

Abstract
Atom‐transfer radical addition (ATRA) reactions have gained a strong foothold in organic synthesis by virtue of their operational simplicity, synthetic versatility, and perfect atom economy. A rich chemical space can be accessed through clever combinations of the simple starting materials. Many variations of this general motif have been reported. However, the vast majority involve the addition of an organic halide across a C=C double bond, resulting in the formation of 1,2‐bifunctional products. This report introduces a significant expansion of this general reactivity concept to give 1,3‐bifunctional adducts through the combination of 1,1‐ATRA to a carbenoid and 1,2‐ATRA to an alkyne. Both processes operate under mild conditions (RT, 5 h) with the same commercial catalyst (CoBr2, dppbz).
Keywords: alkynes, atom-transfer radical addition, carbenes, cobalt, diazo compounds
2+1 ATRA: A three‐component radical addition reaction between alkyl halides, TMSCHN2, and alkynes under mild conditions (RT, 5 h) with a commercial catalyst (CoBr2, dppbz) has been developed. This constitutes a significant extension of conventional (two‐component) atom‐transfer radical addition (ATRA) reactions through the incorporation of a carbenoid precursor, which results in an overall 1,3‐functionalization.
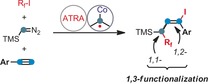
The availability of stable and convenient precursors to carbenes and carbenoids has enabled the development of versatile insertion, addition, cyclization, and cross‐coupling reactions based on such C1 building blocks.1, 2 In particular, metal‐catalyzed transformations of diazo compounds have found wide application in complex molecule syntheses.2 While most metal–carbene complexes engage in two‐electron reaction pathways with C−H, X−H, and π‐bonds, the 3d transition metal cobalt has been demonstrated to exhibit pronounced radical reactivity at the carbenoid Co‐CR2 entity.3 This has mostly been studied with porphyrin–cobalt(II) complexes, which effectively translate the open‐shell configuration of the metal ion through coordination of a carbene ligand into a C‐centered radical (Scheme 1, top).3c–3g The facile generation of such carbene–cobalt complexes with radical reactivity from available diazo compounds has recently been utilized in various new synthetic methods, for example, cyclo‐propanations,3a, 3c, 4 C−H insertions,5 and radical C−C and C−X couplings.6 Despite these recent developments, the chemistry of metallocarbenoids in general, and their combination with radical reactivity modes in particular, are still highly under‐utilized in comparison with the established chemistry of the unsaturated C2 building blocks alkenes and alkynes. We wished to challenge this state of affairs by merging the reactivity of cobalt carbenoids with a reaction manifold that is tightly associated with the use of alkenes and alkynes, the atom transfer radical addition (ATRA). This class of reactions, pioneered by Kharasch over 70 years ago,7 has become one of the most powerful and versatile methods for the direct 1,2‐difunctionalization of alkenes and alkynes in a single operation (Scheme 1, middle). Variations of the general motif have enabled the modular synthesis of building blocks with high levels of (chemo‐, regio‐, stereo‐) selectivity. The early developments of radical‐initiated ATRA reactions7, 8, 9 have recently been complemented with alternative mechanisms operating through metal‐catalyzed single‐electron transfer (SET)10 and photoredox catalysis.11 The adoption of mild reaction conditions has enabled the preparation of densely functionalized halogenated hydrocarbons with great structural diversity and synthetic utility. For example, halofluoroalkylations of alkynes through an ATRA mechanism from inexpensive fluoroalkyl halides have resulted in the synthesis of fluorinated alkenes that exhibit specific solubility, bioavailability, and metabolic properties.11b, 12, 13 We surmised that an extension of the general concept of ATRA reactions with C2 starting materials with an additional C1 building block (=C2+1) would tremendously enhance the chemical space that can be accessed by this transformation. Documented herein are the synthetic and mechanistic details of a C1‐extended ATRA reaction that presents tangible advances over the current state‐of‐the‐art that could not have been predicted: The generation of alkyl radicals and carbenoids can be effected by the same simple cobalt catalyst under identical conditions. The conventional 1,2‐difunctionalization of alkynes by ATRA reaction can be successfully expanded to an overall 1,3‐difunctionalization of the hydrocarbon skeleton (Scheme 1, bottom).
Scheme 1.
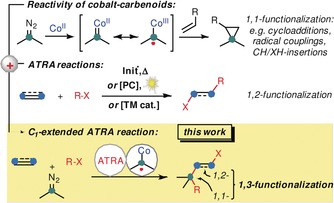
Merging cobalt carbenoid reactivity with metal‐catalyzed ATRA reactions to give a cobalt‐catalyzed C1‐expanded ATRA reaction.
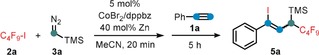
Following our recent report of a cobalt‐catalyzed ATRA reaction of alkynes and alkyl halides,14 we commenced our investigations into a C1‐extended ATRA process with the reaction between phenylacetylene (1 a), n‐perfluorobutyl iodide (2 a), and the commercially available carbene precursor trimethylsilyldiazo‐methane (3 a). Under very similar conditions as reported for the cobalt‐catalyzed ATRA reaction (Scheme 2 a),14 the three‐component reaction was performed with catalytic CoBr2 and dppbz (1,2‐bis(diphenylphosphino)benzene) in the presence of zinc as co‐catalyst. To our delight, the ATRA adduct 4 a was obtained in 27 % yield while the C1‐elongated alkene 5 a was the major product with 51 % yield (Z/E=4:1, Scheme 2 b). The desired carbene‐insertion adduct 5 a constitutes a 1,3‐difunctionalized allylsilane that is formally derived from 1,1‐addition to the carbenoid and 1,2‐addition to the alkyne. Further optimization of the conditions showed that formation of the undesired ATRA product 4 a was suppressed by subsequent addition of 1 a to the alkylation reaction between 2 a and 3 a (Scheme 2 c). Variations of the general conditions showed that other solvents (acetone, DCM, DMF, THF) and ligands (dppe=1,2‐bis(diphenyl‐phosphino)ethane; 2,2′‐bipyridine) gave lower selectivity (with minor formation of 4 a, see Table 1, entries 3–7). CoCl2 and CoBr2 showed similar reactivity, while there was no reaction with cobalt(II) phthalocyanine3 (CoPc, entries 9, 10). Interestingly, other 3d transition metals were not catalytically active (entry 11). With the optimized set of conditions, we explored the substrate scope and limitations of this cobalt‐catalyzed three‐component ATRA modification. Various aryl acetylenes effectively underwent the carbene‐extended ATRA reaction to provide the desired (1‐iodo‐1‐aryl‐3‐yl)trimethylsilanes (5) with yields up to 90 % (Scheme 3 a). Lower yields and Z/E selectivity were obtained with electron‐withdrawing substituents (5 g–5 i, 5 o). A remarkable range of functional groups was tolerated by the mild reaction conditions, including nitrile, ketone, ester, aldehyde, hydroxyl, thiophenyl, and halide groups. The compatibility with halogen atoms (5 d, 5 e, 5 m) and the boronic ester (5 j) provides ample opportunities for further arene manipulations. Steric hindrance of ortho‐substituted arylacetylenes had little influence on the reactivity but effected higher Z/E selectivity (5 p, 5 q, 5 r (50:1), 5 t). Reaction of 3‐ethynylpyridine afforded a mixture of 5 v and the butadiene 6 v derived from TMSF elimination. Work‐up of the inseparable reaction mixture with CsF gave the clean 1,4‐dihalo‐1,3‐butadiene derivative 6 v in 68 % yield (Scheme 3 b). The preferential formation of the Z‐iodoalkene moiety is governed by the minimization of steric repulsion in both the anti‐ and syn‐elimination pathways. Furthermore, alkyl‐substituted alkynes could also be employed: 1‐Octyne, trimethylsilyl acetylene, methyl propiolate, and 5‐cyanopentyne underwent carbene‐ATRA reactions at 60 °C, albeit with lower yields (5 w–5 y). The internal 1‐phenyl‐1‐propyne, allylbenzene, and styrene were unreactive. The cobalt‐catalyzed carbene‐extended ATRA reaction was then applied to other fluoroalkyl halides (Br, I; see Scheme 4 a). With ethyl halodifluoroacetates, a stereochemical switch was observed, from the major formation of the Z adduct (7 e, with X=I) to the E isomer (7 f, X=Br; E/Z isomers were characterized by NOESY NMR). This may be a due to a slower final Br atom abstraction (vs. I) and the increased steric bulkiness in the corresponding transition state (cf. the high E selectivity in ATRA with t‐BuI15 and our earlier studies14). With less electrophilic non‐fluorinated alkyl halides, ATRA between the alkyl halide and TMSCHN2 exclusively took place while the subsequent alkyne insertion did not proceed. Consequently, 1‐halo‐1‐trimethylsilyl alkanes were isolated in good yields in the absence of alkyne (8 a–8 e, Scheme 4 b). Sterically more bulky and less electrophilic alkyl halides underwent only minimal or no conversion under the standard conditions (Scheme 4 c). Benzyl bromide underwent rapid radical dimerization to give bibenzyl under the reaction conditions. The commercial carbene precursor ethyl diazoacetate was unreactive. The synthetic utility of this new ATRA method was demonstrated in gram‐scale preparations and isolations of the desired adducts (Scheme 5). 1,1‐ATRA to TMSCHN2 afforded 8 a in 79 % yield (0.69 g), while a three‐component ATRA involving 1,2‐alkyne insertion gave 5 a in even higher yield (84 %, 0.90 g). Combination of the latter reaction with a subsequent TMSF elimination allowed the isolation of the 1‐iodo‐4‐fluorobutadiene 6 a in a one‐pot reaction (81 %, 0.71 g). The synthetic utility of the ATRA adduct 6 a was demonstrated in selective cross‐coupling reactions to give functionalized dienyne and 1,1‐diaryldiene derivatives. Substitution of both halides was accomplished with thiol and base (Scheme 5, bottom).
Scheme 2.
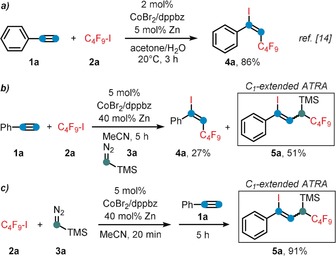
Key observations of the one‐pot ATRA reaction and C1‐extension.
Table 1.
Variation of reaction conditions.
|
Entry |
Deviations from given conditions |
5 a [%][b] |
|---|---|---|
|
1 |
– |
91 (82[c]) |
|
2 |
30 mol % Zn |
61 |
|
3 |
acetone instead of CH3CN |
55[d] |
|
4 |
CH2Cl2 instead of CH3CN |
21 |
|
5 |
DMF or THF instead of CH3CN, respectively |
0[e] |
|
6 |
dppe instead of dppbz |
24[f] |
|
7 |
2,2′‐bipyridine instead of dppbz |
0 |
|
8 |
without CoBr2 or Zn or dppbz, respectively |
0 each |
|
9 |
CoCl2 instead of CoBr2 |
88 |
|
10 |
CoPc instead of CoBr2/dppbz |
0 |
|
11 |
CrCl3/FeBr2/NiBr2/Cp2TiCl2 instead of CoBr2 |
0 |
|
12 |
1 a/2 a/3 a=1.5:2:1 (1.1:1.4:1) |
92 (76) |
Conditions: 1 a (0.2 mmol), 2 a (0.26 mmol), 3 a (0.34 mmol, 2 m in n‐hexane), CoBr2 (5 mol %), dppbz (5 mol %), Zn (40 mol %) in 0.8 mL MeCN. [b] GC yields vs. internal n‐C12H26. [c] Yield of isolated product. [d] 24 % of 4 a as byproduct. [e] 1 equiv Zn. [f] 8 % of 4 a as byproduct.
Scheme 3.
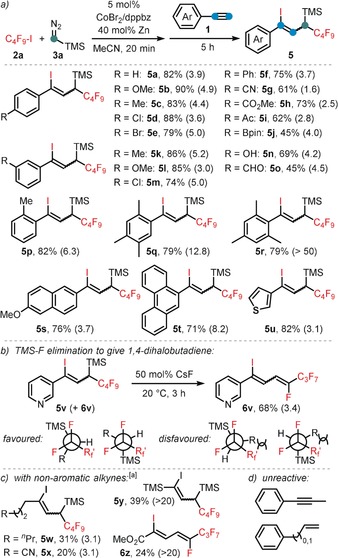
Substrate scope with respect to the alkyne. Conditions: 2 a (0.26 mmol), 3 a (0.34 mmol, 2 m in hexane), CoBr2 (5 mol %), dppbz (5 mol %), Zn (40 mol %), 0.8 mL MeCN, 20 min at 20 °C; then addition of 1 (0.2 mmol), 5 h. Yields of isolated product are given; Z/E ratios in parentheses (determined by GC‐FID).[a] Zn (80 mol %), reaction at 60 °C after alkyne addition.
Scheme 4.
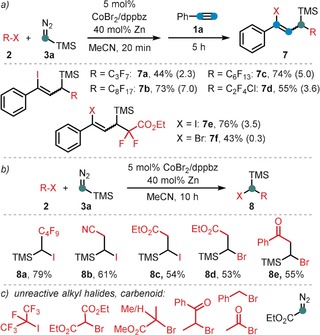
Substrate scope with respect to the alkyl halide. Conditions: 2 (0.26 mmol), 3 a (0.34 mmol, 2 m in hexane), CoBr2 (5 mol %), dppbz (5 mol %), Zn (40 mol %), 0.8 mL MeCN, 20 min at 20 °C; then addition of 1 a (0.2 mmol), 5 h. Yields of isolated product are given; Z/E ratios in parentheses (by GC‐FID).
Scheme 5.
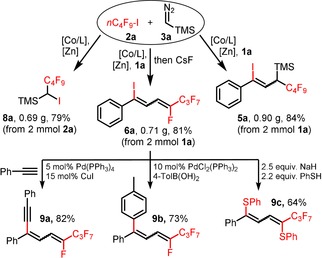
Gram‐scale syntheses of ATRA products and follow‐up reactions.
In an effort to study the mechanism of this new ATRA reaction, we performed a set of key experiments (Scheme 6, R f=nC4F9). From the exploration of the substrate scope (Schemes 4 b and 5), it became apparent that the two‐component ATRA reaction between 2 a and 3 a afforded the 1,1‐adduct 8 a, which itself is an intermediate of the carbene‐extended ATRA reaction with alkynes. Consistently, the two routes toward 5 a starting from 2 a and 8 a gave nearly identical yields under similar conditions [Eq. (1), Scheme 6]. The higher yield of the 1,2‐ATRA of 8 a to the alkyne with higher Zn loading (10 vs. 5 mol %) suggests that additional Zn may facilitate the reductive regeneration of the catalytically active CoI from CoII. The presence of radical traps led to complete inhibition of the alkyne insertion as expected for an atom‐transfer radical addition [TEMPO=2,2,6,6‐tetramethyl‐1‐oxyl‐piperidine, Eq. (2)]. With triethylphosphite, which could act as a vinyl radical trap,16 the phosphonate adduct indeed was detected. The formation of the intermediate 8 a was not observed in the absence of either the [Co] catalyst or Zn [Eq. (3)]. The use of TiCl3 as an alternative SET initiator17 gave traces of 8 a [Eq. (4)]. This observation supports the notion that the cobalt catalyst is not only involved in the generation of the reactive radical species from 2 a but also in decomposing the carbenoid 3 a. Pre‐formation of a CoI solution from 5 mol % CoBr2/dppbz and 1 equiv Zn and sub‐sequent filtration of residual solid Zn gave a catalyst that enabled the 1,1‐ATRA reaction between 2 a and 3 a in 61 % yield [Eq. (5)]. Again, a higher Zn/Co ratio afforded higher yields, most likely as a consequence of rapid regeneration of the CoI catalyst species. On the basis of the collected synthetic and mechanistic data and previous reports,6, 14 we postulate a reaction mechanism that can be categorized into three parts: 1) formation and regeneration of the active CoI catalyst by Zn‐mediated reduction of CoII (Scheme 7, ①); 2) cobalt‐catalyzed 1,1‐ATRA reaction of the alkyl halide to the diazomethane derivative (Scheme 7, ②); 3) 1,2‐ATRA reaction of the key intermediate to the alkyne (Scheme 7, ③). The overall mechanism commences with the reduction of the pre‐catalyst mixture CoBr2/dppbz with Zn to give the postulated active catalyst [CoI(dppbz)2X] (A, with X=Br or I). SET from A to the electrophilic R f‐I results in the CoII complex B and the free radical R f .. CoII‐mediated decomposition of TMSCHN2 affords the CoIII carbene C, which exhibits pronounced radical character at the α‐carbon so that rapid recombination with R f . affords the alkyl cobalt(III) intermediate D.6a The enhanced steric hindrance within this sec‐alkyl ligand and the open‐shell configuration at CoIII very likely effect facile homolysis to the radical‐cation pair E. Iodine atom abstraction with the starting material R f‐I furnishes 1,1‐adduct F, free radical R f ., and CoII complex B. Non‐productive radical dimerization of the sec‐radical TMS‐CH(.)‐R f was observed by GC‐MS [see Eq. (2) and (5), Scheme 6]. In this catalytic cycle [Scheme 7, ②], the [Co] catalyst fulfills two roles: SET activation of R f‐I and carbene‐complex formation from TMSCHN2. The ensuing 1,2‐ATRA reaction of the 1,1‐adduct F with the alkyne [Scheme 7, ③] is initiated by SET from A to F, which gives the sec‐alkyl radical (as in E). Radical addition to the alkyne affords the vinyl radical G, which engages in radical chain transfer with another molecule of intermediate F and furnishes the desired three‐component adduct.
Scheme 6.
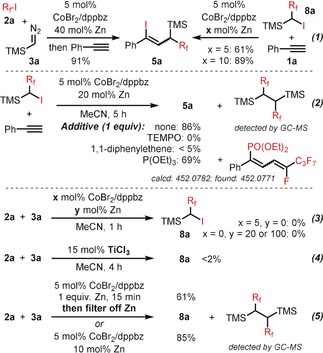
Selected key mechanistic experiments (R f=nC4F9). GC yields are given.
Scheme 7.
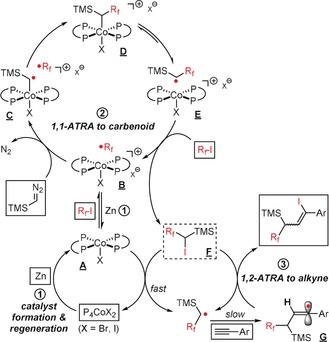
Proposed reaction mechanism.
In summary, a new three‐component radical addition has been developed that significantly expands the scope, versatility, and utility of metal‐catalyzed ATRA reactions through the use of a commercial carbene precursor. The resultant functionalized carbon skeleton is assembled by successive 1,1‐halofluoroalkylation and 1,2‐addition to alkynes in a one‐pot operation.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was generously supported by a consolidator grant of the European Research Council (CoG 683150).
G. Wu, J. Börger, A. Jacobi von Wangelin, Angew. Chem. Int. Ed. 2019, 58, 17241.
In memory of Professor Dieter Enders
Contributor Information
Dr. Guojiao Wu, Email: guojiao.wu@uni-hamburg.de.
Prof. Dr. Axel Jacobi von Wangelin, Email: axel.jacobi@uni-hamburg.de.
References
- 1.
- 1a. Zollinger H., Diazo Chemistry I and II, Wiley-VCH, Weinheim, 1995; [Google Scholar]
- 1b. Doyle M. P., McKervey M. A., Ye T., Modern Catalytic Methods for Organic Synthesis with Diazo Compounds, Wiley, New York, 1998; [Google Scholar]
- 1c. Moss R. A., Doyle M. P., Contemporary Carbene Chemistry, Wiley, Hoboken, 2013. [Google Scholar]
- 2.Recent reviews:
- 2a. Davies H. M. L., Manning J. R., Nature 2008, 451, 417–424; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Davies H. M. L., Denton J. R., Chem. Soc. Rev. 2009, 38, 3061–3071; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Doyle M. P., Duffy R., Ratnikov M., Zhou L., Chem. Rev. 2010, 110, 704–724; [DOI] [PubMed] [Google Scholar]
- 2d. Davies H. M. L., Lian Y., Acc. Chem. Res. 2012, 45, 923–935; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2e. Zhu S.-F., Zhou Q.-L., Acc. Chem. Res. 2012, 45, 1365–1377; [DOI] [PubMed] [Google Scholar]
- 2f. Zhao X., Zhang Y., Wang J., Chem. Commun. 2012, 48, 10162–10173; [DOI] [PubMed] [Google Scholar]
- 2g. Xiao Q., Zhang Y., Wang J., Acc. Chem. Res. 2013, 46, 236–247; [DOI] [PubMed] [Google Scholar]
- 2h. Guo X., Hu W., Acc. Chem. Res. 2013, 46, 2427–2440; [DOI] [PubMed] [Google Scholar]
- 2i. Ford A., Miel H., Ring A., Slattery C. N., Maguire A. R., McKervey M. A., Chem. Rev. 2015, 115, 9981–10080; [DOI] [PubMed] [Google Scholar]
- 2j. Cheng Q.-Q., Deng Y., Lankelma M., Doyle M. P., Chem. Soc. Rev. 2017, 46, 5425–5433; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2k. Xia Y., Qiu D., Wang J., Chem. Rev. 2017, 117, 13810–13889. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Ikeno T., Iwakura I., Yamada T., J. Am. Chem. Soc. 2002, 124, 15152–15153; [DOI] [PubMed] [Google Scholar]
- 3b. Ikeno T., Iwakura I., Yabushita S., Yamada T., Org. Lett. 2002, 4, 517–520; [DOI] [PubMed] [Google Scholar]
- 3c. Dzik W. I., Xu X., Zhang X. P., Reek J. N. H., de Bruin B., J. Am. Chem. Soc. 2010, 132, 10891–10902; [DOI] [PubMed] [Google Scholar]
- 3d. Belof J. L., Cioce C. R., Xu X., Zhang X. P., Space B., Woodcock H. L., Organometallics 2011, 30, 2739–2746; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3e. Dzik W. I., Zhang X. P., de Bruin B., Inorg. Chem. 2011, 50, 9896–9903; [DOI] [PubMed] [Google Scholar]
- 3f. Lu H., Dzik W. I., Xu X., Wojtas L., de Bruin B., Zhang X. P., J. Am. Chem. Soc. 2011, 133, 8518–8521; [DOI] [PubMed] [Google Scholar]
- 3g. Cui X., Xu X., Wojtas L., Kim M. M., Zhang X. P., J. Am. Chem. Soc. 2012, 134, 19981–19984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Selected examples:
- 4a. Zhu S.-F., Xu X., Perman J. A., Zhang X. P., J. Am. Chem. Soc. 2010, 132, 12796–12799; [DOI] [PubMed] [Google Scholar]
- 4b. Cui X., Xu X., Lu H.-J., Zhu S.-F., Wojtas L., Zhang X. P., J. Am. Chem. Soc. 2011, 133, 3304–3307; [DOI] [PubMed] [Google Scholar]
- 4c. Xu X., Zhu S.-F., Cui X., Wojtas L., Zhang X. P., Angew. Chem. Int. Ed. 2013, 52, 11857–11861; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12073–12077; [Google Scholar]
- 4d. Wang Y., Wen X., Cui X., Wojtas L., Zhang X. P., J. Am. Chem. Soc. 2017, 139, 1049–1052; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4e. Hu Y., Lang K., Tao J.-R., Marshall M. K., Cheng Q.-G., Cui X., Wojtas L., Zhang X. P., Angew. Chem. Int. Ed. 2019, 58, 2670–2674; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 2696–2700. [Google Scholar]
- 5.Selected examples:
- 5a. Paul N. D., Mandal S., Otte M., Cui X., Zhang X. P., de Bruin B., J. Am. Chem. Soc. 2014, 136, 1090–1096; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Cui X., Xu X., Jin L.-M., Wojtas L., Zhang X. P., Chem. Sci. 2015, 6, 1219–1224; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Wang Y., Wen X., Cui X., Zhang X. P., J. Am. Chem. Soc. 2018, 140, 4792–4796; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5d. Wen X., Wang Y., Zhang X. P., Chem. Sci. 2018, 9, 5082–5086; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5e. te Grotenhuis C., van den Heuvel N., van der Vlugt J. I., de Bruin B., Angew. Chem. Int. Ed. 2018, 57, 140–145; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 146–151. [Google Scholar]
- 6.
- 6a. Zhang J., Jiang J., Xu D., Luo Q., Wang H., Chen J., Li H., Wang Y., Wan X., Angew. Chem. Int. Ed. 2015, 54, 1231–1235; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1247–1251; For some similar transformation beside the carbene-cobalt complexes: [Google Scholar]
- 6b. Jiang J., Liu J., Yang L., Shao Y., Cheng J., Bao X., Wan X., Chem. Commun. 2015, 51, 14728–14731; [DOI] [PubMed] [Google Scholar]
- 6c. Ling J., Zhang J., Zhao Y., Xu Y., Wang H., Lv Y., Ji M., Ma L., Ma M., Wan X., Org. Biomol. Chem. 2016, 14, 5310–5316; [DOI] [PubMed] [Google Scholar]
- 6d. Wang N.-N., Hao W.-J., Zhang T.-S., Li G., Wu Y.-N., Tu S.-J., Jiang B., Chem. Commun. 2016, 52, 5144–5147; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6e. Li H., Zhao Y., Ma L., Ma M., Jiang J., Wan X., Chem. Commun. 2017, 53, 5993–5996; [DOI] [PubMed] [Google Scholar]
- 6f. Wang H., Li H., Zheng Y., Lian P., Wan X., Chem. Eur. J. 2019, 25, 2195–2198. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Kharasch M. S., Jensen E. V., Urry W. H., Science 1945, 102, 128; [DOI] [PubMed] [Google Scholar]
- 7b. Kharasch M. S., Skell P. S., Fisher P., J. Am. Chem. Soc. 1948, 70, 1055–1059. [Google Scholar]
- 8.
- 8a. Curran D. P., Bosch E., Kaplan J., Newcomb M., J. Org. Chem. 1989, 54, 1826–1831; [Google Scholar]
- 8b. Curran D. P., Chen M.-H., Spleterz E., Seong C. M., Chang C.-T., J. Am. Chem. Soc. 1989, 111, 8872–8878; [Google Scholar]
- 8c. Curran D. P., Seong C. M., J. Am. Chem. Soc. 1990, 112, 9401–9403. [Google Scholar]
- 9.
- 9a. Baciocchi E., Muraglia E., Tetrahedron Lett. 1994, 35, 2763–2766; [Google Scholar]
- 9b. Yorimitsu H., Nakamura T., Shinokubo H., Oshima K., J. Org. Chem. 1998, 63, 8604–8605; [Google Scholar]
- 9c. Yorimitsu H., Nakamura T., Shinokubo H., Oshima K., Omoto K., Fujimoto H., J. Am. Chem. Soc. 2000, 122, 11041–11047. [Google Scholar]
- 10.Recent reviews:
- 10a. Pintauer T., Matyjaszewski K., Chem. Soc. Rev. 2008, 37, 1087–1097; [DOI] [PubMed] [Google Scholar]
- 10b. Muñoz-Molina J. S., Belderrain T. R., Pérez P. J., Eur. J. Inorg. Chem. 2011, 3155–3164; [Google Scholar]
- 10c. Studer A., Curran D. P., Angew. Chem. Int. Ed. 2016, 55, 58–102; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 58–106. [Google Scholar]
- 11.Selected reports:
- 11a. Nguyen J. D., Tucker J. W., Konieczynska M. D., Stephenson C. R. J., J. Am. Chem. Soc. 2011, 133, 4160–4163; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Wallentin C.-J., Nguyen J. D., Finkbeiner P., Stephenson C. R. J., J. Am. Chem. Soc. 2012, 134, 8875–8884; [DOI] [PubMed] [Google Scholar]
- 11c. Pirtsch M., Paria S., Matsuno T., Isobe H., Reiser O., Chem. Eur. J. 2012, 18, 7336–7340; [DOI] [PubMed] [Google Scholar]
- 11d. Arceo E., Montroni E., Melchiorre P., Angew. Chem. Int. Ed. 2014, 53, 12064–12068; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 12260–12264; Reviews: [Google Scholar]
- 11e. Prier C. K., Rankic D. A., MacMillan D. W. C., Chem. Rev. 2013, 113, 5322–5363; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11f. Staveness D., Bosque I., Stephenson C. R. J., Acc. Chem. Res. 2016, 49, 2295–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Müller K., Faeh C., Diederich F., Science 2007, 317, 1881–1886; [DOI] [PubMed] [Google Scholar]
- 12b. Hagmann W. K., J. Med. Chem. 2008, 51, 4359–4369; [DOI] [PubMed] [Google Scholar]
- 12c. O'Hagan D., Chem. Soc. Rev. 2008, 37, 308–319; [DOI] [PubMed] [Google Scholar]
- 12d. Gouverneur V., Müller K., Fluorine in Pharmaceutical and Medicinal Chemistry: From Biophysical Aspects to Clinical Applications, Imperial College Press, London, 2012. [Google Scholar]
- 13.
- 13a. Iqbal N., Jung J., Park S., Cho E. J., Angew. Chem. Int. Ed. 2014, 53, 539–542; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 549–552; [Google Scholar]
- 13b. Xu T., Cheung C., Hu X., Angew. Chem. Int. Ed. 2014, 53, 4910–4914; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5010–5014; [Google Scholar]
- 13c. Belhomme M.-C., Dru D., Xiong H.-Y., Chard D., Besset T., Poisson T., Pannecoucke X., Synthesis 2014, 46, 1859–1870; [Google Scholar]
- 13d. Li G., Cao Y.-X., Luo C.-G., Su Y.-M., Li Y., Lan Q., Wang X.-S., Org. Lett. 2016, 18, 4806–4809; [DOI] [PubMed] [Google Scholar]
- 13e. Barata-Vallejo S., Cooke M. V., Postigo A., ACS Catal. 2018, 8, 7287–7307, and references therein. [Google Scholar]
- 14. Wu G., Jacobi von Wangelin A., Chem. Sci. 2018, 9, 1795–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Ichinose Y., Matsunaga S.-I., Fugami K., Oshima K., Utimoto K., Tetrahedron Lett. 1989, 30, 3155–3158; [Google Scholar]
- 15b. Curran D. P., Kim D., Tetrahedron 1991, 47, 6171–6188. [Google Scholar]
- 16.
- 16a. Jiao X.-Y., Bentrude W. G., J. Am. Chem. Soc. 1999, 121, 6088–6089; [Google Scholar]
- 16b. Cheung C. W., Zhurkin F. E., Hu X., J. Am. Chem. Soc. 2015, 137, 4932–4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Hu C.-M., Qiu Y.-L., J. Fluorine Chem. 1991, 55, 109–111; [Google Scholar]
- 17b. Hu C.-M., Qiu Y.-L., Chin. J. Chem. 1992, 10, 439–444; [Google Scholar]
- 17c. Davis C. R., Burton D. J., Yang Z.-Y., J. Fluorine Chem. 1995, 70, 135–140. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary