analytical tools should be documented. Implementing these recommendations should, at term, promote translation in nutritional sciences and realize the statement made in the introduction of this review: “Tell me what you digest and absorb, and I will tell you what you are.”
Conflict of Interest
The authors declare no conflict of interest.
Walther B., Lett A. M., Bordoni A., Tomás‐Cobos L., Nieto J. A., Dupont D., Danesi F., Shahar D. R., Echaniz A., Re R., Fernandez A. S., Deglaire A., Gille D., Schmid A., Vergères G., GutSelf: Interindividual Variability in the Processing of Dietary Compounds by the Human Gastrointestinal Tract. Mol. Nutr. Food Res. 2019, 63, 1900677 10.1002/mnfr.201900677
References
- 1. Fisher M. F. K., The Physiology of Taste: Meditations on Transcendental Gastronomy (translation of Jean Anthelme Brillat‐Savarin's 1825 book) Random House USA Inc, New York: 2009. [Google Scholar]
- 2. Moran N. E., Mohn E. S., Hason N., J. W. Erdman, Jr. , Johnson E. J., Adv Nutr. 2018, 9, 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. EMEA , 2006, CPMP/ICH/289/95.
- 4. Remond D., Shahar D. R., Gille D., Pinto P., Kachal J., Peyron M. A., Dos Santos C. N., Walther B., Bordoni A., Dupont D., Tomas‐Cobos L., Vergeres G., Oncotarget 2015, 6, 13858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bohn T., Desmarchelier C., Dragsted L. O., Nielsen C. S., Stahl W., Rühl R., Keijer J., Borel P., Mol. Nutr. Food Res. 2017, 61, 1600685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Almeida A. F., Borge G. I. A., Piskula M., Tudose A., Tudoreanu L., Valentova K., Williamson G., Santos C. N., Compr. Rev. Food Sci. Food Saf. 2018, 17, 714. [DOI] [PubMed] [Google Scholar]
- 7. Freeman H. J., World J. Gastrointest. Pharmacol. Ther. 2015, 6, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kiela P. R., Ghishan F. K., Best Pract. Res. Clin. Gastroenterol. 2016, 30, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. He L., Vasiliou K., Nebert D. W., Hum. Genomics 2009, 3, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bröer S., Physiol. Rev. 2008, 88, 249. [DOI] [PubMed] [Google Scholar]
- 11. Dupont D., Le Feunteun S., Marze S., Souchon I., Innovative Food Sci. Emerging Technol. 2017. [Google Scholar]
- 12. Lassauzay C., Peyron M. A., Albuisson E., Dransfield E., Woda A., Eur. J. Oral Sci. 2000, 108, 484. [DOI] [PubMed] [Google Scholar]
- 13. Rémond D., Machebeuf M., Yven C., Buffière C., Mioche L., Mosoni L., Mirand P. P., Am. J. Clin. Nutr. 2007, 85, 1286. [DOI] [PubMed] [Google Scholar]
- 14. Pennings B., Groen B. B. L., van Dijk J. W., de Lange A., Kiskini A., Kuklinski M., Senden J. M. G., van Loon L. J. C., Am. J. Clin. Nutr. 2013, 98, 121. [DOI] [PubMed] [Google Scholar]
- 15. Food and Agricultural Organization of the United Nations , Dietary Protein Quality Evaluation in Human Nutrition. FAO Food and Nutrition Paper, Rome, Italy: 2013, 92, 79. [PubMed] [Google Scholar]
- 16. Fuller M. F., Tome D., J. AOAC Int. 2005, 88, 923. [PubMed] [Google Scholar]
- 17. FAO/WHO/UNU , WHO Technical Report 2007.
- 18. Gilani G. S., Cockell K. A., Sepehr E., J. AOAC Int. 2005, 88, 967. [PubMed] [Google Scholar]
- 19. Evenepoel P., Geypens B., Luypaerts A., Hiele M., Ghoos Y., Rutgeerts P., J. Nutr. 1998, 128, 1716. [DOI] [PubMed] [Google Scholar]
- 20. Bos C., Airinei G., Mariotti F., Benamouzig R., Berot S., Evrard J., Fenart E., Tome D., Gaudichon C., J. Nutr. 2007, 137, 594. [DOI] [PubMed] [Google Scholar]
- 21. Boutrou R., Gaudichon C., Dupont D., Jardin J., Airinei G., Marsset‐Baglieri A., Benamouzig R., Tome D., Leonil J., Am. J. Clin. Nutr. 2013, 97, 1314. [DOI] [PubMed] [Google Scholar]
- 22. Le Veen H. H., Hallinger L., J. Clin. Invest. 1947, 26, 761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Janowitz H. D., Hollander F., J. Clin. Invest. 1952, 31, 338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rick W., Internist 1970, 11, 110. [PubMed] [Google Scholar]
- 25. Norman A., Strandvik B., Ojamae O., Acta Paediatr. 1972, 61, 571. [DOI] [PubMed] [Google Scholar]
- 26. Font M. L., Jimenez‐Vidal M., Bisceglia L., Di Perna M., de Sanctis L., Rousaud F., Zelante L., Palacin M., Nunes V., J. Med. Genet. 2005, 42, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Langen H., von Kietzell D., Byrd D., Arslan‐Kirchner M., Vester U., Stuhrmann M., Dork T., Saar K., Reis A., Schmidtke J., Brodehl J., Pediatr. Nephrol. 2000, 14, 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kleta R., Romeo E., Ristic Z., Ohura T., Stuart C., Arcos‐Burgos M., Dave M. H., Wagner C. A., Camargo S. R., Inoue S., Matsuura N., Helip‐Wooley A., Bockenhauer D., Warth R., Bernardini I., Visser G., Eggermann T., Lee P., Chairoungdua A., Jutabha P., Babu E., Nilwarangkoon S., Anzai N., Kanai Y., Verrey F., Gahl W. A., Koizumi A., Nat. Genet. 2004, 36, 999. [DOI] [PubMed] [Google Scholar]
- 29. Zucchelli M., Torkvist L., Bresso F., Halfvarson J., Hellquist A., Anedda F., Assadi G., Lindgren G. B., Svanfeldt M., Janson M., Noble C. L., Pettersson S., Lappalainen M., Paavola‐Sakki P., Halme L., Farkkila M., Turunen U., Satsangi J., Kontula K., Lofberg R., Kere J., Amato D' M., Inflamm. Bowel Dis. 2009, 15, 1562. [DOI] [PubMed] [Google Scholar]
- 30. Durand E., Boutin P., Meyre D., Charles M. A., Clement K., Dina C., Froguel P., Diabetes 2004, 53, 2483. [DOI] [PubMed] [Google Scholar]
- 31. Suviolahti E., Oksanen L. J., Ohman M., Cantor R. M., Ridderstrale M., Tuomi T., Kaprio J., Rissanen A., Mustajoki P., Jousilahti P., Vartiainen E., Silander K., Kilpikari R., Salomaa V., Groop L., Kontula K., Peltonen L., Pajukanta P., J. Clin. Invest. 2003, 112, 1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stunkard A. J., Messick S., J. Psychosom. Res. 1985, 29, 71. [DOI] [PubMed] [Google Scholar]
- 33. Lawson O. J., Williamson D. A., Champagne C. M., DeLany J. P., Brooks E. R., Howat P. M., Wozniak P. J., Bray G. A., Ryan D. H., Obes. Res. 1995, 3, 153. [DOI] [PubMed] [Google Scholar]
- 34. Binder H. J., Reuben A., in Medical Physiology: A Cellular and Molecular Approach (Eds: Boron W. F., E. L. Boulpaep), Saunders/Elsevier, Philadelphia, PA: 2009, pp. 949. [Google Scholar]
- 35. Ratnayake W. M., Galli C., Ann. Nutr. Metab. 2009, 55, 8. [DOI] [PubMed] [Google Scholar]
- 36. Gilbertson T. A., Curr. Opin. Neurobiol. 1998, 8, 447. [DOI] [PubMed] [Google Scholar]
- 37. Newman L., Haryono R., Keast R., Nutrients 2013, 5, 1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mattes R. D., Physiol. Behav. 2011, 105, 27. [DOI] [PubMed] [Google Scholar]
- 39. Bourlieu C., Menard O., Bouzerzour K., Mandalari G., Macierzanka A., Mackie A. R., Dupont D., Crit. Rev. Food Sci. Nutr. 2014, 54, 1427. [DOI] [PubMed] [Google Scholar]
- 40. Whitcomb D. C., Lowe M. E., Dig. Dis. Sci. 2007, 52, 1. [DOI] [PubMed] [Google Scholar]
- 41. Lehner R., Kuksis A., Prog. Lipid Res. 1996, 35, 169. [DOI] [PubMed] [Google Scholar]
- 42. Friedman H. I., Nylund B., Am. J. Clin. Nutr. 1980, 33, 1108. [DOI] [PubMed] [Google Scholar]
- 43. Senior J. R., J. Lipid Res. 1964, 5, 495. [PubMed] [Google Scholar]
- 44. Tso P., Adv. Lipid Res. 1985, 21, 143. [DOI] [PubMed] [Google Scholar]
- 45. Feltrin K. L., Little T. J., Meyer J. H., Horowitz M., Smout A. J., Wishart J., Pilichiewicz A. N., Rades T., Chapman I. M., Feinle‐Bisset C., Am. J. Physiol.: Regul., Integr. Comp. Physiol. 2004, 287, R524. [DOI] [PubMed] [Google Scholar]
- 46. Hussain M. M., Curr. Opin. Lipidol. 2014, 25, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schaffer J. E., Lodish H. F., Cell 1994, 79, 427. [DOI] [PubMed] [Google Scholar]
- 48. Gajda A. M., Storch J., Prostaglandins, Leukotrienes Essent. Fatty Acids 2015, 93, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dash S., Xiao C., Morgantini C., Lewis G. F., Annu. Rev. Nutr. 2015, 35, 265. [DOI] [PubMed] [Google Scholar]
- 50. Brown J. M., Rudel L. L., Yu L., Biochem. J. 2007, 406, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Duan L. P., Wang H. H., Wang D. Q., J. Lipid Res. 2004, 45, 1312. [DOI] [PubMed] [Google Scholar]
- 52. Wang D. Q., Annu. Rev. Physiol. 2007, 69, 221. [DOI] [PubMed] [Google Scholar]
- 53. Iqbal J., Boutjdir M., Rudel L. L., Hussain M. M., J. Lipid Res. 2014, 55, 2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hussain M. M., Rava P., Walsh M., Rana M., Iqbal J., Nutr. Metab. 2012, 9, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yao Z., McLeod R. S., Biochim. Biophys. Acta 1994, 1212, 152. [DOI] [PubMed] [Google Scholar]
- 56. Tepper B. J., Nurse R. J., Physiol. Behav. 1997, 61, 949. [DOI] [PubMed] [Google Scholar]
- 57. Kamphuis M. M., Saris W. H., Westerterp‐Plantenga M. S., Br. J. Nutr. 2003, 90, 199. [DOI] [PubMed] [Google Scholar]
- 58. Mattes R. D., Chem. Senses 2009, 34, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Galindo M. M., Voigt N., Stein J., van Lengerich J., Raguse J. D., Hofmann T., Meyerhof W., Behrens M., Chem. Senses 2012, 37, 123. [DOI] [PubMed] [Google Scholar]
- 60. Stewart J. E., Feinle‐Bisset C., Golding M., Delahunty C., Clifton P. M., Keast R. S., Br. J. Nutr. 2010, 104, 145. [DOI] [PubMed] [Google Scholar]
- 61. Stewart J. E., Newman L. P., Keast R. S., Clin. Nutr. 2011, 30, 838. [DOI] [PubMed] [Google Scholar]
- 62. Mattes R. D., Am. J. Clin. Nutr. 1993, 57, 373. [DOI] [PubMed] [Google Scholar]
- 63. Stewart J. E., Keast R. S., Int. J. Obes. 2012, 36, 834. [DOI] [PubMed] [Google Scholar]
- 64. Mattes R. D., Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Neyraud E., Palicki O., Schwartz C., Nicklaus S., Feron G., Arch. Oral Biol. 2012, 57, 556. [DOI] [PubMed] [Google Scholar]
- 66. Stewart J. E., Seimon R. V., Otto B., Keast R. S., Clifton P. M., Feinle‐Bisset C., Am. J. Clin. Nutr. 2011, 93, 703. [DOI] [PubMed] [Google Scholar]
- 67. Tucker R. M., Laguna L., Quinn R., Mattes R. D., Chemosens. Percept. 2013, 6, 78. [Google Scholar]
- 68. Reed D. R., Bachmanov A. A., Beauchamp G. K., Tordoff M. G., Price R. A., Behav. Genet. 1997, 27, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Keller K. L., Liang L. C., Sakimura J., May D., van Belle C., Breen C., Driggin E., Tepper B. J., Lanzano P. C., Deng L., Chung W. K., Obesity 2012, 20, 1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pepino M. Y., Love‐Gregory L., Klein S., Abumrad N. A., J. Lipid Res. 2012, 53, 561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tanaka T., Ngwa J. S., van Rooij F. J., Zillikens M. C., Wojczynski M. K., Frazier‐Wood A. C., Houston D. K., Kanoni S., Lemaitre R. N., Luan J., Mikkila V., Renstrom F., Sonestedt E., Zhao J. H., Chu A. Y., Qi L., Chasman D. I., de Oliveira Otto M. C., Dhurandhar E. J., Feitosa M. F., Johansson I., Khaw K. T., Lohman K. K., Manichaikul A., McKeown N. M., Mozaffarian D., Singleton A., Stirrups K., Viikari J., Ye Z., et al., Am. J. Clin. Nutr. 2013, 97, 1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chen M. M., Hale C., Stanislaus S., Xu J., Veniant M. M., J. Endocrinol. 2018, 237, 139. [DOI] [PubMed] [Google Scholar]
- 73. Reed D. R., Xia M. B., Adv. Nutr. 2015, 6, 353S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Running C. A., Mattes R. D., Tucker R. M., Prog. Lipid Res. 2013, 52, 438. [DOI] [PubMed] [Google Scholar]
- 75. Armand M., Hamosh M., DiPalma J. S., Gallagher J., Benjamin S. B., Philpott J. R., Lairon D., Hamosh P., Am. J. Clin. Nutr. 1995, 62, 74. [DOI] [PubMed] [Google Scholar]
- 76. Wermter A. K., Scherag A., Holter K., Reichwald K., Lichtner P., Siegfried W., Blundell J., Lawton C., Whybrow S., Stubbs J., Arch J. R., Meitinger T., Platzer M., Hinney A., Hebebrand J., Obes. Facts 2009, 2, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bisgaier C. L., Glickman R. M., Annu. Rev. Physiol. 1983, 45, 625. [DOI] [PubMed] [Google Scholar]
- 78. Madsen J. L., Dig. Dis. Sci. 1992, 37, 1548. [DOI] [PubMed] [Google Scholar]
- 79. Maillot F., Garrigue M. A., Pinault M., Objois M., Theret V., Lamisse F., Hoinard C., Antoine J. M., Lairon D., Couet C., Diab. Metab. 2005, 31, 69. [DOI] [PubMed] [Google Scholar]
- 80. Clarysse S., Tack J., Lammert F., Duchateau G., Reppas C., Augustijns P., J. Pharm. Sci. 2009, 98, 1177. [DOI] [PubMed] [Google Scholar]
- 81. Rideout T. C., Harding S. V., Jones P. J., Fan M. Z., Vasc. Health Risk Manag. 2008, 4, 1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Simons L. A., Gayst S., Balasubramaniam S., Ruys J., Atherosclerosis 1982, 45, 101. [DOI] [PubMed] [Google Scholar]
- 83. Plosch T., Kruit J. K., Bloks V. W., Huijkman N. C., Havinga R., Duchateau G. S., Lin Y., Kuipers F., J. Nutr. 2006, 136, 2135. [DOI] [PubMed] [Google Scholar]
- 84. Ellegard L., Bosaeus I., Andersson H., Eur. J. Clin. Nutr. 2000, 54, 306. [DOI] [PubMed] [Google Scholar]
- 85. Alvaro A., Rosales R., Masana L., Vallve J. C., J. Nutr. Biochem. 2010, 21, 518. [DOI] [PubMed] [Google Scholar]
- 86. Yang F., Chen G., Ma M., Qiu N., Zhu L., Li J., Lipids Health Dis. 2018, 17, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lobo M. V., Huerta L., Ruiz‐Velasco N., Teixeiro E., de la Cueva P., Celdran A., Martin‐Hidalgo A., Vega M. A., Bragado R., J. Histochem. Cytochem. 2001, 49, 1253. [DOI] [PubMed] [Google Scholar]
- 88. Lynes M. D., Widmaier E. P., Life Sci. 2011, 88, 384. [DOI] [PubMed] [Google Scholar]
- 89. McKimmie R. L., Easter L., Weinberg R. B., Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Siddiqi S., Saleem U., Abumrad N. A., Davidson N. O., Storch J., Siddiqi S. A., C. M. Mansbach, 2nd , J. Lipid Res. 2010, 51, 1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Weizman Z., Durie P. R., Kopelman H. R., Vesely S. M., Forstner G. G., Gut 1986, 27, 1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Yamamoto Y., Hiasa Y., Murakami H., Ikeda Y., Yamanishi H., Abe M., Matsuura B., Onji M., Am. J. Clin. Nutr. 2012, 96, 90. [DOI] [PubMed] [Google Scholar]
- 93. Galman C., Angelin B., Rudling M., Gastroenterology 2005, 129, 1445. [DOI] [PubMed] [Google Scholar]
- 94. Steiner C., Othman A., Saely C. H., Rein P., Drexel H., von Eckardstein A., Rentsch K. M., PLoS One 2011, 6, e25006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Maury E., Ramsey K. M., Bass J., Circ. Res. 2010, 106, 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Beglinger C., Degen L., Physiol. Behav. 2004, 83, 617. [DOI] [PubMed] [Google Scholar]
- 97. Liou A. P., Lu X., Sei Y., Zhao X., Pechhold S., Carrero R. J., Raybould H. E., Wank S., Gastroenterology 2011, 140, 903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Little T. J., Feinle‐Bisset C., Physiol. Behav. 2011, 104, 613. [DOI] [PubMed] [Google Scholar]
- 99. Little T. J., Isaacs N. J., Young R. L., Ott R., Nguyen N. Q., Rayner C. K., Horowitz M., Feinle‐Bisset C., Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G958. [DOI] [PubMed] [Google Scholar]
- 100. Wang H. H., Afdhal N. H., Gendler S. J., Wang D. Q., Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G547. [DOI] [PubMed] [Google Scholar]
- 101. Kawamata Y., Fujii R., Hosoya M., Harada M., Yoshida H., Miwa M., Fukusumi S., Habata Y., Itoh T., Shintani Y., Hinuma S., Fujisawa Y., Fujino M., J. Biol. Chem. 2003, 278, 9435. [DOI] [PubMed] [Google Scholar]
- 102. Poole D. P., Godfrey C., Cattaruzza F., Cottrell G. S., Kirkland J. G., Pelayo J. C., Bunnett N. W., Corvera C. U., Neurogastroenterol. Motil. 2010, 22, 814, e227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Camilleri M., Shin A., Busciglio I., Carlson P., Acosta A., Bharucha A. E., Burton D., Lamsam J., Lueke A., Donato L. J., Zinsmeister A. R., Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Lee R. G., Willingham M. C., Davis M. A., Skinner K. A., Rudel L. L., J. Lipid. Res. 2000, 41, 1991. [PubMed] [Google Scholar]
- 105. Buhman K. K., Accad M., Novak S., Choi R. S., Wong J. S., Hamilton R. L., Turley S., R. V. Farese, Jr. , Nat. Med. 2000, 6, 1341. [DOI] [PubMed] [Google Scholar]
- 106. Willner E. L., Tow B., Buhman K. K., Wilson M., Sanan D. A., Rudel L. L., R. V. Farese, Jr. , Proc. Natl. Acad. Sci. USA 2003, 100, 1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. He X., Lu Y., Saha N., Yang H., Heng C. K., Hum. Genet. 2005, 118, 393. [DOI] [PubMed] [Google Scholar]
- 108. He X., Leow K. Y., Yang H., Heng C. K., Gene 2015, 566, 236. [DOI] [PubMed] [Google Scholar]
- 109. Geschonke K., Klempt M., Lynch N., Schreiber S., Fenselau S., Schrezenmeir J., Ann. N. Y. Acad. Sci. 2002, 967, 548. [DOI] [PubMed] [Google Scholar]
- 110. Agren J. J., Valve R., Vidgren H., Laakso M., Uusitupa M., Arterioscler., Thromb., Vasc. Biol. 1998, 18, 1606. [DOI] [PubMed] [Google Scholar]
- 111. Huggins K. W., Boileau A. C., Hui D. Y., Am. J. Physiol. Endocrinol. Metab. 2002, 283, E994. [DOI] [PubMed] [Google Scholar]
- 112. Richmond B. L., Boileau A. C., Zheng S., Huggins K. W., Granholm N. A., Tso P., Hui D. Y., Gastroenterology 2001, 120, 1193. [DOI] [PubMed] [Google Scholar]
- 113. Lecompte S., Szabo de Edelenyi F., Goumidi L., Maiani G., Moschonis G., Widhalm K., Molnar D., Kafatos A., Spinneker A., Breidenassel C., Dallongeville J., Meirhaeghe A., Borel P., Am. J. Clin. Nutr. 2011, 93, 644. [DOI] [PubMed] [Google Scholar]
- 114. Borel P., Mol. Nutr. Food Res. 2012, 56, 228. [DOI] [PubMed] [Google Scholar]
- 115. Abumrad N. A., Davidson N. O., Physiol. Rev. 2012, 92, 1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Kindel T., Lee D. M., Tso P., Atheroscler. Suppl. 2010, 11, 11. [DOI] [PubMed] [Google Scholar]
- 117. Xie Y., Newberry E. P., Young S. G., Robine S., Hamilton R. L., Wong J. S., Luo J., Kennedy S., Davidson N. O., J. Biol. Chem. 2006, 281, 4075. [DOI] [PubMed] [Google Scholar]
- 118. Sharp D., Blinderman L., Combs K. A., Kienzle B., Ricci B., Wager‐Smith K., Gil C. M., Turck C. W., Bouma M. E., Rader D. J., et al., Nature 1993, 365, 65. [DOI] [PubMed] [Google Scholar]
- 119. Rubin D., Schneider‐Muntau A., Klapper M., Nitz I., Helwig U., Folsch U. R., Schrezenmeir J., Doring F., Hum. Mutat. 2008, 29, 123. [DOI] [PubMed] [Google Scholar]
- 120. Pan S. L., Luo X. Q., Lu Z. P., Lu S. H., Luo H., Liu C. W., Hu C. Y., Yang M., Du L. L., Song Z., Pang G. F., Wu H. Y., Huang J. B., Peng J. H., Yin R. X., Lipids Health Dis. 2012, 11, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Ledmyr H., Karpe F., Lundahl B., McKinnon M., Skoglund‐Andersson C., Ehrenborg E., J. Lipid. Res. 2002, 43, 51. [PubMed] [Google Scholar]
- 122. Tian Y., Li H., Wang S., Yan J., Chen Z., Li Z., Feng H., Zhou H., Ouyang D., Lipids 2015, 50, 275. [DOI] [PubMed] [Google Scholar]
- 123. Hsiao P. J., Lee M. Y., Wang Y. T., Jiang H. J., Lin P. C., Yang Y. H., Kuo K. K., BMC Med. Genet. 2015, 16, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Kozlitina J., Smagris E., Stender S., Nordestgaard B. G., Zhou H. H., Tybjaerg‐Hansen A., Vogt T. F., Hobbs H. H., Cohen J. C., Nat. Genet. 2014, 46, 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Hare O' E. A., Yang R., Yerges‐Armstrong L. M., Sreenivasan U., McFarland R., Leitch C. C., Wilson M. H., Narina S., Gorden A., Ryan K. A., Shuldiner A. R., Farber S. A., Wood G. C., Still C. D., Gerhard G. S., Robishaw J. D., Sztalryd C., Zaghloul N. A., Hepatology 2017, 65, 1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Levy E., J. Lipid Res. 2015, 56, 945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Desmarchelier C., Martin J. C., Planells R., Gastaldi M., Nowicki M., Goncalves A., Valero R., Lairon D., Borel P., J. Clin Endocrinol Metabol. 2014, 99, E484. [DOI] [PubMed] [Google Scholar]
- 128. De Smet E., Mensink R. P., Plat J., Mol. Nutr. Food Res. 2012, 56, 1058. [DOI] [PubMed] [Google Scholar]
- 129. Morton G. M., Lee S. M., Buss D. H., Lawrance P., J. Hum. Nutr. Diet. 1995, 8, 429. [Google Scholar]
- 130. R. E. Ostlund, Jr. , Annu. Rev. Nutr. 2002, 22, 533. [DOI] [PubMed] [Google Scholar]
- 131. Kempen H. J., de Knijff P., Boomsma D. I., van der Voort H. A., Gevers Leuven J. A., Havekes L., Metabolism 1991, 40, 604. [DOI] [PubMed] [Google Scholar]
- 132. Miettinen T. A., Tilvis R. S., Kesaniemi Y. A., Am. J. Epidemiol. 1990, 131, 20. [DOI] [PubMed] [Google Scholar]
- 133. Berge K. E., von Bergmann K., Lutjohann D., Guerra R., Grundy S. M., Hobbs H. H., Cohen J. C., J. Lipid. Res. 2002, 43, 486. [PubMed] [Google Scholar]
- 134. Houweling A. H., Vanstone C. A., Trautwein E. A., Duchateau G. S., Jones P. J., Eur. J. Clin. Nutr. 2009, 63, 543. [DOI] [PubMed] [Google Scholar]
- 135. De Castro‐Oros I., Pampin S., Cofan M., Mozas P., Pinto X., Salas‐Salvado J., Rodriguez‐Rey J. C., Ros E., Civeira F., Pocovi M., Clin. Nutr. 2011, 30, 239. [DOI] [PubMed] [Google Scholar]
- 136. Bayly G. R., in Clinical Biochemistry: Metabolic and Clinical Aspects (Eds: Marshall W., Lapsley M., Day A., R. Ayling), Churchill Livingstone, London: 2014, pp. 702. [Google Scholar]
- 137. Chan Y. M., Varady K. A., Lin Y., Trautwein E., Mensink R. P., Plat J., Jones P. J., Nutr. Rev. 2006, 64, 385. [DOI] [PubMed] [Google Scholar]
- 138. Gylling H., Hallikainen M., Pihlajamaki J., Agren J., Laakso M., Rajaratnam R. A., Rauramaa R., Miettinen T. A., J. Lipid Res. 2004, 45, 1660. [DOI] [PubMed] [Google Scholar]
- 139. Plat J., Bragt M. C., Mensink R. P., J. Lipid Res. 2005, 46, 68. [DOI] [PubMed] [Google Scholar]
- 140. Teupser D., Baber R., Ceglarek U., Scholz M., Illig T., Gieger C., Holdt L. M., Leichtle A., Greiser K. H., Huster D., Linsel‐Nitschke P., Schafer A., Braund P. S., Tiret L., Stark K., Raaz‐Schrauder D., Fiedler G. M., Wilfert W., Beutner F., Gielen S., Grosshennig A., Konig I. R., Lichtner P., Heid I. M., Kluttig A., El Mokhtari N. E., Rubin D., Ekici A. B., Reis A., Garlichs C. D., Hall A. S., Matthes G., Wittekind C., Hengstenberg C., Cambien F., Schreiber S., Werdan K., Meitinger T., Loeffler M., Samani N. J., Erdmann J., Wichmann H. E., Schunkert H., Thiery J., Circ. Cardiovasc. Genet. 2010, 3, 331. [DOI] [PubMed] [Google Scholar]
- 141. Jakulj L., Vissers M. N., Tanck M. W., Hutten B. A., Stellaard F., Kastelein J. J., Dallinga‐Thie G. M., J. Lipid Res. 2010, 51, 3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Miwa K., Inazu A., Kobayashi J., Higashikata T., Nohara A., Kawashiri M., Katsuda S., Takata M., Koizumi J., Mabuchi H., Clin. Sci. 2005, 109, 183. [DOI] [PubMed] [Google Scholar]
- 143. Cohen J. C., Pertsemlidis A., Fahmi S., Esmail S., Vega G. L., Grundy S. M., Hobbs H. H., Proc. Natl. Acad. Sci. USA 2006, 103, 1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. SACN, S. A. C. o. N. , Scientific Advisory Committee on Nutrition 2015.
- 145. Food and Agricultural Organization of the United Nations , Carbohydrates in Human Nutrition, Food and Nutrition Paper, Rome: 1997, 66, 140. [Google Scholar]
- 146. Kaiser S., Healthy Eating | SF Gate 2018.
- 147. Freeman C. R., Zehra A., Ramirez V., Wiers C. E., Volkow N. D., Wang G. J., Front. Biosci. 2018, 23, 2255. [DOI] [PubMed] [Google Scholar]
- 148. Livesey G., Taylor R., Livesey H., Liu S., Am. J. Clin. Nutr. 2013, 97, 584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Zeevi D., Korem T., Zmora N., Israeli D., Rothschild D., Weinberger A.,Ben‐Yacov O., Lador D., Avnit‐Sagi T., Lotan‐Pompan M., Suez J., Mahdi J. A., Matot E., Malka G., Kosower N., Rein M., Zilberman‐Schapira G., Dohnalova L., Pevsner‐Fischer M., Bikovsky R., Halpern Z., Elinav E., Segal E., Cell 2015, 163, 1079. [DOI] [PubMed] [Google Scholar]
- 150. Zhu Y., Hsu W. H., Hollis J. H., Physiol. Behav. 2014, 133, 136. [DOI] [PubMed] [Google Scholar]
- 151. Ranawana V., Clegg M. E., Shafat A., Henry C. J., Nutr. Res. 2011, 31, 452. [DOI] [PubMed] [Google Scholar]
- 152. Ranawana V., Monro J. A., Mishra S., Henry C. J., Nutr. Res. 2010, 30, 246. [DOI] [PubMed] [Google Scholar]
- 153. Nakajima K., World J. Diabet. 2016, 7, 112. [Google Scholar]
- 154. Santos J. L., Saus E., Smalley S. V., Cataldo L. R., Alberti G., Parada J., Gratacos M., Estivill X., J. Nutrigenet. Nutrigenomics 2012, 5, 117. [DOI] [PubMed] [Google Scholar]
- 155. Carpenter D., Dhar S., Mitchell L. M., Fu B., Tyson J., Shwan N. A., Yang F., Thomas M. G., Armour J. A., Hum. Mol. Genet. 2015, 24, 3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Vionnet N., Münger L. H., Freiburghaus C., Burton K. J., Pimentel G., Pralong F. P., Badertscher R., Vergères G., Am. J. Clin. Nutr. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Deng Y., Misselwitz B., Dai N., Fox M., Nutrients 2015, 7, 8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Sibley E., Am. J. Pharmacogenomics 2004, 4, 239. [DOI] [PubMed] [Google Scholar]
- 159. Lukito W., Malik S. G., Surono I. S., Wahlqvist M. L., Asia. Pac. J. Clin. Nutr. 2015, 24, S1. [DOI] [PubMed] [Google Scholar]
- 160. Iizuka K., Nutrients 2017, 9. [Google Scholar]
- 161. Lee H. J., Cha J. Y., BMB Rep. 2018, 51, 429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Vega‐Lopez S., Ausman L. M., Griffith J. L., Lichtenstein A. H., Diabetes Care. 2007, 30, 1412. [DOI] [PubMed] [Google Scholar]
- 163. Seidelmann S. B., Feofanova E., Yu B., Franceschini N., Claggett B., Kuokkanen M., Puolijoki H., Ebeling T., Perola M., Salomaa V., Shah A., Coresh J., Selvin E., MacRae C. A., Cheng S., Boerwinkle E., Solomon S. D., J. Am. Coll. Cardiol. 2018, 72, 1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Holscher H. D., Gut Microbes 2017, 8, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Flint H. J., Nutr. Rev. 2012, 70 Suppl 1, S10. [DOI] [PubMed] [Google Scholar]
- 166. El Kaoutari A., Armougom F., Gordon J. I., Raoult D., Henrissat B., Nat. Rev. Microbiol. 2013, 11, 497. [DOI] [PubMed] [Google Scholar]
- 167. Xiao S., Fei N., Pang X., Shen J., Wang L., Zhang B., Zhang M., Zhang X., Zhang C., Li M., Sun L., Xue Z., Wang J., Feng J., Yan F., Zhao N., Liu J., Long W., Zhao L., FEMS Microbiol. Ecol. 2014, 87, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Wu G. D., Chen J., Hoffmann C., Bittinger K., Chen Y. Y., Keilbaugh S. A., Bewtra M., Knights D., Walters W. A., Knight R., Sinha R., Gilroy E., Gupta K., Baldassano R., Nessel L., Li H., Bushman F. D., Lewis J. D., Science 2011, 334, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Pallister T., Spector T. D., J. R. Soc. Med. 2016, 109, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Roberfroid M., Gibson G. R., Hoyles L., McCartney A. L., Rastall R., Rowland I., Wolvers D., Watzl B., Szajewska H., Stahl B., Guarner F., Respondek F., Whelan K., Coxam V., Davicco M. J., Leotoing L., Wittrant Y., Delzenne N. M., Cani P. D., Neyrinck A. M., Meheust A., Br. J. Nutr. 2010, 104 Suppl 2, S1. [DOI] [PubMed] [Google Scholar]
- 171. Hamaker B. R., Tuncil Y. E., J. Mol. Biol. 2014, 426, 3838. [DOI] [PubMed] [Google Scholar]
- 172. Hahn A., Ströhle A., Wolters M., Ernährung ‐ Physiologische Grundlagen, Prävention, Therapie, Wissenschaftliche Verlagsgesellschaft mbH, Stuttgart: 2005. [Google Scholar]
- 173. Gropper S. S., Smith J. L., Carr T. P., Advanced Nutrition and Human Metabolism Cengage Learning 2016. [Google Scholar]
- 174. Said H. M., Biochem. J. 2011, 437, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Shearer M. J., Fu X., Booth S. L., Adv Nutr 2012, 3, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Hollander D., Muralidhara K., Rim E., Am. J. Physiol. 1976, 230, 251. [DOI] [PubMed] [Google Scholar]
- 177. Rose R. C., A. M. Hoyumpa, Jr. , Allen R. H., H. M. Middleton, 3rd , Henderson L. M., Rosenberg I. H., Fed. Proc. 1984, 43, 2423. [PubMed] [Google Scholar]
- 178. Yamanashi Y., Takada T., Kurauchi R., Tanaka Y., Komine T., Suzuki H., J. Atheroscler. Thromb. 2017, 24, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Stover P. J., Food Nutr. Bull. 2007, 28, S101. [DOI] [PubMed] [Google Scholar]
- 180. Mathias M. G., Coelho‐Landell C. A., Scott‐Boyer M. P., Lacroix S., Morine M. J., Salomao R. G., Toffano R. B. D., Almada M., Camarneiro J. M., Hillesheim E., de Barros T. T., Camelo‐Junior J. S., Campos Gimenez E., Redeuil K., Goyon A., Bertschy E., Leveques A., Oberson J. M., Gimenez C., Carayol J., Kussmann M., Descombes P., Metairon S., Draper C. F., Conus N., Mottaz S. C., Corsini G. Z., Myoshi S. K. B., Muniz M. M., Hernandes L. C., Venancio V. P., Antunes L. M. G., da Silva R. Q., Laurito T. F., Rossi I. R., Ricci R., Jorge J. R., Faga M. L., Quinhoneiro D. C. G., Reche M. C., Silva P. V. S., Falquetti L. L., da Cunha T. H. A., Deminice T. M. M., Tambellini T. H., de Souza G. C. A., de Oliveira M. M., Nogueira‐Pileggi V., Matsumoto M. T., Priami C., Kaput J., Monteiro J. P., Mol. Nutr. Food Res. 2018, 62, e1700613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Block G., Mangels A. R., Patterson B. H., Levander O. A., Norkus E. P., Taylor P. R., J. Am. Coll. Nutr. 1999, 18, 628. [DOI] [PubMed] [Google Scholar]
- 182. Michels A. J., Hagen T. M., Frei B., Annu. Rev. Nutr. 2013, 33, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Shaghaghi M. A., Kloss O., Eck P., Adv Nutr 2016, 7, 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Cahill L. E., El Sohemy A., J. Nutrigenet. Nutrigenomics 2009, 2, 292. [DOI] [PubMed] [Google Scholar]
- 185. Said H. M., Nexo E., Physiology of the Gastrointestinal Tract (6th Edition), Academic Press; 2018, pp. 1201. [Google Scholar]
- 186. Said H. M., Chatterjee N., Haq R. U., Subramanian V. S., Ortiz A., Matherly L. H., Sirotnak F. M., Halsted C., Rubin S. A., Am. J. Physiol.: Cell Physiol. 2000, 279, C1889. [DOI] [PubMed] [Google Scholar]
- 187. Cummings D., Dowling K. F., Silverstein N. J., Tanner A. S., Eryilmaz H., Smoller J. W., Roffman J. L., Nutrients 2017, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Zinck J. W., de Groh M., MacFarlane A. J., Am. J. Clin. Nutr. 2015, 101, 1295. [DOI] [PubMed] [Google Scholar]
- 189. Lee Y.‐Y., Wei Y.‐C., Tian Y.‐F., Sun D.‐P., Sheu M.‐J., Yang C.‐C., Lin L.‐C., Lin C.‐Y., Hsing C.‐H., Li W.‐S., Li C.‐F., Hsieh P.‐L., Lin C.‐Y., J. Cancer 2017, 8, 1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Maqbool M. A., Aslam M., Akbar W., Iqbal Z., J. Agric. Basic Sci. 2017, 02, 9. [Google Scholar]
- 191. Harrison E. H., Kopec R. E., Physiology of the Gastrointestinal Tract (6th Edition), Academic Press, Cambridge, MA: 2018, pp. 1133. [Google Scholar]
- 192. Goncalves A., Roi S., Nowicki M., Dhaussy A., Huertas A., Amiot M. J., Reboul E., Food Chem. 2015, 172, 155. [DOI] [PubMed] [Google Scholar]
- 193. Harrison E. H., Biochim. Biophys. Acta 2012, 1821, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Cooperstone J. L., Goetz H. J., Riedl K. M., Harrison E. H., Schwartz S. J., Kopec R. E., Am. J. Clin. Nutr. 2017, 106, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Borel P., Desmarchelier C., Nutrients 2017, 9, 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Folli C., Viglione S., Busconi M., Berni R., Biochem. Biophys. Res. Commun. 2005, 336, 1017. [DOI] [PubMed] [Google Scholar]
- 197. Hendrickson S. J., Hazra A., Chen C., Eliassen A. H., Kraft P., Rosner B. A., Willett W. C., Am. J. Clin. Nutr. 2012, 96, 1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Borel P., Caillaud D., Cano N. J., Crit. Rev. Food Sci. Nutr. 2015, 55, 1193. [DOI] [PubMed] [Google Scholar]
- 199. Maurya V. K., Aggarwal M., J. Food Sci. Technol. 2017, 54, 3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Gallagher J. C., Endocrinol. Metab. Clin. North Am. 2013, 42, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Desmarchelier C., Borel P., Goncalves A., Kopec R., Nowicki M., Morange S., Lesavre N., Portugal H., Reboul E., J. Nutr. 2016, 146, 2421. [DOI] [PubMed] [Google Scholar]
- 202. He H. Y., Liu M. Z., Zhang Y. L., Zhang W., Genomics, Proteomics Bioinf. 2017, 15, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Jacobs E. T., Van Pelt C., Forster R. E., Zaidi W., Hibler E. A., Galligan M. A., Haussler M. R., Jurutka P. W., Cancer Res. 2013, 73, 2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. McCullough M. L., Bostick R. M., Mayo T. L., Annu. Rev. Nutr. 2009, 29, 111. [DOI] [PubMed] [Google Scholar]
- 205. Desmarchelier C., Borel P., Physiology of the Gastrointestinal Tract (6th Edition), Academic Press, Cambridge, MA 2018, pp. 1181. [Google Scholar]
- 206. Borel P., Desmarchelier C., Int. J. Mol. Sci. 2016, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Borel P., Desmarchelier C., Nowicki M., Bott R., Tourniaire F., Antioxid. Redox Signaling 2015, 22, 669. [DOI] [PubMed] [Google Scholar]
- 208. Yamanashi Y., Takada T., Suzuki H., Pharmacogenet. Genomics 2009, 19, 884. [DOI] [PubMed] [Google Scholar]
- 209. Guardiola M., Ribalta J., Gomez‐Coronado D., Lasuncion M. A., de Oya M., Garces C., Atherosclerosis 2010, 212, 543. [DOI] [PubMed] [Google Scholar]
- 210. Tamargo Menéndez J., Delpón E. E., in: Tresguerres J. A. F., Ariznavarreta C., Cachofeiro V., Cardinali D., Escriche E., Gil‐Loyzaga P., Lahera Juliá V., Mora Teruel F., Romano Pardo M., Tamargo Menéndez J. (Eds.), Fisiología humana, MacGraw Hill‐Interamericana de España, Madrid: 2005, pp. 485. [Google Scholar]
- 211. Anderson J. M., News Physiol. Sci. 2001, 16, 126. [DOI] [PubMed] [Google Scholar]
- 212. Marks J., Pflügers Arch. 2019, 471, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Mäkelä S., Kere J., Holmberg C., Hoglund P., Hum. Mutat. 2002, 20, 425. [DOI] [PubMed] [Google Scholar]
- 214. Nijenhuis T., Vallon V., van der Kemp A. W., Loffing J., Hoenderop J. G., Bindels R. J. J. Clin. Invest. 2005, 115, 1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Medici V., Weiss K. H., Handb. Clin. Neurol. 2017, 142, 35. [DOI] [PubMed] [Google Scholar]
- 216. Kimura T., Kambe T., Int. J. Mol. Sci. 2016, 17, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Kayaalti Z., Aliyev V., Soylemezoglu T., Toxicol. Appl. Pharmacol. 2011, 256, 1. [DOI] [PubMed] [Google Scholar]
- 218. Abrams S. A., Griffin I. J., Hawthorne K. M., Chen Z., Gunn S. K., Wilde M., Darlington G., Shypailo R. J., Ellis K. J., J. Bone Miner. Res. 2005, 20, 945. [DOI] [PubMed] [Google Scholar]
- 219. Whitfield J. B., Cullen L. M., Jazwinska E. C., Powell L. W., Heath A. C., Zhu G., Duffy L. D., Martin N. G., Am. J. Hum. Genet. 2000, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Mithen R., Int. J. Vitam. Nutr. Res. 2007, 77, 205. [DOI] [PubMed] [Google Scholar]
- 221. Fairweather‐Tait S. J., Harvey L., Heath A. L. M., Roe M., Genes & Nutrition 2007, 2, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Thomson A. B., Chopra A., Clandinin M. T., Freeman H., World J. Gastroenterol. 2012, 18, 3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Radio F. C., Majore S., Aurizi C., Sorge F., Biolcati G., Bernabini S., Giotti I., Torricelli F., Giannarelli D., De Bernardo C., Grammatico P., Blood Cells Mol. Dis. 2015, 55, 71. [DOI] [PubMed] [Google Scholar]
- 224. Rivers C. A., Barton J. C., Gordeuk V. R., Acton R. T., Speechley M. R., Snively B. M., Leiendecker‐Foster C., Press R. D., Adams P. C., McLaren G. D., Dawkins F. W., McLaren C. E., Reboussin D. M., Blood Cells Mol. Dis. 2007, 38, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Constantine C. C., Anderson G. J., Vulpe C. D., McLaren C. E., Bahlo M., Yeap H. L., Gertig D. M., Osborne N. J., Bertalli N. A., Beckman K. B., Chen V., Matak P., McKie A. T., Delatycki M. B., Olynyk J. K., English D. R., Southey M. C., Giles G. G., Hopper J. L., Allen K. J., Gurrin L. C., Br. J. Haematol. 2009, 147, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Montalbetti N., Simonin A., Kovacs G., Hediger M. A., Mol. Aspects Med. 2013, 34, 270. [DOI] [PubMed] [Google Scholar]
- 227. Bashiardes S., Godneva A., Elinav E., Segal E., Curr. Opin. Biotechnol. 2018, 51, 57. [DOI] [PubMed] [Google Scholar]
- 228. Meyer K. A., Bennett B. J., Curr. Diab. Rep. 2016, 16, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Cho C. E., Taesuwan S., Malysheva O. V., Bender E., Tulchinsky N. F., Yan J., Sutter J. L., Caudill M. A., Mol. Nutr. Food Res. 2017, 61. [DOI] [PubMed] [Google Scholar]
- 230. Korpela K., Flint H. J., Johnstone A. M., Lappi J., Poutanen K., Dewulf E., Delzenne N., de Vos W. M., Salonen A., PLoS One 2014, 9, e90702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Bos C., Mahé S., Gaudichon C., Benamouzig R., Gausserès N., Luengo C., Ferrière F., Rautureau J., Tomé D., Br. J. Nutr. 1999, 81, 221. [PubMed] [Google Scholar]
- 232. Gaudichon C., Mahé S., Benamouzig R., Luengo C., Fouillet H., Daré S., van Oycke M., Ferrière F., Rautureau J., Tomé D., J. Nutr. 1999, 129, 890. [DOI] [PubMed] [Google Scholar]
- 233. Deglaire A., Bos C., Tome D., Moughan P. J., Br. J. Nutr. 2009, 102, 1752. [DOI] [PubMed] [Google Scholar]
- 234. Oberli M., Marsset‐Baglieri A., Airinei G., Sante‐Lhoutellier V., Khodorova N., Remond D., Foucault‐Simonin A., Piedcoq J., Tome D., Fromentin G., Benamouzig R., Gaudichon C., J. Nutr. 2015, 145, 2221. [DOI] [PubMed] [Google Scholar]
- 235. Bos C., Juillet B., Fouillet H., Turlan L., Dare S., Luengo C., Tounda N' R., Benamouzig R., Gausseres N., Tome D., Gaudichon C., Am. J. Clin. Nutr. 2005, 81, 87. [DOI] [PubMed] [Google Scholar]
- 236. Mariotti F., Mahé S., Luengo C., Benamouzig R., Tomé D., Am. J. Clin. Nutr. 2000, 72, 954. [DOI] [PubMed] [Google Scholar]
- 237. Gausserès N., Mahé S., Benamouzig R., Luengo C., Ferrière F., Rautureau J., Tomé D., J. Nutr. 1997, 127, 1160. [DOI] [PubMed] [Google Scholar]
- 238. Mariotti F., Pueyo M. E., Tomé D., Bérot S., Benamouzig R., Mahé S., J. Nutr. 2001, 131, 1706. [DOI] [PubMed] [Google Scholar]
- 239. Mariotti F., Pueyo M. E., Tomé D., Mahé S., Br. J. Nutr. 2002, 87, 315. [DOI] [PubMed] [Google Scholar]
- 240. FAO/WHO/UNU, 1985.
- 241. Bodwell C. E., Satterlee L. D., Hackler L. R., Am. J. Clin. Nutr. 1980, 33, 677. [DOI] [PubMed] [Google Scholar]
- 242. Wisker E., Bach Knudsen K. E., Daniel M., Feldheim W., Eggum B. O., J. Nutr. 1996, 126, 481. [DOI] [PubMed] [Google Scholar]
- 243. Duell E. J., Lujan‐Barroso L., Llivina C., Munoz X., Jenab M., Boutron‐Ruault M. C., Clavel‐Chapelon F., Racine A., Boeing H., Buijsse B., Canzian F., Johnson T., Dalgard C., Overvad K., Tjonneland A., Olsen A., Sanchez S. C., Sanchez‐Cantalejo E., Huerta J. M., Ardanaz E., Dorronsoro M., Khaw K. T., Travis R. C., Trichopoulou A., Trichopoulos D., Rafnsson S., Palli D., Sacerdote C., Tumino R., Panico S., Grioni S., Bueno‐de‐Mesquita H. B., Ros M. M., Numans M. E., Peeters P. H., Johansen D., Lindkvist B., Johansson M., Johansson I., Skeie G., Weiderpass E., Duarte‐Salles T., Stenling R., Riboli E., Sala N., Gonzalez C. A., Genes Nutr. 2013, 8, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Zanon‐Moreno V., Ciancotti‐Olivares L., Asencio J., Sanz P., Ortega‐Azorin C., Pinazo‐Duran M. D., Corella D., Mol. Vision 2011, 17, 2997. [PMC free article] [PubMed] [Google Scholar]
- 245. Lahner E., Gentile G., Purchiaroni F., Mora B., Simmaco M., Annibale B., Dig. Liver Dis. 2015, 47, 285. [DOI] [PubMed] [Google Scholar]
- 246. Tanner S. M., Li Z., Bisson R., Acar C., Öner C., Öner R., Çetin M., Abdelaal M. A., Ismail E. A., Lissens W., Krahe R., Broch H., Gräsbeck R., Chapelle, A. D. L. , Hum. Mutat. 2004, 23, 327. [DOI] [PubMed] [Google Scholar]
- 247. DeVos L., Chanson A., Liu Z., Ciappio E. D., Parnell L. D., Mason J. B., Tucker K. L., Crott J. W., Am. J. Clin. Nutr. 2008, 88, 1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Stanisławska‐Sachadyn A., Mitchell L. E., Woodside J. V., Buckley P. T., Kealey C., Young I. S., Scott J. M., Murray L., Boreham C. A., McNulty H., Strain J. J., Whitehead A. S., Ann. Hum. Genet. 2009, 73, 484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Borel P., Preveraud D., Desmarchelier C., Nutr. Rev. 2013, 71, 319. [DOI] [PubMed] [Google Scholar]
- 250. Borel P., Desmarchelier C., Nowicki M., Bott R., Free Radic. Biol. Med. 2015, 83, 238. [DOI] [PubMed] [Google Scholar]