

Abstract
Introduction
Emicizumab is a recombinant humanized bispecific monoclonal antibody mimicking the cofactor function of activated factor VIII.
Aim
In this multicentre, open‐label study (HOHOEMI), we evaluated the efficacy, safety and pharmacokinetics of emicizumab in Japanese paediatric patients aged <12 years with severe haemophilia A without factor VIII (FVIII) inhibitors.
Methods
Emicizumab was administered subcutaneously, with four loading doses of 3 mg/kg every week followed by maintenance doses of 3 mg/kg every 2 weeks (Q2W) or 6 mg/kg every 4 weeks (Q4W) in 6 and 7 patients, respectively.
Results
All patients completed at least 24 weeks of treatment. Baseline ages ranged from 4 months to 10 years, and all patients had been treated with FVIII prophylaxis prior to enrolment except a 4‐month‐old patient untreated with FVIII previously. In the respective Q2W and Q4W cohorts, 2/6 and 5/7 patients experienced no treated bleeding events, and annualized bleeding rates for treated bleeding events were 1.3 (95% confidence interval [CI], 0.6‐2.9) and 0.7 (95% CI, 0.2‐2.6). All caregivers preferred emicizumab to the patient's previous treatment. Only one related adverse event (injection site reaction) was observed. There were no thromboembolic events or thrombotic microangiopathy. Individual trough plasma concentrations of emicizumab were within the variability observed in preceding adult/adolescent studies. All patients tested negative for anti‐emicizumab antibodies.
Conclusions
Emicizumab administered Q2W or Q4W was efficacious and safe in paediatric patients with severe haemophilia A without inhibitors. This study was registered at http://www.clinicaltrials.jp (JapicCTI‐173710).
Keywords: bispecific antibody, emicizumab, haemophilia A, non‐inhibitor, paediatrics, prophylaxis
1. INTRODUCTION
Haemophilia A is a lifelong bleeding disorder characterized by a congenital deficiency or dysfunction of factor VIII (FVIII). The standard of care for patients with severe haemophilia A without neutralizing antibodies against FVIII (‘FVIII inhibitors’) is intravenous administration of FVIII products.1 However, despite the regular prophylaxis with FVIII given once or more times per week, the majority of patients are still at the risk of bleeding.2, 3 Frequent intravenous infusions and vascular access are burdensome, particularly for paediatric patients and their caregivers. Moreover, FVIII inhibitors develop in up to approximately 30% of patients with severe haemophilia A receiving FVIII,4, 5 which renders FVIII products ineffective and complicates treatment of such patients.
Emicizumab (HEMLIBRA®; Chugai Pharmaceutical Co., Ltd.) is a recombinant humanized bispecific monoclonal antibody that binds to factor IX, activated factor IX (FIXa), factor X (FX) and activated factor X to, by bridging FIXa and FX, mimic the cofactor function of activated FVIII.6 In adult and adolescent patients with or without inhibitors, clinically meaningful efficacy of emicizumab for bleeding prevention was demonstrated with a subcutaneous maintenance dose of 1.5 mg/kg every week (QW) in the HAVEN 1 and HAVEN 3 studies,7, 8 and similar efficacy profiles were confirmed with less frequent subcutaneous maintenance doses of 3 mg/kg every 2 weeks (Q2W) and 6 mg/kg every 4 weeks (Q4W) in the HAVEN 3 and HAVEN 4 studies, respectively.8, 9 Another study (HAVEN 2) was conducted in paediatric patients with inhibitors.10 However, there was no clinical experience of emicizumab in paediatric patients without inhibitors.
The HOHOEMI study reported herein is the first study for emicizumab in paediatric patients without inhibitors, in which we evaluated the efficacy, safety and pharmacokinetics of the Q2W and Q4W regimens of emicizumab.
2. MATERIALS AND METHODS
This study was conducted at four centres in Japan, beginning in October 2017, in compliance with the Declaration of Helsinki and the ICH Guideline for Good Clinical Practice. The study protocol was approved by the institutional review board at each centre. Patients' legally authorized representatives provided written informed consent for study participation, and patients aged 3 years or older provided assent where possible. This study was registered at http://www.clinicaltrials.jp (JapicCTI‐173710).
2.1. Patients
Eligible participants were <12 years old weighing over 3 kg and had severe congenital haemophilia A without FVIII inhibitors. Patients tested negative for inhibitors (<0.6 BU/mL) within the 8 weeks prior to enrolment. Documentation of bleeding episodes and treatment with coagulation factors (including confirmation of no history of treatment with coagulation factors) was required for the 12 weeks prior to enrolment for patients <2 years old and for the 24 weeks prior to enrolment for patients ≥2 years old. Key exclusion criteria included complication of a bleeding disorder other than congenital haemophilia A, thromboembolic diseases within the past 12 months and high risk of thrombotic microangiopathy (TMA) based on previous or familial history of TMA (eg thrombotic thrombocytopenic purpura, atypical haemolytic uraemic syndrome).
2.2. Study design
This multicentre, open‐label, non‐randomized study was designed to evaluate the efficacy, safety and pharmacokinetics of emicizumab administered subcutaneously at a maintenance dose of 3 mg/kg Q2W or 6 mg/kg Q4W in paediatric patients with haemophilia A without inhibitors. Both the Q2W and Q4W cohorts received a loading dose of 3 mg/kg QW subcutaneously for the first 4 weeks before maintenance dosing (Figure 1). We planned for a minimum of six patients to be enrolled in each cohort. Patient enrolment in the Q2W cohort preceded that in the Q4W cohort. Patients who had received FVIII prophylaxis prior to enrolment were permitted to continue FVIII prophylaxis until receiving the second loading dose of emicizumab. FVIII products were administered for breakthrough bleeding as necessary.
Figure 1.
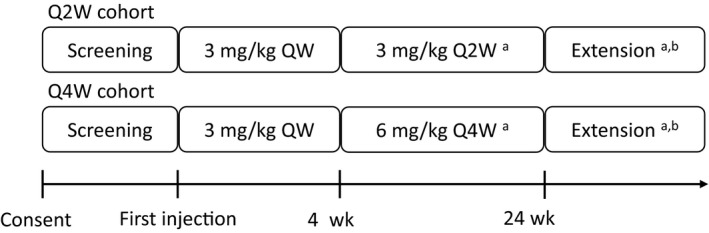
Study design. QW, every week; Q2W, every 2 weeks; Q4W, every 4 weeks. The study design is open‐label, non‐randomized. Patients in the Q4W cohort were enrolled after the completion of enrolment in the Q2W cohort. aPatients who experienced ≥2 bleeding events treated with coagulation factors during the last 8 wk of the first 12 wk of treatment were eligible for up‐titrating the maintenance dose to 3 mg/kg QW. After the first 12 wk of treatment, patients who experienced ≥2 bleeding events treated with coagulation factors during any consecutive 12 wk were eligible for the dose up‐titration. bPatients with sustained clinical benefit during the first 24 wk of treatment could continue emicizumab prophylaxis afterwards
Patients with sustained clinical benefit during the first 24 weeks of treatment could continue emicizumab prophylaxis afterwards. Patients who met the criteria for insufficient bleeding control after receiving emicizumab prophylaxis for at least 12 weeks were offered the opportunity to up‐titrate the maintenance dose to 3 mg/kg QW.
2.3. Outcome measures
Efficacy outcomes included annualized bleeding rates (ABRs) to evaluate the effects of emicizumab prophylaxis on bleeding frequency. The standardized definition of a bleeding event11 was applied (Appendix S1).
Caregivers were asked to record the types and durations of physical activities of patients for scheduled weeks. The types of activities were classified into three categories reflecting the risk of acute injury or collision (low, middle and high) that children could experience while participating in the activity.12 Caregivers were also asked to indicate whether they preferred emicizumab prophylaxis over the patient's previous haemophilia treatment after the first 16 weeks of treatment; caregivers who preferred emicizumab were asked to select influencing factors and the top three factors in order of perceived importance (EmiPref survey8).
Safety outcomes included adverse events (AEs), physical examination findings, vital signs and laboratory test abnormalities. Plasma emicizumab concentrations and anti‐emicizumab antibody positivity were determined as previously described.13, 14 FVIII inhibitors were measured by a clotting time‐based Bethesda assay with emicizumab in plasma samples neutralized by adding two anti‐emicizumab idiotype monoclonal antibodies ex vivo.15
2.4. Statistical analysis
The primary analysis was planned to be performed when the last patient had completed at least 24 weeks of treatment or was withdrawn from the study, and this manuscript reports the results of the primary analysis as of the data cut‐off date of 18 July 2018.
This study was designed to accumulate experience with emicizumab Q2W and Q4W administration in paediatric patients with haemophilia A without inhibitors and support regulatory approval. Considering the purpose and the limited number of patients, the sample size required to assess efficacy, safety and pharmacokinetics was set to at least six patients in each cohort, a total of at least 12 patients. No statistical hypothesis tests were planned. Patient‐level ABRs were calculated as 365.25 times the number of bleeding events divided by the number of days treated in each patient (hereinafter called ‘calculated ABRs’). Cohort‐level ABRs as a representative value for each cohort were estimated using a negative binomial regression model considering the difference of the length of the treatment period among patients (hereinafter called ‘model‐based ABRs’). Model‐based ABRs were used for the primary analysis of the HAVEN 1‐4 studies, and we compared the results based on model‐based ABRs with those of the HAVEN 1‐4 studies. SAS software version 9.2 (SAS Institute Inc) was used for the analyses.
3. RESULTS
3.1. Study population
A total of 13 Japanese male paediatric patients with severe haemophilia A without inhibitors participated in this study, with 6 and 7 patients in the Q2W and Q4W cohorts, respectively (Table 1). The median age (range) at baseline was 6.6 (1.5‐10.7) years and 4.1 (0.3‐8.1) years in the Q2W and Q4W cohorts, respectively. All patients had been treated with FVIII prophylaxis prior to enrolment except one patient aged 4 months in the Q4W cohort who had never received FVIII previously (previously untreated patient; PUP). The prior FVIII prophylaxis was administered about 2 or 3 times a week in 11 patients and once a week in one patient who refused frequent intravenous injection (Table S1). Each cohort included one patient previously treated with immune tolerance induction (ITI) therapy (7.4 and 0.4 years before enrolment). Only one patient in the Q2W cohort had a target joint.
Table 1.
Baseline demographic and clinical characteristics
| Characteristics | Q2W cohort (N = 6) | Q4W cohort (N = 7) |
|---|---|---|
| Age (y), median (range) | 6.6 (1.5‐10.7) | 4.1 (0.3‐8.1) |
| 0 to <2 y, no. (%) | 1 (16.7) | 2 (28.6) |
| 2 to <6 y, no. (%) | 2 (33.3) | 2 (28.6) |
| 6 to <12 y, no. (%) | 3 (50.0) | 3 (42.9) |
| Weight (kg), median (range) | 19.5 (10.9‐35.6) | 15.7 (6.6‐25.6) |
| Patients without FVIII inhibitors, no. (%) | 6 (100) | 7 (100) |
| Patients treated with FVIII prophylaxis prior to enrolment, no. (%) | 6 (100) | 6 (85.7) |
| Short acting, no.a | 5 | 5 |
| Long acting, no.a | 2 | 1 |
| Previously untreated patients (PUPs), no. (%) | 0 (0) | 1 (14.3) |
| Patients previously treated with ITI therapy, no. (%) | 1 (16.7) | 1 (14.3) |
| Patients with target jointb, no. (%) | 1 (16.7) | 0 (0) |
Abbreviations: FVIII, factor VIII; ITI, immune tolerance induction; Q2W, every 2 weeks; Q4W, every 4 weeks.
Multiple choices were allowed.
Target joints were defined as joints in which at least three bleeding events had occurred within the 24 wk prior to enrolment; target joints were not identified in patients <2 y old owing to the lack of historical data collection on bleeding episodes and treatment with coagulation factors during the 24 wk prior to enrolment.
As of the data cut‐off date, all 13 patients were continuing emicizumab prophylaxis, and no patients had dose up‐titration. The median (range) of treatment duration was 39.1 (36.4‐40.3) weeks and 32.1 (24.1‐36.4) weeks in the Q2W and Q4W cohorts, respectively.
3.2. Efficacy
During the on‐treatment period, 2/6 patients in the Q2W cohort and 5/7 patients in the Q4W cohort, including a PUP aged 4 months, had no treated bleeds. In the Q2W cohort, two patients each had two treated bleeds and two patients each had one treated bleed. In the Q4W cohort, one patient had two treated bleeds and one patient had one treated bleed. Out of the six treated bleeds in the Q2W cohort, one treated bleed was a spontaneous joint bleed, and the other five treated bleeds were traumatic including three joint bleeds occurring in three patients. In the Q4W cohort, all three treated bleeds were traumatic and were not joint bleeds. All nine treated bleeding events were successfully managed by episodic treatment with FVIII; eight treated bleeding events were managed with a single dose of FVIII (32.5‐64.7 IU/kg), and the other which occurred in a joint was managed with FVIII given once daily for 5 days (31.4‐32.6 IU/kg). One patient who had had a target joint (left knee) prior to enrolment had no bleeds at the joint. Model‐based ABRs for treated bleeding events were 1.3 (95% confidence interval [CI], 0.6‐2.9) and 0.7 (95% CI, 0.2‐2.6) in the Q2W and Q4W cohorts, respectively (Table 2).
Table 2.
Model‐based and calculated ABRs during emicizumab prophylaxis
| Patients without bleeding, no. (%) | Patients with bleeding, no. (total number of bleeds) | Model‐based ABRsa (95% CI) | Calculated ABRsb mean, median (range) | |
|---|---|---|---|---|
| Q2W cohort, N = 6, median (range) efficacy period: 39.9 (37.9‐41.4) wk | ||||
| Treated bleeds | 2 (33.3%) | 4 (6) | 1.3 (0.6‐2.9) | 1.3, 1.4 (0.0‐2.5) |
| Treated spontaneous bleeds | 5 (83.3%) | 1 (1) | 0.2 (0.0‐1.6) | 0.2, 0.0 (0.0‐1.3) |
| Treated joint bleeds | 2 (33.3%) | 4 (4) | 0.9 (0.3‐2.3) | 0.9, 1.3 (0.0‐1.4) |
| Treated target joint bleeds | 6 (100%) | 0 (0) | NE | 0.0, 0.0 (0.0‐0.0) |
| All bleeds | 0 (0%) | 6 (64) | 14.1 (7.6‐26.2) | 14.2, 10.7 (2.5‐35.0) |
| Q4W cohort, N = 7, median (range) efficacy period: 34.1 (24.1‐37.1) wk | ||||
| Treated bleeds | 5 (71.4%) | 2 (3) | 0.7 (0.2‐2.6) | 0.7, 0.0 (0.0‐3.1) |
| Treated spontaneous bleeds | 7 (100%) | 0 (0) | NE | 0.0, 0.0 (0.0‐0.0) |
| Treated joint bleeds | 7 (100%) | 0 (0) | NE | 0.0, 0.0 (0.0‐0.0) |
| Treated target joint bleeds | 7 (100%) | 0 (0) | NE | 0.0, 0.0 (0.0‐0.0) |
| All bleeds | 1 (14.3%) | 6 (100) | 21.8 (9.2‐51.8) | 21.7, 13.8 (0.0‐80.5) |
Abbreviations: ABR, annualized bleeding rate; CI, confidence interval; NE, not estimable; Q2W, every 2 weeks; Q4W, every 4 weeks.
Model‐based ABRs were derived from a negative binomial regression model including the on‐treatment period as an offset.
Calculated ABRs were derived for each patient, and the summary statistics were derived from the individual calculated ABRs.
For 5/6 patients in the Q2W cohort and all seven patients in the Q4W cohort, calculated ABRs for treated bleeding events in the on‐treatment period decreased from those in the pretreatment period or remained at zero (Figure S1).
Model‐based ABRs for all bleeding events in the on‐treatment period were 14.1 (95% CI, 7.6‐26.2) and 21.8 (95% CI, 9.2‐51.8) in the Q2W and Q4W cohorts, respectively. Non‐treated bleeding events included one muscle bleed and did not include joint bleeds. The remaining bleeds were bleeds at other sites such as subcutaneous tissue or the nose. The majority of the non‐treated bleeding events were traumatic.
The numbers of patients who engaged in moderate‐risk activities was higher after the first week than during the first week. Although no patients engaged in high‐risk activities during the first week, there were a few such patients after the first week. The mean time spent participating in activities with moderate or high risk increased after the first week (Figure 2). No obvious changes in the numbers of patients and the mean time for low‐risk activities were observed during the on‐treatment period (data not shown).
Figure 2.
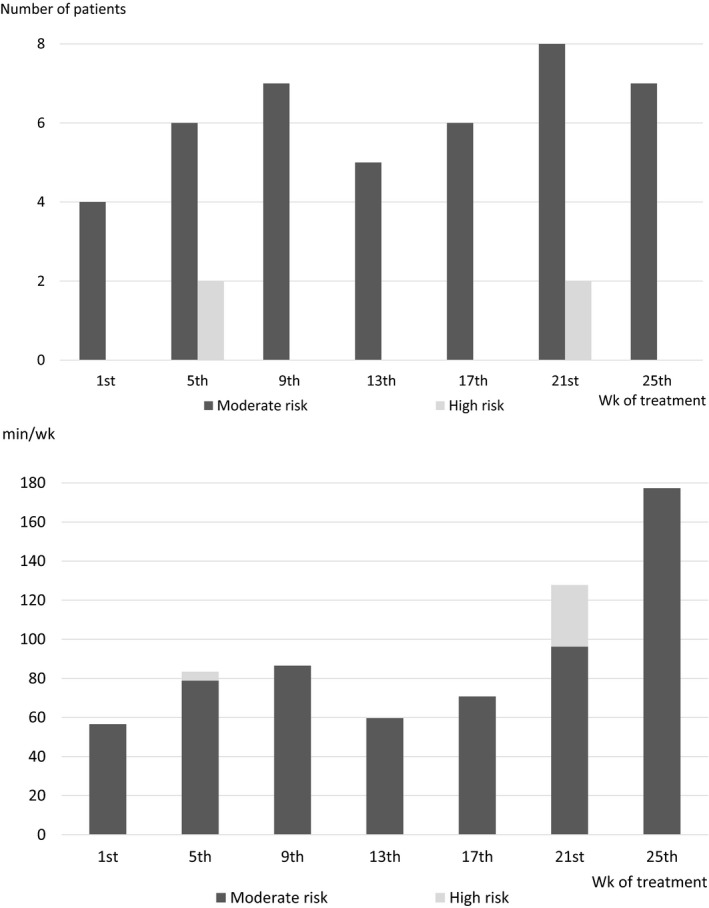
Number of patients participating in physical activity and mean time spent on physical activity. The upper panel shows the numbers of patients participating in each category of physical activity among all 13 enrolled patients in the indicated weeks. The lower panel shows the arithmetic mean times spent participating in each category of physical activity among all 13 enrolled patients in the indicated weeks. If a patient did no activity, time was set to zero for that patient. Activities with low risk include, for example, walking and swimming during which acute injury or collision is considered unlikely. Activities with moderate risk include, for example, soccer and basketball during which acute injury or collision is possible but not likely. Activities with high risk include, for example, rugby and wrestling during which acute injury or collision is likely. The baseline activity level was defined as the activity level during the first week of emicizumab prophylaxis. The data of activities with low risk are not shown in the panels
All caregivers completed the preference survey after the first 16 weeks of treatment, and all reported a preference for emicizumab prophylaxis over the patient's previous haemophilia treatment. All caregivers selected ‘the frequency of treatments was lower’ and ‘route of administration was easier’ as reasons which influenced their preference. The reasons most frequently ranked as the most important for their preference were ‘the frequency of treatments was lower’ (from 5 caregivers, 38.5%), and ‘effect on other activities (work, school, sports and social interactions) was less’ (from 3 caregivers, 23.1%; Figure 3).
Figure 3.
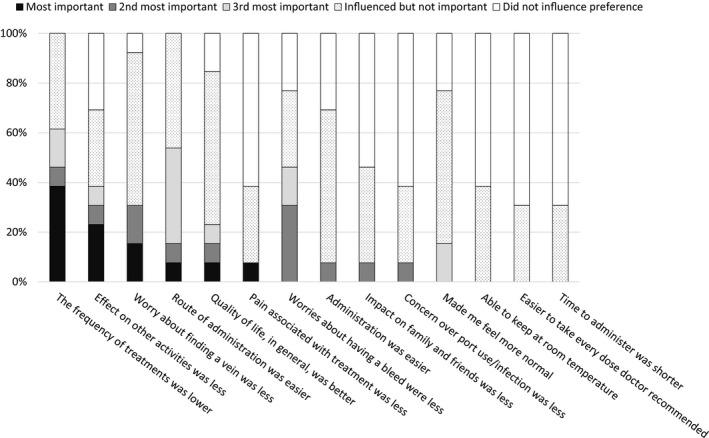
Reasons for caregivers' preference for emicizumab. All caregivers preferred emicizumab prophylaxis to the patient's previous haemophilia treatment. Each reason for the preference was ranked by caregivers. The proportions of the rankings given for each reason are presented here. Of note, the responses from the caregiver of a 4‐month‐old patient untreated with FVIII previously were based on the caregiver's experience of treatment for the patient's elder brother with severe haemophilia A
3.3. Safety
All patients experienced at least one AE, and a total of 133 AEs were reported; 62 and 71 events in the respective Q2W and Q4W cohorts. AEs that occurred in at least two patients are shown in Table 3. The most frequently reported AEs were contusion in 10 patients (76.9%), nasopharyngitis in five patients (38.5%), and excoriation and fall in four patients (30.8%) each. Only one event of injection site reaction was considered related to emicizumab by the investigators; it was of moderate intensity, occurred in the Q2W cohort 38.1 weeks after initiation of emicizumab prophylaxis and resolved without any treatment. There were no AEs that were of severe intensity or were life‐threatening, led to discontinuation of emicizumab prophylaxis, or resulted in dose reduction or interruption. No thromboembolic events, TMA or systemic hypersensitivity reactions were observed.
Table 3.
Adverse events reported in at least two patients
|
Q2W cohort N = 6 |
Q4W cohort N = 7 |
Total N = 13 |
|
|---|---|---|---|
| Total patients with ≥1 AE, no. (%) | 6 (100) | 7 (100) | 13 (100) |
| Total number of AEs | 62 | 71 | 133 |
| AEs reported in at least two patients, no. (%) | |||
| Contusion | 4 (66.7) | 6 (85.7) | 10 (76.9) |
| Nasopharyngitis | 2 (33.3) | 3 (42.9) | 5 (38.5) |
| Excoriation | 2 (33.3) | 2 (28.6) | 4 (30.8) |
| Fall | 1 (16.7) | 3 (42.9) | 4 (30.8) |
| Ligament sprain | 2 (33.3) | 1 (14.3) | 3 (23.1) |
| Influenza | 1 (16.7) | 2 (28.6) | 3 (23.1) |
| Oral contusion | 2 (33.3) | 1 (14.3) | 3 (23.1) |
| Bite | 1 (16.7) | 1 (14.3) | 2 (15.4) |
| Procedural pain | — | 2 (28.6) | 2 (15.4) |
| Scratch | 1 (16.7) | 1 (14.3) | 2 (15.4) |
| Wound | 1 (16.7) | 1 (14.3) | 2 (15.4) |
| Gastroenteritis | — | 2 (28.6) | 2 (15.4) |
| Upper respiratory tract infection | — | 2 (28.6) | 2 (15.4) |
| Diarrhoea | 1 (16.7) | 1 (14.3) | 2 (15.4) |
| Stomatitis | 2 (33.3) | — | 2 (15.4) |
| Arthralgia | 1 (16.7) | 1 (14.3) | 2 (15.4) |
| Eczema | 1 (16.7) | 1 (14.3) | 2 (15.4) |
Abbreviations: AE, adverse event; Q2W, every 2 weeks; Q4W, every 4 weeks.
Post‐traumatic pain (accompanying bleeding) and soft tissue haemorrhage (occurred subcutaneously) were reported as serious adverse events (SAEs) in one patient each in the Q2W and Q4W cohorts, both of which were traumatic bleeds and considered unrelated to emicizumab by the investigators. The two patients were hospitalized for the management of the bleeds and discharged from the hospital after wound healing without treatment with coagulation factors. The causal relationship between emicizumab and both SAEs was ruled out by the investigators.
Three patients underwent minor surgeries without safety issues. Two patients had one tooth extraction each; one patient was managed with a single preoperative preventative dose of FVIII (40.0 IU/kg), and the other patient was managed without preventative doses of FVIII. Neither required FVIII treatment after the surgeries. One patient had removal of an implanted central venous port device with a single preoperative preventative dose of FVIII (64.9 IU/kg) and received a single postoperative preventative dose of FVIII (64.9 IU/kg) after the surgery.
3.4. Pharmacokinetics and immunogenicity
Trough plasma concentrations of emicizumab averaged 48.7 and 48.4 μg/mL at the completion of the loading dose (4 weeks after treatment initiation) in the Q2W and Q4W cohorts, respectively (Figure 4). Mean steady‐state trough concentrations were maintained at approximately 35 and 30 μg/mL in the Q2W and Q4W cohorts, respectively, with individual trough concentrations ranging from 20.9 to 50.5 μg/mL and from 13.4 to 55.2 μg/mL, respectively, from 12 weeks after treatment initiation onwards.
Figure 4.
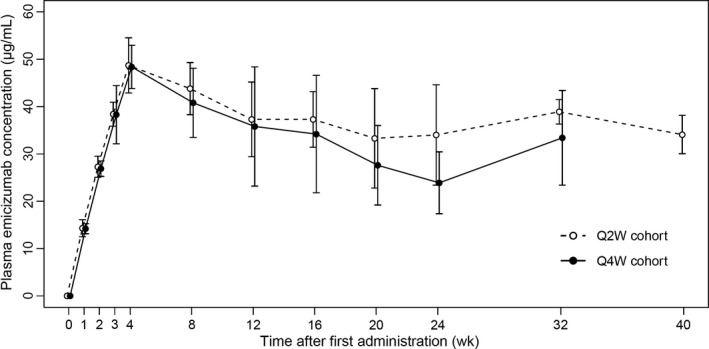
Time courses of trough plasma concentrations of emicizumab. Q2W, every 2 weeks; Q4W, every 4 weeks. Circles indicate the means, and bars on or under the circles indicate the standard deviations
All 13 patients tested negative for anti‐emicizumab antibodies. None of 11 examined patients (two with and nine without history of ITI therapy) had recurrent or de novo development of FVIII inhibitors or developed clinically relevant FVIII inhibitors during the study. No samples for FVIII inhibitor measurement were available from two patients (1.8 and 0.3 years old; both in the Q4W cohort and no history of ITI therapy) during the study due to predefined restrictions on blood sampling volume for ethical considerations.
4. DISCUSSION
In this HOHOEMI study, seven patients (53.8%) including a PUP aged 4 months had no treated bleeding events; the PUP was kept free from exposure to FVIII even during the study. The ABRs for treated bleeding events were low in both the Q2W and Q4W cohorts without clear differences between the cohorts (1.3 [95% CI, 0.6‐2.9] and 0.7 [95% CI, 0.2‐2.6], respectively). Importantly, these ABRs were comparable with those in adult and adolescent patients receiving the same Q2W or Q4W regimens in preceding studies (HAVEN 3 and HAVEN 4); 1.3 (95% CI, 0.8‐2.3) for the Q2W regimen and 2.4 (95% CI, 1.4‐4.3) for the Q4W regimen.8, 9
The results of the preference survey also supported the favourable characteristics of the Q2W and Q4W regimens of emicizumab. The most frequent reason selected as the most important for the preference was that the frequency of treatments was lower, which was expected as a potential benefit from the long half‐life and was in line with the results of HAVEN 3 and HAVEN 4.16 The second most frequent reason was less effect on other activities (work, school, sports and social interactions). This may imply that emicizumab prophylaxis reduces worries about bleeding and other physical or mental complications that patients may experience during such activities, which can in turn provide patients with a positive attitude towards those activities. In addition, lower frequency of treatment should provide a more relaxing schedule of daily life which may allow more activities. These interpretations are supported by the results of the physical activity survey which revealed an increase in patients engaging in activities with moderate or high risk during the on‐treatment period while maintaining ABRs at a low level.
The mean trough plasma concentrations of emicizumab during the 4‐week loading period were comparable with those in adult and adolescent patients receiving the same Q2W or Q4W regimen in HAVEN 3 and HAVEN 48, 9 for both cohorts. During the subsequent maintenance period, the mean steady‐state trough concentrations for the Q2W and Q4W cohorts were slightly lower than those for the Q2W dosing patients in HAVEN 3 and for the Q4W dosing patients in HAVEN 4, respectively. However, the individual trough concentrations were all within the minimum‐to‐maximum ranges of these adult/adolescent studies. In addition, the trough concentrations provided by the Q2W and Q4W regimens (eg ≥30 μg/mL at steady state) may be high enough to achieve almost the maximal effect of emicizumab. The ABRs for treated bleeding events were low in both cohorts, and they were not clearly different between the cohorts and even from those of HAVEN 3 and HAVEN 4, despite the mean steady‐state trough concentrations being slightly different. This absence of further reduction in ABRs depending on the trough concentrations is in line with a recent quantitative analysis indicating that the relationship between plasma emicizumab concentrations and ABRs reaches almost a plateau at above approximately 30 μg/mL 17; this appears similar to a reported relationship between FVIII activity and ABRs indicating that the risk of joint bleeds is minimized at 10‐15 IU/dL and higher.18 Taken together, these findings support the appropriateness of applying the Q2W and Q4W regimens in paediatric patients without inhibitors.
In this study, use of 2 anti‐emicizumab idiotype monoclonal antibodies enabled measurement of FVIII inhibitors in plasma samples involving emicizumab. Alternative approaches include use of a chromogenic Bethesda assay with reagents containing bovine FIXa and FX, which is insensitive to and can avoid interference by emicizumab.19
The main limitations to this study are the small numbers of patients in each cohort and the open‐label, non‐randomized, sequential‐cohort study design, which make it difficult to obtain robust results of efficacy and safety profiles of emicizumab.
5. CONCLUSION
This study showed remarkable efficacy and favourable safety of the Q2W and Q4W regimens of emicizumab in children with severe haemophilia A without inhibitors, including a PUP aged 4 months. The emicizumab exposure observed in this study was within the variability observed in the preceding adult/adolescent studies. These results confirm the appropriateness of applying the Q2W and Q4W regimens of emicizumab in paediatric patients with haemophilia A without inhibitors.
DISCLOSURES
This study was sponsored by Chugai Pharmaceutical Co., Ltd. M. Shima received research funding from Chugai Pharmaceutical Co., Ltd., F. Hoffmann‐La Roche Ltd., Bioverativ Inc, Shire Plc, CSL Behring, KM Biologics Co., Ltd. and Novo Nordisk A/S; consulting fee from Chugai Pharmaceutical Co., Ltd.; payment for lectures on speaker's bureau from Chugai Pharmaceutical Co., Ltd., Bioverativ Inc, Bayer AG and Sysmex corporation; is listed as an entity's board of directors or advisory committee member for Chugai Pharmaceutical Co., Ltd., F. Hoffmann‐La Roche Ltd., BioMarin Pharmaceutical Inc, Bayer AG and Sanofi SA; and is an inventor of patents related to anti‐FIXa/FX bispecific antibodies. K. Nogami received research funding from Chugai Pharmaceutical Co., Ltd., F. Hoffmann‐La Roche Ltd., Shire Plc, Bioverativ Inc, Novo Nordisk A/S and Bayer AG; consulting fee from Chugai Pharmaceutical Co., Ltd.; payment for lectures on speaker's bureau from Chugai Pharmaceutical Co., Ltd., Shire Plc, Bioverativ Inc, Novo Nordisk A/S and Bayer AG; is listed as an entity's board of directors or advisory committee member for Chugai Pharmaceutical Co., Ltd. and F. Hoffmann‐La Roche Ltd.; and is an inventor of patents related to anti‐FIXa/FX bispecific antibodies. S. Nagami is an employee of Chugai Pharmaceutical Co., Ltd. and holds stock in Chugai Pharmaceutical Co., Ltd. S. Yoshida is an employee of Chugai Pharmaceutical Co., Ltd. K. Yoneyama is an employee of Chugai Pharmaceutical Co., Ltd. and is an inventor of patents related to anti‐FIXa/FX bispecific antibodies. A. Ishiguro received research funding from Chugai Pharmaceutical Co., Ltd., F. Hoffmann‐La Roche Ltd., Novo Nordisk A/S, Pfizer Inc, KM Biologics Co., Ltd. and Teijin Pharma Ltd.; consulting fee from Novo Nordisk A/S; and payment for lectures on speaker's bureau from Chugai Pharmaceutical Co., Ltd., Novo Nordisk A/S and Eisai Co., Ltd. T. Suzuki received research funding from Chugai Pharmaceutical Co., Ltd., F. Hoffmann‐La Roche Ltd., Novo Nordisk A/S, BioMarin Pharmaceutical Inc, Shire Plc, Octapharma AG, Bayer AG, Pfizer Inc and Bioverativ Inc; and payment for lectures on speaker's bureau from Chugai Pharmaceutical Co., Ltd., Novo Nordisk A/S, Shire Plc, Bayer AG, Pfizer Inc, Bioverativ Inc, CSL Behring, KM Biologics Co., Ltd., Nihon Pharmaceutical Co., Ltd., Sekisui Medical Co., Ltd., Kyowa Hakko Kirin Co. Ltd., LSI Medience Corporation, Sysmex Corporation and Werfen. M. Taki received research funding from Chugai Pharmaceutical Co., Ltd., F. Hoffmann‐La Roche Ltd., Bioverativ Inc, CSL Behring and Novo Nordisk A/S; payment for lectures on speaker's bureau from Chugai Pharmaceutical Co., Ltd., CSL Behring, Bayer AG, Shire Plc, Bioverativ Inc and Novo Nordisk A/S; and is listed as an entity's board of directors or advisory committee member for Chugai Pharmaceutical Co., Ltd., Novo Nordisk A/S, Bioverativ Inc and Bayer AG.
AUTHOR CONTRIBUTIONS
M. Shima and S. Nagami designed the study, interpreted the data and wrote the manuscript. K. Nogami, A. Ishiguro, T. Suzuki and M. Taki collected and interpreted the data. S. Yoshida and K. Yoneyama designed the study, analysed and interpreted the data, and wrote the manuscript.
Supporting information
ACKNOWLEDGEMENTS
The authors thank all the patients and their family members; Dr Toshiaki Oka at Sapporo Tokushukai Hospital; investigators and staff at participating medical institutions; and colleagues at Chugai Pharmaceutical Co., Ltd, especially Akiko Matsusaki and Taisei Matsumoto. Medical writing and editorial assistance were provided by Takashi Funatogawa, Chugai Pharmaceutical Co., Ltd. Qualified researchers may request access to individual patient‐level data through the clinical study data request platform (http://www.clinicalstudydatarequest.com). For further details on Chugai's Data Sharing Policy and how to request access to related clinical study documents, see here (http://www.chugai-pharm.co.jp/english/profile/rd/ctds_request.html).
Shima M, Nogami K, Nagami S, et al. A multicentre, open‐label study of emicizumab given every 2 or 4 weeks in children with severe haemophilia A without inhibitors. Haemophilia. 2019;25:979–987. 10.1111/hae.13848
REFERENCES
- 1. Srivastava A, Brewer AK, Mauser‐Bunschoten EP, et al. Guidelines for the management of hemophilia. Haemophilia. 2013;19:e1‐e47. [DOI] [PubMed] [Google Scholar]
- 2. Valentino LA, Mamonov V, Hellmann A, et al. A randomized comparison of two prophylaxis regimens and a paired comparison of on‐demand and prophylaxis treatments in hemophilia A management. J Thromb Haemost. 2012;10:359‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mahlangu J, Powell JS, Ragni MV, et al. Phase 3 study of recombinant factor VIII Fc fusion protein in severe hemophilia A. Blood. 2014;123:317‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fischer K, Lewandowski D, Marijke van den Berg H, Janssen MP. Validity of assessing inhibitor development in haemophilia PUPs using registry data: the EUHASS project. Haemophilia. 2012;18:e241‐e246. [DOI] [PubMed] [Google Scholar]
- 5. Peyvandi F, Mannucci PM, Garagiola I, et al. A randomized trial of factor VIII and neutralizing antibodies in hemophilia A. N Engl J Med. 2016;374:2054‐2064. [DOI] [PubMed] [Google Scholar]
- 6. Kitazawa T, Esaki K, Tachibana T, et al. Factor VIIIa‐mimetic cofactor activity of a bispecific antibody to factors IX/IXa and X/Xa, emicizumab, depends on its ability to bridge the antigens. Thromb Haemost. 2017;117:1348‐1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Oldenburg J, Mahlangu JN, Kim B, et al. Emicizumab prophylaxis in hemophilia A with inhibitors. N Engl J Med. 2017;377:809‐818. [DOI] [PubMed] [Google Scholar]
- 8. Mahlangu J, Oldenburg J, Paz‐Priel I, et al. Emicizumab prophylaxis in patients who have hemophilia A without inhibitors. N Engl J Med. 2018;379:811‐822. [DOI] [PubMed] [Google Scholar]
- 9. Pipe SW, Shima M, Lehle M, et al. Efficacy, safety, and pharmacokinetics of emicizumab prophylaxis given every 4 weeks in people with haemophilia A (HAVEN 4): a multicentre, open‐label, non‐randomised phase 3 study. Lancet Haematol. 2019;6(6):e295‐e305. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 10. Young G, Liesner R, Sidonio RF Jr, et al. Emicizumab prophylaxis provides flexible and effective bleed control in children with hemophilia Α with inhibitors: results from the HAVEN 2 study. Blood. 2018;132(Suppl 1):632. [Google Scholar]
- 11. Blanchette VS, Key NS, Ljung LR, et al. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2014;12:1935‐1939. [DOI] [PubMed] [Google Scholar]
- 12. Broderick CR, Herbert RD, Latimer J, van Doorn N. Patterns of physical activity in children with haemophilia. Haemophilia. 2013;19:59‐64. [DOI] [PubMed] [Google Scholar]
- 13. Uchida N, Sambe T, Yoneyama K, et al. A first‐in‐human phase 1 study of ACE910, a novel factor VIII‐mimetic bispecific antibody, in healthy subjects. Blood. 2016;127:1633‐1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Paz‐Priel I, Chang T, Asikanius E, et al. Immunogenicity of emicizumab in people with hemophilia A (PwHA): results from the HAVEN 1–4 studies. Blood. 2018;132(Suppl 1):633. [Google Scholar]
- 15. Nogami K, Soeda T, Matsumoto T, Kawabe Y, Kitazawa T, Shima M. Routine measurements of factor VIII activity and inhibitor titer in the presence of emicizumab utilizing anti‐idiotype monoclonal antibodies. J Thromb Haemost. 2018;16:1383‐1390. [DOI] [PubMed] [Google Scholar]
- 16. Jimenez‐Yuste V, Shima M, Paz‐Priel I, et al. Preference for emicizumab over prior factor treatments: results from the HAVEN 3 and HAVEN 4 studies. Blood. 2018;132(Suppl 1):1187. [Google Scholar]
- 17. Jonsson F, Schmitt C, Petry C, Mercier F, Frey N, Retout S. Exposure‐response modeling of emicizumab for the prophylaxis of bleeding in hemophilia A patients with and without inhibitors against factor VIII. Res Pract Thromb Haemost. 2019;3(Suppl 1):315. Abstract PB0325.31294318 [Google Scholar]
- 18. den Uijl IE, Fischer K, Van Der Bom JG, Grobbee DE, Rosendaal FR, Plug I. Analysis of low frequency bleeding data: the association of joint bleeds according to baseline FVIII activity levels. Haemophilia. 2011;17:41‐44. [DOI] [PubMed] [Google Scholar]
- 19. Adamkewicz JI, Chen DC, Paz‐Priel I. Effects and interferences of emicizumab, a humanised bispecific antibody mimicking activated factor VIII cofactor function, on coagulation assays. Thromb Haemost. 2019;119(07):1084‐1093. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials