

Abstract
Replicative senescence, which is induced by telomere shortening, underlies the loss of regeneration capacity of organs and is ultimately detrimental to the organism. At the same time, it is required to protect organisms from unlimited cell proliferation that may arise from numerous stimuli or deregulations. One important feature of replicative senescence is its high level of heterogeneity and asynchrony, which promote genome instability and senescence escape. Characterizing this heterogeneity and investigating its sources are thus critical to understanding the robustness of replicative senescence. Here we review the different aspects of senescence driven by telomere attrition that are subject to variation in Saccharomyces cerevisiae, the current understanding of the molecular processes at play, and the consequences of heterogeneity in replicative senescence.
Keywords: DNA damage checkpoint, heterogeneity, replicative senescence, telomerase, telomere
Here, we review the different facets of replicative senescence triggered by telomere erosion that are subject to variation in Saccharomyces cerevisiae. We decompose the molecular pathways involved in the generation of heterogeneity and discuss the consequences, in particular, as a threat to replicative senescence robustness. Many concepts discussed here likely apply to other eukaryotic cells in which telomerase is repressed physiologically.
Box: On population doublings and generations.
It is important to note that proliferation potential is defined for a population of cells, and the unit should be population doublings, which is equal to Log2(Nf/Ni) = Log2(Nf) – Log2(Ni), where Ni and Nf are the initial and final numbers of cells in the population, respectively. Because the structure of the population is heterogeneous (i.e., not all cells within the population divide at the same rate and some are arrested or dead), the number of population doublings can deviate substantially from the number of generations or divisions that individual cells actually undergo. Proliferation potential measured in population doublings is also different from the average number of divisions or generations, as cells compete and the fittest are selected. Conversion between population doublings and the average number of divisions or generations is in most cases nontrivial and non‐linear. Only in the case of a perfectly homogeneous population of cells in terms of generation time is the number of population doublings equivalent to the number of divisions or generations. To determine the actual number of divisions or generations cells undergo, we have to use more precise approaches, such as lineage tracking using microfluidics devices.
1. INTRODUCTION
Telomerase elongates telomeres, or the ends of linear chromosomes, and without it telomeres shorten with each cell division. As telomeres shorten, they are no longer able to prevent the ends of chromosomes from being recognized as accidental chromosomal breaks. As a consequence, cells permanently activate the DNA damage checkpoint and enter replicative senescence (d'Adda di Fagagna et al., 2003; Enomoto, Glowczewski, & Berman, 2002; Ijpma & Greider, 2003). This signalling cascade explains the correlation between average telomere length in humans and age: the number of cells in replicative senescence accumulates with age in somatic tissue of primates, in which telomerase expression is downregulated (Hastie et al., 1990; Jeyapalan, Ferreira, Sedivy, & Herbig, 2007). In turn, cancer precursor cells are rare cells that have bypassed replicative senescence (Shay & Wright, 2010). Hence, telomeres act as a molecular alarm clock for the enumeration of generations, and the homeostasis of many organs in humans depends on proper telomere shortening and establishment of replicative senescence. Yet, the predictive power of measuring biological age or cancer risk by telomere length is limited by heterogeneity in the phenotype of replicative senescence (Blackburn, 2000; Karlseder, Smogorzewska, & de Lange, 2002; Suram & Herbig, 2014). Even at the level of individual cells, there is great variation in the onset of senescence in response to telomere shortening. Thus, decomposing the sources and consequences of cell‐to‐cell variation inherent to telomeres and senescence is necessary to uncover the molecular basis of telomere control over the proliferation limit of cells. Budding yeast, in which the phenotype of telomerase inactivation has been studied at length, constitutes a sound model to contribute to such an aim.
Replicative senescence was initially described as the proliferation limit of primary human diploid cells cultivated in vitro (Hayflick, 1965; Hayflick & Moorhead, 1961). This finite lifetime was shown to be an intrinsic property of cells and not a technical issue related to in vitro culture. This discovery suggested that proliferation limits at the cellular level could underlie organismal and tissue ageing. Already at that time, important variation in the onset of senescence, which took place over a period of 1–3 months in these experiments, was observed (Jones, Whitney, & Smith, 1985; Smith & Hayflick, 1974; Smith & Whitney, 1980). Two decades later, a similar heterogeneous proliferation limit was reported in Saccharomyces cerevisiae mutants defective in telomere elongation (Lundblad & Szostak, 1989). At the population level, replicative senescence was described as a progressive decrease in growth rate and concomitant increase in cell death, but the variability in each of these two parameters at the level of the single cell was already appreciated. Although a defect in telomere maintenance clearly caused senescence, one could only speculate about the origin of the heterogeneity, and, for example, rapid telomere shortening, increased oxidative stress, and altered gene expression due to genome‐wide changes in chromatin structure were readily proposed to contribute to heterogeneity in senescence (Bahar et al., 2006; Passos et al., 2007).
We now have a more detailed picture of the molecular mechanisms at play when telomeres are not maintained, in particular in budding yeast, where they have been investigated at length (Wellinger & Zakian, 2012). In S. cerevisiae, telomerase can be inactivated by the deletion of the gene encoding one of its subunits (EST1, EST2, EST3, or TLC1), expression of a dominant negative form, repression of its expression, or a mutation affecting its catalytic activity. Without telomerase activity, and starting typically from an average length of 300–350 bp, telomeres shorten by an average of 3–4 bp per generation because the polymerases are unable to fully replicate linear DNA. This end replication problem is asymmetric in that only one of the two newly replicated telomeres is shorter than the parental telomere. On average, and when one considers successive divisions, the bulk of the telomeres shorten. When telomeres become too short, they trigger a response that is in many ways similar to the DNA damage response, activating the DNA damage checkpoint and arresting the cell in the G2/M transition of the cell cycle, which provides a mechanistic definition of replicative senescence (Teixeira, 2013). In budding yeast, although all 32 telomeres of haploid cells get progressively shorter as the cells divide, one critically short telomere is sufficient to trigger replicative senescence (Abdallah et al., 2009; Xu, Duc, Holcman, & Teixeira, 2013). At the onset of senescence, the average telomere length is ~100–120, but the shortest telomere is significantly shorter, possibly as short as ~20 bp based on mathematical modelling (Bourgeron, Xu, Doumic, & Teixeira, 2015). However, the exact state and threshold length for the shortest telomere are not known, and whether the threshold is clearcut or encompasses a probabilistic range of lengths remains to be investigated. Critically short telomeres being an atypical signal, the robustness of the ensuing DNA damage response and the cell fate decision might differ from other models of DNA damage. For example, repair pathways are mostly inhibited at functional telomeres but can be activated when telomeres are no longer maintained by telomerase (Claussin & Chang, 2015). This is exemplified in the extreme case of postsenescence survivors, which are able to maintain telomeres by recombination‐based mechanisms (Le, Moore, Haber, & Greider, 1999; Lundblad & Blackburn, 1993; Teng, Chang, McCowan, & Zakian, 2000). How short or damaged telomeres activate and maintain the DNA damage checkpoint, how cells decide to repair them, and how repair contributes to heterogeneity in replicative senescence are important questions that remain to be addressed in detail.
Here we review the different facets of heterogeneity in replicative senescence, starting with variation in proliferative potential. We then discuss the mechanisms in budding yeast known to affect and to control, at least partially, the extent of heterogeneity in senescence. Finally, we argue that heterogeneity has important consequences for senescence and other related phenomena. There are numerous points of comparison between replicative senescence in budding yeast and mammalian cells. We propose that many aspects discussed in this review could be conceptually applied to human senescence and genome instability in the early stages of tumorigenesis.
Another ageing phenomenon in budding yeast, referred to as mother cell ageing and corresponding to the limited number of daughter cells a single mother cell can produce, also imposes a proliferation limit called the replicative life span, which is unrelated to telomeres and telomerase (Denoth Lippuner, Julou, & Barral, 2014). Although mother cell ageing and replicative senescence induced by telomere shortening share some properties, including heterogeneity (Knorre, Azbarova, Galkina, Feniouk, & Severin, 2018), they have distinct mechanistic mechanisms.
2. VARIATION IN PROLIFERATION POTENTIAL
Proliferation potential (Box) is the most obvious measure for which replicative senescence appears to be heterogeneous. It was initially defined for primary human fibroblasts as the total number of population doublings achieved over several passages until the culture is no longer able to undergo at least one population doubling in 2 weeks (Smith & Whitney, 1980). In telomerase‐negative budding yeast cultures, a similar definition is used: Proliferation potential represents the total number of population doublings achieved in culture from the moment telomerase is inactivated until the emergence of postsenescence survivors, which efficiently take over the culture at later stages (Lundblad & Blackburn, 1993; Lundblad & Szostak, 1989; Singer & Gottschling, 1994). Around the time survivors emerge, cell cultures reach their lowest growth rate and highest death rate.
2.1. Interclonal variation in proliferation potential
Independent telomerase‐negative single cells, such as telomerase‐negative spores derived from a heterozygous diploid, display distinct proliferation potentials when propagated in culture (Enomoto, Glowczewski, & Berman, 2002; Lundblad & Szostak, 1989; Ritchie, Mallory, & Petes, 1999; Singer & Gottschling, 1994). The observed variation is striking and much greater than variation in common growth that arises naturally between biological replicates. Enomoto and colleagues noted that, in this experimental setting, “this variation was a property associated with each individual spore because early or late senescence of specific spore progeny was reproducible” (Enomoto, Glowczewski, & Berman, 2002). Thus, most interclonal variation in proliferation potential has a biological origin and is determined from the moment telomerase is inactive.
At the population level, replicative senescence is the result of the combination of a reduced rate of cell division and an increased rate of cell death. It is important to note that these two phenomena can be observed even shortly after telomerase inactivation and become widespread in the population later on (Enomoto, Glowczewski, & Berman, 2002; Lundblad & Szostak, 1989; Ritchie, Mallory, & Petes, 1999). Interclonal variation in proliferation potential can thus be explained by differences in either the timing or relative frequency of longer cell cycles and cell death.
2.2. Intraclonal variation in proliferation potential
Although the proliferation potential of a telomerase‐negative culture initiated from a single spore is reproducible when the initial colony is used to inoculate multiple cultures, some heterogeneity in growth is also observed within a culture. Because this heterogeneity appears in cultures inoculated by a single telomerase‐negative clone, it is referred to as intraclonal variation. For instance, when telomerase‐negative cells are restreaked on plates, especially at later passages, the colonies that form are of different sizes, which indicates that individual cells from the same culture have different growth potentials (Enomoto, Glowczewski, & Berman, 2002; Lundblad & Szostak, 1989). In addition, the edges of the colonies are not smooth, which suggests that even within a colony, different cell lineages have distinct growth and mortality rates.
When a pair of telomerase‐negative mother and daughter cells are separated by micromanipulation and grown independently, minor differences in proliferation potential between the two resulting cultures are observed (Xu, Duc, Holcman, & Teixeira, 2013). They are, however, much smaller than the differences observed between pairs of telomerase‐negative spores of the same tetrad (Figure 1a,b). In contrast, a similar experiment performed in human diploid fibroblasts revealed substantial intraclonal variation (Jones, Whitney, & Smith, 1985), which suggests that other factors, independent of the initial telomere content, must become prominent during the course of senescence of human fibroblasts ex vivo (Figure 1c).
Figure 1.
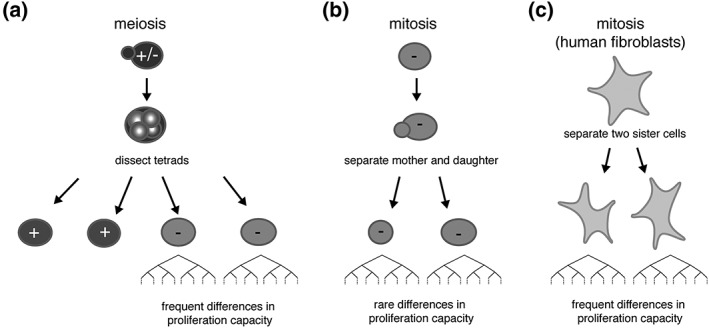
(a) Differences in proliferation observed in pairs of sister telomerase‐negative cells after budding yeast meiosis, (b) budding yeast mitosis, and (c) human fibroblast mitosis. In theory, the lengths of telomeres in a pair of mother and daughter cells after mitosis are similar (but not equal, see Figure 3). (a and b) In contrast, telomere lengths are more frequently different among meiotic products because of independent segregation of chromosomes in meiosis. Proliferation potential in the progenies of telomerase‐negative pairs of cells from a meiotic product is thus more variable than for telomerase‐negative pairs of mitotically divided mother and daughter cells (Enomoto, Glowczewski, & Berman, 2002; Xu, Duc, Holcman, & Teixeira, 2013). (c) In contrast, in human fibroblasts, the onset of senescence is variable among pairs of daughter cells derived from mitosis (Jones, Whitney, & Smith, 1985). This suggests that replicative senescence of human cells is accompanied by events that might amplify heterogeneity. – and + indicate telomerase negative and telomerase positive, respectively.
2.3. Heterogeneity at the level of single cells and individual lineages
Because proliferation potential is an aggregate endpoint measure of the whole history of a population of cells, it does not provide information on the dynamics of cell division that generate interclonal and intraclonal heterogeneity. For instance, any of the following explanations, or a combination of them, can account for the differences in colony size among subcloned telomerase‐negative yeast cells:
the average growth rates in the two colonies might differ;
the mortality rate might be heterogeneous within a colony, higher on average in one colony than the other;
cell cycle arrest might be more frequent in one colony than the other;
cells might reach permanent arrest (senescence) earlier on average in one colony than the other; or
the lag phase in the first cell division might be longer in one colony than the other.
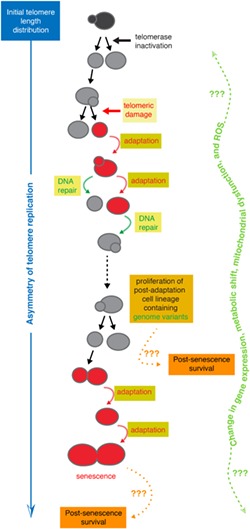
Previous studies have already pointed at several of these possibilities (Enomoto, Glowczewski, & Berman, 2002; Ijpma & Greider, 2003; Lundblad & Szostak, 1989; Ritchie, Mallory, & Petes, 1999). However, to fully characterize the types of heterogeneity responsible for macroscopic variation in colony size, we need to use micromanipulation methods or microfluidics‐based time‐resolved single‐cell approaches (Churikov, Charifi, Simon, & Geli, 2014; Ijpma & Greider, 2003; Lundblad & Szostak, 1989; Xie et al., 2015; Xu et al., 2015). In particular, tracking multiple independent lineages in microfluidics circuits has led to a comprehensive and detailed analysis of the dynamics of cell division from telomerase inactivation to permanent cell cycle arrest and cell death. Important variation in the number of generations an individual lineage undergoes before terminal arrest (i.e., replicative senescence) and cell death has been observed, ranging from ~10 to ~70 (median ± SD = 31 ± 13 generations, in a strain with an initial average telomere length of ~260 bp; Coutelier et al., 2018; Xu et al., 2015). Although many cell lineages switch abruptly from active cell division to two or three prolonged cell cycles before cell death, others exhibit intermittent and stochastic periods of cell cycle arrest, called nonterminal arrests, followed by the resumption of normal cell cycles even shortly after telomerase inactivation (Churikov, Charifi, Simon, & Geli, 2014; Xie et al., 2015; Xu et al., 2015; Figure 2). The mechanisms implicated in these nonterminal arrests are discussed below.
Figure 2.
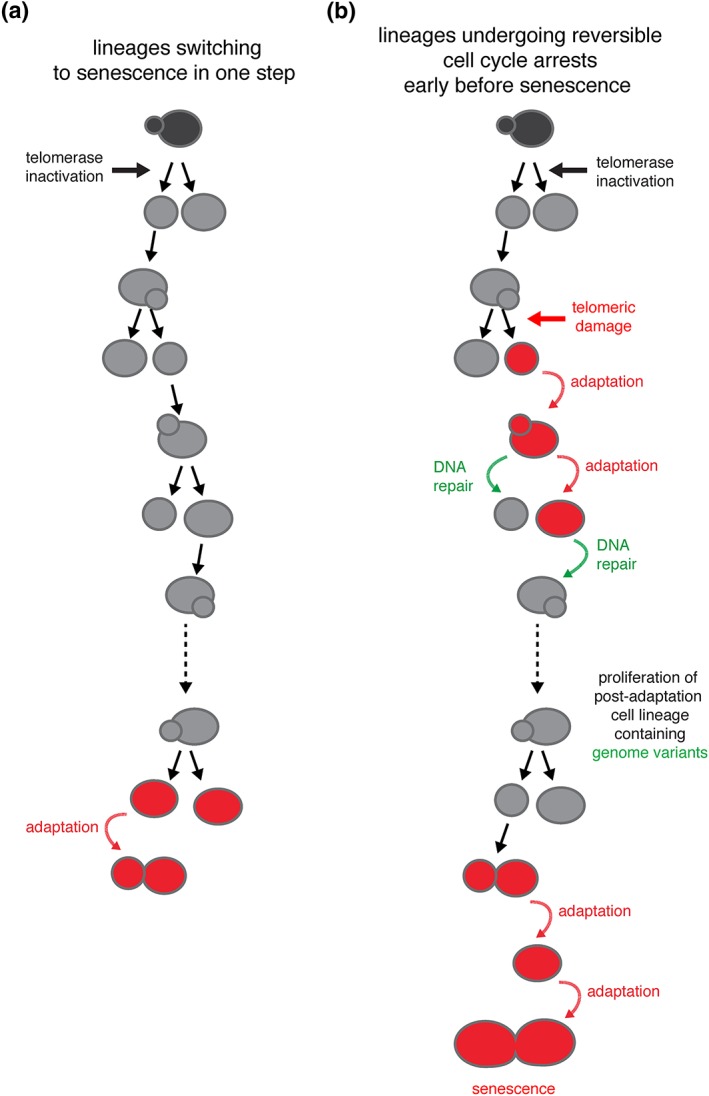
Heterogeneity at the level of single‐cell lineages. (a) Many telomerase‐negative cell lineages, defined here as consecutive cell divisions upon telomerase inactivation until irreversible cell cycle arrest, undergo senescence in a single switch from proliferative to arrested state. (b) In contrast, other cell lineages are likely subject to accidental damage at telomeres that triggers the DNA damage checkpoint and DNA repair mechanisms. The frequent failure of repair mechanisms, combined with the relative ease of bypassing checkpoints by adaptation, then leads to a cascade of genome instability and a multiplicity of cell fates. Modified from Coutelier et al., (2018)
The combination of these two types of heterogeneity, namely variations in cell cycle durations and in the total number of generations until the onset of senescence or cell death, might be sufficient to account for all previously reported aspects of heterogeneity in the growth of telomerase‐negative cell cultures and colonies.
3. SOURCES OF HETEROGENEITY
As stated above, replicative senescence combines several aspects that arrest macroscopic growth, including nonterminal arrests, increased cell cycle durations, and increased cell death. The variable timing and frequency of these events contribute greatly to the differences in heterogeneity we have reviewed. Although they are all consequences of telomerase inactivation, understanding the molecular mechanisms associated with each of these phenomena will help decipher the sources of heterogeneity in senescence. At the core of this issue is the question of whether there exists a unifying molecular definition of replicative senescence with a set sequence of molecular and cellular events or whether telomerase inactivation unleashes multiple pathways that are called replicative senescence only from a phenomenological point of view.
3.1. Variation in telomere length
The dynamics of telomere shortening by the end replication problem and elongation by telomerase create intrinsic heterogeneity in telomere length distribution (Shampay & Blackburn, 1988; Teixeira, Arneric, Sperisen, & Lingner, 2004). The average shortening rate due to the end replication problem is around 3–4 bp per generation and appears to be independent of telomere length (Lundblad & Szostak, 1989; Marcand, Brevet, & Gilson, 1999; Nugent, Hughes, Lue, & Lundblad, 1996; Singer & Gottschling, 1994). The measured value is consistent with the average shortening rate being approximately half of the length of the 3′‐overhang (Larrivee, LeBel, & Wellinger, 2004; Soudet, Jolivet, & Teixeira, 2014). In contrast, the probability of telomere elongation by telomerase is not constant, and shorter telomeres tend to be preferred as substrates of telomerase (Britt‐Compton, Capper, Rowson, & Baird, 2009; Hemann, Strong, Hao, & Greider, 2001; Marcand, Gilson, & Shore, 1997; Teixeira, Arneric, Sperisen, & Lingner, 2004). Telomerase processivity can be quite variable at the molecular level but does not depend on the length of undamaged telomeres (Chang, Arneric, & Lingner, 2007; Teixeira, Arneric, Sperisen, & Lingner, 2004). The combination of all of these molecular processes leads to a wide distribution of telomere lengths at steady state (Xu, Duc, Holcman, & Teixeira, 2013). Although steady‐state telomere length distribution is regulated, variation is observed across natural isolates of S. cerevisiae and Saccharomyces paradoxus (Liti et al., 2009) and in response to external stress (Romano et al., 2013). However, telomere length does not appear to directly affect cellular fitness when telomerase is expressed (Harari, Zadok‐Laviel, & Kupiec, 2017). Telomerase inactivation and the initiation of replicative senescence reveal the consequences of variation in telomere length for cell proliferation.
At the molecular level, replicative senescence involves a DNA damage checkpoint arrest and may be triggered by a limited set of short telomeres reaching a critical threshold (Baird, Rowson, Wynford‐Thomas, & Kipling, 2003; Hemann, Strong, Hao, & Greider, 2001; Ijpma & Greider, 2003; Kaul, Cesare, Huschtscha, Neumann, & Reddel, 2011; Zou, Sfeir, Gryaznov, Shay, & Wright, 2004). This mechanism is compatible with the observation that independent lineages of telomerase‐negative cells enter senescence after a variable number of generations. Indeed, the average length of each telomere varies between different clonal populations, which indicates that the telomere length distribution of the founder cell is unique (Shampay & Blackburn, 1988; Xu, Duc, Holcman, & Teixeira, 2013). Once telomerase is inactivated, the number of generations needed for a unique set of telomeres to shorten until they reach a critica length should thus differ for each lineage derived from a single cell.
The number of critically short telomeres required for robust and permanent arrest can significantly affect the asynchrony of senescence. Although four or five short telomeres are required to stop proliferation in mammalian cells in culture (Kaul, Cesare, Huschtscha, Neumann, & Reddel, 2011), budding yeast requires just a single one. In this model, strains engineered to have a single critically short telomere in the absence of telomerase were used to show that this telomere specifically accumulates single‐stranded DNA, recruits DNA damage response factors, and substantially accelerates senescence (Abdallah et al., 2009; Fallet et al., 2014; Khadaroo et al., 2009), in accordance with the notion that a single short telomere is sufficient to trigger senescence. Numerical simulations suggest that the shortest telomere in a cell displays more variation in length than longer telomeres. By shortening in telomere‐negative lineages, this shortest telomere triggers replicative senescence in an asynchronous manner and substantially more asynchronously in cultures inoculated by a pair of sister cells obtained by meiosis compared to a pair of mother and daughter cells resulting from mitosis (see Figure 1; Xu, Duc, Holcman, & Teixeira, 2013). Combined with the variability in length of the shortest telomere, the random partitioning of newly replicated telomeres, which display a slight asymmetry in length, provides another layer of intraclonal heterogeneity (Figure 3; Bourgeron, Xu, Doumic, & Teixeira, 2015; Eugene, Bourgeron, & Xu, 2017; Soudet, Jolivet, & Teixeira, 2014).
Figure 3.
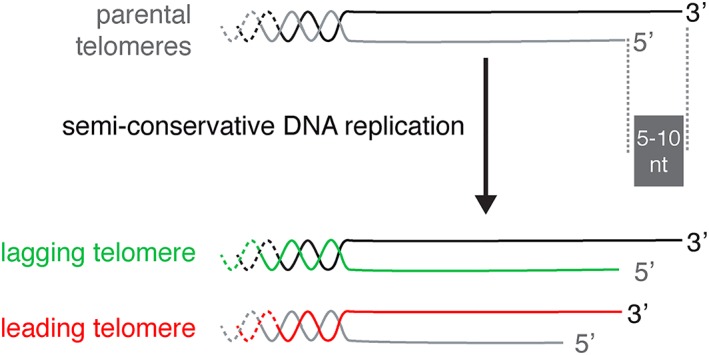
The DNA end replication problem and progressive telomere shortening contain an intrinsic mechanism that generates length asymmetry (modified from Soudet, Jolivet, & Teixeira, 2014). Telomeres end with a 3′‐overhang of 5–10 nucleotides in Saccharomyces cerevisiae. Passage of the replication fork leads to two newly replicated telomeres of different lengths. The telomere replicated by the lagging strand machinery naturally bears a 3′‐overhang by the removal of the last RNA primer of the Okazaki fragment, whereas the telomere replicated by the leading strand machinery requires additional resection and fill‐in steps to regenerate a 3′‐overhang
Thus, the initial telomere length distribution and in particular the variation in length of the shortest telomere are major sources of heterogeneity in replicative senescence, both at the population level (described as interclonal and intraclonal variation in proliferation potential) and at the level of the single cell (in the number of generations undergone by individual lineages). It would be interesting to study the contribution of telomere length distribution to heterogeneity in replicative senescence by taking advantage of the recently described yeast strains with chromosomes fused together into only one or two chromosomes (Luo, Sun, Cormack, & Boeke, 2018; Shao et al., 2018), where telomere length would be much more limited in diversity.
3.2. Activation of the DNA damage checkpoint, DNA repair, and adaptation
The nonterminal arrests observed in individual lineages of telomerase‐negative cells is due to activation of the DNA damage checkpoint, as evidenced using a fluorescent reporter for Rad9 phosphorylation status and by its partial suppression in a mec1∆ sml1∆ mutant (Coutelier et al., 2018; Xu et al., 2015). Although nonterminal arrests can be observed early after telomerase inactivation (<10 generations), they increase in frequency over generations, and their appearance is delayed in strains bearing longer initial telomeres, which suggests that they are correlated with telomere length. In addition, they are likely unrelated to potential noncanonical functions of telomerase, as they are also observed using a catalytic point mutant of Est2 (est2‐D670A). Thus, we hypothesize that nonterminal arrests are caused by a critically short, damaged, or dysfunctional telomere sensed by the DNA damage checkpoint. The molecular nature of the telomeric damage is still elusive, but the early timing of some nonterminal arrests seems inconsistent with progressive telomere shortening due to the end replication problem. Telomere breaks caused by replication defects and fork stalling might be a plausible alternative possibility. Because replication through the repetitive G‐rich telomeric sequences is difficult (Ivessa, Zhou, Schulz, Monson, & Zakian, 2002; Makovets, Herskowitz, & Blackburn, 2004; Miller, Rog, & Cooper, 2006; Sfeir et al., 2009), occasional replication errors at telomeres would fit the apparent stochastic nature of nonterminal arrests. This is supported by data that implicate multiple DNA and RNA processing enzymes and replication checkpoints in the onset of senescence (Azam et al., 2006; Balk et al., 2013; Fallet et al., 2014; Gao, Moss, Parke, Tatum, & Lustig, 2014; Grandin & Charbonneau, 2007; Lafuente‐Barquero et al., 2017), as discussed in Teixeira (2013).
Although end joining acts at telomeres (Mateos‐Gomez et al., 2015; Teixeira, 2013), homology‐directed repair (HDR) is the most studied mechanism of repair acting at telomeres. HDR has mostly been examined in the context of alternative telomere maintenance mechanisms in postsenescence survivors and can be a source of rapid telomere shortening or lengthening in a single cell cycle and consequently a source of heterogeneity in senescence (Cesare & Reddel, 2010; Le, Moore, Haber, & Greider, 1999; Li & Lustig, 1996; Lundblad & Blackburn, 1993; Lustig, 2003; Martens, Chavez, Poon, Schmoor, & Lansdorp, 2000). However, there is also strong evidence that in telomerase‐positive cells, as well as before the onset of replicative senescence in telomerase‐negative cells, HDR factors can be recruited to telomeres. This recruitment is not exclusive to telomeres progressively shortened as a result of the DNA end replication problem, as rapid telomere shortening events are detected in budding yeast strains with very long telomeres (Li & Lustig, 1996). The trigger for repair at telomeres might then result from other stochastic damage, such as replication defects.
At a mechanistic level, telomeres may be subject to sister chromatid exchange, intratelomeric or intertelomeric recombination, break‐induced replication, rolling circle replication initiated by an intra‐telomeric loop, or translocation, as shown in multiple models, including the yeasts S. cerevisiae and Kluyveromyces lactis (Claussin & Chang, 2015; Lustig, 2003; Natarajan & McEachern, 2002). Repair at telomeres is strongly repressed under normal conditions to avoid genome instability. Thus, only a subset of cells would respond to activation of the DNA damage checkpoint by attempting to repair. The cell fate decision to repair is not well‐understood and may depend on the exact molecular nature of the telomere damage and on the presence of factors limiting resection, an essential processing step for recombination, such as Cdc13, Stn1, or the Ku complex (Claussin & Chang, 2015; Westmoreland, Mihalevic, Bernstein, & Resnick, 2018). After homology‐dependent repair of a damaged telomere, the cell presumably inactivates the DNA damage checkpoint and resumes proliferation until the next arrest. If not repaired, the telomeric damage signal should remain and the cell may stay permanently arrested in senescence or undergo adaptation after a longer period of time (see below). Therefore, repair mechanisms and their inherent inefficiency at telomeres contribute to heterogeneity in growth in telomerase‐negative cells.
Arrested cells that do not successfully repair the damage may eventually adapt to DNA damage, a process that allows for cell division despite the presence of unrepaired DNA damage (Lee, Moore, et al., 1998; Sandell & Zakian, 1993; Toczyski, Galgoczy, & Hartwell, 1997). Telomerase‐negative cells that activate the DNA damage checkpoint are also able to adapt after extended arrest (Coutelier et al., 2018; Coutelier & Xu, 2019). Adapted cells retain a partially active checkpoint, which indicates that the initially sensed damage is still present and makes them prone to further repair or adaptation. It is important to note that after adaptation, a cell lineage still has substantial proliferation potential, which might be explained by the fact that telomeric damage is most often not deleterious or by subsequent repair of the damage (Coutelier et al., 2018). Thus, in addition to repair, adaptation is an important mechanism of cell growth for telomerase‐negative cells that experience cell cycle arrest.
Taken together, progressive telomere shortening, rapid telomere shortening, and lengthening linked to repair mechanisms and the status of the DNA damage checkpoint are the key players controlling cell proliferation when telomerase is inactive in budding yeast. Collectively, these parameters can explain most cell‐to‐cell intraclonal and interclonal variation that increases as telomeres shorten. But beyond the dynamics of cell division, these mechanisms are also intimately related to another aspect of heterogeneity in senescence: genome instability and postsenescence survival.
4. CELL DIVERSITY IN SENESCENCE
4.1. Diversity in gene expression and beyond
Like replicative senescent mammalian cells, budding yeast also experiences a deregulation of gene expression and a reorganization of chromatin when telomerase is inactivated (Nautiyal, DeRisi, & Blackburn, 2002; Niederer, Papadopoulos, & Zappulla, 2016; Platt et al., 2013). Whether this deregulated gene expression is a consequence or a cause of the heterogeneity induced by the mechanisms discussed in this review, such as a permanent activation of the DNA damage checkpoint or prolonged G2/M arrest, remains to be investigated. What seems clear is that many genes specifically expressed in senescence are expressed at very low levels. If only a few cells express each gene, each senescent cell likely expresses a distinct combinatorial set of changes, and this likely contributes to phenotypic heterogeneity.
An interesting finding regarding phenotypic changes in senescent budding yeast cultures concerns the activation of genes in response to environmental stresses and in oxidative phosphorylations and alterations in mitochondrial morphology (Nautiyal, DeRisi, & Blackburn, 2002). Mitochondrial dysfunction and oxidative stress at telomeres contribute to senescence and its heterogeneity in mammalian systems (Ahmed & Lingner, 2018; Chen, Ozanne, & Hales, 2005; Passos et al., 2007; Sahin et al., 2011). Thus, potential alterations in the metabolic programme of senescent cells could be studied in budding yeast as well. Note that because S. cerevisiae can grow by either respiration or fermentation, one can expect to disentangle the causes and effects among telomeres and mitochondrial activities and uncover the origin of oxidative stress in the absence of telomerase. That said, potential mitochondrial dysfunction and the concomitant increase in reactive oxygen species in replicative senescent cells is indicative of alterations to the structure of the telomere and generalized genome instability (Fouquerel et al., 2019; Lu & Liu, 2010; Lu, Vallabhaneni, Yin, & Liu, 2013). This is expected to generate yet another level of diversity among populations of senescent cells: genome instability.
4.2. Senescence‐specific genome instability
Global chromosomal instability increases in telomerase‐negative cells over time (Hackett, Feldser, & Greider, 2001; Ijpma & Greider, 2003; Lundblad & Szostak, 1989). An ~10‐fold increase in the mutation rate of reporter genes has been reported and is accompanied by additional chromosomal rearrangements (Coutelier et al., 2018; Hackett, Feldser, & Greider, 2001; Hackett & Greider, 2003; Meyer & Bailis, 2007). The single‐stranded DNA exposed at telomeres when they become critically short or dysfunctional may initiate this genome instability (Fallet et al., 2014; Garvik, Carson, & Hartwell, 1995; Hackett & Greider, 2003). However, the mechanisms behind widespread genome instability due to exposed single‐stranded telomeric DNA are still unclear. We propose that when telomeres are critically short, the accumulation of single‐stranded DNA activates the DNA damage checkpoint and triggers repair mechanisms that generate mutations and chromosomal rearrangements. When repair fails, adaptation to the checkpoint allows for the propagation of molecular structures—such as resected telomeres, terminally deleted telomeres, repair intermediates, stalled and collapsed replication forks, or fused telomeres—that would otherwise maintain an active checkpoint and would not be inherited by the progeny. The resolution of these structures over several cell divisions may subsequently lead to widespread complex chromosomal rearrangements (Beyer & Weinert, 2016; Coutelier et al., 2018; Hackett, Feldser, & Greider, 2001; Hackett & Greider, 2003; Piazza & Heyer, 2019). Adaptation could also contribute to genome instability by forcing defective and asymmetric mitosis, leading to aneuploid cells (Bender et al., 2018; Galgoczy & Toczyski, 2001; Kaye et al., 2004).
Overall, genome instability contributes to the genetic diversity of senescent cells. In mammals, telomerase inactivation also leads to a similar mutator phenotype (Blasco et al., 1997; Chin et al., 1999; Lee, Blasco, et al., 1998), constitutes a potential source of genome variants at work in the early stages of tumorigenesis, and would substantiate the link between cancer incidence and ageing in humans.
4.3. Postsenescence survival
The vast majority of telomerase‐negative cells enters replicative senescence and die, but in most organisms studied, a subpopulation can fuse; recombine their telomeres, subtelomeres, or other genetic elements; and escape growth arrest (Begnis et al., 2018; Cesare & Reddel, 2010; Kachouri et al., 2009; Lundblad & Blackburn, 1993; Nakamura, Cooper, & Cech, 1998). These postsenescence survivors quickly outcompete slow‐growing and arrested senescent cells and take over the culture. The emergence of survivors is an extreme illustration of the phenotypic heterogeneity generated by telomerase‐negative cells and is at the basis of telomerase‐independent tumour growth, accounting for at least 5% of cancers (Barthel et al., 2017; Cesare & Reddel, 2010). First, the time of appearance of survivors in a given telomerase‐negative culture is highly stochastic and variable. Second, different types of survivors display a wide variety of growth rates, which may not be constant over time. Third, the elongation of chromosomal ends by the acquisition of subtelomeric elements and telomere sequences is also an unstable dynamic process, and a culture may contain subpopulations with different patterns of telomere and subtelomere recombination.
The exact molecular nature of the telomeres when survivors emerge is not clear. In budding yeast, the highly complex mixture found in a survivor culture hints at very heterogeneous telomeric states, defined by telomere length (Chang, Dittmar, & Rothstein, 2011; Grandin & Charbonneau, 2009; Lebel et al., 2009), recruitment of factors involved in telomere processing (Claussin & Chang, 2015), and the presence of RNA–DNA hybrids (Balk, Dees, Bender, & Luke, 2014; Misino, Bonetti, Luke‐Glaser, & Luke, 2018; Yu, Kao, & Lin, 2014). The checkpoint status of the cell might also affect the generation of survivors (Tsai, Tseng, Chang, Lin, & Teng, 2002). The highly variable and dynamic nature of survivors in terms of timing, cellular growth rate, and molecular pathways seems therefore intrinsically related to the heterogeneity of replicative senescence itself.
5. CONCLUSIONS
Characterization of the diversity of deregulated processes affecting heterogeneity is more advanced in the field of mother cell ageing, with mechanisms such as the accumulation of extrachromosomal rDNA circles, asymmetric segregation of protein aggregates, or dysfunctional mitochondria and vacuole being well‐documented (Denoth Lippuner, Julou, & Barral, 2014; Knorre, Azbarova, Galkina, Feniouk, & Severin, 2018). In the field of telomere‐induced replicative senescence, an exhaustive characterization of the contribution of deregulated cellular processes would help in formulating an integrative model of replicative senescence that would encompass all levels of heterogeneity.
In sum, although telomerase inactivation deterministically initiates the process, the route to replicative senescence is filled with hurdles that generate cell‐to‐cell variation and subsequent intraclonal and interclonal variation. Deciphering the causes of heterogeneity in replicative senescence would improve understanding of the robustness of the senescent state and the mechanisms at work in escaping it, at both the molecular and systems levels, not only in pathological situations like cancer but also from the perspective of evolution.
ACKNOWLEDGEMENTS
We thank Aurèle Piazza and Héloïse Coutelier for their critical reading of the manuscript. We wish to thank the Teixeira lab members for fruitful discussions. Work in the M.T.T. and Z.X. labs is supported by the Fondation de la Recherche Medicale (“équipe labellisée” to M.T.T.) and by the French National Research Agency (ANR) as part of the Investissements d'Avenir Program (LabEx Dynamo ANR‐11‐LABX‐0011‐01), ANR‐16‐CE12‐0026 to M.T.T. and ANR‐17‐CE20‐0002‐01 to Z.X.
Xu Z, Teixeira MT. The many types of heterogeneity in replicative senescence. Yeast. 2019;36:637–648. 10.1002/yea.3433
The copyright line for this article was changed on 3 September 2019 after original online publication.
Contributor Information
Zhou Xu, Email: zhou.xu@sorbonne-universite.fr.
Maria Teresa Teixeira, Email: teresa.teixeira@ibpc.fr.
REFERENCES
- Abdallah, P. , Luciano, P. , Runge, K. W. , Lisby, M. , Geli, V. , Gilson, E. , & Teixeira, M. T. (2009). A two‐step model for senescence triggered by a single critically short telomere. Nature Cell Biology, 11, 988–993. 10.1038/ncb1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed, W. , & Lingner, J. (2018). PRDX1 and MTH1 cooperate to prevent ROS‐mediated inhibition of telomerase. Genes & Development, 32, 658–669. 10.1101/gad.313460.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam, M. , Lee, J. Y. , Abraham, V. , Chanoux, R. , Schoenly, K. A. , & Johnson, F. B. (2006). Evidence that the S. cerevisiae Sgs1 protein facilitates recombinational repair of telomeres during senescence. Nucleic Acids Research, 34, 506–516. 10.1093/nar/gkj452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar, R. , Hartmann, C. H. , Rodriguez, K. A. , Denny, A. D. , Busuttil, R. A. , Dolle, M. E. , … Vijg, J. (2006). Increased cell‐to‐cell variation in gene expression in ageing mouse heart. Nature, 441, 1011–1014. 10.1038/nature04844 [DOI] [PubMed] [Google Scholar]
- Baird, D. M. , Rowson, J. , Wynford‐Thomas, D. , & Kipling, D. (2003). Extensive allelic variation and ultrashort telomeres in senescent human cells. Nature Genetics, 33, 203–207. 10.1038/ng1084 [DOI] [PubMed] [Google Scholar]
- Balk, B. , Dees, M. , Bender, K. , & Luke, B. (2014). The differential processing of telomeres in response to increased telomeric transcription and RNA‐DNA hybrid accumulation. RNA Biology, 11, 95–100. 10.4161/rna.27798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balk, B. , Maicher, A. , Dees, M. , Klermund, J. , Luke‐Glaser, S. , Bender, K. , & Luke, B. (2013). Telomeric RNA‐DNA hybrids affect telomere‐length dynamics and senescence. Nature Structural & Molecular Biology, 20, 1199–1205. 10.1038/nsmb.2662 [DOI] [PubMed] [Google Scholar]
- Barthel, F. P. , Wei, W. , Tang, M. , Martinez‐Ledesma, E. , Hu, X. , Amin, S. B. , … Verhaak, R. G. (2017). Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nature Genetics, 49, 349–357. 10.1038/ng.3781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begnis, M. , Apte, M. S. , Masuda, H. , Jain, D. , Wheeler, D. L. , & Cooper, J. P. (2018). RNAi drives nonreciprocal translocations at eroding chromosome ends to establish telomere‐free linear chromosomes. Genes & Development, 32, 537–554. 10.1101/gad.311712.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender, K. , Vydzhak, O. , Klermund, J. , Busch, A. , Grimm, S. , & Luke, B. (2018). Checkpoint adaptation in repair‐deficient cells drives aneuploidy and resistance to genotoxic agents. BioArchiv. [DOI] [PubMed] [Google Scholar]
- Beyer, T. , & Weinert, T. (2016). Ontogeny of unstable chromosomes generated by telomere error in budding yeast. PLoS Genetics, 12, e1006345 10.1371/journal.pgen.1006345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn, E. H. (2000). Telomere states and cell fates. Nature, 408, 53–56. 10.1038/35040500 [DOI] [PubMed] [Google Scholar]
- Blasco, M. A. , Lee, H. W. , Hande, M. P. , Samper, E. , Lansdorp, P. M. , DePinho, R. A. , & Greider, C. W. (1997). Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell, 91, 25–34. 10.1016/S0092-8674(01)80006-4 [DOI] [PubMed] [Google Scholar]
- Bourgeron, T. , Xu, Z. , Doumic, M. , & Teixeira, M. T. (2015). The asymmetry of telomere replication contributes to replicative senescence heterogeneity. Scientific Reports, 5, 15326 10.1038/srep15326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britt‐Compton, B. , Capper, R. , Rowson, J. , & Baird, D. M. (2009). Short telomeres are preferentially elongated by telomerase in human cells. FEBS Letters, 583, 3076–3080. 10.1016/j.febslet.2009.08.029 [DOI] [PubMed] [Google Scholar]
- Cesare, A. J. , & Reddel, R. R. (2010). Alternative lengthening of telomeres: models, mechanisms and implications. Nature Reviews. Genetics, 11, 319–330. 10.1038/nrg2763 [DOI] [PubMed] [Google Scholar]
- Chang, M. , Arneric, M. , & Lingner, J. (2007). Telomerase repeat addition processivity is increased at critically short telomeres in a Tel1‐dependent manner in Saccharomyces cerevisiae. Genes & Development, 21, 2485–2494. 10.1101/gad.1588807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, M. , Dittmar, J. C. , & Rothstein, R. (2011). Long telomeres are preferentially extended during recombination‐mediated telomere maintenance. Nature Structural & Molecular Biology, 18, 451–456. 10.1038/nsmb.2034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J.‐H. , Ozanne, S. E. , & Hales, C. N. (2005). Heterogeneity in premature senescence by oxidative stress correlates with differential DNA damage during the cell cycle. DNA Repair, 4, 1140–1148. 10.1016/j.dnarep.2005.06.003 [DOI] [PubMed] [Google Scholar]
- Chin, L. , Artandi, S. E. , Shen, Q. , Tam, A. , Lee, S. L. , Gottlieb, G. J. , … DePinho, R. A. (1999). p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell, 97, 527–538. 10.1016/S0092-8674(00)80762-X [DOI] [PubMed] [Google Scholar]
- Churikov, D. , Charifi, F. , Simon, M. N. , & Geli, V. (2014). Rad59‐facilitated acquisition of Y′ elements by short telomeres delays the onset of senescence. PLoS Genetics, 10, e1004736 10.1371/journal.pgen.1004736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claussin, C. , & Chang, M. (2015). The many facets of homologous recombination at telomeres. Microb Cell, 2, 308–321. 10.15698/mic2015.09.224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutelier, H. , & Xu, Z. (2019). Adaptation in replicative senescence: A risky business. Current Genetics, 65, 711–716. 10.1007/s00294-019-00933-7 [DOI] [PubMed] [Google Scholar]
- Coutelier, H. , Xu, Z. , Morisse, M. C. , Lhuillier‐Akakpo, M. , Pelet, S. , Charvin, G. , … Teixeira, M. T. (2018). Adaptation to DNA damage checkpoint in senescent telomerase‐negative cells promotes genome instability. Genes & Development, 32, 1499–1513. 10.1101/gad.318485.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Adda di Fagagna, F. , Reaper, P. M. , Clay‐Farrace, L. , Fiegler, H. , Carr, P. , Von Zglinicki, T. , … Jackson, S. P. (2003). A DNA damage checkpoint response in telomere‐initiated senescence. Nature, 426, 194–198. [DOI] [PubMed] [Google Scholar]
- Denoth Lippuner, A. , Julou, T. , & Barral, Y. (2014). Budding yeast as a model organism to study the effects of age. FEMS Microbiology Reviews, 38, 300–325. 10.1111/1574-6976.12060 [DOI] [PubMed] [Google Scholar]
- Enomoto, S. , Glowczewski, L. , & Berman, J. (2002). MEC3, MEC1, and DDC2 are essential components of a telomere checkpoint pathway required for cell cycle arrest during senescence in Saccharomyces cerevisiae. Molecular Biology of the Cell, 13, 2626–2638. 10.1091/mbc.02-02-0012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugene, S. , Bourgeron, T. , & Xu, Z. (2017). Effects of initial telomere length distribution on senescence onset and heterogeneity. Journal of Theoretical Biology, 413, 58–65. 10.1016/j.jtbi.2016.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallet, E. , Jolivet, P. , Soudet, J. , Lisby, M. , Gilson, E. , & Teixeira, M. T. (2014). Length‐dependent processing of telomeres in the absence of telomerase. Nucleic Acids Research, 42, 3648–3665. 10.1093/nar/gkt1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouquerel, E. , Barnes, R. P. , Uttam, S. , Watkins, S. C. , Bruchez, M. P. , & Opresko, P. L. (2019). Targeted and persistent 8‐oxoguanine base damage at telomeres promotes telomere loss and crisis. Molecular Cell, 75, 117–130.e6. 10.1016/j.molcel.2019.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galgoczy, D. J. , & Toczyski, D. P. (2001). Checkpoint adaptation precedes spontaneous and damage‐induced genomic instability in yeast. Molecular and Cellular Biology, 21, 1710–1718. 10.1128/MCB.21.5.1710-1718.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, H. , Moss, D. L. , Parke, C. , Tatum, D. , & Lustig, A. J. (2014). The Ctf18RFC clamp loader is essential for telomere stability in telomerase‐negative and mre11 mutant alleles. PLoS ONE, 9, e88633 10.1371/journal.pone.0088633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garvik, B. , Carson, M. , & Hartwell, L. (1995). Single‐stranded DNA arising at telomeres in cdc13 mutants may constitute a specific signal for the RAD9 checkpoint. Molecular and Cellular Biology, 15, 6128–6138. 10.1128/MCB.15.11.6128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandin, N. , & Charbonneau, M. (2007). Mrc1, a non‐essential DNA replication protein, is required for telomere end protection following loss of capping by Cdc13, Yku or telomerase. Molecular Genetics and Genomics, 277, 685–699. 10.1007/s00438-007-0218-0 [DOI] [PubMed] [Google Scholar]
- Grandin, N. , & Charbonneau, M. (2009). Telomerase‐ and Rad52‐independent immortalization of budding yeast by an inherited‐long‐telomere pathway of telomeric repeat amplification. Molecular and Cellular Biology, 29, 965–985. 10.1128/MCB.00817-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett, J. A. , Feldser, D. M. , & Greider, C. W. (2001). Telomere dysfunction increases mutation rate and genomic instability. Cell, 106, 275–286. 10.1016/S0092-8674(01)00457-3 [DOI] [PubMed] [Google Scholar]
- Hackett, J. A. , & Greider, C. W. (2003). End resection initiates genomic instability in the absence of telomerase. Molecular and Cellular Biology, 23, 8450–8461. 10.1128/MCB.23.23.8450-8461.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harari, Y. , Zadok‐Laviel, S. , & Kupiec, M. (2017). Long telomeres do not affect cellular fitness in yeast. MBio, 8 10.1128/mBio.01314-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie, N. D. , Dempster, M. , Dunlop, M. G. , Thompson, A. M. , Green, D. K. , & Allshire, R. C. (1990). Telomere reduction in human colorectal carcinoma and with ageing. Nature, 346, 866–868. 10.1038/346866a0 [DOI] [PubMed] [Google Scholar]
- Hayflick, L. (1965). The limited in vitro lifetime of human diploid cell strains. Experimental Cell Research, 37, 614–636. 10.1016/0014-4827(65)90211-9 [DOI] [PubMed] [Google Scholar]
- Hayflick, L. , & Moorhead, P. S. (1961). The serial cultivation of human diploid cell strains. Experimental Cell Research, 25, 585–621. 10.1016/0014-4827(61)90192-6 [DOI] [PubMed] [Google Scholar]
- Hemann, M. T. , Strong, M. A. , Hao, L. Y. , & Greider, C. W. (2001). The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell, 107, 67–77. 10.1016/S0092-8674(01)00504-9 [DOI] [PubMed] [Google Scholar]
- Ijpma, A. S. , & Greider, C. W. (2003). Short telomeres induce a DNA damage response in Saccharomyces cerevisiae. Molecular Biology of the Cell, 14, 987–1001. 10.1091/mbc.02-04-0057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivessa, A. S. , Zhou, J. Q. , Schulz, V. P. , Monson, E. K. , & Zakian, V. A. (2002). Saccharomyces Rrm3p, a 5′ to 3′ DNA helicase that promotes replication fork progression through telomeric and subtelomeric DNA. Genes & Development, 16, 1383–1396. 10.1101/gad.982902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyapalan, J. C. , Ferreira, M. , Sedivy, J. M. , & Herbig, U. (2007). Accumulation of senescent cells in mitotic tissue of aging primates. Mechanisms of Ageing and Development, 128, 36–44. 10.1016/j.mad.2006.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, R. B. , Whitney, R. G. , & Smith, J. R. (1985). Intramitotic variation in proliferative potential: Stochastic events in cellular aging. Mechanisms of Ageing and Development, 29, 143–149. 10.1016/0047-6374(85)90014-4 [DOI] [PubMed] [Google Scholar]
- Kachouri, R. , Dujon, B. , Gilson, E. , Westhof, E. , Fairhead, C. , & Teixeira, M. T. (2009). Large telomerase RNA, telomere length heterogeneity and escape from senescence in Candida glabrata. FEBS Letters, 583, 3605–3610. 10.1016/j.febslet.2009.10.034 [DOI] [PubMed] [Google Scholar]
- Karlseder, J. , Smogorzewska, A. , & de Lange, T. (2002). Senescence induced by altered telomere state, not telomere loss. Science, 295, 2446–2449. 10.1126/science.1069523 [DOI] [PubMed] [Google Scholar]
- Kaul, Z. , Cesare, A. J. , Huschtscha, L. I. , Neumann, A. A. , & Reddel, R. R. (2011). Five dysfunctional telomeres predict onset of senescence in human cells. EMBO Reports, 13, 52–59. 10.1038/embor.2011.227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye, J. A. , Melo, J. A. , Cheung, S. K. , Vaze, M. B. , Haber, J. E. , & Toczyski, D. P. (2004). DNA breaks promote genomic instability by impeding proper chromosome segregation. Current Biology, 14, 2096–2106. 10.1016/j.cub.2004.10.051 [DOI] [PubMed] [Google Scholar]
- Khadaroo, B. , Teixeira, M. T. , Luciano, P. , Eckert‐Boulet, N. , Germann, S. M. , Simon, M. N. , … Lisby, M. (2009). The DNA damage response at eroded telomeres and tethering to the nuclear pore complex. Nature Cell Biology, 11, 980–987. 10.1038/ncb1910 [DOI] [PubMed] [Google Scholar]
- Knorre, D. A. , Azbarova, A. V. , Galkina, K. V. , Feniouk, B. A. , & Severin, F. F. (2018). Replicative aging as a source of cell heterogeneity in budding yeast. Mechanisms of Ageing and Development, 176, 24–31. 10.1016/j.mad.2018.09.001 [DOI] [PubMed] [Google Scholar]
- Lafuente‐Barquero, J. , Luke‐Glaser, S. , Graf, M. , Silva, S. , Gomez‐Gonzalez, B. , Lockhart, A. , … Luke, B. (2017). The Smc5/6 complex regulates the yeast Mph1 helicase at RNA‐DNA hybrid‐mediated DNA damage. PLoS Genetics, 13, e1007136 10.1371/journal.pgen.1007136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrivee, M. , LeBel, C. , & Wellinger, R. J. (2004). The generation of proper constitutive G‐tails on yeast telomeres is dependent on the MRX complex. Genes & Development, 18, 1391–1396. 10.1101/gad.1199404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le, S. , Moore, J. K. , Haber, J. E. , & Greider, C. W. (1999). RAD50 and RAD51 define two pathways that collaborate to maintain telomeres in the absence of telomerase. Genetics, 152, 143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebel, C. , Rosonina, E. , Sealey, D. C. , Pryde, F. , Lydall, D. , Maringele, L. , & Harrington, L. A. (2009). Telomere maintenance and survival in saccharomyces cerevisiae in the absence of telomerase and RAD52. Genetics, 182, 671–684. 10.1534/genetics.109.102939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, H. W. , Blasco, M. A. , Gottlieb, G. J. , Horner, J. W. 2nd , Greider, C. W. , & DePinho, R. A. (1998). Essential role of mouse telomerase in highly proliferative organs. Nature, 392, 569–574. 10.1038/33345 [DOI] [PubMed] [Google Scholar]
- Lee, S. E. , Moore, J. K. , Holmes, A. , Umezu, K. , Kolodner, R. D. , & Haber, J. E. (1998). Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell, 94, 399–409. 10.1016/S0092-8674(00)81482-8 [DOI] [PubMed] [Google Scholar]
- Li, B. , & Lustig, A. J. (1996). A novel mechanism for telomere size control in Saccharomyces cerevisiae. Genes & Development, 10, 1310–1326. 10.1101/gad.10.11.1310 [DOI] [PubMed] [Google Scholar]
- Liti, G. , Carter, D. M. , Moses, A. M. , Warringer, J. , Parts, L. , James, S. A. , … Louis, E. J. (2009). Population genomics of domestic and wild yeasts. Nature, 458, 337–341. 10.1038/nature07743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, J. , & Liu, Y. (2010). Deletion of Ogg1 DNA glycosylase results in telomere base damage and length alteration in yeast. The EMBO Journal, 29, 398–409. 10.1038/emboj.2009.355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, J. , Vallabhaneni, H. , Yin, J. , & Liu, Y. (2013). Deletion of the major peroxiredoxin Tsa1 alters telomere length homeostasis. Aging Cell, 12, 635–644. 10.1111/acel.12085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundblad, V. , & Blackburn, E. H. (1993). An alternative pathway for yeast telomere maintenance rescues est1‐senescence. Cell, 73, 347–360. 10.1016/0092-8674(93)90234-H [DOI] [PubMed] [Google Scholar]
- Lundblad, V. , & Szostak, J. W. (1989). A mutant with a defect in telomere elongation leads to senescence in yeast. Cell, 57, 633–643. 10.1016/0092-8674(89)90132-3 [DOI] [PubMed] [Google Scholar]
- Luo, J. , Sun, X. , Cormack, B. P. , & Boeke, J. D. (2018). Karyotype engineering by chromosome fusion leads to reproductive isolation in yeast. Nature, 560, 392–396. 10.1038/s41586-018-0374-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustig, A. J. (2003). Clues to catastrophic telomere loss in mammals from yeast telomere rapid deletion. Nature Reviews. Genetics, 4, 916–923. 10.1038/nrg1207 [DOI] [PubMed] [Google Scholar]
- Makovets, S. , Herskowitz, I. , & Blackburn, E. H. (2004). Anatomy and dynamics of DNA replication fork movement in yeast telomeric regions. Molecular and Cellular Biology, 24, 4019–4031. 10.1128/MCB.24.9.4019-4031.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcand, S. , Brevet, V. , & Gilson, E. (1999). Progressive cis‐inhibition of telomerase upon telomere elongation. The EMBO Journal, 18, 3509–3519. 10.1093/emboj/18.12.3509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcand, S. , Gilson, E. , & Shore, D. (1997). A protein‐counting mechanism for telomere length regulation in yeast. Science, 275, 986–990. 10.1126/science.275.5302.986 [DOI] [PubMed] [Google Scholar]
- Martens, U. M. , Chavez, E. A. , Poon, S. S. , Schmoor, C. , & Lansdorp, P. M. (2000). Accumulation of short telomeres in human fibroblasts prior to replicative senescence. Experimental Cell Research, 256, 291–299. 10.1006/excr.2000.4823 [DOI] [PubMed] [Google Scholar]
- Mateos‐Gomez, P. A. , Gong, F. , Nair, N. , Miller, K. M. , Lazzerini‐Denchi, E. , & Sfeir, A. (2015). Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature, 518, 254–257. 10.1038/nature14157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, D. H. , & Bailis, A. M. (2007). Telomere dysfunction drives increased mutation by error‐prone polymerases Rev1 and ζ in Saccharomyces cerevisiae. Genetics, 175, 1533–1537. 10.1534/genetics.106.068130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, K. M. , Rog, O. , & Cooper, J. P. (2006). Semi‐conservative DNA replication through telomeres requires Taz1. Nature, 440, 824–828. 10.1038/nature04638 [DOI] [PubMed] [Google Scholar]
- Misino, S. , Bonetti, D. , Luke‐Glaser, S. , & Luke, B. (2018). Increased TERRA levels and RNase H sensitivity are conserved hallmarks of post‐senescent survivors in budding yeast. Differentiation, 100, 37–45. 10.1016/j.diff.2018.02.002 [DOI] [PubMed] [Google Scholar]
- Nakamura, T. M. , Cooper, J. P. , & Cech, T. R. (1998). Two modes of survival of fission yeast without telomerase. Science, 282, 493–496. 10.1126/science.282.5388.493 [DOI] [PubMed] [Google Scholar]
- Natarajan, S. , & McEachern, M. J. (2002). Recombinational telomere elongation promoted by DNA circles. Molecular and Cellular Biology, 22, 4512–4521. 10.1128/MCB.22.13.4512-4521.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nautiyal, S. , DeRisi, J. L. , & Blackburn, E. H. (2002). The genome‐wide expression response to telomerase deletion in Saccharomyces cerevisiae. Proceedings of the National Academy of Sciences of the United States of America, 99, 9316–9321. 10.1073/pnas.142162499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederer, R. O. , Papadopoulos, N. , & Zappulla, D. C. (2016). Identification of novel noncoding transcripts in telomerase‐negative yeast using RNA‐seq. Scientific Reports, 6, 19376 10.1038/srep19376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent, C. I. , Hughes, T. R. , Lue, N. F. , & Lundblad, V. (1996). Cdc13p: A single‐strand telomeric DNA‐binding protein with a dual role in yeast telomere maintenance. Science, 274, 249–252. 10.1126/science.274.5285.249 [DOI] [PubMed] [Google Scholar]
- Passos, J. F. , Saretzki, G. , Ahmed, S. , Nelson, G. , Richter, T. , Peters, H. , … von Zglinicki, T. (2007). Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere‐dependent senescence. PLoS Biology, 5, e110 10.1371/journal.pbio.0050110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza, A. , & Heyer, W. D. (2019). Homologous recombination and the formation of complex genomic rearrangements. Trends in Cell Biology, 29, 135–149. 10.1016/j.tcb.2018.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt, J. M. , Ryvkin, P. , Wanat, J. J. , Donahue, G. , Ricketts, M. D. , Barrett, S. P. , … Johnson, F. B. (2013). Rap1 relocalization contributes to the chromatin‐mediated gene expression profile and pace of cell senescence. Genes & Development, 27, 1406–1420. 10.1101/gad.218776.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie, K. B. , Mallory, J. C. , & Petes, T. D. (1999). Interactions of TLC1 (which encodes the RNA subunit of telomerase), TEL1, and MEC1 in regulating telomere length in the yeast Saccharomyces cerevisiae. Molecular & Cellular Biology, 19, 6065–6075. 10.1128/MCB.19.9.6065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano, G. H. , Harari, Y. , Yehuda, T. , Podhorzer, A. , Rubinstein, L. , Shamir, R. , … Kupiec, M. (2013). Environmental stresses disrupt telomere length homeostasis. PLoS Genetics, 9, e1003721 10.1371/journal.pgen.1003721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin, E. , Colla, S. , Liesa, M. , Moslehi, J. , Muller, F. L. , Guo, M. , … Depinho, R. A. (2011). Telomere dysfunction induces metabolic and mitochondrial compromise. Nature, 470, 359–365. 10.1038/nature09787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandell, L. L. , & Zakian, V. A. (1993). Loss of a yeast telomere: Arrest, recovery, and chromosome loss. Cell, 75, 729–739. 10.1016/0092-8674(93)90493-A [DOI] [PubMed] [Google Scholar]
- Sfeir, A. , Kosiyatrakul, S. T. , Hockemeyer, D. , MacRae, S. L. , Karlseder, J. , Schildkraut, C. L. , & de Lange, T. (2009). Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell, 138, 90–103. 10.1016/j.cell.2009.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shampay, J. , & Blackburn, E. H. (1988). Generation of telomere‐length heterogeneity in Saccharomyces cerevisiae. Proceedings of the National Academy of Sciences of the United States of America, 85, 534–538. 10.1073/pnas.85.2.534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao, Y. , Lu, N. , Wu, Z. , Cai, C. , Wang, S. , Zhang, L. L. , … Qin, Z. (2018). Creating a functional single‐chromosome yeast. Nature, 560, 331–335. 10.1038/s41586-018-0382-x [DOI] [PubMed] [Google Scholar]
- Shay, J. W. , & Wright, W. E. (2010). Telomeres and telomerase in normal and cancer stem cells. FEBS Letters., 584, 3819–3825. 10.1016/j.febslet.2010.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer, M. S. , & Gottschling, D. E. (1994). TLC1: template RNA component of Saccharomyces cerevisiae telomerase. Science, 266, 404–409. 10.1126/science.7545955 [DOI] [PubMed] [Google Scholar]
- Smith, J. R. , & Hayflick, L. (1974). Variation in the life‐span of clones derived from human diploid cell strains. The Journal of Cell Biology, 62, 48–53. 10.1083/jcb.62.1.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, J. R. , & Whitney, R. G. (1980). Intraclonal variation in proliferative potential of human diploid fibroblasts: Stochastic mechanism for cellular aging. Science, 207, 82–84. 10.1126/science.7350644 [DOI] [PubMed] [Google Scholar]
- Soudet, J. , Jolivet, P. , & Teixeira, M. T. (2014). Elucidation of the DNA end‐replication problem in Saccharomyces cerevisiae. Molecular Cell, 53, 954–964. 10.1016/j.molcel.2014.02.030 [DOI] [PubMed] [Google Scholar]
- Suram, A. , & Herbig, U. (2014). The replicometer is broken: Telomeres activate cellular senescence in response to genotoxic stresses. Aging Cell, 13, 780–786. 10.1111/acel.12246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira, M. T. (2013). Saccharomyces cerevisiae as a model to study replicative senescence triggered by telomere shortening. Frontiers in Oncology, 3, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira, M. T. , Arneric, M. , Sperisen, P. , & Lingner, J. (2004). Telomere length homeostasis is achieved via a switch between telomerase‐extendible and ‐nonextendible states. Cell, 117, 323–335. 10.1016/S0092-8674(04)00334-4 [DOI] [PubMed] [Google Scholar]
- Teng, S. C. , Chang, J. , McCowan, B. , & Zakian, V. A. (2000). Telomerase‐independent lengthening of yeast telomeres occurs by an abrupt Rad50p‐dependent, Rif‐inhibited recombinational process. Molecular Cell, 6, 947–952. 10.1016/S1097-2765(05)00094-8 [DOI] [PubMed] [Google Scholar]
- Toczyski, D. P. , Galgoczy, D. J. , & Hartwell, L. H. (1997). CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell, 90, 1097–1106. 10.1016/S0092-8674(00)80375-X [DOI] [PubMed] [Google Scholar]
- Tsai, Y. L. , Tseng, S. F. , Chang, S. H. , Lin, C. C. , & Teng, S. C. (2002). Involvement of replicative polymerases, Tel1p, Mec1p, Cdc13p, and the Ku complex in telomere‐telomere recombination. Molecular and Cellular Biology, 22, 5679–5687. 10.1128/MCB.22.16.5679-5687.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellinger, R. J. , & Zakian, V. A. (2012). Everything you ever wanted to know about Saccharomyces cerevisiae telomeres: Beginning to end. Genetics, 191, 1073–1105. 10.1534/genetics.111.137851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westmoreland, J. W. , Mihalevic, M. J. , Bernstein, K. A. , & Resnick, M. A. (2018). The global role for Cdc13 and Yku70 in preventing telomere resection across the genome. DNA Repair (Amst), 62, 8–17. 10.1016/j.dnarep.2017.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, Z. , Jay, K. A. , Smith, D. L. , Zhang, Y. , Liu, Z. , Zheng, J. , … Blackburn, E. H. (2015). Early telomerase inactivation accelerates aging independently of telomere length. Cell, 160, 928–939. 10.1016/j.cell.2015.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Z. , Duc, K. D. , Holcman, D. , & Teixeira, M. T. (2013). The length of the shortest telomere as the major determinant of the onset of replicative senescence. Genetics, 194, 847–857. 10.1534/genetics.113.152322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Z. , Fallet, E. , Paoletti, C. , Fehrmann, S. , Charvin, G. , & Teixeira, M. T. (2015). Two routes to senescence revealed by real‐time analysis of telomerase‐negative single lineages. Nature Communications, 6, 7680 10.1038/ncomms8680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, T. Y. , Kao, Y. W. , & Lin, J. J. (2014). Telomeric transcripts stimulate telomere recombination to suppress senescence in cells lacking telomerase. Proceedings of the National Academy of Sciences of the United States of America, 111, 3377–3382. 10.1073/pnas.1307415111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou, Y. , Sfeir, A. , Gryaznov, S. M. , Shay, J. W. , & Wright, W. E. (2004). Does a sentinel or a subset of short telomeres determine replicative senescence? Molecular Biology of the Cell, 15, 3709–3718. 10.1091/mbc.e04-03-0207 [DOI] [PMC free article] [PubMed] [Google Scholar]