

Abstract
Receptor‐interacting protein kinase 3 (RIP3) is the core regulator that switches cell death from apoptosis to necrosis. However, its role in tumor immunity is unknown. In this study, decreased RIP3 expression was observed in patients with hepatocellular carcinoma (HCC), which correlates with myeloid‐derived suppressor cell (MDSC) accumulation. Moreover, RIP3 is a prognosis factor for patients with HCC. We further found that RIP3 knockdown results in an increase of MDSCs and a decrease of interferon gamma–positive (IFN‐γ+) cluster of differentiation 8–positive (CD8+) tumor‐infiltrating lymphocytes (IFN‐γ+ CD8+ T cells) in hepatoma tissues, thus promoting immune escape and HCC growth in immunocompetent mice. By phosphorylating P65Ser536 and promoting phosphorylated P65Ser536 nuclear translocation, RIP3 knockdown increases the expression of chemokine (C‐X‐C motif) ligand 1 (CXCL1) in HCC cells. RIP3 knockdown induces MDSC recruitment through the CXCL1–chemokine (C‐X‐C motif) receptor 2 (CXCR2) axis. Furthermore, a CXCR2 antagonist substantially suppresses MDSC chemotaxis and HCC growth in RIP3 knockout mice. Conclusion: RIP3 deficiency is an essential factor directing MDSC homing to HCC and promoting CXCL1/CXCR2‐induced MDSC chemotaxis to facilitate HCC immune escape and HCC progression; blocking the CXCL1–CXCR2 chemokine axis may provide an immunological therapeutic approach to suppress progression of RIP3 deficiency HCC.
Abbreviations
- Cas9
CRISPR‐associated 9
- CD
cluster of differentiation
- ChIP
chromatin immunoprecipitation
- CM
conditioned media
- CXCL1
chemokine (C‐X‐C motif) ligand 1
- CXCR2
chemokine (C‐X‐C motif) receptor 2
- DEN
diethylnitrosamine
- ELISA
enzyme‐linked immunosorbent assay
- Gr‐1
granulocyte receptor 1
- GSK’872
N‐5‐benzothiazolyl‐6‐[(1‐methylethyl)sulfonyl]‐4‐quinolinamine
- HCC
hepatocellular carcinoma
- HLA‐DR
human leukocyte antigen DR
- IFN‐γ
interferon gamma
- IgG
immunoglobulin G
- IKBα
NFKB inhibitor alpha
- JSH‐23
4‐methyl‐1‐N‐(3‐phenylpropyl)benzene‐1,2‐diamine
- KO
knockout
- Luc
luciferase
- Ly6
lymphocyte antigen 6
- MDSC
myeloid‐derived suppressor cell
- MT
mutated
- p‐
phosphorylated
- PBS
phosphate‐buffered saline
- RIP3
receptor‐interacting protein kinase 3
- sgRNA
single‐guide RNA
- shNC
short hairpin negative control
- shRIP3
short hairpin RIP3
- Treg
regulatory T cell
- WT
wild type
Hepatocellular carcinoma (HCC) is one of the leading causes of cancer‐related deaths worldwide.1 Lack of effective treatments results in poor prognosis of HCC, with 5‐year survival rates of 3%‐11%.2 Thus, exploring the pathogenesis and developing therapeutic targets are urgently warranted for this deadly cancer.
Immune escape is one of the major hallmarks of HCC.3, 4 It is well recognized that HCC is a typical inflammation‐related cancer and usually progresses within a preexisting chronic inflammatory, fibrotic, or cirrhotic liver and therefore is accompanied by infiltration of immune cells.5 Immune escape is often accomplished by activation of immune checkpoint signals such as programmed cell death receptor 1 and its ligand and recruitment of immunosuppressive cells such as myeloid‐derived suppressor cells (MDSCs).6, 7, 8 MDSCs represent a heterogeneous population of myeloid progenitors that accumulate in the tumor microenvironment and serve as powerful proinflammatory mediators in suppressing T‐cell functions to contribute to immune evasion.9 Activated MDSCs express a source of secreted cytokines and enzymes, which repress T cells and natural killer cells but activate regulatory T cells (Tregs).10, 11, 12 However, the hepatic oncogenic signaling that drives MDSC recruitment and its molecular mechanisms remain poorly defined.
Receptor‐interacting protein kinase 3 (RIP3) is a core regulator of necroptosis.13 Studies have reported that RIP3 is essential for liver injury, inflammatory hepatocarcinogenesis, and cholestasis.14, 15 However, its role in HCC immunity remains largely unknown. In this study, we found that hepatic RIP3 deficiency promotes chemokine (C‐X‐C motif) ligand 1 (CXCL1)/chemokine (C‐X‐C motif) receptor 2 (CXCR2)–induced MDSC recruitment and reduces the infiltration of interferon gamma–positive (IFN‐γ+) cluster of differentiation 8–positive (CD8+) T cells to facilitate immune escape and progression. By phosphorylating P65Ser536 and promoting phosphorylated (p‐) P65Ser536 nuclear translocation, RIP3 knockdown increases the expression of CXCL1 in HCC cells. Furthermore, antagonizing CXCR2 substantially suppresses MDSC chemotaxis and HCC growth in RIP3 knockout (KO) mice. These findings reveal the mechanism of RIP3‐mediated immune evasion, which provides an effective therapeutic target in HCC immunotherapy.
Materials and Methods
Establishment of Orthotopic HCC Models
For orthotopic implantation, 2 × 106 Hepa1‐6, Huh‐7, or MHCC97H cells were resuspended in 50 μL serum‐free Roswell Park Memorial Institute 1640/Matrigel mixture. Under anesthesia, mice (five in each group) were orthotopically inoculated in the left hepatic lobe with a microsyringe through an 8‐mm transverse incision in the upper abdomen. When the mice were sacrificed, their livers were dissected for flow cytometry and histological evaluation. Animal studies were approved by the local regulatory agency (Laboratory Animal Research Center of the Fourth Military Medical University). All animal protocols are approved by governmental and institutional guidelines for animal welfare.
Establishment of RIP3 KO Mice
RIP3 KO mice were constructed using a clustered regularly interspaced short palindromic repeats (CRISP)/CRISPR‐associated 9 (CAS9) system in our laboratory. A single‐guide RNA (sgRNA) vector against exon 3 of RIP3 was designed and constructed. Then, the sgRNA and Cas9 were transcribed by T7 RNA polymerase in vitro. Cas9 mRNA and sgRNA were mixed and microinjected into fertilized eggs of C57BL/6 mice. Gene sequencing was used to detect mutations of RIP3. Quantitative PCR and western blot were used to detect the expression of RIP3.
MDSC Transwell Invasion Assay
In vitro migration of murine MDSCs was evaluated in 24‐well plates with Transwell polycarbonate‐permeable supports (8.0 μm; Merck Millipore). Freshly isolated splenic MDSCs (3 × 105, >90% purity) were seeded on the upper chambers of the inserts, supplemented with 10 ng/mL granulocyte‐macrophage colony‐stimulating factor and 10 ng/mL interleukin‐4, after incubation with a CXCR2 antagonist, SB265610, for 1 hour. The conditioned media (CM) from Hepa1‐6–short hairpin RIP3 (shRIP3) or Hepa1‐6 cells treated with N‐5‐benzothiazolyl‐6‐[(1‐methylethyl)sulfonyl]‐4‐quinolinamine (GSK’872) were placed in the lower chamber. After incubation for 24 hours, the MDSCs in the bottom compartment were counted.
Preparation of Single‐Cell Suspensions
Prior to magnetic bead cell sorting and flow‐cytometric analysis, single‐cell suspensions were prepared. Briefly, mice were anesthetized and perfused with Hank’s buffer without Ca2+ from the portal vein, followed by Hank’s buffer with Ca2+ and Mg2+ containing 0.2 mg/mL of collagenase IV (C5138; Sigma‐Aldrich). The liver and tumor were dissected and finely minced into pieces with the gentleMACS Dissociator (Miltenyi Biotech) in perfusion buffer containing 0.2 mg/mL of collagenase IV and 10 μg/mL of deoxyribonuclease (DNase) I (D5025; Sigma‐Aldrich). A single‐cell suspension was obtained by passing through a 70‐μm cell mesh and resuspending in Hank’s buffer. Primary hepatocytes were pelleted by low‐speed centrifugation at 50g for 3 minutes. Supernatants were further centrifuged at 400g for 10 minutes to obtain myeloid cells. Murine splenic cells were isolated as described.16
Flow‐Cytometric Analysis
For murine samples, mice with tumors were put under anesthesia, and tumors were collected. For human samples, tumor cells were collected from patients with HCC at the time of surgery. This study was approved by the Ethics Committee of the Fourth Military Medical University, and informed consent was acquired from all patients. Cells were incubated with fragment crystallizable (Fc) block for 10 minutes at room temperature and stained with fluorophore‐conjugated antibodies for 45 minutes at 4°C in staining buffer (phosphate‐buffered saline [PBS] with 0.5% bovine serum albumin and 2 mM ethylene diamine tetraacetic acid). For intracellular IFN‐γ staining, a Cytofix/Cytoperm Fixation/Permeabilization Kit (554714; BD Biosciences) was used. Matched isotype antibodies were used as controls. Data were acquired using a fluorescence‐activated cell sorting LSRFortessa (BD Biosciences) and analyzed with FlowJo software (BD Biosciences).
MDSC Isolation by Magnetic Bead Cell Sorting
Isolation of CD11b+ granulocyte receptor 1–positive (Gr‐1+) total MDSCs, CD11b+ lymphocyte antigen 6 complex locus G‐negative (Ly6G‐) lymphocyte antigen 6 complex locus C‐high (Ly6Chi) Mo‐MDSCs, and CD11b+Ly6ClowLy6Ghi Gr‐MDSCs was performed by using mouse MDSC isolation kit (130‐094‐538; Miltenyi Biotech, Auburn, CA) according to the manufacturer’s instruction. Briefly, after Fc receptor blockade, cells were stained with biotin‐conjugated Gr‐1 or Ly6G antibody and further labeled with antibiotin microbeads. Labeled cells were passed through the separation columns (Miltenyi Biotech) for magnetic cell separation. Retained cells were analyzed for the CD11b+Gr‐1+/CD11b+Ly6G–Ly6Chi/CD11b+Ly6ClowLy6Ghi population to assess MDSC purity (>90%) by flow cytometry.
Gene Microarray Imaging and Data Analysis
The array data were analyzed for summarization, normalization, and quality control using GeneSpring software V13 (Agilent). To select differentially expressed genes, we used threshold values of ≥2‐fold and ≤−2‐fold change and a Benjamini‐Hochberg‐corrected P value of 0.05. The data were log2‐transformed and median‐centered by genes using the Adjust Data function of CLUSTER 3.0 software, then further analyzed with hierarchical clustering with average linkage. Finally, we performed tree visualization using Java Treeview (Stanford University School of Medicine, Stanford, CA).
RT2 Profiler PCR Array
The RT2 profiler PCR array was performed for screening hepatoma‐derived cytokines/chemokines by use of the Mouse Cytokines and Chemokines PCR Array (PAMM‐150Z; SABiosciences, Hilden, Germany). The RT2 profiler PCR array was probed by the Profiler PCR Array System and SYBR green/fluorescein quantitative PCR Master Mix (SABiosciences) in QuantStudio 7 Flex Real‐Time PCR System (Applied Biosystems, Carlsbad, CA) in accordance with the manufacturer’s protocol. Pan‐cytokine/chemokine gene expression was analyzed at the PCR Array Data Analysis Web Portal, which automatically performs all 2‐△Ct‐based fold‐change calculations from the specific uploaded raw threshold cycle data.
Chromatin Immunoprecipitation Assay
For chromatin immunoprecipitation (ChIP), 1 × 107 cells were harvested for each condition. Huh‐7 cells and Huh‐7–shRIP3 cells were washed twice in PBS, crosslinked with formaldehyde, lysed with sodium dodecyl sulfate buffer, and sonicated. Sheared DNA was precleared with protein A‐agarose/salmon sperm DNA beads and rotated for 2 hours at 4°C (17‐371, EZ‐ChIP Kit; Merck Millipore), immunoprecipitated with P65 antibody (8242; Cell Signaling Technology) and immunoglobulin G, and rotated overnight at 4°C. Agarose beads were incubated with antibody–protein–DNA complex and washed with a low‐salt immune complex wash buffer, a high‐salt immune complex wash buffer, and a lithium chloride immune complex wash buffer according to the manufacturer’s protocol. DNA–protein complexes were eluted in 1% sodium dodecyl sulfate and 0.1 M NaHCO3 elution buffer and decrosslinked with 0.2 M NaCl overnight at 65°C. DNA was purified using phenol chloroform extraction and ethanol precipitation, eluted in DNase/ribonuclease‐free water, and used for quantitative RT‐PCR analysis.
Dual Luciferase Reporter Assay
HEK‐293 cells were transfected with firefly luciferase (Luc) reporter gene constructs and human renilla Luc–cytomegalovirus vector (Promega). The CXCL1 (NM_001511) promoter region was cloned by gene synthesis according to the coding sequence and inserted into pGL4.10 Luc vector (Promega). For the Luc assay, cells were grown to 60%‐80% confluence. CXCL1 promoter–reporter gene constructs were then cotransfected with the following vectors using Lipofectamine 2000 (Invitrogen) as recommended by the manufacturer: RIP3‐pcDNA3.1, P65 (wild type [WT])‐pcDNA3.1, and P65 (mutated [MT])‐pcDNA3.1. The amount of total DNA in each transfection was normalized by the addition of an empty vector to the reaction mixture. The cells were then prepared for the Luc assays using the Dual Luciferase Reporter Assay System (Promega). Relative activity was defined as the ratio of firefly Luc activity to renilla Luc activity.
Statistical Analysis
Statistical analyses were performed by SPSS 23.0 (SPSS, Inc.) and GraphPad Prism 6 (GraphPad Software, Inc.). The independent Student t test was used to compare data between two groups. Spearman’s rank correlation was employed to analyze the association between immune parameters and RIP3 expression. Overall survival was calculated using Kaplan‐Meier analysis and compared by the log‐rank test. A two‐tailed P value < 0.05 was considered statistically significant.
Additional Materials and Methods
Additional materials and methods are included in the Supporting Information.
Results
Tumor‐Infiltrating CD11B+CD33+ Human Leukocyte Antigen‐DR− MDSCs in Patients With HCC Exert Potent Autologous CD8+ T‐Cell Suppression
We first investigated the abundance of MDSCs (CD11b+CD33+ human leukocyte antigen [HLA]‐DR– immature myeloid cells) in HCC patients by flow cytometry. Significantly higher frequencies of MDSCs in tumors, matched nontumor liver tissues, and peripheral blood of patients were observed compared with peripheral blood of healthy donors (Fig. 1A). Moreover, tumor‐infiltrating MDSCs encompassed similar levels of CD14+ monocytic and CD15+ granulocytic subsets (Fig. 1B). In contrast, the frequencies of CD8+ T cells in tumors, matched nontumor liver tissues, and peripheral blood were significantly lower than those of healthy donors (Fig. 1C). We further conducted an autologous CD8+ T‐cell proliferation assay to investigate the immunosuppressive activity of the tumor‐infiltrating MDSCs. Potent inhibition of CD8+ T‐cell proliferation induced by CD3/CD28 and interleukin‐2 was observed, particularly by coculture with tumor‐infiltrating MDSCs, which confirmed their immunosuppressive capacities (Fig. 1D). This result demonstrates that MDSCs may exert potent autologous CD8+ T‐cell suppression to promote immune escape in HCC.
Figure 1.
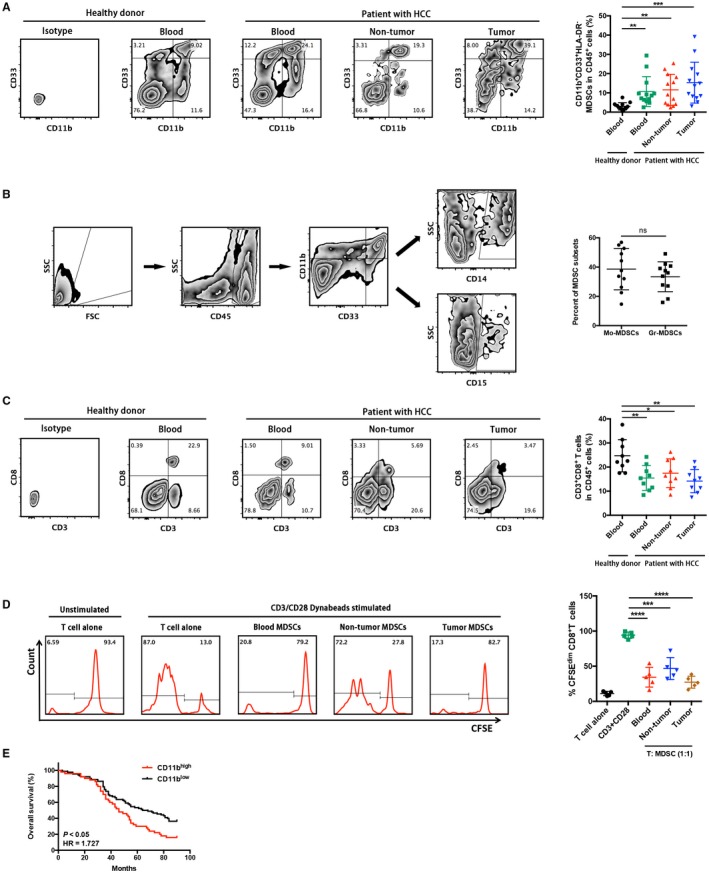
Tumor‐infiltrating CD11b+CD33+HLA‐DR− MDSCs in patients with HCC exert potent autologous CD8+ T‐cell suppression. (A) Representative CD11b+CD33+ MDSC zebra plots are shown in blood, nontumor, and tumor tissues from patients with HCC and healthy controls following a leukocyte gate (CD45+HLA‐DR−). Cells stained by isotype antibody were used as fluorescence baseline control. CD11b+CD33+HLA‐DR− cells were further analyzed and presented in 13 patients with HCC and 13 healthy donors. **P < 0.01, ***P < 0.001. (B) Gating strategies of MDSC subsets in flow cytometry. Similar levels of CD14+ monocytic and CD15+ granulocytic subsets were observed in HCC tissues. (C) Corresponding CD3+CD8+ T‐cell proportions in CD45+ leukocytes were determined. *P < 0.05, **P < 0.01. (D) Autologous T‐cell proliferation assay. The percentage of 5(6)‐carboxyfluorescein succinimidyl ester low population represents the proportion of proliferating CD3+CD8+ T cells. Representative flow‐cytometric data and a statistical diagram are shown. ***P < 0.001, ****P < 0.0001. (E) Kaplan‐Meier overall survival curves of patients with HCC with high or low expression (stratified by median) of CD11b. Abbreviations: CFSE, 5(6)‐carboxyfluorescein succinimidyl ester; FSC, forward scatter; SSC, side scatter.
Next, we further investigated whether overexpression of MDSC markers was associated with prognosis of HCC patients. Kaplan‐Meier analysis revealed that patients with CD11bhi expression significantly correlated with shorter overall survival rates (Fig. 1E).
Inverse Expression of RIP3 and MDSC Markers in HCC Correlates With Poor Prognosis of Patients
Given the anti‐inflammatory role of RIP3 in hepatocarcinogenesis, we next investigated the relationship between RIP3 and MDSCs in clinical samples by immunohistochemistry in 138 pairs of tumor and nontumor liver tissues. We found that the expression level of RIP3 was decreased in HCC tumor tissues in contrast to that in normal livers, while up‐regulation of the myeloid cell marker CD11b was observed (Fig. 2A). The mRNA and protein expression levels of RIP3 were also decreased in human HCC cell lines compared with normal liver cell lines (Fig. 2B). We next performed immunohistochemistry for RIP3 and CD11b in paired human HCC tissues. Correlation analyses showed that RIP3 negatively correlated with CD11b (Fig. 2C). We next evaluated RIP3 expression in HCC patient databases with Oncomine, which showed a consistent decrease of RIP3 in HCC tissue (Fig. 2D). Next, we further investigated whether expression level of RIP3 was associated with clinical characteristics and prognosis of HCC patients. The expression of RIP3 correlated significantly with tumor–node–metastasis stages (P < 0.05, χ2 test; Table 1). Consistent with our observation, the relationships between RIP3 and MDSCs were also identified by quantitative real‐time PCR using an independent cohort of 27 patients (Fig. 2E), therefore demonstrating the clinical significance of inverse RIP3 expression and MDSC accumulation in HCC. Kaplan‐Meier analysis revealed that patients with RIP3low expression significantly correlated with shorter overall survival rates (Fig. 2F).
Figure 2.
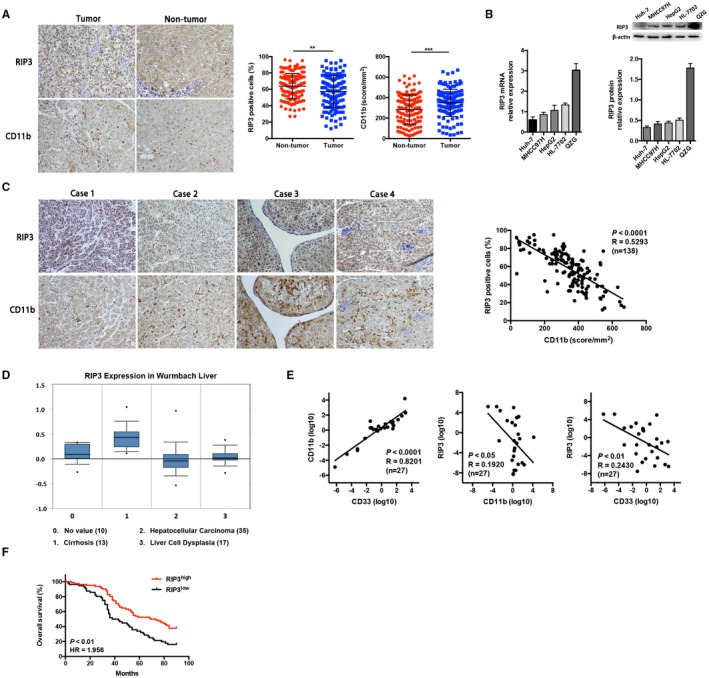
Inverse expression of RIP3 and MDSC markers in HCC correlates with poor prognosis of patients. (A) Representative pictures of RIP3 and CD11b immunohistochemical staining in nontumor and tumor tissues of patients with HCC are shown (n = 138, ×100 magnification). **P < 0.01, ***P < 0.001. (B) The expression level of RIP3 was decreased in human HCC cell lines compared with normal liver cell lines by quantitative real‐time PCR and western blot. Human HCC cell lines were Huh‐7, MHCC97H, and HepG2; human normal liver cell lines were HL‐7702 and QZG. (C) Representative pictures of RIP3 and CD11b immunohistochemical staining in tumors of patients with HCC are shown (×100 magnification). The association between CD11b and RIP3 scores is shown in 138 patients with HCC. (D) A consistent decrease of RIP3 in HCC tissue was shown in the Wurmbach Liver Database in Oncomine. (E) Correlation analysis between RIP3 and MDSCs by quantitative real‐time PCR using an independent cohort of 27 patients. (F) Kaplan‐Meier overall survival curves of patients with HCC with high or low expression (stratified by median) of RIP3. Abbreviation: HR, hazard ratio.
Table 1.
Correlation Between RIP3 Expression and Clinical Parameters in HCC Patients
| Parameters | Cases | RIP3 Expression Level | P | |
|---|---|---|---|---|
| <Median | >Median | |||
| Gender | 0.541 | |||
| Male | 115 | 52 | 63 | |
| Female | 23 | 12 | 11 | |
| Age (years) | 0.427 | |||
| <55 | 74 | 32 | 42 | |
| ≥55 | 64 | 32 | 32 | |
| Child grade | 0.334 | |||
| A | 46 | 24 | 22 | |
| B | 92 | 40 | 52 | |
| Tumor diameter | 0.847 | |||
| <5 cm | 96 | 44 | 52 | |
| ≥5 cm | 42 | 20 | 22 | |
| Tumor numbers | 0.579 | |||
| <3 | 85 | 41 | 44 | |
| ≥3 | 53 | 23 | 30 | |
| TNM stage | 0.012 | |||
| I‐II | 72 | 26 | 46 | |
| III‐IV | 66 | 38 | 28 | |
| T stage | 0.340 | |||
| I+II | 75 | 32 | 43 | |
| III+IV | 63 | 32 | 31 | |
| M stage | 0.139 | |||
| M0 | 103 | 44 | 59 | |
| M1 | 35 | 20 | 15 | |
Bold indicates significance.
Abbreviation: TNM, tumor–node–metastasis.
Hepatic RIP3 Deficiency Drives MDSC Recruitment to Promote Tumor Progression
To investigate whether hepatic RIP3 deficiency induces the accumulation of MDSCs to promote HCC progression, we established HCC‐bearing murine models by direct intrahepatic injection of HCC cells into the liver capsule (Supporting Fig. S1A). To examine whether MDSCs were present in our mouse HCC models, we orthotopically inoculated human HCC cell lines, MHCC97H and Huh‐7, and a mouse HCC cell line, Hepa1‐6, into immune‐deficient (xenograft, BALB/c nude) and immune‐competent (syngeneic, C57BL/6) mice and detected the dissociated tumors with mouse MDSC markers CD11b and Gr‐1. Flow‐cytometric analysis showed that MDSCs were more abundant in all of the mice with orthotopic tumors than those without xenograft. A significantly larger number of MDSCs could be observed in tumors derived from Hepa1‐6 cells (Fig. 3A).
Figure 3.
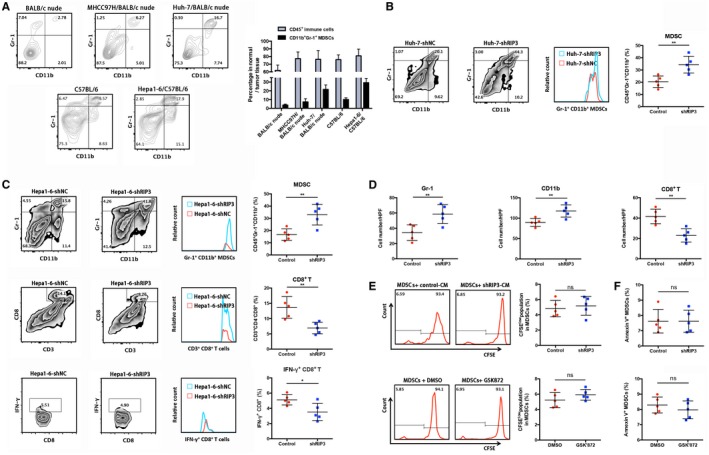
Hepatic RIP3 deficiency drives MDSC recruitment to promote tumor progression. (A) Different human and mouse HCC cell lines were orthotopically implanted into the left lobes of the livers of BALB/c nude or C57BL/6 mice. Harvested tumors were dissociated, and the percentages of CD11b+Gr+ MDSCs were evaluated by flow‐cytometric analysis. (B) Representative CD11b+Gr‐1+ MDSC zebra plots are shown in tumor tissues from orthotopic HCC models (n = 5, nude mice) of Huh‐7–shNC and Huh‐7–shRIP3 following a leukocyte gate (CD45+).
**P < 0.01. (C) Representative CD11b+Gr‐1+ MDSCs, CD3+CD8+ T cells, and IFN‐γ+CD8+ T cell zebra plots are shown in tumor tissues from orthotopic HCC models (n = 5, C57BL/6) of Hepa1‐6–shNC and Hepa1‐6–shRIP3 following a leukocyte gate (CD45+). *P < 0.05, **P < 0.01. (D) Immunostained cell count in Hepa1‐6–shNC and Hepa1‐6–shRIP3 orthotopic tumors from immunocompetent mice (n = 5). Gr‐1+ (left), CD11b+ (middle), and CD8+ (right). **P < 0.01. (E) The percentage of 5(6)‐carboxyfluorescein succinimidyl ester low population represents the proportion of proliferating MDSCs. Representative flow‐cytometric data and a statistical diagram are shown. (F) The percentage of Annexin V+ MDSCs represents the proportion of apoptotic MDSCs. A statistical diagram is shown. Abbreviations: CFSE, 5(6)‐carboxyfluorescein succinimidyl ester; DMSO, dimethyl sulfoxide; HPF, high power field; ns, nonsignificant.
To evaluate the effects of RIP3 on MDSC recruitment, we implanted Huh‐7–shRIP3 cells into BALB/c nude mice and found that knockdown of RIP3 drastically increased MDSC infiltration (Fig. 3B). To better simulate the immune‐competent niche, we next orthotopically inoculated murine Hepa1‐6 cells into C57BL/6 mice and found that knockdown of RIP3 in HCC cells increased the number of MDSCs in the tumors as represented by CD11b+Gr‐1+ myeloid progenitors (upper panel, Fig. 3C) but reduced the CD8+ T‐cell population (middle panel, Fig. 3C). Moreover, the percentage of IFN‐γ‐positive CD8+ tumor‐infiltrating lymphocytes significantly decreased in Hepa1‐6–shRIP3 models, suggesting that RIP3 deficiency increases MDSC infiltration and suppresses antitumor immunity (lower panel, Fig. 3C). Microscopically, Hepa1‐6–shRIP3 tumors were associated with decreased CD8+ tumor‐infiltrating lymphocytes and increased infiltration by Gr‐1‐positive or CD11b‐positive MDSCs, which was consistent with flow‐cytometric analyses of immune cells in orthotopic tumors (Fig. 3D). Next, we investigated whether the accumulation of MDSCs was derived from an increase of proliferation or a decrease of apoptosis. No significant increase of MDSC proliferation was observed in Hepa1‐6–shRIP3‐bearing mice or in GSK’872 (a specific RIP3 inhibitor)‐treated mice (Fig. 3E). Similarly, no significant decrease was observed in an MDSC apoptosis assay (Fig. 3F). These results suggest that hepatic RIP3 deficiency promoted MDSC‐enhanced chemotaxis but not increased proliferation or decreased apoptosis.
To further directly investigate whether hepatic RIP3 deficiency recruits MDSCs in vitro, we established a coculture system with Transwell membranes and performed a chemotaxis assay. We placed the CM generated from Hepa1‐6–short hairpin negative control (shNC) and Hepa1‐6–shRIP3 cells in the bottom wells of the Matrigel‐coated Transwell inserts as chemoattractants. The upper chambers were seeded with freshly isolated splenic MDSCs from HCC‐bearing mice (Fig. 4A). Chemotaxis assays showed that the migration index of MDSCs was significantly increased following coculture with Hepa1‐6–shRIP3 cells (Fig. 4B). Consistent with these findings, blockade of RIP3 by its specific antagonist GSK’872 also increased the migration index of MDSCs compared with placebo (Fig. 4C). Taken together, these results suggest that hepatic RIP3 deficiency promoted MDSC chemotaxis.
Figure 4.
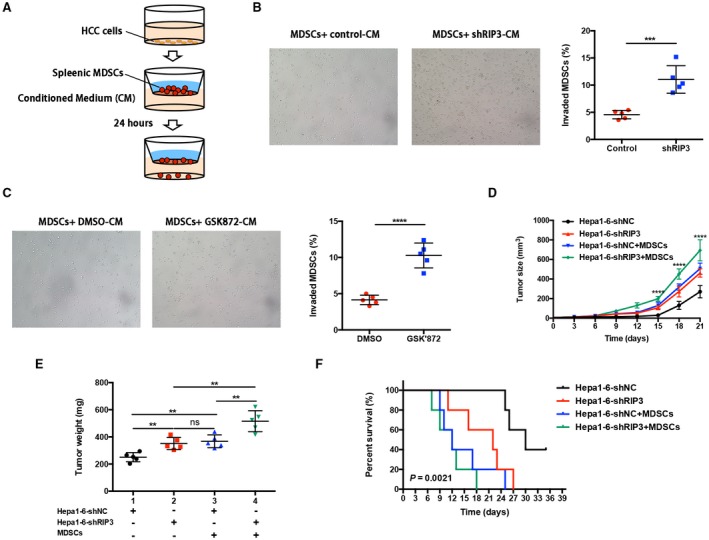
Hepatic RIP3 deficiency drives MDSC recruitment to promote tumor progression. (A) Matrigel invasion assay of MDSCs. (B) CM collected from Hepa1‐6–shNC and Hepa1‐6–shRIP3 clones or (C) from Hepa1‐6–shNC added by GSK’872 or dimethyl sulfoxide were placed in the lower chambers. Freshly isolated splenic MDSCs were seeded in the upper chambers and allowed to invade for 24 hours. Total numbers of invaded MDSCs found in the lower chambers were counted. ***P < 0.001, ****P < 0.0001. (D) HCC and MDSC coinoculation experiment. C57BL/6 mice were subcutaneously inoculated with 2 × 105 mouse HCC cells (Hepa1‐6) alone, 2 × 105 Hepa1‐6, and 3 × 104 MDSCs (HCC+MDSCs) (n = 5 for each group). Tumor size was monitored with a caliper, and the tumor growth curve was plotted. ****P < 0.0001. (E) Tumor weight after sacrifice. **P < 0.01. (F) The probability of overall survival was significantly different in four HCC and MDSC coinoculation groups. Abbreviation: ns, nonsignificant.
To investigate whether RIP3 deficiency promotes tumor growth through accumulation of tumor‐infiltrating MDSCs in vivo, we adoptively transferred tumor‐infiltrating MDSCs and HCC cells into murine subcutis and compared the tumor growth rate and tumor weight among (1) Hepa1‐6–shNC, (2) Hepa1‐6–shRIP3, (3) Hepa1‐6–shNC and tumor‐infiltrating MDSCs, (4) Hepa1‐6–shRIP3 and tumor‐infiltrating MDSCs (5:1 ratio) (Supporting Fig. S1B). By adoptive transfer, we found that coinjection of RIP3 knockdown HCC cells and MDSCs intensely promoted the growth rate (Fig. 4D) and tumor weight (Fig. 4E) of HCC. Meanwhile, the survival analysis revealed that mice bearing Hepa1‐6–shRIP3 cells combined with MDSCs achieved the poorest survival rates compared with other groups (Fig. 4F). Compared with mice bearing Hepa1‐6–shNC cells, the tumor growth rate was significantly higher in mice bearing Hepa1‐6–shNC cells combined with MDSCs, and the survival rate was significantly lower. Similarly, compared with mice bearing Hepa1‐6–shRIP3 cells, the tumor growth rate was significantly higher in mice bearing Hepa1‐6–shRIP3 cells combined with MDSCs, and the survival rate was significantly lower. Taken together, these data converged to demonstrate that hepatic RIP3 deficiency promoted MDSC recruitment and HCC progression.
RIP3 Deficiency Up‐Regulates CXCL1 and Induces MDSC Chemotaxis through the Cognate Receptor CXCR2
The MDSC invasion assay showed that CM from RIP3 knockdown cells possesses effective chemotactic ability for MDSCs. We established RIP3 KO mice with a C57BL/6 background. RIP3 KO mice and littermates were injected with a single dose of diethylnitrosamine (DEN; 25 mg/kg) on postnatal day 14. We regularly sacrificed one cohort of mice after DEN injection every 2 months to monitor the development of HCC. By gene ontology term cluster analysis using an Agilent microarray, we found that the genes and pathways involving chemotaxis and cell movement were significantly dysregulated in RIP3 KO mouse tumor tissues induced by DEN (left panel, Fig. 5A). To further identify the responsible chemokines or cytokines regulated by RIP3, we used a PCR array to quantify the expression of a panel of cytokines and chemokines in Hepa1‐6 cells. Of all the factors evaluated, CXCL1 was one of the most abundantly expressed chemokines compared with any others induced by RIP3 deficiency (right panel, Fig. 5A). To further investigate the correlation between RIP3 and CXCL1 in human HCC samples, we examined the expression levels of RIP3 and CXCL1 and found a significant negative correlation (Fig. 5B). The fact that CXCL1 was induced by RIP3 deficiency was confirmed by quantitative real‐time PCR and enzyme‐linked immunosorbent assay (ELISA) in two human HCC cell lines, Huh‐7 and MHCC97H (Fig. 5C).
Figure 5.
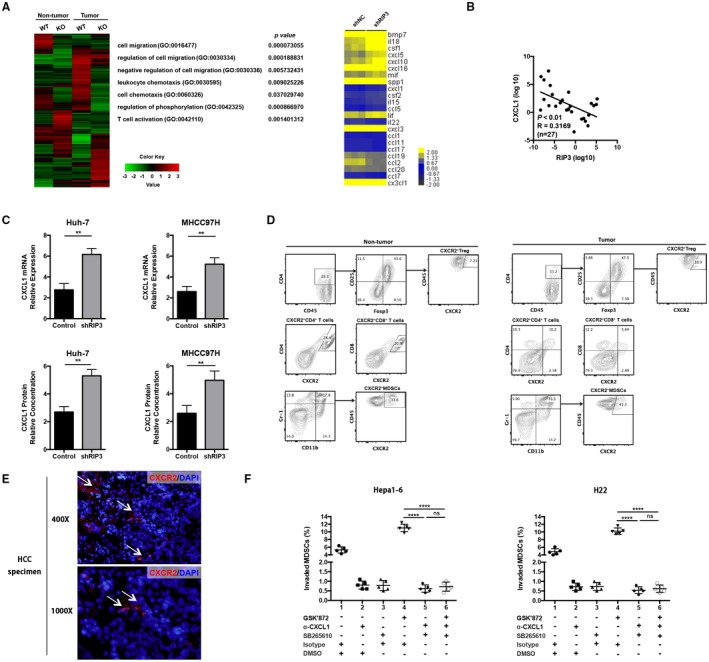
RIP3 deficiency up‐regulates CXCL1 and induces MDSC chemotaxis through the cognate receptor CXCR2. (A) Gene microarray imaging (left panel) and RT2 profiler PCR array (right panel) were performed for screening dysregulated hepatoma‐derived genes and pathways. The top 10 variable genes were listed. (B) Correlation analysis between RIP3 and CXCL1 by quantitative real‐time PCR using an independent cohort of 27 patients. (C) RT‐PCR and ELISA of human HCC cell lines Huh‐7‐shNC and Huh‐7‐shRIP3 (left panel) and MHCC97H‐shNC and MHCC97H‐shRIP3 (right panel). **P < 0.01. (D) Representative flow‐cytometric images of CXCR2+ cells in Tregs, CD4+ T cells, CD8+ T cells, and MDSCs. (E) Representative immunofluorescence of CXCR2+ cells in human HCC tissues. Upper, ×400; lower, ×1,000. (F) Chemotaxis response of murine MDSCs treated with GSK’872, α‐CXCL1, and SB265610 in the presence of Hepa1‐6 or H22 CM, respectively. Left, Hepa1‐6; right, H22. Abbreviations: DAPI, 4′,6‐diamidino‐2‐phenylindole; DMSO, dimethyl sulfoxide; GO, gene ontology; ns, nonsignificant.
To further delineate the possible crosstalk between HCC cells and MDSCs, we next investigated whether MDSCs express CXCR2, the cognate receptor for CXCL1. Flow‐cytometric analysis showed that the expression level of CXCR2 of MDSCs was more abundant than that of Tregs, CD4+ T cells, and CD8+ T cells in both tumor and nontumor tissues (Fig. 5D). Immunofluorescence analysis also showed that CXCR2+ cells existed in HCC tumors (Fig. 5E). We further investigated the chemotactic ability of CXCL1 and CXCR2 for MDSCs by chemotaxis assay. We found that CXCL1 was capable of recruiting MDSCs, whereas the chemotactic ability was drastically deprived by treatment with CXCL1 neutralizing antibody. A CXCR2 antagonist, SB265610, also significantly inhibited MDSC chemotaxis (Fig. 5F). Taken together, RIP3 deficiency up‐regulates CXCL1 and induces MDSC chemotaxis through the cognate receptor CXCR2.
RIP3 Deficiency Induces Nuclear Translocation of p‐P65SER536 and the Binding of p‐P65SER536 to the CXCL1 Promoter to Increase the Transcription of CXCL1
We next elucidated the molecular mechanism underlying RIP3‐mediated CXCL1 production. We first predicted the potential transcriptional factors binding to the CXCL1 promoter by using Genomatix and identified nuclear factor kappa B (NF‐κB, P65) as the most potent candidate. Indeed, knockdown of RIP3 markedly increased the phosphorylation level of P65 at Ser536 (p‐P65Ser536) in both Huh‐7 and MHCC97H cells, while the phosphorylation level of NFKB inhibitor alpha (IKBα) as well as the basal levels of P50 and P65 did not change (Fig. 6A). These results indicate that RIP3 deficiency may induce serine phosphorylation of P65 in HCC cells. Transcription factors might play roles through their translocation from the endochylema to the nucleus. Therefore, we treated Huh‐7–shRIP3 and MHCC97H–shRIP3 cells with 4‐methyl‐1‐N‐(3‐phenylpropyl)benzene‐1,2‐diamine (JSH‐23), a small molecule compound that interferes with NF‐κB nuclear translocation. We found that JSH‐23 abrogated both RIP3‐meidated CXCL1 transcription and expression (upper panel, Fig. 6B). Consistent data were obtained when we treated the Huh‐7 and MHCC97H cells with both JSH‐23 and GSK’872 (lower panel, Fig. 6B). Moreover, we performed immunofluorescence staining for p‐P65 and found that the p‐P65 fluorescence was strong and predominantly localized to the nucleus in GSK’872‐treated HCC cells (Fig. 6C). In addition, cytoplasmic and nuclear proteins were extracted and analyzed. These results confirmed that P65 phosphorylation significantly increased in the nucleus of RIP3 knockdown HCC cells (Fig. 6D). Collectively, these results indicate that RIP3 deficiency induces P65 serine phosphorylation and nuclear localization in human HCC cells.
Figure 6.
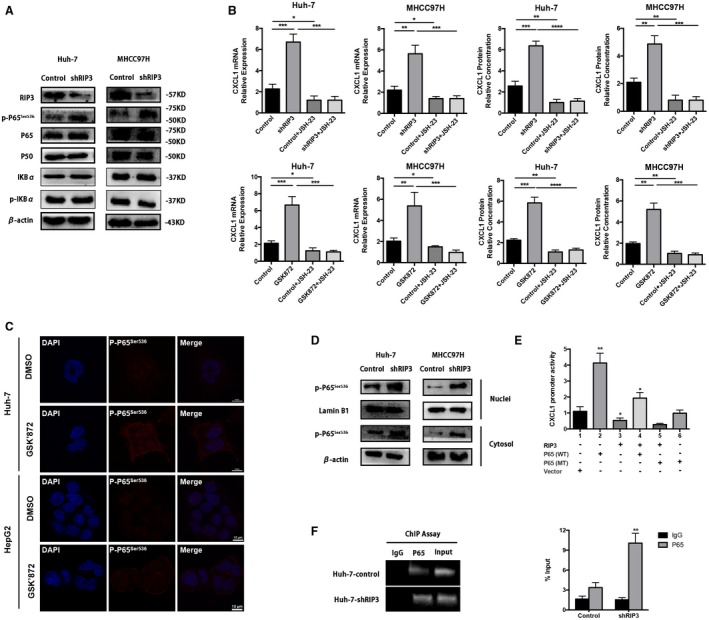
RIP3 deficiency induces nuclear translocation of p‐P65Ser536 and the binding of p‐P65Ser536 to the CXCL1 promoter to increase the transcription of CXCL1. (A) Western blotting of RIP3, p‐P65Ser536, P65, P50, IKBα, and p‐IKBα from Huh‐7 and MHCC97H cells. β‐Actin was used as control. (B) RT‐PCR and ELISA of human HCC cell lines treated by JSH‐23. Upper panel, Huh‐7–shNC, Huh‐7–shRIP3, MHCC97H–shNC, and MHCC97H–shRIP3; lower panel, Huh‐7, Huh‐7‐GSK’872, MHCC97H, and MHCC97H‐GSK’872. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. (C) Immunofluorescence of p‐P65Ser536 of Huh‐7 or HepG2 treated by GSK’872 at 30 μM concentration for 24 hours. (D) P65 phosphorylation levels were tested in the nuclear and cytoplasmic fractions of Huh‐7–shRIP3 and MHCC97H–shRIP3 cells. Protein expression levels in the nuclear and cytosolic fractions were normalized to lamin B1 and β‐actin, respectively. (E) The activity of the CXCL1 promoter was tested by dual Luc reporter assay in HEK‐293 cells. The CXCL1 promoter linked to the Luc promoter gene was cotransfected with either RIP3‐pcDNA3.1, P65 (WT)‐pcDNA3.1, Ser536 P65 (MT)‐pcDNA3.1 expression plasmids, or pcDNA3.1 empty vector, alone and in combination. At 48 hours after transfection, Luc activity was measured. *P < 0.05, **P < 0.01. (F) ChIP assay using antibody against P65 was performed in Huh‐7 cells. No antibody was used as a blank control. Normal mouse IgG was used as a negative control, and input indicates 5% input DNA, a positive amplification control. The fragment amplified by the CXCL1 promoter‐specific primers contains the P65 binding sequence, whereas the control PCR that generated the fragment does not contain any P65 binding sequence. Quantitative ChIP was also performed. **P < 0.01. Abbreviations: DAPI, 4′,6‐diamidino‐2‐phenylindole; DMSO, dimethyl sulfoxide.
To detect whether RIP3 deficiency–promoted CXCL1 induction was mediated through P65, a CXCL1 (–1099/+101)/Luc promoter construct was cotransfected with RIP3 or P65 expression plasmids, alone or in combination. We observed that P65 alone stimulated CXCL1 (–1099/+101)/Luc reporter gene activity but that RIP3 alone suppressed the CXCL1 (–1099/+101) promoter activity. However, the combination of both RIP3 and P65 resulted in a decrease of CXCL1 (–1099/+101)/Luc reporter gene activity compared with P65 alone (lanes 2‐4, Fig. 6E). This result may indicate that RIP3 and P65 perform functionally inverse roles in enhancing the CXCL1 promoter activity. The inverse functions suggest a molecular link between RIP3 and P65 in CXCL1 induction. The serine phosphorylation motif at residues 536 of P65 is critical for transcriptional activity of P65. To further address whether P65 is required for RIP3‐mediated CXCL1 induction, we cotransfected the CXCL1 (–1099/+101)/Luc promoter construct and the RIP3 expression plasmid together with either P65 (WT) or Ser536 mutated P65 (MT) expression plasmid. Compared with RIP3 cotransfected with P65 (WT), RIP3 cotransfected with P65 (MT) suppressed CXCL1 promoter activation (Fig. 6E, lanes 4 and 5). The inhibitory effect of P65 (MT) on CXCL1 promoter activation may indicate that RIP3‐mediated CXCL1 induction is induced by P65.
To directly elucidate the effects of RIP3 on the binding of P65 to the CXCL1 promoter, ChIP assays were performed in Huh‐7 cells. We found that knockdown of RIP3 markedly increased binding activity of P65 to the CXCL1 promoter (Fig. 6F). These findings suggest that RIP3 deficiency may increase the binding activity of P65 to the CXCL1 promoter. Taken together, RIP3 deficiency induces nuclear translocation of p‐P65Ser536 and the binding of p‐P65Ser536 to the CXCL1 promoter to increase the transcription of CXCL1.
Combination of RIP3 and CXCR2 Blockade Inhibits MDSC Chemotaxis to the Tumor
We have so far demonstrated that RIP3 deficiency induces MDSC chemotaxis by up‐regulating the cognate ligand of CXCR2, to suppress CD8+ T‐cell antitumor immunity to promote HCC immune escape. Next, we used HCC‐bearing mouse models to analyze the in vivo function of MDSC blockade with a CXCR2 antagonist. Hepa1‐6 cells stably overexpressing RIP3 (Hepa1‐6–RIP3) and control vector‐transfected Hepa1‐6 cells (Hepa1‐6–control) were subcutaneously inoculated in C57BL/6 mice to establish HCC‐bearing mouse models. MDSC depletion in vivo using an anti‐Gr‐1‐antibody inhibited tumor growth in Hepa1‐6–control tumor‐bearing mice but not in Hepa1‐6–RIP3 tumor‐bearing mice (Fig. 7A). Flow‐cytometric analysis showed that the frequencies of tumor‐infiltrating MDSCs decreased and that the ratio of CD8+ T cells to MDSCs increased in anti‐Gr‐1 antibody‐treated Hepa1‐6–control tumors (Fig. 7B). However, MDSC depletion by anti‐Gr‐1 antibody did not affect the frequencies of tumor‐infiltrating MDSCs of Hepa1‐6–RIP3 tumor‐bearing mice (Fig. 7C). Furthermore, blockade of MDSC chemotaxis in vivo using a CXCR2 antagonist (SB265610) markedly inhibited tumor growth in Hepa1‐6–control tumor‐bearing mice but not in Hepa1‐6–RIP3 tumor‐bearing mice (Fig. 7D). We also found that the frequencies of tumor‐infiltrating MDSCs decreased and that the ratio of CD8+ T cells to MDSCs increased in SB265610‐treated Hepa1‐6–control tumors (Fig. 7E). Notably, there were no significant differences in the frequencies of MDSCs and the ratio of CD8+ T cells to MDSCs when Hepa1‐6–RIP3 tumor‐bearing mice were treated with the CXCR2 antagonist (Fig. 7F). These results suggest that the antagonizing CXCR2 may reverse immunosuppression by inhibiting MDSC chemotaxis to the tumor, thereby facilitating antitumor immunity of CD8+ T cells to inhibit tumor immune escape.
Figure 7.
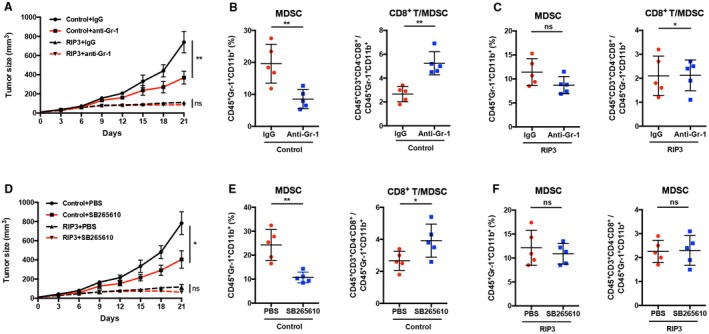
Blockade of CXCR2 reverses MDSC chemotaxis mediated by RIP3 deficiency to the tumor. (A) Tumor growth in mice subcutaneously injected with Hepa‐1‐6–control cells or Hepa‐1‐6–RIP3 cells, treated with anti‐Gr‐1 antibody (2 mg/kg) or IgG twice a week from day 1 after tumor inoculation. P values represent significance between two groups at day 21 (n = 5). (B) Flow‐cytometric analyses of subcutaneous Hepa‐1‐6–control cells treated with anti‐Gr‐1 antibody or IgG at day 21. Percentage of positive cells relative to total cells is plotted. MDSCs (left) and CD8+ T cells/MDSCs (right) (n = 5). (C) Flow‐cytometric analyses of subcutaneous Hepa‐1‐6–RIP3 cells treated with anti‐Gr‐1 antibody or IgG at day 21. The percentage of positive cells relative to total cell count is plotted. MDSCs (left) and CD8+ T cells/MDSCs (right) (n = 5). (D) Tumor growth in mice subcutaneously injected with Hepa‐1‐6–control cells or Hepa‐1‐6–RIP3 cells, treated with SB265610 (2 mg/kg body weight) or PBS six times a week from day 1 after tumor inoculation. P values represent significance between two groups at day 21 (n = 5). (E) Flow‐cytometric analyses of subcutaneous Hepa‐1‐6–control tumors treated with SB265610 or PBS at day 21. Percentage of positive cells relative to total cells is plotted. MDSCs (left) and CD8+ T cells/MDSCs (right) (n = 5). (F) Flow‐cytometric analyses of subcutaneous Hepa‐1‐6–RIP3 cells treated with SB265610 or PBS at day 21. The percentage of positive cells relative to total cell count is plotted. MDSCs (left) and CD8+ T cells/MDSCs (right) (n = 5). *P < 0.05, **P < 0.01. Abbreviation: ns, nonsignificant.
To further confirm the effects of RIP3 deficiency on MDSC accumulation through up‐regulation of CXCR2 ligand, we established RIP3 KO mouse HCC models induced by DEN. Since postnatal month 8, we treated the mice daily with either PBS or SB265610 to block the CXCL1/CXCR2 crosstalk between hepatoma cells and MDSCs for a consecutive month (Fig. 8A). Blockade of MDSC chemotaxis by antagonizing CXCR2 significantly decreased tumor weight and number in both WT mice and KO mice, respectively. However, no marked difference was observed in SB265610‐treated WT and KO mice (Fig. 8B, C). We also found that the frequencies of tumor‐infiltrating MDSCs decreased, and the ratio of CD8+ T cells to MDSCs increased in both WT mice and KO mice (Fig. 8D). These results indicate that combination of RIP3 and CXCR2 blockade inhibits MDSC chemotaxis to the tumor.
Figure 8.
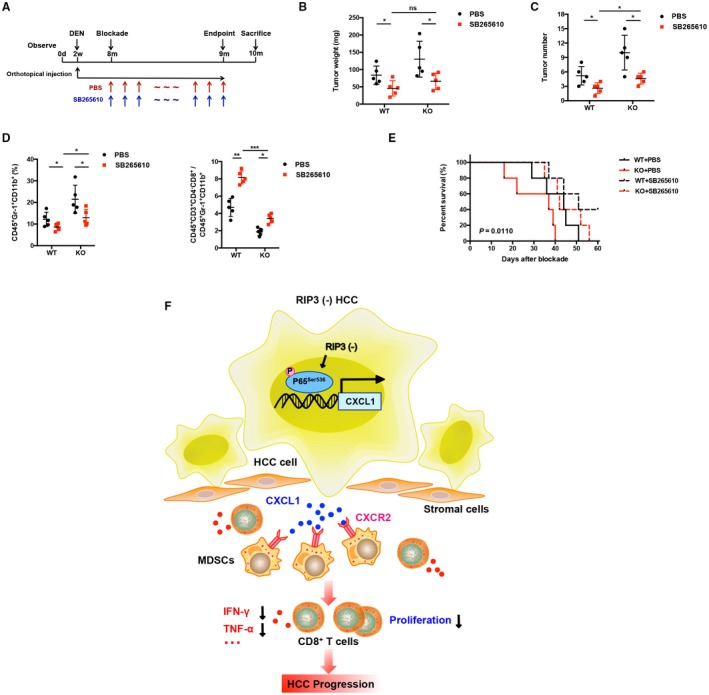
Blockade of CXCR2 reverses MDSC chemotaxis mediated by RIP3 deficiency to the tumor. (A) Flowchart of DEN (25 mg/kg)–induced HCC models in RIP3 KO mice and littermates. SB265610 (2 mg/kg body weight) or PBS was injected intraperitoneally every day since the eighth month after birth. (B) Tumor weight and (C) tumor number were monitored (left, weight; right, number; n = 5). *P < 0.05. (D) Flow‐cytometric analyses of DEN‐induced HCC of RIP3 KO mice or littermates treated by SB265610 or PBS. Percentage of positive cells relative to total cells is plotted. MDSCs (left) and CD8+ T cells/MDSCs (right) (n = 5). *P < 0.05, **P < 0.01, ***P < 0.001. (E) The probability of overall survival was significantly different in DEN‐induced HCC of RIP3 KO mice or littermates treated by SB265610 or PBS. (F) Schematic representation of RIP3 signaling in HCC immune evasion. Hepatic RIP3 deficiency promotes HCC immune escape by up‐regulation of CXCL1 and recruitment of MDSCs through CXCR2, the exclusive cognate receptor of CXCL1. RIP3 inhibits CXCL1 expression by disrupting phosphorylation of P65Ser536 and inhibiting p‐P65Ser536 nuclear translocation, as well as its direct co‐occupancy of CXCL1 promoter. Abbreviation: ns, nonsignificant.
We also investigated the survival rate of mice bearing HCC induced by DEN after treatment with a CXCR2 antagonist (Fig. 8E). We identified the date of SB265610 treatment as the outset and the date of sacrifice as the deadline. With the exception of WT mice treated with SB265610, the other three groups exclusively uniformly succumbed to HCC within 60 days. The KO mice treated with PBS yielded the poorest survival advantages, while treatment with SB265610 in KO mice demonstrated enhanced efficacy, with all mice surviving more than 30 days. And WT mice treated with SB265610 achieved the best survival rates among all the groups. Overall, these data demonstrate that blockade of CXCR2 increases the survival rate in RIP3 KO mice bearing HCC induced by DEN.
Discussion
Cancer immunotherapy can induce durable antitumor responses and long‐term remission in patients with advanced cancers within a wide range of histologies. However, the response rate of cancer immunotherapy is still limited.17, 18, 19, 20 As suppressive myeloid cells have emerged as a major barrier of antitumor immunity in HCC,21 we focused on myeloid cells within the HCC microenvironment and investigated the mechanisms of MDSC recruitment. In this study, we found that RIP3 expression is decreased in HCC patients, which correlates with MDSC infiltration and poor prognosis. RIP3 deficiency is an essential factor directing MDSC homing to HCC. RIP3 deficiency promotes CXCL1/CXCR2‐induced MDSC chemotaxis and reduces the infiltration of IFN‐γ+ CD8+ T cells, thus facilitating immune escape and HCC progression (Fig. 8F).
RIP3 is well acknowledged as a molecular switch from apoptosis to necroptosis, which is triggered by caspase‐8 inhibition. Recently, RIP3 has been reported to play essential roles in liver injury and inflammatory hepatocarcinogenesis. RIP3 protects mice from high‐fat diet–induced liver injury, associated with decreased inflammation and hepatocyte apoptosis, as well as early fibrotic responses.14 RIP3 is activated in chronically inflamed livers, impedes caspase‐8‐dependent chromosomal aberrations, and inhibits HCC growth.15 Loss of RIP3 was common in the majority of cancers, but its significance and molecular mechanisms remain unclear. In this study, we reported the immunological effect of hepatic RIP3 deficiency on the tumor microenvironment in HCC.
MDSCs, a heterogeneous population of myeloid progenitors derived from the bone marrow into tumor sites, represent the major immunosuppressive population that exists only in pathological conditions such as chronic inflammation and cancer.22 MDSCs abundantly accumulate in human HCC, which is recognized as a major mechanism adopted by cancers to escape immune surveillance.23 More importantly, they are functionally regarded as major suppressors of T‐cell cytotoxicity. The generation and accumulation of these immature myeloid cells within the tumor sites inhibit CD8+ T‐cell proliferation and cytotoxicity by depriving amino acids through arginase‐1 expression, releasing oxidizing molecules, and inducing other immunosuppressive cells such as tumor‐associated macrophages and Tregs.22 Expansion of MDSCs is a major mechanism used by cancers to escape immune surveillance. In this study, we demonstrated that MDSCs accumulated in the peripheral blood or tumor tissues and were associated with poor prognosis in patients with HCC. MDSCs accounted for up to about 24% of total immune cells in the peripheral blood and 39% of total immune cells in the tumor. Further, we found that tumor‐infiltrating MDSCs exert potent CD8+ T‐cell suppression in HCC, which is consistent with previous studies.24
In the present study, we found that the expression level of RIP3 is negatively associated with MDSC infiltration in human HCC tissues. Besides, low expression of RIP3 and overexpression of MDSC markers were associated with clinicopathological characteristics and prognosis of HCC patients. Patients with low expression of RIP3 or high infiltration of MDSCs had significantly poor prognosis. Orthotopic HCC models demonstrated that hepatic RIP3 deficiency increases the frequencies of tumor‐infiltrating MDSCs but decreases the frequencies of IFN‐γ+CD8+ T cells. Thus, we next confirmed that hepatic RIP3 deficiency induces MDSC recruitment using the Transwell assay in vitro, not by increased proliferation or decreased apoptosis. Together, these data demonstrate that hepatic RIP3 deficiency promotes MDSC recruitment.
Chemotaxis plays a crucial role in orchestrating the recruitment and trafficking of MDSCs into tumor sites through the interaction of chemokines and their cognate receptors.25, 26, 27 In CXC chemokine subfamilies, CXCL6 and CXCL8 can bind to both CXCR1 and CXCR2, while the other CXC chemokine ligands (CXCL1, CXCL2, CXCL3, CXCL5, and CXCL7) can only bind to and activate the receptor CXCR2, owing to a Glu‐Leu‐Arg (ELR) motif at their NH2 terminus.28 As a typical inflammation‐related cancer, HCC is characterized by aberrant secretion of cytokines and chemokines, as well as recruitment and infiltration of immune cells within tumor sites.5, 29, 30, 31 In our microarray data, chemotaxis‐associated pathways were significantly regulated by RIP3. By cytokine and chemokine PCR array analysis, we characterized RIP3 deficiency as a chemotaxis inducer that drives MDSC recruitment through up‐regulation of CXCL1, which is the major chemokine that attracts MDSCs to the tumor through CXCR2, the exclusive cognate receptor of CXCL1. In addition, we evaluated other immune cells in tumor and nontumor tissue of HCC‐bearing mice. Flow‐cytometric analysis confirmed that the proportions of CXCR2+ MDSCs were significantly higher than CXCR2+ Tregs, CXCR2+CD4+ T cells, and CXCR2+CD8+ T cells in both tumor and nontumor tissues. These results showed that the MDSC population displayed the highest CXCR2 level among the immune cells infiltrating in fresh HCC tissues, which was the dominant subset responsible for immune escape in HCC microenvironment.
Previous studies have demonstrated that Gr‐1 neutralizing antibody can effectively deplete MDSCs in vivo, but absence of Gr‐1 in human urges us to discover more surface markers of MDSCs for clinical implications.32 In this study, we found that blockade of MDSC chemotaxis by antagonizing CXCR2 can be one of the targets for immunotherapy. Moreover, our study shows that RIP3 deficiency is a key driver of MDSC recruitment and provides a strong rationale for the application of CXCR2 antagonists in HCC immunotherapy. The expression level of RIP3 in tumor tissue might be a predictor for clinical prognosis and immunotherapy effect in HCC patients. More importantly, targeting RIP3 in HCC might be considered for rational use of chemotherapeutic and targeted drugs, which is essential for reeducating the immunosuppressive tumor microenvironment and enhancing the antitumor immunity. Accordingly, blocking the RIP3–CXCL1–CXCR2 axis can be an efficacious strategy for HCC patients to prevent tumor immune escape. Larger cohort studies using inhibitors might confirm the role of the RIP3–CXCL1–CXCR2 axis in HCC immunotherapy.
In conclusion, we found that loss of RIP3 is common in most HCC patients and is a driver of MDSC recruitment. RIP3 deficiency promotes CXCL1/CXCR2‐induced MDSC chemotaxis to facilitate immune escape and HCC progression. By phosphorylating P65Ser536 and promoting p‐P65Ser536 nuclear translocation, RIP3 deficiency increases the expression of CXCL1 in HCC cells. A CXCR2 antagonist suppresses MDSC infiltration and delays HCC growth in RIP3 KO mice. Our findings not only illustrate the mechanism of RIP3‐mediated immune evasion by recruiting MDSCs to local inflamed tissues but also show that blocking MDSC recruitment may be a promising immunological therapeutic approach to protect against progression of RIP3 deficiency HCC.
Supporting information
Supported by grants from the National Natural Science Foundation of China (81472700 and 81872316), the Shaanxi Science and Technology New Star Project (2017KJXX‐68), and the National Science and Technology Major Special Project of China (2018ZX09101001).
Potential conflict of interest: Nothing to report.
Contributor Information
Juan Tang, Email: tangjuan1@fmmu.edu.cn.
Zhi‐Nan Chen, Email: znchen@fmmu.edu.cn.
References
Author names in bold designate shared co‐first authorship.
- 1. Islami F, Miller KD, Siegel RL, Fedewa SA, Ward EM, Jemal A. Disparities in liver cancer occurrence in the United States by race/ethnicity and state. CA Cancer J Clin 2017;67:273‐289. [DOI] [PubMed] [Google Scholar]
- 2. Llovet JM, Villanueva A, Lachenmayer A, Finn RS. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat Rev Clin Oncol 2015;12:408‐424. [DOI] [PubMed] [Google Scholar]
- 3. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 4. Liu LZ, Zhang Z, Zheng BH, Shi Y, Duan M, Ma LJ, et al. CCL15 recruits suppressive monocytes to facilitate immune escape and disease progression in hepatocellular carcinoma. Hepatology 2019;69:143‐159. [DOI] [PubMed] [Google Scholar]
- 5. Giannelli G, Rani B, Dituri F, Cao Y, Palasciano G. Moving towards personalised therapy in patients with hepatocellular carcinoma: the role of the microenvironment. Gut 2014;63:1668‐1676. [DOI] [PubMed] [Google Scholar]
- 6. Gabrilovich DI, Nagaraj S. Myeloid‐derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009;9:162‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Veglia F, Perego M, Gabrilovich D. Myeloid‐derived suppressor cells coming of age. Nat Immunol 2018;19:108‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Marvel D, Gabrilovich DI. Myeloid‐derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest 2015;125:3356‐3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li J, Lee Y, Li Y, Jiang Y, Lu H, Zang W, et al. Co‐inhibitory molecule B7 superfamily member 1 expressed by tumor‐infiltrating myeloid cells induces dysfunction of anti‐tumor CD8+ T cells. Immunity 2018;48:773‐786. [DOI] [PubMed] [Google Scholar]
- 10. Turley SJ, Cremasco V, Astarita JL. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat Rev Immunol 2015;15:669‐682. [DOI] [PubMed] [Google Scholar]
- 11. Wan S, Kuo N, Kryczek I, Zou W, Welling TH. Myeloid cells in hepatocellular carcinoma. Hepatology 2015;62:1304‐1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schupp J, Krebs FK, Zimmer N, Trzeciak E, Schuppan D, Tuettenberg A. Targeting myeloid cells in the tumor sustaining microenvironment. Cell Immunol 2017. 10.1016/j.cellimm.2017.10.013. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 13. Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. RIP3, an energy metabolism regulator that switches TNF‐induced cell death from apoptosis to necrosis. Science 2009;325:332‐336. [DOI] [PubMed] [Google Scholar]
- 14. Roychowdhury S, McCullough RL, Sanz‐Garcia C, Saikia P, Alkhouri N, Matloob A, et al. Receptor interacting protein 3 protects mice from high‐fat diet–induced liver injury. Hepatology 2016;64:1518‐1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vucur M, Reisinger F, Gautheron J, Janssen J, Roderburg C, Cardenas DV, et al. RIP3 inhibits inflammatory hepatocarcinogenesis but promotes cholestasis by controlling caspase‐8‐ and JNK‐dependent compensatory cell proliferation. Cell Rep 2013;4:776‐790. [DOI] [PubMed] [Google Scholar]
- 16. Nakatsukasa H, Zhang D, Maruyama T, Chen H, Cui K, Ishikawa M, et al. The DNA‐binding inhibitor Id3 regulates IL‐9 production in CD4+ T cells. Nat Immunol 2015;16:1077‐1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature 2016;539:443‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moynihan KD, Opel CF, Szeto GL, Tzeng A, Zhu EF, Engreitz JM, et al. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat Med 2016;22:1402‐1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sharma P, Allison JP. The future of immune checkpoint therapy. Science 2015;348:56‐61. [DOI] [PubMed] [Google Scholar]
- 20. Pitt JM, Vetizou M, Daillere R, Roberti MP, Yamazaki T, Routy B, et al. Resistance mechanisms to immune‐checkpoint blockade in cancer: tumor‐intrinsic and ‐extrinsic factors. Immunity 2016;44:1255‐1269. [DOI] [PubMed] [Google Scholar]
- 21. Mizukoshi E, Yamashita T, Arai K, Terashima T, Kitahara M, Nakagawa H, et al. Myeloid‐derived suppressor cells correlate with patient outcomes in hepatic arterial infusion chemotherapy for hepatocellular carcinoma. Cancer Immunol Immunother 2016;65:715‐725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gabrilovich DI, Ostrand‐Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 2012;12:253‐268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Kruger C, Manns MP, et al. A new population of myeloid‐derived suppressor cells in hepatocellular carcinoma patients induces CD4+CD25+Foxp3+ T cells. Gastroenterology 2008;135:234‐243. [DOI] [PubMed] [Google Scholar]
- 24. Zhou J, Liu M, Sun H, Feng Y, Xu L, Chan AWH, et al. Hepatoma‐intrinsic CCRK inhibition diminishes myeloid‐derived suppressor cell immunosuppression and enhances immune‐checkpoint blockade efficacy. Gut 2018;67:931‐944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chiu DK, Xu IM, Lai RK, Tse AP, Wei LL, Koh HY, et al. Hypoxia induces myeloid‐derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C‐C motif) ligand 26. Hepatology 2016;64:797‐813. [DOI] [PubMed] [Google Scholar]
- 26. Okuma A, Hanyu A, Watanabe S, Hara E. p16Ink4a and p21Cip1/Waf1 promote tumour growth by enhancing myeloid‐derived suppressor cells chemotaxis. Nat Commun 2017;8:2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ban Y, Mai J, Li X, Mitchell‐Flack M, Zhang T, Zhang L, et al. Targeting autocrine CCL5‐CCR5 axis reprograms immunosuppressive myeloid cells and reinvigorates antitumor immunity. Cancer Res 2017;77:2857‐2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rot A, von Andrian UH. Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol 2004;22:891‐928. [DOI] [PubMed] [Google Scholar]
- 29. Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol 2017;17:559‐572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marra F, Tacke F. Roles for chemokines in liver disease. Gastroenterology 2014;147:577‐594. [DOI] [PubMed] [Google Scholar]
- 31. Prieto J, Melero I, Sangro B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 2015;12:681‐700. [DOI] [PubMed] [Google Scholar]
- 32. Qin H, Lerman B, Sakamaki I, Wei G, Cha SC, Rao SS, et al. Generation of a new therapeutic peptide that depletes myeloid‐derived suppressor cells in tumor‐bearing mice. Nat Med 2014;20:676‐681. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials