

Abstract
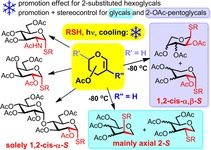
A comprehensive optimization and mechanistic study on the photoinduced hydrothiolation of different d‐ and l‐ hexo‐ and pentoglycals with various thiols was performed, at the temperature range of RT to −120 °C. Addition of thiols onto 2‐substituted hexoglycals proceeded with complete 1,2‐cis‐α‐stereoselectivity in all cases. Hydrothiolation of 2‐substituted pentoglycals resulted in mixtures of 1,2‐cis‐α‐ and ‐β‐thioglycosides of varying ratio depending on the configuration of the reactants. Hydrothiolation of unsubstituted glycals at −80 °C proceeded with excellent yields and, except for galactal, provided the axially C2‐S‐linked isomers with high selectivity. Cooling was always beneficial to the efficacy, increased the yields and in most cases significantly raised the stereoselectivity. The suggested mechanism explains the different conformational preferences of the intermediate carbon‐centered radicals, which is a crucial factor in the stereoselectivity of the reactions.
Keywords: glycal, photoactivation, stereoselective synthesis, thioglycoside, thiyl radical addition
Three types of acetylated glycals were reacted with various thiols under UV‐irradiation at the temperature range of RT to −120 °C. Cooling proved always beneficial to the reaction efficacy, being −80 °C the optimal temperature in most cases. The low‐temperature thiol‐ene coupling of glycals was found to provide an easy access to 1,2‐cis‐α‐1‐thio‐hexosides up to tetrasaccharides, 1,2‐cis‐α‐ and β‐1‐thio‐pentosides and axially C2‐S‐linked glycomimetics.
Introduction
Oligosaccharides, glycoconjugates and their mimetics are investigated in molecular recognition studies,1, 2, 3 drug discovery4 and vaccine development.5 Thioglycosides in which the glycosidic oxygen is replaced by a sulfur atom are metabolically stable analogues of the natural O‐glycosides, justifying their synthesis and application as glycomimetics.6
While many strategies exist for the synthesis of 1,2‐trans‐thio‐linked glycomimetics,7, 8, 9 the formation of 1,2‐cis‐thioglycosides with high stereocontrol is known to be a major challenge.10, 11, 12, 13 The α‐glycosides are abundantly found in nature, and many 1,2‐cis‐α‐linked sugars, including α‐d‐glucose, d‐galactose and d‐glucosamine play crucial roles in various biological processes.14 Therefore, there is a high demand for the stereoselective synthesis of thio‐analogues of these biorelevant sugars.
In this context, the radical mediated thiol‐ene coupling emerged as a powerful method for glycoconjugation15, 16, 17 which, if unsaturated sugars are applied as the alkene partners, opens the way for the stereoselective synthesis of a broad range of thio‐linked glycomimetics.18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30 We demonstrated that the UV‐light‐initiated addition of various thiols including amino acid, peptide, and sugar thiols to 2‐substituted hexoglycals occurs with complete stereoselectivity, providing an easy access to 1,2‐cis‐α‐linked thiodisaccharides and thioglycoconjugates.26, 27, 28, 29, 30 Recently, we have demonstrated that the reaction temperature profoundly influences the efficacy of the reactions by controlling the equilibrium of the reversible propagation step of the radical chain process.28, 29 Our study revealed that cooling promotes while heating inhibits the thiol‐ene couplings of 2‐substituted glycals, and the unfavorable steric and electronic effects resulting in low conversions at room temperature could be overcome by conducting the reactions at −80 °C.
To further investigate the applicability of the low‐temperature photoinitiated thiol‐ene coupling in the stereoselective thioconjugation, we continued to study the reactions of 2‐substituted hexoglycals (1–3), supplemented with the disaccharide glycal 4. Applied thiol partners comprise alkyl thiols (12, 13, 15 and 16), thioacid 14, mercaptoacids (17 and 19) and 1‐thiosugars (18, 20–22) including disaccharide thiol 18 (Figure 1). Moreover, our study was extended to two additional glycal types of both d‐ and l‐configurations, the 2‐acetoxy pentoglycals (5–7), and the unsubstituted hexoglycals (8–11) (Figure 1). Only two reports of thiol‐ene reactions of these glycal types have been published in the literature hitherto which are also limited to room‐temperature reactions of a few d‐isomers.23, 26 Importantly, while the thiol‐ene coupling of 2‐substituted hexoglycals proceeds with exclusive 1,2‐cis‐α‐stereoselectivity, the 2‐substituted pentoglycals were found to react with only moderate diastereoselectivities.26 Moreover, in the case of unsubstituted hexoglycals, although regioselective hydrothiolation has been observed to occur at the C2‐position, the diastereoselectivity and the efficacy of the reaction were strongly varying depending upon the C3 and C4 configurations of the glycals.23
Figure 1.
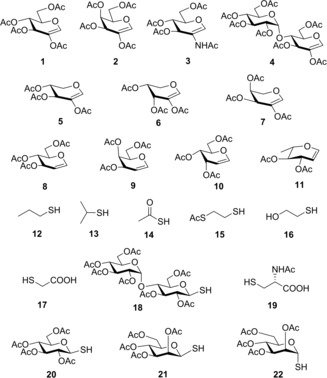
Investigated glycals and thiols.
Herein, we report a systematic study on three types of glycals, shown in Figure 1, by varying not only the experimental parameters of the thiol‐ene couplings such as reaction temperature and ratio of reactants but examining sterical and electronic factors on both the reacting glycal and thiol. The careful analysis of the results of this comprehensive optimization study made it possible to give a viable mechanistic explanation for the highly different levels of stereoselectivity observed on the different types of glycals.
Results and Discussion
Hydrothiolation of 2‐substituted hexoglycals
We have found earlier that while 2‐acetoxy‐d‐glucal 1 readily reacted with thiol‐containing peptides and sugars at room temperature, only moderate conversions have been observed with simple thiols such as tert‐butyl or benzyl mercaptans, and high amount of unreacted 1 was recovered from the reaction mixtures.26, 27 Therefore, first, we studied the temperature‐dependence of the reactions between 1 and thiols 12–16 under the previously established conditions using 3×15 minutes of irradiation at λ max 365 nm in the presence of 3×0.1 equiv of the photoinitiator 2,2‐dimethoxy‐2‐phenylacetophenone (DPAP). Using 5 equiv of alkyl thiols 12 and 13 at room temperature, glycal 1 reacted with low conversions (Table 1, entries 1 and 6). Increasing the thiol excess to 3×5 equivalents a significant progress of the reactions was observed and the corresponding α‐thioglycosides 23 and 24 were formed in 51–54 % yields (Table 1, entries 2 and 7). Performing the reactions at 0 °C with 5 equiv thiol excess an even higher promoting effect was observed in both cases (entries 4 and 9) to afford 23 and 24 with 65 and 68 % yields, respectively. However, further cooling of the reaction mixture proved to be detrimental to the efficacy of the addition reactions (Entries 5 and 10).
Table 1.
Hydrothiolation of 2‐acetoxy‐d‐glucal 1 with various thiols at different temperatures.
|
| ||||
|---|---|---|---|---|
|
Entry |
Thiol |
Product |
Temperature (thiol equiv) |
Yield [%][a] |
|
1 |
12 (3–15 equiv) |
|
rt (5) |
35 |
|
2 |
rt (3×5) |
54 |
||
|
3 |
0 °C (3) |
46 |
||
|
4 |
0 °C (5) |
65 |
||
|
5 |
−40 °C (3) |
18 |
||
|
|
|
|
|
|
|
6 |
13 (3–15 equiv) |
|
rt (5) |
32 |
|
7 |
rt (3×5) |
51 |
||
|
8 |
0 °C (3) |
56 |
||
|
9 |
0 °C (5) |
68 |
||
|
10 |
−40 °C (3) |
50 |
||
|
|
|
|
|
|
|
11 |
14 (6–24 equiv) |
|
rt/−40 °C[b] |
0 |
|
12 |
rt[c] |
9 |
||
|
13 |
−20 °C[b] |
9 |
||
|
14 |
−20 °C[c] |
19 |
||
|
|
|
|
|
|
|
15 |
15 (3 equiv) |
|
rt |
55 |
|
16 |
0 °C |
81 |
||
|
|
|
|
|
|
|
17 |
16 (2 equiv) |
|
rt |
40 |
|
18 |
−80 °C |
70 |
||
|
|
|
|
|
|
|
19 |
17 (2 equiv) |
|
rt |
55 |
|
20 |
−80 °C |
67 |
||
|
|
|
|
|
|
|
21 |
18 (1.5 equiv) |
|
rt |
5 |
|
22 |
0 °C |
10 |
||
|
23 |
−20 °C |
14 |
||
|
24 |
−40 °C |
29 |
||
|
25 |
−80 °C |
58 |
||

[a] Isolated yield; by‐product formation was not observed, the low/moderate yields are the results of the low/moderate conversion of the alkenes; [b] 6 equiv of thiol was used; [c] 3×8 equiv of thiol was used.
Interestingly, thioacetic acid 14 showed a significantly lower reactivity than alkyl thiols, and the isolated yield of the α‐1‐thio‐acetate product 25 did not reach 20 % at any temperature despite using very high thiol excess (Entries 11–14).
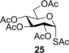
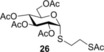
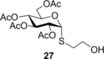
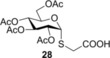
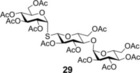
Spacer‐armed α‐thioglycosides such as 26, 27 and 28 can be considered as valuable building blocks in the construction of neoglycoconjugates, glycopeptides and glycodendrimers. Therefore, their synthesis was studied by using functionalized thiols 15, 16 and 17. In all three cases the conversion of the starting glycal and also the isolated yield of the product could be significantly increased by cooling. Importantly, the optimal reaction temperatures were remarkably different, 0 °C for thiol 15 while −80 °C for thiols 16 and 17, producing the corresponding α‐thioglycosides 26, 27 and 28 in 81, 70 and 67 % yields, respectively (Entries 15–20). Applying disaccharide 18 as the thiol partner, a very low conversion of 1 was observed at room temperature to give trisaccharide 29 only with 5 % yield. It was surprising, because we have found earlier that 1‐thioglycoses, for example, 20 and 22, added to 1 efficiently at room temperature providing the corresponding thiodisaccharides in high yields.26 Fortunately, the efficacy of the thiol‐ene coupling between disaccharide thiol 18 and glycal 1 increased gradually in line with the decrease of the reaction temperature and the yield of 29 reached 58 % at −80 °C (Entries 21–25). Noteworthy, in these reactions we had to use DMF as a co‐solvent due to the low solubility of thiol 18 in toluene.
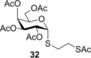
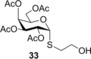
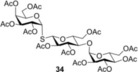
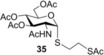
To further explore the mechanism and scope of the reaction the d‐galactose‐derived glycal 2 and 2‐acetamido‐d‐glucal 3 were systematically reacted (Table 2). As already observed in our previous experiments, efficient thiol‐ene couplings of 2 and 3 required lower temperatures than those of the d‐gluco analogue 1.28, 29 Similar trends were observed when thiols 12 or 14 were coupled to 2, being −40 to −80 °C the optimal temperature range of these reactions.(Table 2). While reaction between thiol 12 and glycal 2 occurred with low efficacy at RT and even at 0 °C, a significantly increased conversion was observed at −40 °C giving the corresponding α‐1‐thio‐galactoside 30 in a 56 % yield. However, conducting the reaction at −80 °C again led to a decrease in the conversion. Using thioacetic acid 14 as the thiol partner, the cooling exerted its beneficial promoting effect again. Unfortunately, the best yields of 31, achieved at the −40 to −80 °C temperature range, did not exceed 26 % (Entries 5–9). The reactivity of 2‐acetoxy‐galactal 2 towards thiols 15, 16 and 18 (Entries 10–16) proved to be similar to the ones of 2‐acetoxy‐d‐glucal 1. The optimal reaction temperature was −80 °C with thiols 16 and 18, giving the corresponding α‐thio‐linked products 33 and 34 in high yields. At the same time, the most efficient reaction with thiol 15 was found to occur at 0 °C providing 32 with 69 % yield. These results highlight that the type of the thiol is also a crucial factor in the thiol‐ene coupling. Reactions of 2‐acetamido‐d‐glucal 3 with thiols 15 and 14 showed a similar temperature dependence as the analogous reactions of 2‐acetoxy‐glycals 1 and 2. Noteworthy, 2‐acetamido‐d‐glucal 3 reacted with thioacetic acid 14 with much higher efficacy than either 1 or 2, and the yield of the α‐1‐thio‐acetate 36 reached 63 % at −80 °C. One possible reason behind this is that the 2‐acetamido group is capable to stabilize the carbon centered radical intermediate (vide infra) formed from the acylated thiyl radical better than the 2‐acetoxy group. Interestingly, in the thiol‐ene coupling between glycal 3 and disaccharide thiol 18 the best yield of trisaccharide 37 was found at −40 °C and further cooling to −80 °C led to a drop of the yield from 65 to 33 %.
Table 2.
The effect of the C4 configuration and type of the C2‐substituent of glycals on the thiol‐ene reaction
|
| ||||
|---|---|---|---|---|
|
Entry |
Thiol+Alkene |
Product |
Temperature (thiol equiv) |
Yield [%][a] |
|
1 |
12 (5 equiv) +2 |
|
rt |
28 |
|
2 |
0 °C |
26 |
||
|
3 |
−40 °C |
56 |
||
|
4 |
−80 °C |
22 |
||
|
|
|
|
|
|
|
5 |
14 (6–18 equiv) +2 |
|
rt[b] |
0 |
|
6 |
0 °C[b] |
15 |
||
|
7 |
−40 °C[b] |
22 |
||
|
8 |
−40 °C[c] |
26 |
||
|
9 |
−80 °C[b] |
23 |
||
|
|
|
|
|
|
|
10 |
15 (3 equiv) +2 |
|
rt |
52 |
|
11 |
0 °C |
69 |
||
|
12 |
−40 °C |
41 |
||
|
|
|
|
|
|
|
13 |
16 (2–3 equiv) +2 |
|
rt, (3) |
58 |
|
14 |
−80 °C, (2) |
86 |
||
|
|
|
|
|
|
|
15 |
18 (1.5 equiv) +2 |
|
−40 °C |
35 |
|
16 |
−80 °C |
75 |
||
|
|
|
|
|
|
|
17 |
15 (1.5 equiv) +3 |
|
rt |
46 |
|
18 |
0 °C |
83 |
||
|
19 |
−80 °C |
45 |
||
|
|
|
|
|
|
|
20 |
14 (6–24 equiv) +3 |
|
−20 °C[b] |
36 |
|
21 |
−40 °C[b] |
50 |
||
|
22 |
−40 °C[d] |
56 |
||
|
23 |
−80 °C[c] |
63 |
||
|
|
|
|
|
|
|
24 |
18 (1.3 equiv) +3 |
|
−40 °C |
65 |
|
25 |
−80 °C |
33 |
||
[a] Isolated yield; by‐product formation was not observed, the low/moderate yields are the results of the low/moderate conversion of the alkenes; [b] 6 equiv of thiol was used; [c] 3×6 equiv of thiol was used; [d] 3×8 equiv of thiol was used.
Next, the thiol‐ene coupling of the maltose‐derived 2‐acetoxy glycal 4 was systematically studied as a block synthesis route to thiomimetics of higher oligosaccharides and glycoconjugates (Scheme 1). In general, the reactivity of 4 proved to be slightly lower than the ones of the monosaccharide congeners.26, 27, 28, 29 Reacting 4 with thiols 19, 20 and 18 the best yields of the corresponding thioglycosides 38, 39 and 42 were achieved at the temperature range of −20 to −40 °C, while with 1‐thiomannoses 21 and 22 the most efficient reactions occurred at −80 °C. Comparing the reactions using the β‐d‐gluco‐ and β‐ and α‐d‐mannopyranosyl thiols 20, 21 and 22 reveals that the efficacy of the addition is strongly dependent on the configuration of the thiol. Noteworthy, this phenomenon has been observed in the thiol‐ene couplings of nucleoside exomethylenes.21 The yield in the coupling reaction of disaccharide glycal 4 with disaccharide thiol 18 was significantly lower than the ones observed in the reactions of monosaccharide thiols and glycals, which can partly be attributed to the low solubility of the disaccharides.
Scheme 1.
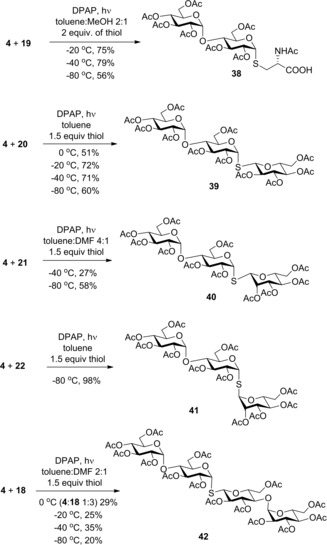
Hydrothiolation reactions of 2‐acetoxy‐maltal 4.
Summarizing this detailed synthetic work makes it possible to get deep insight into the mechanism of the reaction as follows. The thiol‐ene reaction is known to occur through a reversible thiyl addition (propagation) step followed by an irreversible hydrogen abstraction (chain transfer) step by the carbon centered radical intermediate (e.g. 1‐R and 2‐R) formed in the first step (Scheme 2).31, 32, 33 The efficiency of the thiol‐ene reactions highly depends on the stability of the carbon‐centered radical intermediate, which directly influences both the chain‐transfer activation barrier and the reversibility of the propagation step as revealed by recent computational and kinetic studies.33 The stability of the radical intermediate depends on the difference of the activation energy barriers in the forward and reverse steps, which difference is determined by the relative energy levels of the starting compounds (the glycal and the thiyl radical) and the intermediate carbon centered radical. If the activation energy barrier of the reverse propagation step is sufficiently higher than that of the forward one, the equilibrium of the reversible propagation step lies toward the intermediate carbon centered radical and efficient reaction can occur at room temperature. However, if the energy barrier of the reverse reaction is only slightly higher than that of the forward one, the carbon centered radical readily fragments at room temperature in an intramolecular reaction, without forming the final product in an intermolecular way. In the case of the above studied reactions, only few of the intermediates formed were sufficiently stable at room temperature to allow hydrothiolation in an acceptable efficacy. Similar to our recent results,28 we have found that freezing the intermediate radical is an adequate strategy to promote the reaction by preventing its degradation and thereby allowing it to react with a thiol in the hydrogen abstraction step. Although the cooling was beneficial in all reactions, the optimal reaction temperature was found to be varying depending on the configuration and substituents of the glycals, and also on the nature of the thiols. The configuration effect can easily be understood by the example of glucose and galactose (Scheme 2). While glucal 1 is at a higher energy level relative to galactal 2, due to the steric congestion between the equatorial C2 and C3 substituents in the 4H5 half chair conformation of 1, out of the formed carbon centered radical intermediates existing in a 4C1 chair conformation, the gluco configured 1‐R featuring an all‐equatorial substitution pattern has a lower energy than the galacto congener 2‐R. It means that the galactosyl C2 radical intermediate is less stable, more prone to decompose into the starting compounds than the gluco one, consequently, the cooling is more crucial in the galacto case to achieve efficient thiol‐ene couplings.
Scheme 2.
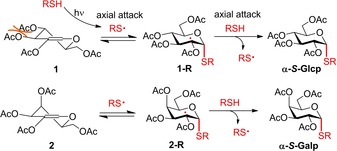
Reversible thiyl addition (propagation) step and irreversible hydrogen abstraction (chain transfer) step in the thiol‐ene couplings of 2‐substituted glycals.
Our results also proved that the nature of the thiols exerts great influence on the efficiency of the addition too. For example, in the case of thiols 12, 13 and 15 the excessive cooling proved to be detrimental to the efficacy of the reaction.34 We assume that the excessive cooling could either over‐stabilize these alkyl thiyl radicals, shifting the equilibrium of the reversible propagation step toward the starting glycals, or stop the reaction via preventing the hydrogen abstraction step by the intermediate carbon centered radicals.
While the addition of thiyl radicals to linear olefins generally exhibits poor stereoselectivity, in the case of substituted or conformationally locked cyclic alkenes the addition occurs preferentially in a trans‐diaxial manner as the result of a kinetically favored axial attack of the thiyl radical onto the cyclic alkene in its half‐chair conformation together with a stereoselective hydrogen abstraction from the thiol into an axial position.35, 36 However, complete diastereoselectivity was rarely observed, because the nature of the thiol, the configuration of the alkene, the molar ratio of the reactants and also the temperature play a role in the stereoselectivity observed.18, 21, 23, 36
We assume that in the case of 2‐substituted hexoglycals, the rapidly reversible nature of the thiyl addition step33 and the exquisite stability of the 4C1 conformation of d‐hexopyranoses together ensure the exclusive 1,2‐cis‐α‐stereoselectivity of the reaction. As it is illustrated in the example of 1 (Scheme 3) the starting hexoglycals exist in a rapid interconversion equilibrium between 4H5 and 5H4 half chair conformations.37 Although thiyl radicals can attack both faces of both conformers, the only productive attack occurs on the bottom face of the 4H5 conformational form leading to the formation of the stable 4C1 conformer of the C2‐centered radical intermediate. This chair conformation has exquisite stability due the equatorial position of the bulky C6 group. (This productive route is highlighted in green.) Upon other possible attacks of thiyl radicals the pyranosyl ring would only flip to high‐energy skew boat and 1C4 conformations which rapidly decompose. In other words: thiyl additions that produce high‐energy C2‐centered radical intermediates are unproductive as the equilibrium of these rapidly reversible steps33 completely shifts toward the starting materials. Our results demonstrate that radical hydrothiolation of all 2‐substituted d‐hexoglycals proceeds via the route above. Analogously, the thiol‐ene couplings of l‐hexo congeners proceed exclusively through the 1C4 conformational form of the radical intermediates.28 As the kinetically preferred axial H‐abstraction can only occur on the radical bearing an equatorially positioned acetoxy group, the possible interconversion of the pyramidal carbon‐centered radical was not taken into account.
Scheme 3.
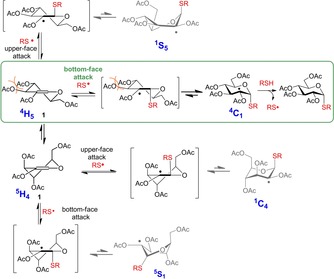
Productive and unproductive attacks of thiyl radicals onto 2‐acetoxy hexopyranosyl glycal 1. The only productive pathway leading to complete 1,2‐cis‐α diastereoselectivity is highlighted in green. The conformational changes of the formed C2‐radicals follow the pseudo‐rotational itinerary of the pyranosyl ring interconversion map.38
Hydrothiolation of 2‐substituted pentoglycals
The picture on the mechanism could be further clarified when hydrothiolation reactions of 2‐acetoxy pentoglycals 5–7 were studied (Scheme 4). Surprisingly, no reaction was observed between the d‐xylose‐derived glycal 5 and 1‐thioglucose 20 at either RT or 0 °C. Conducting the reaction at −40 °C, a moderate conversion of 5 occurred producing a ≈2:1 mixture of two 1,2‐cis‐linked thiodisaccharides, the β‐d‐lyxopyranosyl 43 and the α‐d‐xylopyranosyl 44 with 21 % overall yield. Cooling the reaction to −80 °C, the isolated yield reached 50 % and almost complete diastereoselectivity was observed in favor of the β‐thiolyxoside 43. Reacting 5 with α‐1‐thiomannose 22 at −80 °C complete conversion and almost full stereoselectivity were observed and the 1,2‐cis‐linked β‐d‐lyxo configured thiodisaccharide 45 was isolated with 95 % yield. The structure of this compound was supported by X‐ray measurements (Figure 2). Interestingly, running this reaction at 0 °C an opposite diastereoselectivity was observed providing the α‐d‐xylopyranosyl thioglycoside 46 as the major product along with the β‐d‐lyxo minor product 45 in a 2.5:1 ratio and with 40 % overall yield.
Scheme 4.
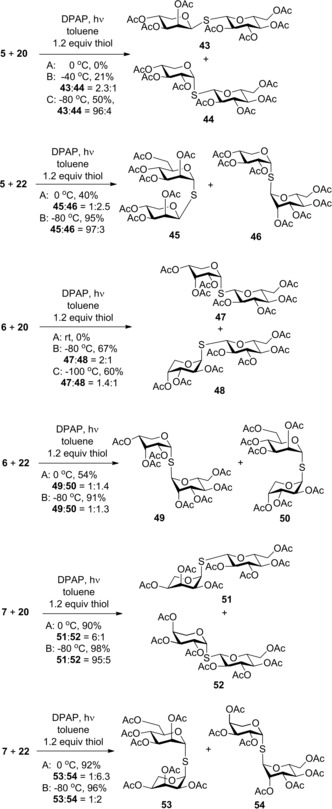
Hydrothiolation reactions of 2‐acetoxy pentoglycals.
Figure 2.
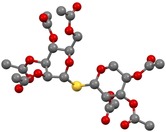
ORTEP view of thiodisaccharide 45, hydrogen atoms are omitted for clarity.
Upon thiol‐ene coupling of the d‐arabinose‐derived glycal 6 with 1‐thioglucose 20 at RT and 0 °C, very low conversions of the glycal were observed again and no product could be isolated. Conducting the reaction at −80 °C gave the α‐d‐ribosyl thiodisaccharide 47 as the major product in 46 % yield along with 21 % yield of the β‐d‐arabinose‐containing minor stereoisomer 48. A further cooling to −100 °C led to a slight decrease in both the yield and stereoselectivity of the addition producing 47 and 48 in a 1.4:1 ratio.
Hydrothiolation of 6 with α‐1‐thiomannose 22 at 0 °C gave a 1.4 to 1 mixture of the corresponding α‐d‐ribosyl and β‐d‐arabinosyl thiodisaccharides 49 and 50 in 54 % yield. By cooling the reaction to −80 °C, the overall yield reached 91 %, however, the stereoselectivity did not increase.
Hydrothiolation of the l‐arabinose‐derived glycal 7 with 1‐thioglucose 20 at −80 °C gave the α‐l‐ribopyranosyl thiodisaccharide 51 almost with complete stereoselectivity in 98 % isolated yield. The reaction proceeded with similar efficacy at 0 °C, however slightly lower stereoselectivity was observed. Reaction between 7 and α‐1‐thiomannose 22 at 0 °C proceeded with complete conversion and high diastereoselectivity, affording the corresponding thiodisaccharides 54 and 53 in a 6.3:1 ratio in favor of the β‐l‐arabinosyl isomer. Surprisingly, at −80 °C a similarly high conversion but a much lower diastereoselectivity (54 to 53 ≈2 to 1) was observed.
The low or no conversions of 2‐acetoxy pentopyranosyl glycals upon hydrothiolation at RT or 0 °C demonstrate that the C2‐centered pentopyranosyl radicals generally have lower stability than the hexopyranosyl congeners (except for the l‐arabino case), therefore again, cooling is crucial for the efficient reactions. Fortunately, good to high conversions could be achieved by running the thiol‐ene couplings at −80 °C. However, the diastereoselectivity showed high dependence on the configuration of the reactants and the temperature. Although in most cases the cooling has increased the stereoselectivity of the additions, the opposite effect was also observed (e.g. in reactions of 6+22 and 7+22).
The lack of complete diastereoselectivity in the pentose series can be explained with the higher conformational flexibility of the pentopyranoses.39 While in the case of hexopyranoses, one of the chair conformations of the intermediate radical has exquisite stability due the bulky C6 group, in the case of pentopyranoses, the 1C4 and 4C1 forms of the radical intermediate have comparable stabilities; therefore, the reaction can proceed through both chair conformations (Scheme 5). Hence, productive attacks can occur on the bottom face of the 4H5 conformational form and on the upper face on the 5H4 conformational form leading to carbon centered radical intermediates existing in 1C4 and 4C1 chair conformations, respectively. We assume that the cooling could tune (generally increase) the stereoselectivity by either shifting the interconversion equilibrium between 4H5 and 5H4 conformations of the starting glycal toward one of the half chair conformers, or by inhibiting the thiyl addition process requiring the higher activation energy.
Scheme 5.
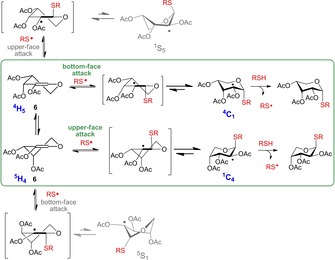
Plausible mechanistic pathway of free‐radical addition of thiols to 2‐acetoxy pentopyranosyl glycals, shown on the example of d‐arabinose‐derived glycal 6. The productive routes resulting exclusively in 1,2‐cis thioglycosides are highlighted in green.
Hydrothiolation of unsubstituted glycals
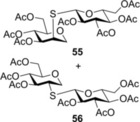
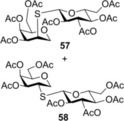
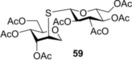
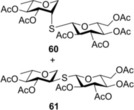
Finally, to push the scope of the reaction further we extended our study to d‐ and l‐glycals without a C2 substituent (Table 3). The room temperature hydrothiolation of peracetylated d‐glycals have been studied by Dondoni and Marra.23 They described that regioselective addition occurred in all cases producing 1‐deoxy‐2‐S‐disaccharides through the corresponding glycosyl radical intermediates. However, the efficiency and diastereoselectivity of the reaction were highly varied depending on the C‐3 and C‐4 configurations of the glycals. For example, while hydrothiolation of d‐glucal 8 using 6 equiv of thiol 20 proceeded efficiently but with very low stereoselectivity, from d‐allal 10, under the same conditions, the altritol derivative 59 was found to be formed with full stereoselectivity, however, only in low (38 %) isolated yield.23 To study the effect of cooling on the efficiency and diastereoselectivity of the additions, the d‐glucose‐, ‐galactose‐, ‐allose‐ and l‐rhamnose‐derived glycals 8, 9, 10 and 11 were reacted with thiol 20 at different temperatures. Performing the reactions at −80 °C, almost complete conversions were observed in all cases and, with the exception of the galacto case, the additions occurred with high to complete diastereoselectivity in favor of the axially C2‐S‐linked products (Table 3). Addition of 20 to 8 and 11 at RT proceeded with low diastereoseletivity, affording a 1.3:1 mixture of the d‐manno (55) and d‐gluco (56) as well as a 2.5:1 mixture of the 6‐deoxy‐l‐manno (l‐rhamno) (60) and 6‐deoxy‐l‐gluco (61) products (Table 3, entries 1 and 10). Conducting the same reactions at −80 °C using only slight thiol excess efficient additions occurred yielding the corresponding axially linked C‐2‐thiodisaccharides 55 and 60 with high stereoselectivity (entries 2 and 11). The configuration of the major product 60 formed from 11 was proven by X‐ray crystallography (Figure 3). In the case of glycal 8, further cooling to −120 °C did not improve the diastereoselectivity of the reaction. Importantly, though the reaction mixture was frozen at −120 °C, the reaction took place. Hydrothiolation of d‐allal 10 occurred with complete diastereoselectivity at both RT and −80 °C providing exclusively the 2‐S‐axial d‐altro‐configured product 59, and by cooling the yield of 59 reached 92 % (entries 8 and 9). However, addition of 1‐thioglucose 20 to the d‐galactal triacetate 9 afforded a diastereomeric mixture of 57 and 58 at any of temperatures studied. Interestingly, the moderate talo‐selectivity observed at 0 °C decreased by cooling and transformed to a very slight galacto selectivity at −80 °C (Table 3, entries 4–7).
Table 3.
Hydrothiolation reactions of d‐ and l‐hexoglycals.
|
| |||||
|---|---|---|---|---|---|
|
Entry |
Alkene +thiol |
Product |
Temp. |
Yield [%][a] |
Ratio A:B |
|
1 |
8+20 (1.2 equiv) |
|
rt |
80 |
1.3:1[b] |
|
2 |
−80 °C |
76 |
8.5:1 |
||
|
3 |
−120 °C |
75 |
6:1 |
||
|
|
|
|
|
|
|
|
4 |
9+20 (1.2 equiv) |
|
0 °C |
67 |
2.3:1 |
|
5 |
−40 °C |
95 |
1.5:1 |
||
|
6 |
−80 °C |
95 |
1:1.2 |
||
|
7 |
−120 °C |
95 |
1:1.2 |
||
|
|
|
|
|
|
|
|
8 |
10+20 (1.2 equiv) |
|
rt |
38 |
1:0[b] |
|
9 |
−80 °C |
92 |
1:0 |
||
|
|
|
|
|
|
|
|
10 |
11+20 (1.2–2.0 equiv) |
|
rt |
66 |
2.5:1[c] |
|
11 |
−80 °C |
81 |
5.5:1[d] |
||

[a] Combined isolated yield; [b] data are taken from Ref. 23, 6 equiv, of thiol was used; [c] 2.0 equiv of thiol was used. [d] 1.2 equiv of thiol was used.
Figure 3.
ORTEP view of 60, hydrogen atoms are omitted for clarity.
The stereochemical outcome of the above additions can be explained by the different conformational preferences of the intermediate C‐1 radicals. While the C‐2‐centered hexopyranosyl radicals preferentially adopt chair conformation independently of the configuration, the conformation of glycosyl radicals is determined by the configuration of the C‐2 substituent.40, 41 It has been demonstrated by Giese and co‐workers on the basis of electron paramagnetic resonance (ESR) spectroscopy study that the most preferred conformation of glycopyranosyl radicals is the one in which the C2‐alkoxy/acyloxy substituent adopts a quasi axial position, because this conformer is stabilized by the so‐called quasi‐homo‐anomeric effect.42, 43, 44 Accordingly, while C‐1 mannosyl radicals exist in 4C1 chair conformation, C‐1 glucosyl radicals preferably flip to the most stable twisted B2,5 conformation—which is practically identical with the 1S5 skew‐boat conformer38—and galactosyl radicals adopt the 4H5 half chair conformation.40, 41, 42, 43, 44, 45 Consequently, products 55, 57 and 59 with an axially linked SR group can be formed through the 4C1 conformational form of the corresponding glycosyl radical via an upper‐face attack by the thiyl radical to the 4H5 conformational form of d‐glycals, and, analogously, compound 60 was formed upon bottom‐face attack by the thiyl radical on l‐glycal 11 in its 5H4 conformation (Scheme 6, line A). Moreover, attacks from opposite direction by thiyl radicals to the glycals in their same half‐chair conformations also proved to be productive in the case of 8, 9 and 11, producing the equatorially S‐linked products 56, 58 and 61 through the corresponding glucosyl and galactosyl radicals in their stable skew‐boat and half‐chair conformations (Scheme 6, line B). Upon attacks of thiyl radicals at either side to the other half‐chair conformation of the starting glycals the pyranosyl ring would only flip to high‐energy skew boat and chair conformations bearing axial C6‐group which decompose rapidly.
Scheme 6.
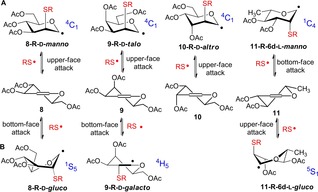
Stable glycopyranosyl radical intermediates formed upon upper‐ and bottom‐face attacks by the thiyl radicals to the 4H5 (d‐series) or 5H4 (l‐series) conformational form of glycals 8–11. The conformational changes follow the pseudo‐rotational itinerary of the pyranosyl ring interconversion map.38
By cooling, the ratio of products formed through the lower‐energy radical intermediates can be raised. Hence, upon hydrothiolation of 8 and 11 at low temperature the diastereoselectivity in favor of the manno‐configured products increased significantly. In the case of galactal 9, the decreased ratio of the talo‐configured product at low temperature can be attributed to the 1,3‐diaxial repulsion between the C2 and C4 substituents in the 4C1 conformation which destabilize the talopyranosyl radical intermediate compared to the 4H5 galacto one. In the case of d‐allal 11, the axial C3‐substituent hinders the attack of thiyl radicals from the bottom face ensuring the complete stereoselectivity of addition occurring on the upper face.
Conclusions
A systematic study on the photoinduced radical mediated hydrothiolation of 2‐substituted hexoglycals, 2‐substituted pentoglycals and unsubstituted hexoglycals were performed, producing 39 thioglycosides up to tetrasaccharide (30 new). We studied the effect of the temperature and the configuration of thiols and glycals on the efficacy and stereochemical outcome of the reactions. In the case of 2‐substituted hexoglycals the reactions proceeded with complete 1,2‐cis‐α‐stereoselectivity in all cases, however, the efficacy of the addition was varying depending on the temperature and on the structure of the reactants. The progression of the reactions could significantly be promoted by cooling, and, except for alkyl thiols, good to high conversions were achieved at −40/−80 °C. In the case of alkyl thiols, applying high thiol excess and running the reactions at RT or 0 °C proved to be the adequate strategy to elicit efficient additions. Hydrothiolation of 2‐substituted pentoglycals led to diastereosiomeric mixtures of 1,2‐cis‐α‐ and 1,2‐cis‐β‐thioglycosides. Noteworthy, the additions occurred with exclusive 1,2‐cis selectivity, no 1,2‐trans‐linked product was observed in any case. Upon additions of different thiosugars to pentoglycals the stereoselectivity and the efficacy of the additions highly depended upon the configuration of both reactants. Although good to excellent yields and in some cases almost complete stereoselectivity were achieved at −80 °C, the stereochemistry of the products was hardly predictable. Hydrothiolation of unsubstituted glycals proceeded with significantly higher efficacy at −80 °C than at RT, and the stereoselectivity could generally also be highly increased in favor of the axially C2‐S‐linked products. The only exception was the d‐galactal, hydrothiolation of which gave the 2‐thio‐d‐galacto and 2‐thio‐d‐talo diastereoisomers in similar amounts independently of the reaction temperature.
In agreement of all synthetic and kinetic observations we assume that the stereoselectivity of the reactions are controlled by the stability of the carbon‐centered radical intermediates of different conformations. The exclusive 1,2‐cis‐α‐stereoselectivity of hydrothiolation of 2‐substituted hexoglycals can be explained by the exquisite stability of the radical intermediate in the 4C1 conformation. In the case of 2‐substituted pentoglycals and unsubstituted hexoglycals the higher conformational flexibility of the intermediate radicals allows the formation of two diastereoisomers, in these cases the low‐reaction temperature proved to be a controlling effect shifting the product ratio towards the stereoisomer which formed through the more stable radical.
Experimental Section
General Information
2,3,4,6‐Tetra‐O‐acetyl‐1,5‐anhydro‐d‐arabino‐hex‐1‐enitol (1),46 2,3,4,6‐tetra‐O‐acetyl‐1,5‐anhydro‐d‐lyxo‐hex‐1‐enitol (2),46 2‐acetamido‐3,4,6‐tri‐O‐acetyl‐1,5‐anhydro‐2‐deoxy‐d‐arabino‐hex‐1‐enitol (3),47 2,3,6‐tri‐O‐acetyl‐4‐O‐(2,3,4,6‐tetra‐O‐acetyl‐α‐d‐glucopyranosyl)‐1,5‐anhydro‐d‐arabino‐hex‐1‐enitol (2‐acetoxy‐maltal peracetate, 4),48 2,3,4‐tri‐O‐acetyl‐1,5‐anhydro‐d‐threo‐pent‐1‐enitol (2‐acetoxy‐3,4‐di‐O‐acetyl‐d‐xylal, 5 49), 2,3,4‐tri‐O‐acetyl‐1,5‐anhydro‐d‐erythro‐pent‐1‐enitol (2‐acetoxy‐3,4‐di‐O‐acetyl‐d‐arabinal) (6),50 2,3,4‐tri‐O‐acetyl‐1,5‐anhydro‐l‐erythro‐pent‐1‐enitol (2‐acetoxy‐3,4‐di‐O‐acetyl‐l‐arabinal, 7),50 3,4,6‐tri‐O‐acetyl‐1,5‐anhydro‐d‐lyxo‐hex‐1‐enitol, (3,4,6‐tri‐O‐acetyl‐d‐galactal, 9),51 3,4,6‐tri‐O‐acetyl‐1,5‐anhydro‐d‐ribo‐hex‐1‐enitol (3,4,6‐tri‐O‐acetyl‐d‐allal, 10),52 3,4,6‐tri‐O‐acetyl‐1,5‐anhydro‐l‐arabino‐hex‐1‐enitol (3,4‐di‐O‐acetyl‐l‐rhamnal, 11),53 mono‐S‐acetyl‐ethanedithiol (15),54 2,3,4,6‐tetra‐O‐acetyl‐α‐d‐glucopyranosyl‐(1→4)‐2,3,6‐tri‐O‐acetyl‐1‐thio‐β‐d‐glucopyranose (18),55 2,3,4,6‐tetra‐O‐acetyl‐1‐thio‐β‐d‐glucopyranose (20),56 2,3,4,6‐tetra‐O‐acetyl‐1‐thio‐β‐d‐mannopyranose (21)55 and 2,3,4,6‐tetra‐O‐acetyl‐1‐thio‐α‐d‐mannopyranose (22)57 were prepared according to the literature procedures. 3,4,6‐Tri‐O‐acetyl‐d‐glucal (8) was purchased from Carbosynth. n‐propyl‐mercaptane (12), iso‐propyl‐mercaptane (13), thioacetic acid (14), mono‐S‐acetyl‐ethanedithiol (15), 2‐mercaptoethanol (16), thioglycolic acid (17), N‐acetyl‐l‐cysteine (19) and initiator 2,2‐dimethoxy‐2‐phenylacetophenone (DPAP) were purchased from Sigma Aldrich Chemical Co. Optical rotations were measured at room temperature with a PerkinElmer 241 automatic polarimeter. TLC was performed on Kieselgel 60 F254 (Merck) with detection by UV‐light (254 nm) and immersing into sulfuric acidic ammonium‐molibdenate solution or 5 % ethanolic sulfuric acid followed by heating. Flash column chromatography was performed on Silica gel 60 (Merck 0.040–0.063 mm), column chromatography was performed on Silica gel 60 (Merck 0.063–0.200 mm). Organic solutions were dried over Na2SO4 or MgSO4, and concentrated in vacuum. One‐ and two‐dimensional 1H, 13C, COSY and HSQC spectra were recorded with Bruker DRX‐360 (1H: 360 MHz; 13C:90 MHz), Bruker DRX‐400 (1H: 400 MHz; 13C:100 MHz) and Avance II 500 (1H: 500.13 MHz; 13C:125.76 MHz) spectrometers at 25 °C. Chemical shifts are referenced to Me4Si (0.00 ppm for 1H) and to the residual solvent signals (CDCl3: 77.1, [D6]DMSO: 39.5, CD3OD: 49.3 for 13C). MALDI‐TOF MS analyses of the compounds were carried out in the positive reflectron mode using a BIFLEX III mass spectrometer (Bruker, Germany) equipped with delayed‐ion extraction. 2,5‐Dihydroxybenzoic acid (DHB) was used as matrix and F3CCOONa as cationising agent in DMF. ESI‐TOF HRMS spectra were recorded by a microTOF‐Q type QqTOFMS mass spectrometer (Bruker) in the positive ion mode using MeOH as the solvent.
The photoinduced reactions were carried out in a borosilicate vessel by irradiation with a Hg‐lamp giving maximum emission at 365 nm.
CCDC https://www.ccdc.cam.ac.uk/services/structures?id=doi:10.1002/chem.201903095 contain the supplementary crystallographic data for this paper. These data are provided free of charge by http://www.ccdc.cam.ac.uk/.
The chemical shifts of the compounds are signed according to Figure 4.
Figure 4.
Denotation of the compounds for the NMR assignation.
General method for photoinitiated free radical thiol addition
The corresponding alkene, thiol and DPAP (DPAP, 0.1 equiv/alkene) were dissolved in the given solvent. The reaction mixture was cooled to the given temperature and was irradiated with UV light for 15 min. After irradiation another 0.1 equiv of DPAP was added and the irradiation continued for another 15 min. The addition of 0.1 equiv of DPAP and the irradiation was repeated one more time. In some cases, the reaction mixtures were frozen by cooling (−80 to −120 °C). In such cases the frozen mixture was thawed before adding the next portion of DPAP. The reactions also took place in frozen mixtures. The solvent was evaporated in vacuo and the crude product was purified by column chromatography or by flash column chromatography.
Propyl 2,3,4,6‐tetra‐O‐acetyl‐1‐thio‐α‐d‐glucopyranoside (23) 25: A: Compound 1 (0.5 mmol, 165 mg) in toluene (3.0 mL) was reacted with thiol 12 (2.5 mmol, 225 μL, 5.0 equiv) at RT. according to the general method. The crude product was purified by flash chromatography (n‐hexane/acetone 75:25) to give compound 23 (72 mg, 35 %) as white crystals. B: Compound 1 (0.5 mmol, 165 mg), thiol 12 (2.5 mmol, 225 μL, 5.0 equiv) and DPAP (0.05 mmol, 14 mg, 0.1 equiv) were dissolved in toluene (3.0 mL). The reaction mixture was irradiated with UV‐light at RT for 15 min. The addition of 5.0 equiv of thiol 12, 0.1 equiv of DPAP and the irradiation was repeated two more times. The solvent was evaporated in vacuo and the crude product was purified to give compound 23 with 54 % yield. C: The reaction was repeated at 0 °C with 3.0 and 5.0 equiv of 12 at 0 °C, with 46 % and 65 % yield, respectively. D: The reaction was repeated at −40 °C with 3.0 equiv of thiol 12 according to the general method to give 23 with 18 % yield. R f=0.47 (n‐hexane/acetone 7:3); m.p. 106–108 °C, lit.25 m.p. 105–106 °C; [α]D 20=+185.7 (c=0.28 in CHCl3), lit.25 [α]D 20=+185; 1H NMR (400 MHz, CDCl3): δ=5.66 (d, J=5.8 Hz, 1 H, H‐1), 5.37 (t, J=9.8 Hz, 1 H, H‐3), 5.12–4.95 (m, 2 H, H‐2, H‐4), 4.44 (ddd, J=10.2, 4.8, 2.3 Hz, 1 H, H‐5), 4.30 (dd, J=12.2, 4.8 Hz, 1 H, H‐6a), 4.08 (dd, J=12.3, 2.2 Hz, 1 H, H‐6b), 2.67–2.36 (m, 2 H, SCH 2), 2.09 (s, 3 H, AcCH 3), 2.07 (s, 3 H, AcCH 3), 2.04 (s, 3 H, AcCH 3), 2.02 (s, 3 H, AcCH 3), 1.63 (q, J=7.3 Hz, 2 H, SCH2CH 2), 0.98 ppm (td, J=7.4, 2.0 Hz, 3 H, SCH2CH2CH 3). 13C NMR (100 MHz, CDCl3): δ=170.6, 169.9, 169.9, 169.6 (4×C, AcCO), 82.1 (1×C, C‐1), 70.8, 70.6, 68.6, 67.6 (4C, skeletal carbons), 62.0 (1C, C‐6), 32.3 (1C, SCH2), 22.8 (1C, SCH2 CH2CH3), 20.8, 20.7, 20.7, 20.6 (4C, AcCH3), 13.4 ppm (1C, CH2CH2 CH3); ESI‐MS: m/z calcd for C17H26NaO9S [M+Na]+ 429.1195, found 429.1190.
(1‐Methylethyl) 2,3,4,6‐tetra‐O‐acetyl‐1‐thio‐α‐d‐glucopyranoside (24): A: Compound 1 (3.0 mmol, 990 mg) was reacted with thiol 13 (9.0 mmol, 845 μL, 3.0 equiv) in toluene (5.0 mL) at 0 °C according to the general method. The crude product was purified by flash chromatography (n‐hexane/acetone 75:25) to give 24 (682 mg, 56 %) as yellow powder. B: The reaction was repeated with 5.0 equiv of 13 at RT to give 24 with 32 % yield. C: The reaction was repeated with 5.0 equiv of 13 at 0 °C to give 24 with 68 % yield. D: The reaction was repeated with 3.0 equiv of 13 at −40 °C to give 24 with 50 % yield. E: Compound 1 (0.5 mmol, 165 mg), thiol 13 (2.5 mmol, 230 μL, 5.0 equiv) and DPAP (0.05 mmol, 14 mg, 0.1 equiv) were dissolved in toluene (3.0 mL). The reaction mixture was irradiated with UV‐light at RT for 15 min. The addition of 5.0 equiv of thiol 13, 0.1 equiv of DPAP and the irradiation was repeated two more times. The solvent was evaporated in vacuo and the crude product was purified to give compound 24 with 51 % yield. R f=0.55 (n‐hexane/acetone 7:3); m.p. 71–73 °C; [α]D 20=+164.2 (c=0.12 in CHCl3); 1H NMR (360 MHz, CDCl3): δ=5.75 (d, J=5.9 Hz, 1 H, H‐1), 5.34 (t, J=9.8 Hz, 1 H, H‐3), 5.11–4.93 (m, 2 H, H‐2, H‐4), 4.46 (ddd, J=10.2, 4.7, 2.3 Hz, 1 H, H‐5), 4.30 (dd, J=12.3, 4.7 Hz, 1 H, H‐6a), 4.07 (dd, J=12.4, 2.3 Hz, 1 H, H‐6b), 3.00 (hept, J=6.8 Hz, 1 H, SCH(CH3)2), 2.08, 2.06, 2.04, 2.02 (4xs, 12 H, 4×AcCH 3), 1.30 ppm (t, J=6.8 Hz, 6 H, 2×SCHCH 3). 13C NMR (90 MHz, CDCl3): δ=170.6, 169.9, 169.6 (4xC, AcCO), 81.1 (1C, C‐1), 70.7, 70.6, 68.7, 67.6 (4C, skeletal carbons), 62.0 (1C, C‐6), 35.0 (1C, SCH(CH3)2), 23.8, 23.5 (2C, 2×iPrCH3), 20.8, 20.7, 20.6 ppm (4C, 4×AcCH3); ESI‐MS: m/z calcd for C17H26NaO9S [M+Na]+ 429.1195, found 429.1189.
2,3,4,6‐Tetra‐O‐acetyl‐1‐S‐acetyl‐1‐thio‐α‐d‐glucopyranose (25) 58: A: Compound 1 (330 mg, 1.0 mmol) was reacted with thiol 14 (429 μL, 6.0 mmol, 6.0 equiv) in toluene (3.0 mL) at −20 °C according to the general method. The crude product was purified by flash column chromatography (n‐hexane/acetone 8:2) to give 25 (37 mg, 9 %) as yellow powder. B: Compound 1 (660 mg, 2.0 mmol), thiol 14 (16.0 mmol, 1.146 mL, 8.0 equiv) and DPAP (0.1 mmol, 25 mg, 0.1 equiv) were dissolved in toluene (3.0 mL). The reaction mixture was cooled to −20 °C and was irradiated with UV‐light for 15 min. The addition of 8.0 equiv of thiol 14, 0.1 equiv of DPAP and the irradiation was repeated two more times. The solvent was evaporated in vacuo and the crude product was purified by flash column chromatography (n‐hexane/acetone 8:2) to give compound 25 with 19 % yield. C: Compound 1 (660 mg, 2.0 mmol), thiol 14 (16.0 mmol, 1.146 mL, 8.0 equiv) and DPAP (0.1 mmol, 25 mg, 0.1 equiv) were dissolved in toluene (3.0 mL). The reaction mixture was irradiated with UV‐light at RT for 15 min. The addition of 8.0 equiv of thiol 14, 0.1 equiv of DPAP and the irradiation was repeated two more times. The solvent was evaporated in vacuo and the crude product was purified to give compound 25 with 9 % yield. D: The reaction was repeated at room temperature and at −40 °C using 6.0 equiv of thiol according to the general method, but no product could be isolated. R f=0.17 (n‐hexane/acetone 8:2); m.p. 129–132 °C, lit.58 m.p. 123–125 °C [α]D 20=+123.6 (c=0.11 in CHCl3), lit.58 [α]D 20=+143.5; 1H NMR (400 MHz, CDCl3): δ=6.22 (d, J=5.2 Hz, 1 H, H‐1), 5.24 (dd, J=10.1, 5.1 Hz, 1 H, H‐2), 5.18 (t, J=9.5 Hz, 1 H), 5.10 (t, J=9.5 Hz, 1 H) (H‐3 and H‐4), 4.28 (dd, J=12.5, 4.1 Hz, 1 H, H‐6a), 4.05 (dd, J=12.5, 2.2 Hz, 1 H, H‐6b), 3.96 (ddd, J=9.9, 4.0, 2.3 Hz, 1 H, H‐5), 2.43 (s, 3 H, AcCH 3), 2.08 (s, 3 H, AcCH 3), 2.03 (s, 3 H, AcCH 3), 2.02 (s, 3 H, AcCH 3), 2.02 ppm (s, 3 H, AcCH 3). 13C NMR (100 MHz, CDCl3): δ=170.7, 170.1, 169.5, 169.4 (4C, 4×AcCO), 80.5 (1C, C‐1), 71.6, 71.3, 69.2, 68.0 (4C, skeletal carbons), 61.7 (1C, C‐6), 31.6 (1C, SAcCH3), 20.8, 20.7, 20.7 ppm (4C, 4×OAcCH3); ESI‐MS: m/z calcd for C16H22NaO10S [M+Na]+ 429.0831, found 429.0826.
(2‐Acetylthio)ethyl 2,3,4,6‐tetra‐O‐acetyl‐1‐thio‐α‐d‐glucopyranoside (26): A: Compound 1 (660 mg, 2.0 mmol) and thiol 15 (6.0 mmol, 816 μL, 3.0 equiv) were reacted in toluene (8.0 mL) at RT according to the general method. The crude product was purified by flash chromatography (n‐hexane/acetone 8:2) to give 26 (512 mg, 55 %) as colorless syrup. B: The reaction was repeated at 0 °C in the same scale with 81 % yield. R f=0.12 (n‐hexane/acetone 8:2). [α]D 20=+164.9 (c=0.41 in CHCl3), 1H NMR (400 MHz, CDCl3): δ=5.73 (d, J=5.8 Hz, 1 H, H‐1), 5.34 (t, J=9.8 Hz, 1 H, H‐3), 5.08–5.00 (m, 2 H, H‐2, H‐4), 4.43 (ddd, J=10.1, 4.9, 2.0 Hz, 1 H, H‐5), 4.29 (dd, J=12.3, 5.0 Hz, 1 H, H‐6a), 4.09 (dd, J=12.3, 1.9 Hz, 1 H, h‐6b), 3.21–3.02 (m, 2 H, SCH 2), 2.85–2.66 (m, 2 H, SCH 2), 2.35 (s, 3 H, AcCH 3), 2.08 (s, 3 H, AcCH 3), 2.07 (s, 3 H, AcCH 3), 2.04 (s, 3 H, AcCH 3), 2.02 ppm (s, 3 H, AcCH 3). 13C NMR (100 MHz, CDCl3): δ=194.9 (1C, SAcCO), 170.5, 169.9, 169.8, 169.6 (4C, 4×AcCO), 82.5 (1C, C‐1), 70.6, 70.4, 68.5, 67.9 (4C, skeletal carbons), 62.0 (1C, C‐6), 30.6 (1C, SAcCH3), 30.4, 29.2, 20.7, 20.7, 20.6, 20.6 ppm (4C, 4×AcCH3), MALDI‐TOF‐MS: m/z calcd for C18H16NaO10S2 [M+Na]+ 489.086, found 489.578.
(2‐Hydroxy)ethyl 2,3,4,6‐tetra‐O‐acetyl‐1‐thio‐α‐d‐glucopyranoside (27): A: Compound 1 (165 mg, 0.5 mmol) and thiol 16 (71 μL, 1.0 mmol, 2.0 equiv) were reacted in toluene (3.0 mL) at −80 °C according to the general method. The crude product was purified by flash column chromatography (n‐hexane/acetone 7:3) to give 27 (142 mg, 70 %) as colourless syrup. B: The reaction was repeated at RT to give 27 with 40 % yield. R f=0.17 (n‐hexane/acetone 7:3); [α]D 20=+89.3 (c=0.43 in CHCl3); 1H NMR (400 MHz, CDCl3): δ=5.71 (d, J=5.8 Hz, 1 H, H‐1), 5.35 (t, J=9.8 Hz, 1 H, H‐3), 5.07–4.99 (m, 2 H, H‐2, H‐4), 4.47 (ddd, J=10.1, 4.9, 1.9 Hz, 1 H, H‐5), 4.26 (dd, J=12.3, 5.2 Hz, 1 H, H‐6a), 4.12 (dd, J=12.3, 1.9 Hz, 1 H, H‐6b), 3.77 (d, J=6.5 Hz, 2 H), 2.86–2.77 (m, 1 H), 2.77–2.68 (m, 1 H), 2.10 (s, 3 H, AcCH 3), 2.08 (s, 3 H, AcCH 3), 2.05 (s, 3 H, AcCH 3), 2.03 (s, 3 H, AcCH 3). 13C NMR (100 MHz, CDCl3): δ=170.8, 170.1, 169.8 84C, 4×AcCO), 82.7 (1C, C‐1), 70.7, 70.3, 68.6, 67.9 (4C, skeletal carbon), 62.1, 61.6 (2C, C‐6, CH2OH), 34.0 (1C, SCH2), 20.8, 20.8, 20.7, 20.7 ppm (4C, 4×AcCH3); ESI‐MS: m/z calcd for C16H24NaO10S [M+Na]+ 431.0988, found 431.0982.
2‐(2,3,4,6‐Tetra‐O‐acetyl‐1‐thio‐α‐d‐glucopyranosyl) acetic acid (28): A: Compound 1 (330 mg, 1.0 mmol) in a mixture of toluene (0.6 mL) and MeOH (1.2 mL) was reacted with thiol 17 (140 μL, 2.0 mmol, 2.0 equiv) at −80 °C according to the general method. The crude product was purified by flash chromatography (CHCl2/MeOH 95:5) to give compound 28 (280 mg, 67 %) as white foam. B: The reaction was repeated at RT to give 28 with 55 % yield. R f=0.46 (CH2Cl2/MeOH 9:1); [α]D 20=+175.6 (c=0.25 in CHCl3), 1H NMR (400 MHz, CDCl3): δ=10.31 (br. s, 1 H, COOH), 5.78 (d, J=5.8 Hz, 1 H, H‐1), 5.37 (t, J=9.8 Hz, 1 H, H‐3), 5.18–5.00 (m, 2 H, H‐2, H‐4), 4.42 (d, J=10.4 Hz, 1 H, H‐5), 4.28 (dd, J=12.5, 4.3 Hz, 1 H, H‐6a), 4.07 (d, J=10.0 Hz, 1 H, H‐6b), 3.44 (d, J=15.6 Hz, 1 H, SCH 2a), 3.26 (d, J=15.7 Hz, 1 H, SCH 2b), 2.10 (s, 3 H, AcCH 3), 2.08 (s, 3 H, AcCH 3), 2.05 (s, 3 H, AcCH 3) 2.02 ppm (s, 3 H, AcCH 3). 13C NMR (100 MHz, CDCl3): δ=174.5 (1C, COOH), 170.9, 170.0, 169.9, 169.7 (4C, 4×AcCO), 82.1 (1C, C‐1), 70.4, 68.3, 68.0 (3C, skeletal carbons), 61.7 (1C, C‐6), 31.5 (1C, CH2COOH), 20.7, 20.6, 20.5 ppm (3C, 3×AcCH3); MALDI‐TOF‐MS: m/z calcd for C16H22NaO11S [M+Na]+: 445.078 found: 445.077.
2,3,4,6‐Tetra‐O‐acetyl‐α‐d‐glucopyranosyl‐(1→4)‐2,3,6‐tri‐O‐acetyl‐1‐thio‐β‐d‐glucopyranosyl‐(1→1)‐2,3,4,6‐tetra‐O‐acetyl‐α‐d‐glucopyranoside (29): A: Compound 1 (66 mg, 0.2 mmol) and thiol 18 (196 mg, 0.3 mmol, 1.5 equiv) were reacted in a mixture of toluene (1.0 mL) and DMF (0.5 mL) at 0 °C according to the general method. The crude product was purified by flash chromatography (CH2Cl2/acetone 94:6) to give compound 29 (20 mg, 10 %) as white foam. B: The reaction was repeated at −20 °C in the same scale with 14 % yield. C: The reaction was repeated at −40 °C in the same scale with 29 % yield. D: The reaction was repeated at −80 °C in the same scale with 58 % yield. E: The reaction was repeated at RT in the same scale with 5 % yield. R f=0.24 (CH2Cl2/acetone 9:1); [α]D 20=+104.4 (c=0.32 in CHCl3), 1H NMR (500 MHz, CDCl3): δ=5.93 (d, J=5.7 Hz, 1 H, H‐1′′), 5.41 (d, J=4.0 Hz, 1 H, H‐1), 5.38–5.30 (m, 1 H), 5.32–5.24 (m, 1 H), 5.23 (t, J=9.0 Hz, 1 H), 5.13 (t, J=9.8 Hz, 1 H), 5.06 (t, J=9.9 Hz, 2 H), 4.99 (dd, J=10.3, 5.7 Hz, 1 H), 4.91 (t, J=9.6 Hz, 1 H), 4.86 (dd, J=10.5, 4.0 Hz, 1 H), 4.60 (d, J=10.1 Hz, 1 H), 4.57 (dd, J=12.4, 2.5 Hz, 1 H), 4.42 (dd, J=12.7, 2.8 Hz, 1 H), 4.33 (dt, J=10.3, 2.6 Hz, 1 H), 4.29–4.24 (m, 2 H), 4.10 (dt, J=12.3, 3.0 Hz, 3 H), 4.05 (dd, J=12.5, 2.4 Hz, 2 H), 4.01–3.94 (m, 3 H), 3.70 (ddd, J=9.8, 4.1, 2.5 Hz, 1 H), 2.15 (s, 3 H), 2.11 (d, J=1.3 Hz, 8 H), 2.04–1.98 ppm (m, 33 H, 11×AcCH3). 13C NMR (126 MHz, CDCl3): δ=170.7, 170.7, 170.5, 170.3, 170.0, 170.0, 169.7, 169.6, 169.6, 169.5 (11C, 11xAcCH3), 95.8, 82.0, 81.8 (3C, C‐1, C‐1′, C‐1′′), 76.3, 72.3, 72.0, 70.5, 70.4, 70.2, 69.5, 68.8, 68.7, 68.1, 68.0 (12C, skeletal carbons), 62.6, 61.6, 61.3 (3C, C‐6, C‐6′, C‐6′′), 21.0, 21.0, 20.9, 20.9, 20.8, 20.8, 20.7 ppm (11C, 11×AcCH3); ESI‐MS: m/z calcd for C40H54NaO26S [M+Na]+ 1005.252, found 1005.250.
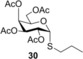
Propyl 2,3,4,6‐tetra‐O‐acetyl‐1‐thio‐α‐d‐galactopyranoside (30): A: Compound 2 (165 mg, 0.5 mmol) and thiol 12 (225 μL, 2.5 mmol, 5.0 equiv) were reacted in toluene (2.0 mL) at RT according to the general method. The crude product was purified by flash chromatography (n‐hexane/acetone 85:15) to give compound 30 (57 mg, 28 %) as white powder. B: The reaction was repeated at 0 °C in the same scale with 26 % yield. C: The reaction was repeated at −40 °C in the same scale with 56 % yield. D: The reaction was repeated at −80 °C in the same scale with 22 % yield. R f=0.30 (CH2Cl2/acetone 9:1); m.p. 77–80 °C; [α]D 20=+167.8 (c=0.27 in CHCl3); 1H NMR (400 MHz, CDCl3): δ=5.72 (d, J=5.2 Hz, 1 H, H‐1), 5.45 (dd, J=3.0, 1.0 Hz, 1 H, H‐3), 5.25 (d, J=5.2 Hz, 1 H, H‐2), 5.23 (d, J=3.1 Hz, 1 H, H‐4), 4.60 (t, J=6.7 Hz, 1 H, H‐5), 4.12 (s, 1 H, H‐6a), 4.10 (d, J=0.6 Hz, 1 H, H‐6b), 2.57 (ddd, J=12.8, 7.7, 6.7 Hz, 1 H, SCH 2a), 2.52–2.43 (m, 1 H, SCH 2b), 2.15 (s, 3 H, AcCH 3), 2.08 (s, 3 H, AcCH 3), 2.05 (s, 3 H, AcCH 3), 2.00 (s, 3 H, AcCH 3), 1.63 (ddd, J=14.8, 7.4, 2.6 Hz, 2 H, CH2CH 2CH3), 0.99 ppm (t, J=7.3 Hz, 3 H, CH2CH2CH 3). 13C NMR (100 MHz, CDCl3): δ=170.4, 170.3, 169.9 (4C, 4×AcCO), 82.4 (1C, C‐1), 68.2, 68.1, 66.5 (3C, skeletal carbons), 61.9 (1C, C‐6), 32.1 (1C, SCH2), 22.9 (1C, CH2 CH2CH3), 20.9, 20.8, 20.7 (4C, 4×AcCH3), 13.6 ppm (1C, CH2CH2 CH3); MALDI‐TOF‐MS: m/z calcd for C17H26NaO9S [M+Na]+ 429.120, found 429.144.
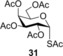
2,3,4,6‐tetra‐O‐acetyl‐1‐S‐acetyl‐1‐thio‐α‐d‐galactopyranose (31) 59: A: Compound 2 (165 mg, 0.5 mmol) and thiol 14 (280 μL, 3.0 mmol, 6.0 equiv) were reacted in a mixture of toluene (1.0 mL) and methanol (1.0 mL) at −80 °C according to the general method. The crude product was purified by flash silica gel chromatography (CH2Cl2/acetone 110:2) to give 31 (46 mg, 23 %) as yellow syrup. B: The reaction was repeated at −40 °C in the same scale with 22 % yield C: The reaction was repeated at 0 °C in the same scale with 15 % yield. D: Compound 2 (3.0 mmol, 990 mg), thiol 14 (18.0 mmol, 1.29 mL, 6.0 equiv) and DPAP (0.3 mmol, 75 mg, 0.1 equiv) were dissolved in toluene (5.0 mL). The reaction mixture was cooled to −40 °C and was irradiated with UV‐light at RT for 15 min. The addition of 6.0 equiv of thiol 14, 0.1 equiv of DPAP and the irradiation was repeated two more times. The solvent was evaporated in vacuo and the crude product was purified to give 31 (250 mg, 26 %) as yellow syrup. E: The reaction was repeated at room temperature using 6.0 equiv of thiol according to the general method, but no product could be isolated. R f=0.29 (CH2Cl2/acetone=98:2); m.p. 119–121 °C (Et2O), lit.59 m.p. 119 °C [α]D 20=+120.9 (c=0.21 in CHCl3), lit.59 [α]D 20=+48; 1H NMR (400 MHz, CDCl3): δ=6.27 (d, J=5.5 Hz, 1 H, H‐1), 5.48 (dd, J=11.0, 5.5 Hz, 1 H, H‐2), 5.44 (dd, J=3.3, 1.2 Hz, 1 H, H‐4), 5.04 (dd, J=11.0, 3.3 Hz, 1 H, H‐3), 4.18 (td, J=6.5, 1.1 Hz, 1 H, H‐5), 4.11–4.03 (m, 2 H, H‐6ab), 2.42 (s, 3 H, SAcCH 3), 2.15 (s, 3 H, AcCH 3), 2.03 (s, 3 H, AcCH 3), 2.02 (s, 3 H, AcCH 3), 2.00 ppm (s, 3 H, AcCH 3). 13C NMR (100 MHz, CDCl3): δ=191.8 (1C, SAcCO), 170.4, 170.2, 170.0, 169.7 (4C, 4×OAcCO), 81.2 (1C, C‐1), 70.4, 69.0, 67.3, 66.5 (4C, skeletal carbons), 61.2 (1C, C‐6), 31.6 (1C, SAcCH3), 20.7, 20.7, 20.7 ppm (4C, 4×AcCH3); MALDI‐TOF‐MS: m/z calcd for C16H22NaO10S [M+Na]+ 429.083, found 429.199.
(2‐Acetylthio)ethyl 2,3,4,6‐tetra‐O‐acetyl‐1‐thio‐α‐d‐galactopyranoside (32): A: Compound 2 (660 mg, 2.0 mmol) and thiol 15 (816 μL, 6.0 mmol, 3.0 equiv) were reacted in toluene (5.0 mL) at room temperature according to the general method. The crude product was purified by flash column chromatography (CH2Cl2/acetone 95/5) to give 32 (473 mg, 52 %) as yellow oil. B: Compound 2 (110 mg, 0.3 mmol) and thiol 15 (136 μL, 0.9 mmol, 3.0 equiv) were reacted in toluene (3.0 mL) at 0 °C according to the general method with 69 % yield. C: The reaction was repeated at −40 °C in the same scale with 41 % yield. R f=0.67 (CH2Cl2/acetone 95:5), [α]D 20=+164.0 (c=0.52 in CHCl3), 1H NMR (400 MHz, CDCl3): δ=5.80 (d, J=5.5 Hz, 1 H, H‐1), 5.46–5.43 (m, 1 H), 5.27 (dd, J=10.8, 5.6 Hz, 1 H, H‐2), 5.19 (dd, J=10.8, 3.2 Hz, 1 H, H‐3), 4.58 (t, J=6.4 Hz, 1 H), 4.12 (d, J=6.4 Hz, 2 H), 3.20–3.02 (m, 2 H, SCH 2), 2.84–2.76 (m, 1 H, SCH 2a), 2.75–2.65 (m, 1 H, SCH 2b), 2.35 (s, 3 H, AcCH 3), 2.15 (s, 3 H, AcCH 3), 2.08 (s, 3 H, AcCH 3), 2.04 (s, 3 H, AcCH 3), 1.99 ppm (s, 3 H, AcCH 3). 13C NMR (100 MHz, CDCl3): δ=194.9, 170.3, 170.1, 170.0, 169.8 (5C, 5xAcCO), 82.8 (1C, C‐1), 68.0, 67.9, 67.8, 66.8 (4C, skeletal carbons), 61.9 (1C, C‐6), 30.6 (1C, SAcCH3), 30.1, 29.2 (2C, 2×SCH2), 20.8, 20.6, 20.6 ppm (4C, 4×OAcCH3). MALDI‐TOF‐MS: m/z calcd for C18H26NaO10S2 [M+Na]+ 489.087, found 489.080.
(2‐Hydroxyethyl) 2,3,4,6‐tetra‐O‐acetyl‐1‐thio‐α‐d‐galactopyranoside (33): A: Compound 2 (1.32 g, 4.0 mmol) and thiol 16 (844 μL, 12.0 mmol, 3.0 equiv) were reacted in toluene (5.0 mL) at RT according to the general method. The crude product was purified by flash column chromatography (n‐hexane/EtOAc 7:3) to give 33 (938 mg, 58 %) as white foam. B: The reaction was repeated with 2.0 equiv of thiol at −80 °C with 86 % yield. R f=0.42 (n‐hexane/EtOAc 1:1), [α]D 20=+180.2 (c=0.47 in CHCl3); 1H NMR (400 MHz, CDCl3): δ=5.77 (d, J=5.2 Hz, 1 H, H‐1), 5.45 (s, 1 H), 5.26 (dd, J=10.7, 5.4 Hz, 1 H, H‐2), 5.19 (dd, J=10.8, 2.3 Hz, 1 H, H‐3), 4.63 (t, J=5.9 Hz, 1 H), 4.13 (d, J=6.1 Hz, 2 H), 3.88 (t, J=5.6 Hz, 1 H), 3.74 (dd, J=17.0, 5.4 Hz, 3 H), 2.88 (t, J=5.9 Hz, 2 H), 2.85–2.79 (m, 1 H), 2.76–2.66 (m, 2 H, SCH 2), 2.16 (s, 3 H, AcCH 3), 2.09 (s, 3 H, AcCH 3), 2.07 (s, 3 H, AcCH 3), 2.00 ppm (s, 3 H, AcCH 3). 13C NMR (100 MHz, CDCl3): δ=170.4, 170.1, 169.8 (4C, 4×AcCO), 82.8 (1C, C‐1), 67.9, 67.8, 67.7, 66.7 (4C, skeletal carbons), 61.8, 61.4, 60.2, 33.3 (1C, SCH2), 20.6, 20.5, 20.4 ppm (4C, 4×AcCH3). MALDI‐TOF‐MS: m/z calcd for C16H24NaO10S [M+Na]+ 431.099, found 431.094.
2,3,4,6‐Tetra‐O‐acetyl‐α‐d‐glucopyranosyl‐(1→4)‐2,3,6‐tri‐O‐acetyl‐1‐thio‐β‐d‐glucopyranosyl‐(1→1)‐2,3,4,6‐tetra‐O‐acetyl‐α‐d‐galactopyranoside (34): A: Compound 2 (83 mg, 0.25 mmol) and thiol 18 (225 mg, 0.375 mmol, 1.5 equiv) were reacted in a mixture of toluene (2.0 mL) and N‐N‐dimethylformamide (1.0 mL) at −40 °C according to the general method. The crude product was purified by flash column chromatography (n‐hexane/acetone 7:3) to give 34 (86 mg, 35 %) as white powder. B: The reaction was repeated at −80 °C to give 34 with 75 % yield. R f=0.15 (n‐hexane/acetone 7:3), m.p.: 78–81 °C, [α]D 20=+116.1 (c=0.33 in CHCl3), 1H NMR (400 MHz, CDCl3): δ=5.99–5.93 (m, 1 H, H‐1), 5.45 (d, J=1.6 Hz, 1 H, H‐1′′), 5.42 (d, J=3.9 Hz, 1 H), 5.39–5.30 (m, 2 H), 5.27–5.18 (m, 2 H), 5.14 (dd, J=11.0, 3.2 Hz, 1 H), 5.06 (t, J=9.9 Hz, 1 H), 4.92 (d, J=9.7 Hz, 1 H), 4.89–4.82 (m, 1 H), 4.62 (d, J=10.0 Hz, 1 H, H‐1′), 4.55 (d, J=2.3 Hz, 1 H), 4.51 (d, J=6.6 Hz, 1 H), 4.26 (ddd, J=16.8, 10.4, 6.7 Hz, 3 H), 4.16 (d, J=3.6 Hz, 1 H), 4.14–4.10 (m, 3 H), 4.09–3.91 (m, 5 H), 3.75–3.69 (m, 1 H), 2.15 (d, J=4.1 Hz, 6 H), 2.11 (s, 3 H), 2.07–1.98 ppm (m, 30 H). 13C NMR (100 MHz, CDCl3): δ=170.6, 170.4, 170.3, 170.3, 170.1, 170.0, 169.9, 169.5 (11C, 11×AcCO), 95.6 (1C, C‐1′′), 82.1, 81.7 (2C, C‐1, C‐1′), 76.6, 76.3, 72.2, 71.9, 70.1, 69.3, 68.6, 68.0, 67.8, 67.3, 67.2 (12C, skeletal carbons), 62.6, 61.5, 60.3 (3C, C‐6, C‐6′, C‐6′′), 20.9, 20.8, 20.7, 20.7 ppm (11C, 11×AcCH3). MALDI‐TOF‐MS: m/z calcd for C40H54NaO26S [M+Na]+ 1005.252, found 1005.318.
(2‐Acetylthio)ethyl 2‐acetamido‐3,4,6‐tri‐O‐acetyl‐2‐deoxy‐1‐thio‐α‐d‐glucopyranoside (35): A: Compound 3 (660 mg, 2.0 mmol), and thiol 15 (408 μL, 3.0 mmol, 1.5 equiv) were reacted in toluene (10 mL) at RT according to the general method. The solvent was evaporated under reduced pressure and the crude product was purified by flash chromatography (CH2Cl2/acetone 9:1) to give 35 (432 mg, 46 %) as white foam. B: The reaction was repeated at 0 °C in the same scale with 83 % yield. C: The reaction was repeated at −80 °C in the same scale with 45 % yield. R f=0.25 (CH2Cl2/acetone 9:1), [α]D 20=+108.5 (c=0.16 in CHCl3), 1H NMR (400 MHz, CDCl3): δ=5.79 (d, J=8.7 Hz, 1 H, NH), 5.50 (d, J=5.4 Hz, 1 H, H‐1), 5.15–4.99 (m, 2 H, H‐2, H‐4), 4.51 (ddd, J=10.7, 8.7, 5.4 Hz, 1 H, H‐5), 4.36 (ddd, J=9.6, 4.8, 2.2 Hz, 1 H), 4.26 (dd, J=12.3, 4.9 Hz, 1 H, H‐6a), 4.09 (dd, J=12.3, 2.3 Hz, 1 H, H‐6b), 3.16 (ddd, J=13.6, 8.6, 6.3 Hz, 1 H), 3.06 (ddd, J=13.6, 8.7, 6.3 Hz, 1 H), 2.81 (qdd, J=13.5, 8.6, 6.3 Hz, 2 H, CH2), 2.34 (s, 3 H, COCH3), 2.08 (s, 3 H, COCH3), 2.04 (s, 3 H, COCH3), 2.03 (s, 3 H, COCH3), 1.96 ppm (s, 3 H, COCH3).13C NMR (100 MHz, CDCl3): δ=195.0, 171.6, 170.7, 170.0, 169.3, (5C, 5×COCH3) 85.1 (C‐1), 71.3, 68.7, 68.2 (C‐5, C‐4, C‐3), 62.1 (C‐6), 52.5 (C‐2), 31.7 (SCH2), 30.7, (SCOCH3), 29.5 (SCH2), 23.3, 20.8, 20.7 ppm (5C, 5×COCH3). MALDI‐TOF‐MS: m/z calcd for C18H27NO9S2 [M+Na]+: 488.102 found, found 488.100.
2‐Acetamido‐2‐deoxy‐3,4,6‐tri‐O‐acetyl‐1‐S‐acetyl‐1‐thio‐α‐d‐glucopyranose (36): A: Compound 3 (329 mg, 1.0 mmol) and thiol 14 (429 μL, 6.0 mmol, 6.0 equiv) were reacted at −20 °C in a mixture of toluene (2.0 mL) and MeOH (1.0 mL) according to the general method. The crude product was purified by flash column chromatography (CH2Cl2/acetone 96/4) to give 36 (146 mg, 36 %) as yellow powder. B: The reaction was repeated at −40 °C in the same scale to give 36 with 50 % yield. C: The reaction was repeated at −40 °C in the same scale using 3×8 equiv of thiol 14 to give 36 with 56 % yield. D: The reaction was repeated at −80 °C in the same scale using 3×8 equiv of thiol 14 to give 36 with 63 % yield. R f=0.28 (CH2Cl2/acetone 9:1); m.p. 140–143 °C; [α]D 20=+101.6 (c=0.25 in CHCl3); 1H NMR (400 MHz, CDCl3): δ=6.07 (d, J=5.1 Hz, 1 H, NHAc), 5.76 (d, J=8.8 Hz, 1H H‐1), 5.15–5.03 (m, 1 H), 4.90–4.78 (m, 1 H), 4.60 (ddd, J=11.2, 8.7, 5.2 Hz, 1 H), 4.19 (dd, J=12.5, 4.1 Hz, 1 H, H‐6a), 3.99 (dd, J=12.5, 2.4 Hz, 1 H, H‐6b), 3.87 (ddd, J=10.1, 4.1, 2.4 Hz, 1 H, H‐5), 2.40 (s, 3 H, SAcCH 3), 2.01 (s, 3 H, OAcCH 3), 1.97 (s, 3 H, OAcCH 3), 1.97 (s, 3 H, OAcCH 3), 1.86 ppm (s, 3 H, NHCAcH 3). 13C NMR (100 MHz, CDCl3): δ=190.7 (1C, SCOCH3), 171.4, 170.6, 169.9, 169.1 (4×C, COCH3), 82.3 (1C, C‐1), 71.9, 71.8, 67.7 (3C, skeletal carbons), 61.7 (1C, C‐6), 51.6 (1C, C‐2), 31.6 (1C, SCOCH3), 23.0 (1C, NHCOCH3), 20.6, 20.6, 20.5 ppm (3C, OCOCH3). ESI‐MS: m/z calcd for C16H23NNaO9S [M+Na]+ 428.0991, found 428.0985.
2‐Acetamido‐2‐deoxy‐3,4,6‐tri‐O‐acetyl‐1‐thio‐α‐d‐glucopyranosyl‐(1→1)‐2,3,6‐tri‐O‐acetyl‐4‐O‐(2,3,4,6‐tetra‐O‐acetyl‐α‐d‐glucopyranosyl)‐β‐d‐glucopyranoside (37): A: Compound 3 (110 mg, 0.3 mmol) and thiol 18 (254 mg, 0.38 mmol, 1.3 equiv) were reacted in a mixture of toluene (2.0 mL) and DMF (1.0 mL) at −80 °C according to the general method. The crude product was purified by flash column chromatography (CH2Cl2/MeOH 95/5) to give 37 (98 mg, 33 %) as colorless syrup. B: The reaction was repeated at −40 °C in the same scale to give 37 with 65 % yield. R f=0.23 (CH2Cl2/MeOH 95:5), [α]D 20=+90.4 (c=0.23 in CHCl3), 1H NMR (400 MHz, CDCl3): δ=5.78 (d, J=8.2 Hz, 1 H, NH), 5.74 (d, J=5.3 Hz, 1 H, H‐1′′), 5.36 (d, J=3.9 Hz, 1 H, H‐1), 5.29 (t, J=10.0 Hz, 1 H), 5.19 (t, J=7.4 Hz, 1 H), 5.14 (t, J=8.1 Hz, 1 H), 5.00 (t, J=9.9 Hz, 1 H, ), 4.97–4.91 (m, 1 H), 4.89 (t, J=8.4 Hz, 1 H), 4.80 (dd, J=10.5, 4.0 Hz, 1 H), 4.59 (d, J=10.0 Hz, 1 H), 4.52 (dd, J=12.1, 1.5 Hz, 1 H), 4.42 (ddd, J=11.5, 8.2, 5.4 Hz, 1 H), 4.33 (dd, J=12.6, 2.4 Hz, 1 H), 4.21 (dd, J=12.8, 3.8 Hz, 1 H), 4.17 (d, J=10.2 Hz, 1 H), 4.06 (dd, J=12.2, 4.0 Hz, 2 H), 4.01(dd, J=5.7, 1.7 Hz, 1 H), 3.97 (d, J=5.6 Hz, 1 H), 3.95–3.87 (m, 2 H), 3.67 (d, J=9.6 Hz, 1 H), 2.10 (s, 3 H, AcCH 3), 2.06 (s, 3 H, AcCH 3), 2.05 (s, 3 H, AcCH 3), 1.97 (dd, J=9.6, 5.2 Hz, 21 H, 7x AcCH 3), 1.88 ppm (s, 3 H, AcCH 3). 13C NMR (100 MHz, CDCl3): δ=171.7, 170.6, 170.5, 170.3, 170.1, 169.9, 169.4, 169.1 (11C, 11×AcCO), 95.7 (1C, C‐1′′), 83.0, 81.7 (2C, C‐1, C‐1′), 76.6, 76.1, 72.1, 71.8, 71.1, 70.0, 69.3, 69.3, 68.5, 67.9, 67.4 (11C, skeletal carbons), 62.4, 61.4, 61.2 (3C, C‐6, C‐6′, C‐6′′), 52.5 (1C, C‐2), 23.0, 20.9, 20.7, 20.7, 20.6 ppm (11C, 11×AcCH3), ESI‐MS: m/z calcd for C40H55NNaO25S [M+Na]+ 1004.2682, found 1004.2680.
N ‐Acetyl‐S‐[4‐O‐(2,3,4,6‐tetra‐O‐acetyl‐α‐d‐glucopyranosyl)‐2,3,6‐tri‐O‐acetyl‐1‐thio‐α‐d‐glucopyranosyl]‐l‐cysteine (38): A: Compound 4 (309 mg, 0.5 mmol), and thiol 19 (160 mg, 1.0 mmol, 2.0 equiv) and DPAP (15 mg, 0.05 mmol, 0.1 equiv) were dissolved in a mixture of toluene (2.0 mL) and MeOH (1.0 mL). The reaction mixture was cooled to −20 °C and was irradiated with UV light (365 nm) for 15 minutes. The addition of DPAP and irradiation were repeated four times more. The solvent was evaporated under reduced pressure. The crude product was purified by flash chromatography (CH2Cl2/MeOH 95:5) to give 38 (292 mg, 75 %) as colorless crystals. B: The reaction was repeated at −40 °C in the same scale to give 38 with 79 % yield. C: The reaction was repeated at −80 °C in the same scale to give 38 with 56 % yield. R f=0.34 (CH2Cl2/MeOH 9:1); m.p. 141–144 °C; [α]D 20=+116.7 (c=0.27 in CHCl3); 1H NMR (400 MHz, DMSO): δ=7.85 (d, J=7.8 Hz, 1 H, NH), 5.54 (d, J=5.3 Hz, 1 H, H‐1′), 5.25 (d, J=2.7 Hz, 1 H, H‐1), 5.20 (t, J=10.2 Hz, 2 H), 5.00 (t, J=9.8 Hz, 1 H), 4.93–4.89 (m, 1 H, H‐2′), 4.87 (dd, J=10.7, 3.1 Hz, 1 H), 4.41 (d, J=11.2 Hz, 1 H), 4.27 (d, J=7.1 Hz, 2 H, H‐α), 4.22–4.13 (m, 2 H), 3.98 (dd, J=18.9, 10.2 Hz, 3 H), 3.03–2.93 (m, 1 H, H‐β), 2.87 (dd, J=12.7, 7.9 Hz, 1 H, H‐β), 2.08 (s, 3 H, AcCH 3), 2.02 (s, 3 H, AcCH 3), 2.01–1.97 (m, 12 H, 4×AcCH 3), 1.96 (s, 3 H, AcCH 3), 1.87 (s, 3 H, AcCH 3). 13C NMR (100 MHz, DMSO): δ=170.3, 170.1, 170.0, 169.6, 169.6, 169.5, 169.2, 169.0 (8C, 8×AcCO), 95.7 (1C, C‐1′), 82.1 (1C, C‐1), 73.7, 71.6, 70.1, 69.5, 68.9, 68.2, 68.1, 67.7 (8C, skeletal carbons), 62.7, 61.4 (2C, C‐6, C‐6′), 53.8 (1C, C‐α), 33.1 (1C, C‐β), 22.8 (1C, NAcCH3), 20.6, 20.5, 20.4, 20.3 (7C, 7×OAcCH3); MALDI‐TOF‐MS: m/z calcd for C31H43NnaO20S [M+Na]+ 804.200, found 804.139.
2,3,4,6‐Tetra‐O‐acetyl‐α‐d‐glucopyranosyl‐(1→4)‐2,3,6‐tri‐O‐acetyl‐1‐thio‐α‐d‐glucopyranosyl‐(1→1)‐2,3,4,6‐tetra‐O‐acetyl‐β‐d‐glucopyranoside (39): A: Compound 4 (309 mg, 0.5 mmol) was dissolved in toluene (2.0 mL) and thiol 20 (273 mg, 0.75 mmol, 1.5 equiv) and DPAP (15 mg, 0.05 mmol, 0.1 equiv) were added. The reaction mixture was cooled to 0 °C and was irradiated with UV light (365 nm) for 15 minutes. The addition of DPAP and irradiation were repeated four times more. The solvent was evaporated under reduced pressure. The crude product was purified by flash chromatography (hexane/acetone 7/3) to give 39 (252 mg, 51 %) as white foam. B: The reaction was repeated at −20 °C to give 39 with 72 % yield. C: The reaction was repeated at −40 °C to give 39 with 71 % yield. D: The reaction was repeated at −80 °C, to give 39 with 60 % yield. R f=0.24 (n‐hexane/acetone 6:4); [α]D 20=+116.7 (c=0.27 in CHCl3); 1H NMR (500 MHz, CDCl3): δ=5.81 (d, J=5.6 Hz, 1 H, H‐1′′), 5.40 (d, J=4.0 Hz, 1 H, H‐1), 5.35 (ddd, J=11.3, 10.2, 9.0 Hz, 2 H), 5.18 (t, J=9.3 Hz, 1 H), 5.08 (ddd, J=17.4, 10.0, 8.6 Hz, 3 H), 4.89–4.82 (m, 2 H), 4.58 (d, J=10.1 Hz, 1 H), 4.47 (dd, J=12.4, 2.6 Hz, 1 H), 4.33 (dt, J=9.7, 2.6 Hz, 1 H), 4.26 (ddd, J=18.7, 12.4, 2.9 Hz, 2 H), 4.19 (t, J=3.1 Hz, 2 H), 4.06–3.99 (m, 2 H), 3.93 (dt, J=10.3, 2.8 Hz, 1 H), 3.74 (ddd, J=10.0, 3.9, 2.6 Hz, 1 H), 2.15, 2.11, 2.09, 2.06, 2.03, 2.02, 2.02, 2.00, 2.00 ppm (9xs, 33 H, 11×AcCH 3). 13C NMR (126 MHz, CDCl3): δ=170.7, 170.7, 170.6, 170.5, 170.2, 169.9, 169.8, 169.6, 169.5, 169.4, 169.1 (11C, AcCO), 95.7 (1C, C‐1), 82.8, 81.8 (2×C, C‐1′, C‐1′′), 76.4, 73.9, 72.4, 72.3, 71.1, 70.1, 69.4, 69.1, 68.5, 68.0, 67.8 (12C, skeletal carbons), 62.3, 61.9, 61.3 (3C, C‐6, C‐6′, C‐6′′), 21.0, 20.8, 20.7 ppm (11C, 11×AcCH3). MALDI‐TOF‐MS: m/z calcd for C40H54NaO26S [M+Na]+ 1005.252, found 1005.220.
2,3,4,6‐Tetra‐O‐acetyl‐α‐d‐glucopyranosyl‐(1→4)‐2,3,6‐tri‐O‐acetyl‐1‐hio‐α‐d‐glucopyranosyl‐(1→1)‐2,3,4,6‐tetra‐O‐acetyl‐β‐d‐mannopyranoside (40): A: Compound 4 (163 mg, 0.25 mmol) and thiol 21 (124 mg, 0.375 mmol, 1.5 equiv) were reacted in a mixture of toluene (2.0 mL) and N‐N‐dimethylformamide (0.5 mL) at −40 °C according to the general method. The crude product was purified by flash chromatography (hexane/acetone 7/3) to give 40 (66 mg, 27 %) as white powder. B: The reaction was repeated at −80 °C to give 40 with 58 % yield. R f=0.26 (n‐hexane/acetone 6:4); m.p. 96–99 °C, [α]D 20=+95.0 (c=0.32 in CHCl3); 1H NMR (400 MHz, CDCl3) δ=5.76 (d, J=5.5 Hz, 1 H), 5.55 (d, J=3.2 Hz, 1 H), 5.44–5.40 (m, 2 H), 5.36 (t, J=10.0 Hz, 1 H), 5.25 (t, J=10.1 Hz, 1 H), 5.10 (d, J=9.8 Hz, 1 H), 5.05 (dd, J=10.5, 3.7 Hz, 1 H), 4.95 (dd, J=10.0, 5.5 Hz, 1 H), 4.85 (dd, J=10.4, 3.8 Hz, 2 H), 4.48 (t, J=12.7 Hz, 2 H), 4.30–4.16 (m, 5 H), 4.09–3.99 (m, 2 H), 3.92 (d, J=10.1 Hz, 1 H), 3.78–3.69 (m, 1 H), 2.24 (s, 3 H, AcCH 3), 2.15 (s, 3 H, AcCH 3), 2.13 (s, 3 H, AcCH 3), 2.10 (s, 3 H, AcCH 3), 2.07 (s, 3 H, AcCH 3), 2.06 (s, 3 H, AcCH 3), 2.03 (s, 9 H, 3xAcCH 3), 2.01 (s, 3 H, AcCH 3), 1.98 ppm (s, 3 H, AcCH 3). 13C NMR (100 MHz, CDCl3): δ=170.7, 170.7, 170.5, 170.4, 170.2, 170.1, 169.8, 169.7, 169.6, 169.4 (11C, 11×AcCO), 95.6 (1C, C‐1′′), 82.2, 82.0 (2C, C‐1, C‐1′), 77.0, 72.3, 72.1, 71.8, 71.2, 70.8, 70.1, 69.2, 68.9, 68.4, 67.8, 65.0 (12C, skeletal carbons), 62.6, 62.1, 61.2 (3C, C‐6, C‐6′, C‐6′′), 20.9, 20.8, 20.8, 20.7, 20.7, 20.7, 20.6, 20.6 ppm (11C, 11×AcCH3); MALDI‐TOF‐MS: m/z calcd for C40H54NaO26S [M+Na]+ 1005.252, found 1005.303.
2,3,4,6‐Tetra‐O‐acetyl‐α‐d‐glucopyranosyl‐(1→4)‐2,3,6‐tri‐O‐acetyl‐1‐hio‐α‐d‐glucopyranosyl‐(1→1)‐2,3,4,6‐tetra‐O‐acetyl‐α‐d‐mannopyranoside (41): Compound 4 (309 mg, 0.5 mmol) and thiol 22 (273 mg, 0.75 mmol, 1.5 mmol) were reacted in toluene (2.0 mL) at −80 °C according to the general method. The crude product was purified by flash chromatography (hexane/acetone 7/3) to give 41 (481 mg, 98 %) as white foam. R f=0.25 (n‐hexane/acetone 6:4), [α]D 20=+187,1 (c=0.21 in CHCl3), 1H NMR (400 MHz, CDCl3): δ=5.70 (d, J=5.6 Hz, 1 H, H‐1′′), 5.46–5.24 (m, 7 H), 5.07 (t, J=9.8 Hz, 1 H), 4.99 (dd, J=9.3, 5.7 Hz, 1 H, H‐2′′), 4.88 (dd, J=10.4, 3.8 Hz, 1 H), 4.47 (d, J=11.9 Hz, 1 H), 4.35 (d, J=9.5 Hz, 1 H), 4.30–4.19 (m, 4 H), 4.01 (ddd, J=28.9, 21.1, 9.8 Hz, 4 H), 2.18 (s, 4 H), 2.12 (s, 3 H, AcCH 3), 2.10 (s, 9 H, 3x AcCH 3), 2.09 (s, 3 H, AcCH 3), 2.07 (s, 3 H, AcCH 3), 2.05 (s, 3 H, AcCH 3), 2.04 (s, 3 H, AcCH 3), 2.03 (s, 3 H, AcCH 3), 2.01 (s, 3 H, AcCH 3). 13C NMR (100 MHz, CDCl3): δ=170.7, 170.6, 170.5, 169.8, 169.7, 169.6 (11C, 11×AcCO), 95.9 (1C, C‐1′′), 79.7, 79.0 (2C, C‐1, C‐1′), 73.2, 72.3, 71.2, 70.3, 70.1, 70.0, 69.4, 69.3, 68.6, 68.0, 66.2 (11C, skeletal carbons), 62.7, 62.4, 61.6 (3C, C‐6, C‐6′, C‐6′′), 20.9, 20.7, 20.7 (11C, 11×AcCH3), MALDI‐TOF‐MS: m/z calcd for C40H54NaO26S [M+Na]+ 1005.252, found 1005.187.
2,3,4,6‐Tetra‐O‐acetyl‐α‐d‐glucopyranosyl‐(1→4)‐2,3,6‐tri‐O‐acetyl‐1‐thio‐α‐d‐glucopyranosyl‐(1→1)‐2,3,6‐tri‐O‐acetyl‐4‐O‐(2,3,4,6‐tetra‐O‐acetyl‐α‐d‐glucopyranosyl)‐β‐d‐glucopyranoside (42): A: Compound 4 (309 mg, 0.5 mmol) and thiol 18 (489 mg, 0.75 mmol, 1.5 equiv) were reacted in a mixture of toluene (2.0 mL) and N,N‐dimethylformamide (1.0 mL) at −20 °C. The crude product was purified by flash chromatography (CH2Cl2/acetone 9/1) to give 42 (158 mg, 25 %) as colorless syrup. B: The reaction was repeated at −40 °C in the same scale to give 42 with 35 % yield. C: The reaction was repeated at −80 °C in the same scale to give 42 with 20 % yield. D: The reaction was repeated at 0 °C in the same scale with 3 equiv of thiol 18 to give 42 with 29 % yield. R f=0.34 (n‐hexane/acetone 6:4), [α]D 20=+144.1 (c=0.19 in CHCl3), 1H NMR (400 MHz, CDCl3): δ=5.80 (d, J=5.6 Hz, 1 H), 5.48–5.28 (m, 6 H), 5.24 (t, J=9.0 Hz, 1 H), 5.12–5.03 (m, 2 H), 4.96–4.80 (m, 4 H), 4.64–4.58 (m, 2 H), 4.49 (d, J=10.6 Hz, 1 H), 4.33–4.21 (m, 5 H), 4.17–4.12 (m, 1 H), 4.10–3.92 (m, 7 H), 3.73 (d, J=9.5 Hz, 1 H), 2.17 (s, 9 H), 2.11 (s, 7 H), 2.07 (s, 3 H), 2.06–1.99 ppm (m, 30 H). 13C NMR (100 MHz, CDCl3): δ=170.8, 170.7, 170.5, 170.3, 170.0, 169.6, 169.5 (14C, 14×AcCO), 95.7 (2C, C‐1′′, C‐1′′′), 82.1 (2C, C‐1, C‐1′), 76.8, 76.3, 72.4, 72.2, 71.0, 70.1, 69.4, 69.2, 68.6, 68.5, 68.0, 68.0 (16C, skeletal carbons), 62.6, 62.3, 61.5, 61.3 (4C, C‐6, C‐6′, C‐6′′, C‐6′′), 21.0, 21.0, 20.9, 20.8, 20.7, 20.6 ppm (14C, 14×AcCH3). MALDI‐TOF‐MS: m/z calcd for C52H70NaO34S [M+Na]+ 1293.337, found 1293.327.
2,3,4‐Tri‐O‐acetyl‐β‐d‐lyxopyranosyl‐(1→1)‐2,3,4,6‐tetra‐O‐acetyl‐1‐thio‐β‐d‐glucopyranoside (43) and 2,3,4‐Tri‐O‐acetyl‐α‐d‐xylopyranosyl‐(1→1)‐2,3,4,6‐tetra‐O‐acetyl‐1‐thio‐β‐d‐glucopyranoside (44): A: Compound 5 (100 mg, 0.387 mmol) and thiol 20 (169 mg, 0.465 mmol, 1.2 equiv) were reacted in toluene (2.7 mL) at −80 °C according to the general method. The crude product was purified by flash chromatography (n‐hexane/acetone 7:3) to give 43 (116 mg, 48 %) as white crystals and 44 (6 mg, 2 %) as colorless syrup. B: The reaction was repeated at 0 °C. The conversion was less than 5 %, only disulfide byproduct could be isolated. C: The reaction was repeated at −40 °C to give a mixture of 43 and 44 with a 2.3:1 ratio with 21 % overall yield.
Data of 43: R f=0.24 (n‐hexane/acetone 7:3); m.p. 74–77 °C, [α]D 20=−97,7 (c=0.27 in CHCl3), 1H NMR (500 MHz, CDCl3): δ=5.46 (t, J=3.5 Hz, 1 H, H‐2), 5.34 (d, J=3.5 Hz, 1 H, H‐1), 5.23 (t, J=9.4 Hz, 1 H, H‐3′), 5.17 (dd, J=7.0, 3.3 Hz, 1 H, H‐3), 5.09 (t, J=9.7 Hz, 1 H, H‐4′), 5.04–4.99 (m, 2 H, H‐4, H‐2′), 4.76 (d, J=10.2 Hz, 1 H, H‐1′), 4.29–4.24 (m, 2 H, H‐6′a, H‐5a), 4.12 (dd, J=12.4, 2.1 Hz, 1 H, H‐6′b), 3.72 (ddd, J=10.0, 4.9, 2.2 Hz, 1 H, H‐5′), 3.52 (dd, J=12.4, 5.6 Hz, 1 H, H‐5b), 2.11 (s, 3 H, COCH3), 2.10 (s, 6 H, 2×COCH3), 2.06 (s, 3 H, COCH3), 2.06 (s, 3 H, COCH3), 2.03 (s, 3 H, COCH3), 2.01 ppm (s, 3 H, COCH3); 13C NMR (126 MHz, CDCl3): δ=170.6, 170.1, 169.8, 169.7, 169.5, 169.4 and 169.3 (6C, 6×COCH3), 81.4 (C‐1), 79.8 (C‐1′), 75.9 (C‐5′), 73.8 (C‐3′), 70.2 (C‐2′), 68.7 (C‐3), 68.2 (C‐4′), 68.0 (C‐2), 67.5 (C‐4), 62.8 (C‐5)*, 62.0 (C‐6′), 20.8, 20.7, 20.6, 20.6 and 20.6 ppm (6C, 6×COCH3). *Note: Peak can only be seen on HSQC spectrum. MALDI‐TOF‐MS: m/z calcd for C25H34NaO16S [M+Na]+ 645.147, found 645.146
Data of 44: R f=0.25 (n‐hexane/acetone 7:3). m.p. 176–179 °C, [α]D 20=+70.5 (c=0.20 in CHCl3), 1H NMR (400 MHz, CDCl3): δ=5.81 (d, J=5.5 Hz, 1 H, H‐1), 5.30 (t, J=9.6 Hz, 1 H), 5.17 (t, J=9.3 Hz, 1 H), 5.12–5.00 (m, 2 H), 4.94 (tt, J=8.8, 4.5 Hz, 2 H), 4.54 (d, J=10.1 Hz, 1 H, H‐1′), 4.15 (qd, J=12.4, 3.7 Hz, 2 H), 3.99 (dd, J=11.6, 10.4 Hz, 1 H), 3.80 (dd, J=11.6, 5.7 Hz, 1 H), 3.72 (ddd, J=10.0, 4.9, 2.4 Hz, 1 H), 2.08 (s, 3 H, AcCH 3), 2.05–2.02 (m, 15 H, 5×AcCH 3), 2.00 ppm (s, 3 H, AcCH3) 13C NMR (100 MHz, CDCl3): δ=170.7, 170.3, 170.0, 169.8, 169.8, 169.4, 169.2 (7×C, AcCO), 82.8, 82.3 (2C, C‐1, C‐1′), 76.2, 74.0, 71.0, 70.9, 69.6, 69.1, 68.1(7C, skeletal carbons), 62.1 (1C, C‐5), 60.0 (1C, C‐6′), 20.8, 20.7 ppm (7C, 7× AcCH3). MALDI‐TOF‐MS: m/z calcd for C37H42NaO5S [M+Na]+ 645.147, found 645.128.
2,3,4‐Tri‐O‐acetyl‐β‐d‐lyxopyranosyl‐(1→1)‐2,3,4,6‐tetra‐O‐acetyl‐1‐thio‐α‐d‐mannopyranoside (45) and 2,3,4‐tri‐O‐acetyl‐α‐d‐xylopyranosyl‐(1→1)‐2,3,4,6‐tetra‐O‐acetyl‐1‐thio‐α‐d‐mannopyranoside (46): A: Compound 5 (100 mg, 0.387 mmol) and thiol 22 (169 mg, 0.465 mmol, 1.2 equiv) were reacted in toluene (2.7 mL), at −80 °C according to the general method. The crude product was purified by column chromatography to give 45 (214 mg, 89 %) as white crystals and an inseparable ≈2:1 mixture of 45 and 46 (12 mg, 6 %). B: The reaction was repeated at 0 °C in the same scale to give an inseparable mixture of 45 and 46 in a ratio of 1:2.5 with 40 % overall yield.
Data of 45: R f=0.26 (n‐hexane/acetone 7:3); m.p. 160–162 °C, [α]D 20=−10.0 (c=0.40 in CHCl3), 1H NMR (500 MHz, CDCl3): δ=5.46 (t, J=3.4 Hz, 1 H, H‐2), 5.39 (s, 1 H, H‐1′), 5.35–5.31 (m, 2 H) and 5.29 (dd, J=10.0, 3.1 Hz, 1 H) (H‐4, H‐3′, H‐2′), 5.19 (d, J=3.2 Hz, 1 H, H‐1), 5.16 (dd, J=7.1, 3.3 Hz, 1 H, H‐3), 5.01 (ddd, J=7.0, 5.9, 3.7 Hz, 1 H, H‐4), 4.35–4.27 (m, 2 H, H‐6′a, H‐5′), 4.23 (dd, J=12.4, 3.6 Hz, 1 H, H‐5a), 4.06 (dd, J=11.9, 1.9 Hz, 1 H, H‐6′b), 3.51 (dd, J=12.4, 5.9 Hz, 1 H, H‐6′b), 2.17 (s, 3 H, COCH3), 2.14 (s, 3 H, COCH3), 2.10 (s, 3 H, COCH3), 2.09 (s, 3 H, COCH3), 2.06 (s, 3 H, COCH3), 2.00 ppm (s, 3 H, COCH3); 13C NMR (126 MHz, CDCl3): δ=170.7, 170.0, 169.9, 169.7, 169.7 (6C, 6×COCH3), 83.6, 83.2 (2C, C‐1, C‐1′), 70.6, 69.8, 69.4, 68.8, 68.7, 67.4, 66.2 (7C, skeleton carbons), 62.1, 60.5 (2C, C‐5, C‐6′), 21.0, 20.9, 20.8, 20.8, 20.7 ppm (6C, 6×COCH3). MALDI‐TOF‐MS: m/z calcd for C25H34NaO16S [M+Na]+ 645.147, found 645.147
Data of 46: NMR data are given from the 1:2.5 mixture of 45 and 46.
1H NMR (400 MHz, CDCl3): δ=5.71 (d, J=5.3 Hz, 1 H, H‐1), 5.37 (s, 1 H, H‐1′), 5.35 (dd, J=3.1, 1.5 Hz, 1 H), 5.34–5.31 (m, 1 H), 5.29 (s, 1 H), 5.28–5.25 (m, 2 H), 5.04–4.98 (m, 2 H), 4.33 (dt, J=13.5, 3.4 Hz, 1 H), 4.29–4.26 (m, 1 H), 4.24 (s, 1 H), 4.23–4.18 (m, 1 H), 4.08 (dd, J=14.2, 10.3 Hz, 2 H), 4.02–3.94 (m, 1 H), 3.89 (dd, J=11.5, 5.7 Hz, 1 H), 2.17 (s, 3 H, COCH 3), 2.11 (s, 3 H, COCH 3), 2.10 (s, 3 H, COCH 3), 2.07 (s, 3 H, COCH 3), 2.06 (s, 3 H, COCH 3), 2.05 (s, 3 H, COCH 3), 2.01 ppm (s, 3 H, COCH 3). 13C NMR (100 MHz, CDCl3): δ=169.9, 169.9, 169.8, 169.6, 169.6 (7C, 7×COCH3), 80.2, 79.0 (2C, C‐1, C‐1′), 70.8, 70.1, 69.8, 69.4, 69.3, 68.6, 66.1 (7C, skeletal carbons), 62.2, 60.1 (2C, C‐5, C‐6′), 20.7, 20.7, 20.6, 20.6 ppm (7C, 7×COCH3).
2,3,4‐Tri‐O‐acetyl‐1‐thio‐α‐d‐ribopyranosyl‐(1→1)‐2,3,4,6‐tetra‐O‐acetyl‐β‐d‐glucopyranoside (47) and 2,3,4‐tri‐O‐acetyl‐1‐thio‐β‐d‐arabinopyranosyl‐(1→1)‐2,3,4,6‐tetra‐O‐acetyl‐β‐d‐glucopyranoside (48): A: Compound 6 (258 mg, 1.0 mmol) and thiol 20 (437 mg 1.2 mmol, 1.2 equiv) in toluene (4.0 mL) were reacted at −80 °C according to the general method. The crude product was purified by flash column chromatography (gradient elution n‐hexane/acetone 75/25→6/4) to give compounds 47 (284 mg, 46 %) and 48 (128 mg, 21 %) both as white syrup. B: The reaction was repeated at −100 °C to give 47 and 48 with 60 % overall yield. The 47:48 ratio was 1.4:1. C: The reaction was repeated at room temperature using 1.5 equiv of thiol, but no product could be isolated.
Data of 47: R f=0.39 (n‐hexane/acetone 6:4), [α]D 20=+52.45 (c=0.10 in CHCl3), 1H NMR (400 MHz, CDCl3): δ=5.60 (d, J=5.1 Hz, 1 H, H‐1), 5.51 (t, J=2.7 Hz, 1 H, H‐3), 5.20–5.14 (m, 1 H, H‐2′), 5.13–5.07 (m„ 3 H, H‐4′, H‐3′, H‐2), 4.99 (ddd, J=9.6, 4.8, 3.1 Hz, 1 H, H‐4), 4.50 (d, J=9.7 Hz, 1 H, H‐1′), 4.22 (dd, J=12.4, 4.9 Hz, 1 H, H‐6′a), 4.14 (dd, J=12.4, 2.4 Hz, 1 H, H‐6′b), 4.08 (dd, J=11.4, 9.8 Hz, 1 H, H‐5a), 3.75 (ddd, J=9.8, 4.8, 2.4 Hz, 1 H, H‐5′), 3.63 (dd, J=11.5, 4.7 Hz, 1 H, H‐5b), 2.15 (s, 3 H, COCH3), 2.09 (s, 3 H, COCH3), 2.05 (s, 6 H, 2×COCH3), 2.03 (s, 3 H, COCH3), 2.03 (s, 3 H, COCH3), 2.00 ppm (s, 3 H, COCH3). 13C NMR (100 MHz, CDCl3): δ=170.7 (COCH3), 170.3 (COCH3), 169.9 (COCH3), 169.6 (COCH3), 169.4 (COCH3), 169.4 (COCH3), 169.0 (COCH3), 83.1 (C‐1′), 80.3 (C‐1), 76.2 (C‐5′), 74.2, 71.1, 68.1, 67.8, 67.5 (5C, C‐4′, C‐3′, C‐3, C‐2′, C‐2), 65.8 (C‐4), 62.1 (C‐6 and C‐6′), 58.2 (C‐5)* 20.9, 20.8, 20.7, 20.6 and 20.6 ppm (7C, 7×COCH3). *Note: Signal can only be seen on HSQC spectrum. MALDI‐TOF‐MS: m/z calcd for C25H34NaO16S+ [M+Na]+ 645.15, found 645.16.
Data of 48: R f=0.47 (n‐hexane/acetone 6:4), [α]D 20=−161.2 (c=0.46 in CHCl3), 1H NMR (400 MHz, CDCl3): δ=5.86 (d, J=5.3 Hz, 1 H, H‐1), 5.38 (dd, J=10.4, 5.4 Hz, 1 H, H‐2), 5.34 (s (broad), 1 H, H‐4), 5.22 (t, J=9.3 Hz, 1 H, H‐3′), 5.14–5.03 (m, 3 H, H‐4′, H‐3, H‐2′), 4.69 (d, J=10.3 Hz, 1 H, H‐1′), 4.28 (dd, J=12.4, 5.2 Hz, 1 H, H‐6′a), 4.16 (d, J=12.8 Hz, 1 H, H‐5a), 4.07 (dd, J=12.3, 1.8 Hz, 1 H, H‐6′b), 3.75 (dd, J=13.1, 2.3 Hz, 1 H, H‐5b), 3.68 (ddd, J=9.7, 5.0, 1.9 Hz, 1 H, H‐5′), 2.15 (s, 3 H, COCH3), 2.10 (s, 3 H, COCH3), 2.08 (s, 3 H, COCH3), 2.07 (s, 3 H, COCH3), 2.02 (s, 6 H, 2xCOCH3), 2.01 ppm (s, 3 H, COCH3). 13C NMR (100 MHz, CDCl3): δ=170.7, 170.2, 170.2, 169.9, 169.4, 169.3 (7C, 7xCOCH3), 80.5, 80.4 (2C, C‐1′, C‐1), 76.1 (C‐5′), 73.9, 70.1, 68.3, 68.3, 67.7, 67.4 (6C, C‐4′, C‐4, C‐3′, C‐3, C‐2′, C‐2), 62.0 (C‐6′), 61.4 (C‐5), 20.9, 20.8, 20.8, 20.7, 20.6 ppm (7C, 7×COCH3), MALDI‐TOF‐MS: m/z calcd for C25H34NaO16S [M+Na]+ 645.15, found 645.17.
2,3,4‐Tri‐O‐acetyl‐1‐thio‐α‐d‐ribopyranosyl‐(1→1)‐2,3,4,6‐tetra‐O‐acetyl‐α‐d‐mannopyranoside (49) and 2,3,4‐tri‐O‐acetyl‐1‐thio‐β‐d‐arabinopyranosyl‐(1→1)‐2,3,4,6‐tetra‐O‐acetyl‐α‐d‐mannopyranoside (50): A: Compound 6 (100 mg, 0.387 mmol) and thiol 22 (170 mg, 0.464 mmol, 1.2 equiv) were reacted in toluene (2.0 mL) at −80 °C according to the general method. The crude product was purified by flash column chromatography (n‐hexane/acetone 7/3) to give a 1:1.3 mixture of 49 and 50 (221 mg, 91 %). A second purification by flash column chromatography (gradient elution CH2Cl2/acetone 100/1→100/2) gave 50 (82 mg, 34 %) and a 2.4:1 mixture of 49:50 (67 mg, 28 %), both as colorless syrup B: The reaction was repeated at 0 °C in the same scale to give 49 and 50 with 54 % overall yield, the 49:50 ratio was 1:1.4.
Data of 49 (NMR data are given from the 2.4:1 mixture of 49:50): R f=0.28 (n‐hexane/acetone 7:3); 1H NMR (400 MHz, CDCl3): δ=5.50 (s, 1 H), 5.42–5.38 (m, 2 H), 5.34–5.28 (m, 3 H), 5.21–5.16 (m, 1 H), 5.03 (dt, J=7.7, 3.9 Hz, 1 H), 4.33–4.21 (m, 3 H), 4.19–4.05 (m, 3 H), 3.68 (dd, J=11.5, 4.3 Hz, 1 H), 2.18 (s, 6 H, 2xAcCH 3), 2.11 (s, 3 H, AcCH 3), 2.11 (s, 3 H, AcCH 3), 2.06 (s, 3 H, AcCH 3), 2.06 (s, 3 H, AcCH 3), 2.01 ppm (s, 3 H, AcCH 3);13C NMR (100 MHz, CDCl3): δ=170.0, 169.9, 169.7, 169.7, 169.5 (7C, 7×AcCO), 79.9, 79.1, (2C, C‐1 & C‐1′)70.6, 69.5, 69.4, 67.5, 67.4, 66.3, 65.9 (7C, skeletal carbons), 62.3 (1C, C‐6′), 21.0, 20.9, 20.7, 20.7 (7C, 7×AcCH3).
Data of 50: R f=0.28 (n‐hexane/acetone 7:3); [α]D 20=−46.9 (c=0.29 in CHCl3); 1H NMR (400 MHz, CDCl3): δ=5.80 (d, J=4.9 Hz, 1 H, H‐1), 5.38–5.35 (m, 1 H, H‐4′), 5.34–5.29 (m, 3 H, H‐1′, H‐2, H‐4), 5.28–5.24 (m, 1 H, H‐3′), 5.19 (dd, J=10.2, 3.3 Hz, 1 H, H‐3), 4.32–4.21 (m, 2 H, H‐6′a and H‐2′), 4.13 (d, J=13.1 Hz, 1 H, H‐5a), 4.08 (d, J=10.4 Hz, 1 H, H‐6′b), 3.76 (dd, J=13.2, 2.6 Hz, 1 H, H‐5b), 2.17 (s, 3 H, AcCH 3), 2.14 (s, 3 H, AcCH 3), 2.12 (s, 3 H, AcCH 3), 2.08 (s, 3 H, AcCH 3), 2.07 (s, 3 H, AcCH 3), 2.04 (s, 3 H, AcCH 3), 2.01 ppm (s, 3 H, AcCH 3). 13C NMR (100 MHz, CDCl3): δ=170.5, 170.2, 170.0, 169.8, 169.7 (7C, 7xAcCO), 84.6, 82.7 (2C, C‐1 and C‐1′), 71.4 (1C, C‐4′), 70.4 (1C, C‐2′), 69.3 (1C, C‐3′), 67.5 (1C, C‐3), 68.2, 68.1, 66.0 (3C, C‐2, C‐4, C‐5′)62.4, 62.2 (2C, C‐5 and C‐6), 20.9, 20.8, 20.7, 20.7, 20.7 ppm (7C, 7×AcCH3). MALDI‐TOF‐MS: m/z calcd for C25H34NaO16S [M+Na]+ 645.146, found 645.149.
2,3,4‐Tri‐O‐acetyl‐α‐l‐ribopyranosyl‐(1→1)‐2,3,4,6‐tetra‐O‐acetyl‐1‐thio‐β‐d‐glucopyranoside (51) and 2,3,4‐tri‐O‐acetyl‐β‐l‐arabinopyranosyl‐(1→1)‐2,3,4,6‐tetra‐O‐acetyl‐1‐thio‐β‐d‐glucopyranoside (52): A: Compound 7 (100 mg, 0.387 mmol) and thiol 20 (169 mg, 0.465 mmol, 1.20 equiv) were reacted in toluene (2.7 mL) at −80 °C according to the general method. The crude product was purified by flash chromatography to give 51 (225 mg, 93 %) and a mixture of 51 and 52 (11 mg, 5 %, 51:52=1:0.41) both as colorless syrup. B: The reaction was repeated at 0 °C to give 51 and 52 with 90 % overall yield, the 51:52 ratio was ≈6:1.
Data of 51: R f=0.16 (n‐hexane/acetone 7:3); [α]D 20=−48,65 (c=0.37 in CHCl3); 1H NMR (500 MHz, CDCl3): δ=5.50 (t, J=2.6 Hz, 1 H, H‐3), 5.48 (d, J=4.8 Hz, 1 H, H‐1), 5.22 (t, J=9.4 Hz, 1 H, H‐3′), 5.16 (dd, J=5.0, 2.9 Hz, 1 H, H‐2), 5.08 (t, J=9.8 Hz, 1 H, H‐4′), 5.05–5.00 (m, 2 H, H‐4, H‐2′), 4.68 (d, J=10.2 Hz, 1 H, H‐1′), 4.27 (dd, J=12.4, 5.3 Hz, 1 H, H‐6′a), 4.10–4.05 (m, J=9.2 Hz, 2 H, H‐6′b, H‐5a), 3.72–3.67 (m, 1 H, H‐5′), 3.66 (dd, J=11.5, 4.7 Hz, 1 H, H‐5b), 2.13 (s, 3 H, COCH3), 2.09 (s, 3 H, COCH3), 2.07 (s, 3 H, COCH3), 2.07 (s, 3 H, COCH3), 2.05 (s, 3 H, COCH3), 2.03 (s, 3 H, COCH3), 2.01 ppm (s, 3 H, COCH3); 13C NMR (126 MHz, CDCl3): δ=170.8, 170.3, 169.9, 169.8, 169.6, 169.6, 169.4 (7C, 7×COCH3), 82.1, 79.8 (2C, C‐1, C‐1′), 76.1, 74.0, 70.4, 68.4, 67.7, 67.6, 65.9 (7C, skeletal carbons), 62.2 (C‐6′), 21.0, 20.8, 20.8, 20.7 ppm (7C, 7×COCH3). MALDI‐TOF‐MS: m/z calcd for C25H34NaO16S [M+Na]+ 645.147, found 645.144.
2,3,4,6‐Tetra‐O‐acetyl‐α‐d‐mannopyranosyl‐(1→1)2,3,4‐tri‐O‐acetyl‐α‐l‐ribopyranoside (53) and 2,3,4,6‐Tetra‐O‐acetyl‐α‐d‐mannopyranosyl‐(1→1)‐2,3,4‐tri‐O‐acetyl‐β‐l‐arabinopyranoside (54): A: Compound 7 (200 mg, 0.75 mmol) and thiol 22 (336 mg, 0.93 mmol, 1.2 equiv) were reacted in toluene (1.0 mL) at −80 °C. The crude product was purified by flash column chromatography (gradient elution n‐hexane/EtOAc 65/35→6/4) to give 53 (150 mg, 32 %) and 54 (298 mg, 64 %) both as a colorless syrup. B: The reaction was repeated at 0 °C to give 53 and 54 with 92 % overall yield, the 53:54 ratio was 1:6.3.
Data of 53: R f=0.24 (n‐hexane/EtOAc 1:1), [α]D 20=−8,0 (c=0.15 in CHCl3), 1H NMR (400 MHz, CDCl3): δ=5.50 (t, J=3.1 Hz, 1 H, H‐3), 5.38 (d, J=5.3, 1 H, H‐1), 5.36‐.28 (m, Hz, 4 H, H‐1′, H‐2′, H‐3′, H‐4′), 5.15 (dd, J=5.0, 3.1 Hz, 1 H, H‐2), 5.01 (ddd, J=8.8, 4.4, 3.1 Hz, 1 H, H‐4), 4.35 (m, 1 H, H‐5′), 4.27 (dd, J=12.2, 5.3 Hz, 1 H, H‐6′a), 4.13–4.05 (m, 2 H, H‐6′b, H‐5a), 3.69 (dd, J=11.6, 4.4 Hz, 1 H, H‐5b), 2.17 (s, 6 H, 2xAcCH 3), 2.11 (s, 3 H, AcCH 3), 2.09 (s, 3 H, AcCH 3), 2.07 (s, 3 H, AcCH 3), 2.05 (s, 3 H, AcCH 3), 2.01 ppm (s, 3 H, AcCH 3). 13C NMR (100 MHz, CDCl3): δ=170.6, 169.9, 169.8, 169.7, 169.6, 169.6, 169.5 (7C, 7xAcCO), 83.8 (1C, C‐1′), 83.3 (1C, C‐1), 70.4, 69.5, 69.3, 66.2 ppm (4C, C‐5′, C2′, C‐3′, C‐4′), MALDI‐TOF‐MS: m/z calcd for C25H34NaO16S [M+Na]+ 645.147, found 645.142.
Data of 54: R f=0.36 (n‐hexane/EtOAc 1:1), [α]D 20=+186,5 (c=0.70 in CHCl3), 1H NMR (400 MHz, CDCl3): δ=5.81 (d, J=5.3 Hz, 1 H, H‐1), 5.37 (d, J=1.5 Hz, 1 H), 5.36–5.33 (m, 2 H), 5.33 (s, 1 H), 5.29–5.25 (m, 1 H), 5.16 (dd, J=10.4, 3.4 Hz, 1 H), 4.30–4.19 (m, 3 H), 4.16–4.08 (m, 2 H), 3.77 (dd, J=13.2, 2.6 Hz, 1 H), 2.17 (s, 3 H, AcCH 3), 2.14 (s, 3 H, AcCH 3), 2.13 (s, 3 H, AcCH 3), 2.11 (s, 3 H, AcCH 3), 2.07 (s, 3 H, AcCH 3), 2.03 (s, 3 H, AcCH 3), 2.01 ppm (s, 3 H, AcCH 3). 13C NMR (100 MHz, CDCl3): δ=170.8, 170.2, 170.0, 169.9, 169.9, 169.8, 169.7 (7C, 7xAcCO), 81.1, 79.2 (2C, C‐1, C‐1′), 71.0, 70.0, 69.4, 68.4, 67.8, 67.6, 66.2 (7C, skeletal carbons), 62.4, 61.8 (2C, C‐6, C‐5′), 21.0, 20.8, 20.8, 20.7 ppm (7C, 7×AcCH3), MALDI‐TOF‐MS: m/z calcd for C25H34NaO16S [M+Na]+ 645.146, found 645.149.
3,4,6‐Tri‐O‐acetyl‐1,5‐anhydro‐2‐thio‐2‐S‐(2,3,4,6‐tetra‐O‐acetyl‐β‐d‐glucopyranosyl)‐d‐mannitol (55) and 3,4,6‐tri‐O‐acetyl‐1,5‐anhydro‐2‐thio‐2‐S‐(2,3,4,6‐tetra‐O‐acetyl‐β‐d‐glucopyranosyl)‐d‐glucitol (56): A: Compound 8 (136 mg, 0.5 mmol) and thiol 20 (218 mg, 0.6 mmol, 1.2 equiv) were reacted in toluene (5.0 mL), at −80 °C according to the general method. The crude product was purified by column chromatography (n‐hexane/acetone 7:3) to give compound 55 (216 mg, 68 %) and 56 (26 mg, 8 %). B: The reaction was carried out at −120 °C in the same scale using 1.2 equiv of thiol 20 to give 55 and 56 in 75 % overall yield with a ratio of 55:56=6:1. 55 [α]D 20=−68.4 (c=0.13 in CHCl3) lit.23 −72.0; 56 [α]D 20=−6.89 (c=0.12 in CHCl3) lit.23 −18.8.
NMR data of 55 and 56 are identical to the ones described in literature.23
3,4,6‐Tri‐O‐acetyl‐1,5‐anhydro‐2‐thio‐2‐S‐(2,3,4,6‐tetra‐O‐acetyl‐β‐d‐glucopyranosyl)‐d‐talitol (57) and 3,4,6‐tri‐O‐acetyl‐1,5‐anhydro‐2‐thio‐2‐S‐(2,3,4,6‐tetra‐O‐acetyl‐β‐d‐glucopyranosyl)‐d‐galactitol (58): A: Compound 9 (310 mg, 1.14 mmol) and thiol 20 (498 mg, 1.37 mmol, 1.2 equiv) were reacted in toluene (5.0 mL) at 0 °C according to the general method. The crude product was purified by flash chromatography (CHCl3/acetone 95/5) to give 57 and 58 with 67 % overall yield (486 mg), with 2.3:1 57:58 ratio, as yellow syrup. B: The reaction was repeated at −40 °C to give 57 and 58 with 95 % overall yield, (57:58=1.5:1). C: The reaction was repeated at −80 °C to give 57 and 58 with 95 % overall yield, (57:58=1:1.2). D: The reaction was repeated at −120 °C to give 57 and 58 with 95 % overall yield,(57:58=1:1.2).
Data of 57: R f=0.15 (CH2Cl2/acetone 95:5), [α]D 20=−48.28 (c=0.29 in CHCl3) lit.‐44.3,58 1H NMR (400 MHz, CDCl3): δ=5.26–5.21 (m, 2 H), 5.19 (d, J=9.3 Hz, 1 H), 5.07 (t, J=7.3 Hz, 1 H), 5.03 (t, J=7.2 Hz, 1 H), 4.49 (d, J=10.0 Hz, 1 H), 4.30–4.22 (m, 2 H), 4.16 (dd, J=12.3, 4.0 Hz, 1 H), 4.12 (d, J=5.0 Hz, 1 H), 4.08 (dd, J=8.3, 3.8 Hz, 1 H), 3.92–3.86 (m, 1 H), 3.83 (dd, J=12.3, 2.7 Hz, 1 H), 3.68 (ddd, J=10.0, 4.7, 2.1 Hz, 1 H, H‐5′), 3.37 (dd, J=6.6, 4.1 Hz, 1 H), 2.12 (s, 3 H, AcCH 3), 2.08 (s, 3 H, AcCH 3), 2.06 (s, 6 H, 2x AcCH 3), 2.02 (s, 3 H, AcCH 3), 2.00 ppm (s, 3 H, AcCH 3). 13C NMR (100 MHz, CDCl3): δ=170.6, 170.2, 169.9, 169.4, 169.3 (6C, 7×AcCO), 83.3 (1C, C‐1′), 76.2, 74.6, 73.9, 69.6, 69.2, 68.1, 67.0 (7C, skeletal carbons), 62.0, 61.7 (2C, C‐1, C‐6′), 42.0 (1C, C‐2), 20.8, 20.8, 20.6, 20.6 ppm (7C, 6×AcCH 3). MALDI‐TOF‐MS: m/z calcd for C26H36NaO16S [M+Na]+ 659.162, found 659.159.
Data of 58: R f=0.24 (CH2Cl2/acetone 95:5), [α]D 20=−27.5 (c=0.12 in CHCl3),1H NMR (400 MHz, CDCl3): δ=5.36 (d, J=2.7 Hz, 1 H), 5.21 (t, J=9.3 Hz, 1 H), 5.06 (t, J=9.7 Hz, 1 H), 4.95–4.89 (m, 1 H), 4.89–4.86 (m, 1 H), 4.69 (d, J=10.2 Hz, 1 H), 4.31 (d, J=4.8 Hz, 1 H), 4.27 (t, J=5.3 Hz, 1 H), 4.24 (d, J=5.2 Hz, 1 H), 4.14 (dd, J=12.4, 2.3 Hz, 1 H), 4.07 (dd, J=6.4, 2.2 Hz, 2 H), 3.80 (t, J=6.5 Hz, 1 H), 3.73 (ddd, J=9.9, 5.1, 2.3 Hz, 1 H), 3.40 (t, J=11.8 Hz, 1 H), 3.28 (dd, J=11.5, 4.8 Hz, 1 H), 2.17 (s, 3 H, AcCH 3), 2.10 (s, 3 H, AcCH 3), 2.05 (s, 3 H, AcCH 3), 2.04 (s, 3 H, AcCH 3), 2.03 (s, 3 H, AcCH 3), 2.02 (s, 3 H, AcCH 3), 2.00 (s, 3 H, AcCH 3). 13C NMR (100 MHz, CDCl3): δ=170.7, 170.6, 170.3, 170.2, 169.4, 169.3 (7C, 7×AcCO), 84.8 (1C, C‐1′), 70.6 (1C, C‐1), 76.0, 75.1, 73.6, 72.5, 70.5, 68.3, 67.4 (7C, skeletal carbons), 62.1 (1C, C‐6′), 42.4 (1C, C‐2), 20.8, 20.8, 20.8, 20.7 ppm (7C, 7×AcCH3). ESI‐MS: m/z calcd for C26H36NaO16S [M+Na]+ 659.162, found 659.159
3,4,6‐Tri‐O‐acetyl‐1,5‐anhydro‐2‐thio‐2‐S‐(2,3,4,6‐tetra‐O‐acetyl‐β‐d‐glucopyranosyl)‐d‐altritol (59): Compound 10 (200 mg, 0.735 mmol), thiol 20 (321 mg, 0.882 mmol, 1.2 equiv) and DPAP (19 mg, 0.073 mmol, 0.1 equiv) were reacted in toluene (5.5 mL), at −80 °C according to the general method. The crude product was purified by column chromatography (n‐hexane/acetone 7/3) to give compound 59 (430 mg, 92 %) as colorless syrup. R f=0.41 (n‐hexane/acetone 6:4); [α]D 20=−14.5 (c=0.29 in CHCl3) lit.23 −8.8; 1H NMR (400 MHz, CDCl3): δ=5.54 (t, J=3.1 Hz, 1 H, H‐3), 5.31 (dd, J=10.3, 2.8 Hz, 1 H, H‐4), 5.23 (t, J=9.3 Hz, 1 H, H‐3′), 5.13 (t, J=9.7 Hz, 1 H, H‐4′), 5.04 (t, J=9.6 Hz, 1 H, H‐2′), 4.79 (d, J=10.1 Hz, 1 H, H‐1′), 4.23 (dd, J=12.3, 2.3 Hz, 1 H, H‐6′a), 4.20 (d, J=4.3 Hz, 1 H, H‐6′b), 4.18 (s, 1 H, H‐6a), 4.17 (s, 1 H, H‐6b), 4.10 (dd, J=12.3, 2.4 Hz, 1 H, H‐1a), 3.97 (d, J=12.3 Hz, 1 H, H‐1b), 3.91 (dt, J=9.9, 3.7 Hz, 1 H, H‐5), 3.79–3.73 (m, 1 H, H‐5′), 3.19 (s, 1 H, H‐2), 2.14 (s, 3 H, COCH3), 2.11 (s, 3 H, COCH3), 2.07 (s, 3 H, COCH3), 2.06 (s, 3 H, COCH3), 2.02 (s, 3 H, COCH3), 2.01 (s, 3 H, COCH3), 2.00 ppm (s, 3 H, COCH3); 13C NMR (100 MHz, CDCl3): δ=170.9, 170.7, 170.2, 169.7, 169.4, 169.3, 169.1 (7C, 7×COCH3), 84.3 (C‐1′), 76.0 (C‐5′), 73.9 (C‐3′), 73.0 (C‐5), 70.5 (C‐3), 70.2 (C‐2′), 68.2 (C‐4′), 67.0 (C‐1), 64.6 (C‐4), 62.9 (C‐6′), 61.8 (C‐6), 45.0 (C‐2), 21.0, 20.8, 20.7, 20.6, 20.6 ppm (7C, 7×COCH3). ESI‐MS: m/z calcd for C26H36NaO16S [M+Na]+ 659.162, found 659.159.
3,4‐Di‐O‐acetyl‐1,5‐anhydro‐6‐deoxy‐2‐thio‐2‐S‐(2,3,4,6‐tetra‐O‐acetyl‐β‐d‐glucopyranosyl)‐l‐manno‐hexitol (60) and 3,4‐di‐O‐acetyl‐1,5‐anhydro‐6‐deoxy‐2‐thio‐2‐S‐(2,3,4,6‐tetra‐O‐acetyl‐β‐d‐glucopyranosyl)‐l‐gluco‐hexitol (61): A: Compound 11 (107 mg, 0.5 mmol), thiol 20 (219 mg, 0.6 mmol, 1.2 equiv) and DPAP (38 mg, 0.05 mmol, 0.1 equiv) were reacted in toluene (3.0 mL), at −80 °C according to the general method. The crude product was purified by flash chromatography (n‐hexane/acetone 75/25) to give compound 60 as white crystals (135 mg, 47 %), 61 as white foam (36 mg, 12 %), and an inseparable 3:1 mixture of 60 and 61 (22 %). B: The reaction was repeated at RT with 2.0 equiv of 20 to give 60 and 61 with 66 % overall yield (60:61=2.5:1).
Data of 60: R f=0.11 (n‐hexane/acetone 7:3), m.p. 176–178 °C, [α]D 20=−0.90, (c=0.11 in CHCl3), 1H NMR (400 MHz, CDCl3): δ=5.18 (t, J=9.3 Hz, 1 H, H‐3′), 5.06 (t, J=9.7 Hz, 1 H, H‐4′), 5.04 (dd, J=9.6, 4.4 Hz, 1 H, H‐3), 4.96 (t, J=9.7 Hz, 1 H) and 4.96 (t, J=9.2 Hz, 1 H) (H‐4 and H‐2′), 4.60 (d, J=10.1 Hz, 1 H, H‐1′), 4.20 (dd, J=12.4, 5.1 Hz, 1 H, H‐6′a), 4.15–4.10 (m, 2 H„ H‐6′b and H‐1a), 3.83 (dd, J=12.3, 1.9 Hz, 1 H, H‐1b), 3.68–3.60 (m, 2 H, H‐5′ and H‐2), 3.42 (dq, J=8.8, 6.2 Hz, 1 H, H‐5), 2.11 (s, 3 H, COCH3), 2.09 (s, 3 H, COCH3), 2.09 (s, 3 H, COCH3), 2.06 (s, 3 H, COCH3), 2.03 (s, 3 H, COCH3), 2.01 (s, 3 H, COCH3), 1.19 ppm (d, J=6.2 Hz, 3 H, 3×H‐6); 13C NMR (100 MHz, CDCl3): δ=170.6, 170.3, 170.2, 169.7, 169.5, 169.3 (6C, 6×COCH3), 84.1 (C‐1′), 76.0, 75.3, 73.8, 71.6, 70.3, 68.1 (7C, skeleton carbons), 70.4 (C‐1), 62.0 (C‐6′), 45.7 (C‐2), 20.9, 20.8, 20.7, 20.7, 20.6, 20.6 (6C, 6×COCH3), 17.6 ppm (C‐6). MALDI‐TOF‐MS: m/z calcd for C24H34NaO14S [M+Na]+ 601.157, found 601.064.
Data of 61: R f=0.15 (n‐hexane/acetone 7:3), [α]D 20=−48.52 (c=0.27 in CHCl3), 1H NMR (400 MHz, CDCl3): δ=5.21 (t, J=9.3 Hz, 1 H, H‐3′), 5.05 (t, J=9.8 Hz, 1 H, H‐4′), 5.00–4.93 (m, 2 H, H‐3, H‐2′), 4.75 (t, J=9.3 Hz, 1 H, H‐4), 4.60 (d, J=10.0 Hz, 1 H, H‐1′), 4.22 (dd, J=12.3, 5.4 Hz, 1 H, H‐6′a), 4.17–4.09 (m, 2 H, H‐6′b, H‐1a), 3.72 (ddd, J=10.0, 5.3, 2.4 Hz, 1 H, H‐5′), 3.48–3.40 (m, 2 H, H‐5, H‐1b), 3.11 (td, J=11.4, 4.9 Hz, 1 H, H‐2), 2.10 (s, 3 H, COCH3), 2.05 (s, 3 H, COCH3), 2.04 (s, 3 H, COCH3), 2.03 (s, 3 H, COCH3), 2.03 (s, 3 H, COCH3), 2.00 (s, 3 H, COCH3), 1.19 ppm (d, J=6.1 Hz, 3 H); 13C NMR (100 MHz, CDCl3): δ=170.7, 170.4, 170.2, 170.0, 169.5, 169.2 (6C, 6×COCH3), 81.6 (C‐1′), 76.0, 75.1, 75.0, 73.9, 72.2, 70.1, 68.2 (7C, skeleton carbons), 70.8 (C‐1′), 62.2 (C‐6′), 44.5 (C‐2), 20.8, 20.8, 20.7, 20.7 ppm (6C, 6×COCH3), 17.9 (C‐6). MALDI‐TOF‐MS: m/z calcd for C24H34NaO14S [M+Na]+ 601.157, found 601.064.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was funded by the National Research, Development and Innovation Office of Hungary (TÉT 15 IN‐1‐2016‐0071 and OTKA K109208), Gedeon Richter's Talentum Foundation (1103 Budapest, Gyömrői út 19–21.), the “Debrecen Venture Catapult Program” (EFOP‐3.6.1‐16‐2016‐00022 and EFOP‐3.6.3‐VEKOP‐16‐2017‐00009) and the ÚNKP‐18‐3 New National Excellence Program of the Ministry of Human Capacities. The research was also supported by the EU and co‐financed by the European Regional Development Fund under the projects GINOP‐2.3.2‐15‐2016‐00008, GINOP‐2.3.3‐15‐2016‐00021 and GINOP‐2.3.3‐15‐2016‐00004.
V. Kelemen, M. Bege, D. Eszenyi, N. Debreczeni, A. Bényei, T. Stürzer, P. Herczegh, A. Borbás, Chem. Eur. J. 2019, 25, 14555.
References
- 1. Wang Z., Chinoy Z. S., Ambre S. G., Peng W., McBride R., de Vries R. P., Glushka J., Paulson J. C., Boons G.-J., Science 2013, 341, 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Cecioni S., Imberty A., Vidal S., Chem. Rev. 2015, 115, 525–561; [DOI] [PubMed] [Google Scholar]
- 2b. Jančaříková G., Herczeg M., Fujdiarová E., Houser J., Kövér K. E., Borbás A., Wimmerová M., Csávás M., Chem. Eur. J. 2018, 24, 4055–4068; [DOI] [PubMed] [Google Scholar]
- 2c. Goyard D., Baldoneschi V., Varrot A., Fiore M., Imberty A., Richichi B., Renaudet O., Nativi C., Bioconjugate Chem. 2018, 29, 83–88; [DOI] [PubMed] [Google Scholar]
- 2d. Cabanettes A., Perkams L., Spies C., Unverzagt C., Varrot A., Angew. Chem. Int. Ed. 2018, 57, 10178–11018; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10335–10338; [Google Scholar]
- 2e. Diercks T., Infantino A. S., Unione L., Jiménez-Barbero J., Oscarson S., Gabius H.-J., Chem. Eur. J. 2018, 24, 15761–15765. [DOI] [PubMed] [Google Scholar]
- 3. Sattin S., Bernardi A., Carbohydrate Chemistry, Vol. 41, RSC Publications, Cambridge, 2016, pp. 1–25. [Google Scholar]
- 4.
- 4a. Ernst B., Magnani J. L., Nat. Rev. Drug Discovery 2009, 8, 661–667; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Cipolla L., Araújo A. C., Bini D., Gabrielli L., Russo L., Shaikh N., Expert Opin. Drug Discovery 2010, 5, 721–737. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Fernández-Tejada A., Cañada F. J., Jiménez-Barbero J., Chem. Eur. J. 2015, 21, 10616–10628; [DOI] [PubMed] [Google Scholar]
- 5b. Wei M.-M., Wang Y.-S., Ye X.-S., Med. Res. Rev. 2018, 38, 1003–1026; [DOI] [PubMed] [Google Scholar]
- 5c. Anish C., Schumann B., Pereira C. L., Seeberger P. H., Chem. Biol. 2014, 21, 38–50; [DOI] [PubMed] [Google Scholar]
- 5d. Kaploneka P., Khana N., Reppe K., Schumann B., Emmadi M., Lisboa M. P., Xu F.-F., Calow A. D. J., Parameswarappa S. G., Witzenrath M., Pereira C. L., Seeberger P. H., Proc. Natl. Acad. Sci. USA 2018, 115, 13353–13358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Driguez H., ChemBioChem 2001, 2, 311–318. [DOI] [PubMed] [Google Scholar]
- 7. Pachamuthu K., Schmidt R. R., Chem. Rev. 2006, 106, 160–187. [DOI] [PubMed] [Google Scholar]
- 8. Szilágyi L., Varela O., Current Org. Chem. 2006, 10, 1745–1770. [Google Scholar]
- 9. Jana M., Misra A. K., J. Org. Chem. 2013, 78, 2680–2686. [DOI] [PubMed] [Google Scholar]
- 10. Ellis D., Norman S. E., Osborn H. M. I., Tetrahedron 2008, 64, 2832–2854. [Google Scholar]
- 11. Crich D., Li H., J. Org. Chem. 2000, 65, 801–805. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Dere R. T., Wang Y., Zhu X., Org. Biomol. Chem. 2008, 6, 2061–2063; [DOI] [PubMed] [Google Scholar]
- 12b. Wang H., Zhu X., Org. Biomol. Chem. 2014, 12, 7119–7126. [DOI] [PubMed] [Google Scholar]
- 13. Tardieu D., Céspedes Dávila M. F., Hazelard D., Compain P., Synthesis 2018, 50, 3927–3930. [Google Scholar]
- 14.Selected recent publications for the synthesis of oligosaccharides containing 1,2-cis-α-O-glycosidic bonds:
- 14a. Yu F., Li J., DeMent P. M., Tu Y.-J., Schlegel H. B., Nguyen H. M., Angew. Chem. Int. Ed. 2019, 58, 6957–6961; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 7031–7035; [Google Scholar]
- 14b. Qin C., Schumann B., Zou X., Pereira C. L., Tian G., Hu J., Seeberger P. H., Yin J., J. Am. Chem. Soc. 2018, 140, 3120–3127; [DOI] [PubMed] [Google Scholar]
- 14c. Zou X., Qin C., Claney L., Pereira C. L., Tian G., Hu J. J., Seeberger P. H., Yin J., Chem. Eur. J. 2018, 24, 2868–2872; [DOI] [PubMed] [Google Scholar]
- 14d. Tanaka M., Nakagawa A., Nishi N., Iijima K., Sawa R., Takahashi D., Toshima K., J. Am. Chem. Soc. 2018, 140, 3644–3651. [DOI] [PubMed] [Google Scholar]
- 15. Dondoni A., Angew. Chem. Int. Ed. 2008, 47, 8995–8997; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 9133–9135. [Google Scholar]
- 16. Dondoni A., Marra A., Chem. Soc. Rev. 2012, 41, 573–586. [DOI] [PubMed] [Google Scholar]
- 17. McSweeney L., Dénès F., Scanlan E. M., Eur. J. Org. Chem. 2016, 2080–2095. [Google Scholar]
- 18. Fiore M., Marra A., Dondoni A., J. Org. Chem. 2009, 74, 4422–4425. [DOI] [PubMed] [Google Scholar]
- 19. Lázár L., Csávás M., Tóth M., Somsák L., Borbás A., Chem. Pap. 2015, 69, 889–895. [Google Scholar]
- 20. József J., Juhász L., Illyés T. Z., Csávás M., Borbás A., Somsák L., Carbohydr. Res. 2015, 413, 63–69. [DOI] [PubMed] [Google Scholar]
- 21. Bege M., Bereczki I., Herczeg M., Kicsák M., Eszenyi D., Herczegh P., Borbás A., Org. Biomol. Chem. 2017, 15, 9226–9233. [DOI] [PubMed] [Google Scholar]
- 22. József J., Juhász L., Somsák L., New J. Chem. 2019, 43, 5670–5686. [Google Scholar]
- 23. Staderini S., Chambery A., Marra A., Dondoni A., Tetrahedron Lett. 2012, 53, 702–704. [Google Scholar]
- 24. Lázár L., Juhász L., Batta G., Borbás A., Somsák L., New J. Chem. 2017, 41, 1284–1292. [Google Scholar]
- 25. Araki Y., Matsuura K., Ishido Y., Kushida K., Chem. Lett. 1973, 2, 383–386. [Google Scholar]
- 26. Lázár L., Csávás M., Herczeg M., Herczegh P., Borbás A., Org. Lett. 2012, 14, 4650–4653. [DOI] [PubMed] [Google Scholar]
- 27. Lázár L., Csávás M., Hadházi Á., Herczeg M., Tóth M., Somsák L., Barna T., Herczegh P., Borbás A., Org. Biomol. Chem. 2013, 11, 5339–5350. [DOI] [PubMed] [Google Scholar]
- 28. Eszenyi D., Kelemen V., Balogh F., Bege M., Csávás M., Herczegh P., Borbás A., Chem. Eur. J. 2018, 24, 4532–4536. [DOI] [PubMed] [Google Scholar]
- 29. Eszenyi D., Lázár L., Borbás A., McCourt R. in Carbohydrate Chemistry: Proven Synthetic Methods, Vol. 4 (Eds.: P. Kováč, C. Vogel, P. Murphy), CRC Press, Boca Raton, 2017, pp. 33–44. [Google Scholar]
- 30. Lázár L., Borbás A., Somsák L., Carbohydr. Res. 2018, 470, 8–12. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Posner T., Ber. Dtsch. Chem. Ges. 1905, 38, 646–657; [Google Scholar]
- 31b. Kharasch M. S., Read A. T., Mayo F. R., Chem. Ind. 1938, 57, 752; [Google Scholar]
- 31c. Griesbaum K., Angew. Chem. Int. Ed. Engl. 1970, 9, 273–287; [Google Scholar]; Angew. Chem. 1970, 82, 276–290. [Google Scholar]
- 32.
- 32a. Cramer N. B., Reddy S. K., Cole M., Hoyle C., Bowman C. N., J. Polym. Sci. Part A 2004, 42, 5817–5826; [Google Scholar]
- 32b. Hoyle C. E., Bowman C. N., Angew. Chem. Int. Ed. 2010, 49, 1540–1573; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 1584–1617; [Google Scholar]
- 32c. Cramer N. B., Reddy S. K., O'Brien A. K., Bowman C. N., Macromolecules 2003, 36, 7964–7969. [Google Scholar]
- 33. Northrop B. H., Coffey R. N., J. Am. Chem. Soc. 2012, 134, 13804–13817. [DOI] [PubMed] [Google Scholar]
- 34.Recently we observed that hydrothiolation of sugar exomethylenes with higher alkyl thiols also occurred with higher efficacy at RT and 0 °C than at −80 °C, see: Bege M., Kiss A., Kicsák M., Bereczki I., Baksa V., Király G., Szemán-Nagy G., Szigeti M. Z., Herczegh P., Borbás A., Molecules 2019, 24, 2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dénès F., Pichowicz M., Povie G., Renaud P., Chem. Rev. 2014, 114, 2587–2693. [DOI] [PubMed] [Google Scholar]
- 36.
- 36a. LeBel N. A., DeBoer A., J. Am. Chem. Soc. 1967, 89, 2784; [Google Scholar]
- 36b. LeBel N. A., Czaja R. F., DeBoer A., J. Org. Chem. 1969, 34, 3112–3126; [Google Scholar]
- 36c. Bordwell F. G., Landis P. S., Whitney G. S., J. Org. Chem. 1965, 30, 3764. [Google Scholar]
- 37. Rico M., Santoro J., Org. Magn. Res. 1976, 8, 49–55. [Google Scholar]
- 38.
- 38a. Stoddart J. F., Stereochemistry of Carbohydrates, Wiley, New York, 1971; [Google Scholar]
- 38b. Kuszmann J. in The Organic Chemistry of Sugars, (Eds.: D. E. Levy, P. Fügedi), CRC Press, Boca Raton, 2006, pp. 25–52; [Google Scholar]
- 38c. Bérces A., Whitfield D. M., Nukada T., Tetrahedron 2001, 57, 477–491; [Google Scholar]
- 38d. Cremer D., Pople J. A., J. Am. Chem. Soc. 1975, 97, 1354–1358; [Google Scholar]
- 38e. Jeffrey G. A., Yates J. H., Carbohydr. Res. 1979, 74, 319–322. [Google Scholar]
- 39. Horton D., Durette P. L., J. Org. Chem. 1971, 36, 2658–2669. [Google Scholar]
- 40. Giese B., Angew. Chem. Int. Ed. Engl. 1989, 28, 969–1146; [Google Scholar]; Angew. Chem. 1989, 101, 993–1004. [Google Scholar]
- 41. Pérez-Martín I., Suárez E. in Encyclopedia in Radicals in Chemistry, Biology and Materials, Wiley, Chichester, 2012, pp. 1131–1174. [Google Scholar]
- 42. Dupuis J., Giese B., Rüegge D., Fischer H., Korth H.-G., Sustmann R., Angew. Chem. Int. Ed. Engl. 1984, 23, 896–898; [Google Scholar]; Angew. Chem. 1984, 96, 887–888. [Google Scholar]
- 43. Korth H.-G., Sustmann R., Dupuis J., Giese B., J. Chem. Soc. Perkin Trans. 2 1986, 1453–1459. [Google Scholar]
- 44. Giese B., Dupuis J., Angew. Chem. Int. Ed. Engl. 1983, 22, 622–623; [Google Scholar]; Angew. Chem. 1983, 95, 633–634. [Google Scholar]
- 45. Chandra H., Symons M. C. R., Korth H.-G., Sustmann R., Tetrahedron Lett. 1987, 28, 1455–1458. [Google Scholar]
- 46. Davuluri R., Lerner L. M., Carbohydr. Res. 1972, 22, 345–350. [Google Scholar]
- 47.
- 47a. González A. G., Vadlamani G., Mark B. L., Withers S. G., Chem. Commun. 2016, 52, 7943–7946; [DOI] [PubMed] [Google Scholar]
- 47b. Zhang L., Muthana M. M., Yu H., McArthur J. B., Qu J., Chen X., Carbohydr. Res. 2016, 419, 18–28; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47c. Pravdić N., Franjić-Mihalić I., Danilov B., Carbohydr. Res. 1975, 45, 302–306. [Google Scholar]
- 48.
- 48a. Nakagawa T., Lichtenthaler F. W., Tetrahedron: Asymmetry 2011, 22, 740–744; [Google Scholar]
- 48b. Lainé V., Coste-Sarguet A., Gadelle A., Defaye J., Perly B., Djedaini-Pilard F., J. Chem. Soc. Perkin Trans. 2 1995, 1479–1487. [Google Scholar]
- 49. Capaccio C. A. I., Varela O., J. Org. Chem. 2001, 66, 8859–8866. [DOI] [PubMed] [Google Scholar]
- 50. Capaccio C. A. I., Varela O., J. Org. Chem. 2002, 67, 7839–7846. [DOI] [PubMed] [Google Scholar]
- 51. Somsák L., Németh I., J. Carbohydr. Chem. 1993, 12, 679–684. [Google Scholar]
- 52. Xiao R., Dane E. L., Zeng J., McKnight C. J., Grinstaff M. W., J. Am. Chem. Soc. 2017, 139, 14217–14223. [DOI] [PubMed] [Google Scholar]
- 53. Hsu M.-Y., Liu Y.-P-, Lam S., Lin S.-C., Wang C.-C., Beilstein J. Org. Chem. 2016, 12, 1758–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wiesler W. T., Caruthers M. H., J. Org. Chem. 1996, 61, 4272–4281. [DOI] [PubMed] [Google Scholar]
- 55. Ghosh T., Santra A., Misra A. K., Beilstein J. Org. Chem. 2013, 9, 974–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Horton D., Methods Carbohydr. Chem. 1963, 2, 433–437. [Google Scholar]
- 57. Matta K. L., Girotra R. N., Barlow J. J., Carbohydr. Res. 1975, 43, 101–109. [DOI] [PubMed] [Google Scholar]
- 58. Gadelle A., Defaye J., Pedersen C., Carbohydr. Res. 1990, 200, 497–498. [Google Scholar]
- 59. Blanc-Muasser M., Vigne L., Driguez H., Tetrahedron Lett. 1990, 31, 3869–3870. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary