

Abstract
Background
The current randomized, double‐blind, phase 2 study assessed the efficacy and safety profile of a single intravenous administration of fosnetupitant, a neurokinin 1 receptor antagonist prodrug, for the prevention of chemotherapy‐induced nausea and vomiting in Japanese patients receiving cisplatin‐based chemotherapy.
Methods
Patients scheduled to receive cisplatin (at a dose of ≥70 mg/m2)‐based regimens were randomly assigned to receive fosnetupitant at a dose of 81 mg or 235 mg or placebo in combination with palonosetron at a dose of 0.75 mg and dexamethasone. The primary endpoint was complete response (CR; no vomiting and no rescue medication) during the overall phase (0‐120 hours). The overall CR rate was compared between each dose of fosnetupitant and the placebo group adjusting for the stratification factors of sex and age class (age <55 years vs age ≥55 years). Safety was assessed, with special attention given to events that potentially were suggestive of infusion site reactions.
Results
A total of 594 patients were randomized. Of these, 194 patients, 195 patients, and 195 patients, respectively, in the placebo and fosnetupitant 81‐mg and 235‐mg dose groups were evaluable for efficacy. The overall CR rate was 54.7% for the placebo group, 63.8% for the fosnetupitant 81‐mg dose group (adjusted difference, 9.1%; 95% CI, ‐0.4% to 18.6% [P = .061]), and 76.8% for the fosnetupitant 235‐mg dose group (adjusted difference, 22.0%; 97.5% CI, 11.7% to 32.3% [P < .001]). Safety profiles were comparable between the 3 groups. The incidence of infusion site reactions related to fosnetupitant was ≤1% in each dose group.
Conclusions
Fosnetupitant at a dose of 235 mg provided superior prevention of chemotherapy‐induced nausea and vomiting among patients receiving cisplatin‐based chemotherapy compared with the control group, and with a satisfactory safety profile.
Keywords: chemotherapy‐induced nausea and vomiting, fosnetupitant, highly emetogenic chemotherapy, injection site reaction, neurokinin 1 receptor antagonist, phase 2
Short abstract
Fosnetupitant at a dose of 235 mg combined with palonosetron and dexamethasone was demonstrated to be effective in the prevention of emesis during the first 0 to 120 hours after the administration of cisplatin. The results of the current study suggest that fosnetupitant at a dose of 235 mg also may improve the percentage of patients with no nausea in the delayed phase (24 ‐ 120 hours after cisplatin administration), which is an unmet medical need of patients with cancer who are receiving chemotherapy.
Introduction
Chemotherapy‐induced nausea and vomiting (CINV) is a common side effect related to cancer treatment, thereby making the prevention of CINV very important. Antiemetic guidelines have recommended the use of combinations of 3 or 4 prophylactic agents, including dexamethasone, 5‐hydroxytryptamine 3 (5‐HT3) receptor antagonists (RAs), neurokinin 1 (NK1) RAs, and olanzapine for the prevention of CINV in patients receiving highly emetogenic chemotherapy (HEC).1, 2, 3 With NK1 RAs, the use of an intravenous formulation can improve compliance compared with an oral formulation because intravenous agents are administered only on the day of chemotherapy. However, fosaprepitant, which is an intravenously administered NK1 RA, may cause infusion site reactions (ISRs).4 To expand the treatment options for the prevention of CINV, new agents are required.
Fosnetupitant is a phosphorylated prodrug of netupitant, which has high selectivity and an affinity for the NK1 receptor. Netupitant is marketed as NEPA (a fixed oral combination of netupitant at a dose of 300 mg and palonosetron at a dose of 0.5 mg) worldwide, except in Japan. The optimal dose of fosnetupitant is 235 mg (corresponding to a 260‐mg dose of fosnetupitant chloride hydrochloride), which is the bioequivalent of netupitant at a dose of 300 mg in terms of area under the exposure time curve.5 Fosnetupitant in combination with palonosetron (at a dose of 0.25 mg intravenously), NEPA for injection, was approved for the prevention of acute and delayed CINV associated with HEC in the United States based on bioequivalence data and on a phase 3 safety study in initial and repeated cycles in >400 patients.6
A phase 1 study in healthy adults in Japan demonstrated that fosnetupitant was well tolerated up to a dose of 353 mg and confirmed that fosnetupitant rapidly converts to netupitant after administration.7 A phase 2 study of oral netupitant in Japanese patients receiving HEC assessed the dose‐response relationship using 3 doses (30 mg, 100 mg, and 300 mg). However, the dose‐response relationship and efficacy of netupitant could not be clearly demonstrated because a placebo was not included for comparison.8 Therefore, we considered that the inclusion of a placebo arm was needed to confirm the efficacy of fosnetupitant.
The primary endpoint of the current study was to determine the fosnetupitant dose superior to placebo. To the best of our knowledge, the current study represents the first efficacy study in the development of fosnetupitant as a single agent. To this aim, the efficacy and safety of a single dose of fosnetupitant of 81 mg (corresponding to fosnetupitant chloride hydrochloride at a dose of 90 mg) or 235 mg was compared with placebo plus palonosetron at a dose of 0.75 mg and dexamethasone in Japanese patients receiving cisplatin‐based HEC.
Materials and Methods
Study Design
The current multicenter, randomized, double‐blind, placebo‐controlled, parallel group phase 2 study was conducted at 74 institutions in Japan. Study sites are listed in Supporting Table S1. The study was approved by the institutional review board of each participating institution and was performed in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice guidelines. All patients provided written informed consent before study enrollment.
This trial was registered with Clinical Trial Registration (number Japic CTI‐163355).
Patients
Patients aged ≥20 years who had a confirmed malignant solid tumor, were scheduled to receive cisplatin at a dose of ≥70 mg/m2, and had received no chemotherapy or prior low or minimally emetogenic chemotherapy regimen were eligible. Patients were required to have an Eastern Cooperative Oncology Group performance status of 0 or 1 and adequate hematologic, hepatic, and renal function.
Patients were excluded if they had gastrointestinal stenosis; any vomiting, retching, or nausea within 24 hours prior to enrollment; severe complications, infection, or diabetes mellitus that could be associated with difficulties with the administration of dexamethasone; or hypersensitivity to NK1 RAs, 5‐HT3 RAs, or dexamethasone. Patients who had received a cytochrome P450 3A4 inhibitor or inducer, had received an opioid analgesic, had undergone surgery, or had undergone radiotherapy within 7 days before registration and pregnant and nursing women also were excluded.
Treatment
Patients were randomly assigned using the minimization method, stratified by sex and age class (age <55 years vs age ≥55 years), to receive placebo or fosnetupitant at a dose of 81 mg or 235 mg. The study drug and palonosetron at a dose of 0.75 mg were administered intravenously approximately 60 minutes prior to the administration of cisplatin and infused for 30 minutes on day 1. The dose of palonosetron of 0.75 mg is approved in Japan based on the study.9 Dexamethasone (at a dose of 9.9 mg for the fosnetupitant group and 13.2 mg for the placebo group) was administered intravenously 60 minutes prior to the administration of cisplatin. On days 2 to 4, dexamethasone at a dose of 6.6 mg was administered intravenously in the morning. Because dexamethasone exposure is increased when given in combination with fosnetupitant, the dose of dexamethasone in the groups of patients receiving fosnetupitant was reduced to achieve dexamethasone exposure similar to that in the placebo group.
Rescue medication was permitted at the discretion of the investigators for the treatment of vomiting or nausea during the 168 hours after the initiation of cisplatin. Aprepitant, fosaprepitant, palonosetron, and/or dexamethasone were not permitted as rescue medications.
During the study, treatment assignment was masked from all patients, investigators, and study personnel except for the pharmacists who were preparing the study drugs at the institutions, who were prohibited from divulging any information regarding drug assignment.
Endpoints
The primary endpoint of the current study was the percentage of patients with a complete response (CR; no emesis and no rescue medication) during the overall phase (0‐120 hours after cisplatin administration). The secondary endpoints were the percentages of patients with CR during the acute (0‐24 hours) and delayed (24‐120 hours) phases, and during 24 to 168 hours after cisplatin administration as well as complete protection (CR plus no more than mild nausea); total control (CR plus no nausea); and no vomiting, no nausea, and no significant nausea during the acute, delayed, and overall phases as well as during 24 to 168 hours after the initiation of cisplatin. Other secondary endpoints were the rates of adverse events (AEs) and the frequency of ISRs.
Assessment
All patients were hospitalized for an 8‐day observation period after the administration of cisplatin. They recorded episodes of nausea and vomiting every 24 hours in a diary. An emetic episode was defined as ≥1 continuous episodes of vomiting and retching. The severity of nausea was measured using a 4‐point Likert scale (none, mild, moderate, or severe).
AEs were assessed by the investigators according to the National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.03). ISRs were evaluated after study drug administration in patients without central venous access devices.
Pharmacokinetics
Blood samples were obtained for the evaluation of fosnetupitant and netupitant pharmacokinetics (PKs) from at least 10 patients in each group at the following time points: before dosing and at 0.25, 0.5, 0.75, 1, 2, 3, 5, 8, 24, 48, 72, 120, and 168 hours after administration of the study drug. Plasma samples were prepared by centrifugation of the blood samples. Fosnetupitant and netupitant concentrations in the plasma samples were determined using validated liquid chromatography/tandem mass spectrometry methods. The PK parameters shown in Supporting Table S2 were calculated from the obtained plasma concentration profiles.
Statistical Analysis
The primary objective of the current study was to demonstrate the superiority of fosnetupitant over placebo in terms of the percentage of patients who achieved a CR during the overall phase in the full analysis set, which comprised all patients who received cisplatin, the study drug, palonosetron, and dexamethasone on day 1. For the primary efficacy analysis, the overall CR rates were estimated using the logistic regression model with the stratification factors of sex and age class (age <55 years vs age ≥55 years) as covariates by marginal standardization10 and compared between each fosnetupitant dose group and the placebo group at an overall 2‐sided significance level of 5% by statistical testing assuming normality. Testing was adjusted for multiplicity using the Hochberg method to maintain the overall 2‐sided significance level at 5%. Under the assumption that overall CR rates were 65% for fosnetupitant and 50% for placebo based on a previous phase 2 study of fosnetupitant,11 a phase 2 study of netupitant conducted in Japan,8 and a phase 3 study of palonosetron,9 a sample size of 185 evaluable patients per group (585 in total, considering the possibility of dropouts) was determined to ensure a power of at least 80% for each comparison in the primary analysis.
Safety was analyzed in the as‐treated population, which included all patients who received the study drug.
PK parameters were determined for fosnetupitant and netupitant. Each PK parameter was evaluated in the fosnetupitant groups.
Statistical analysis was performed using SAS statistical software (version 9.4; SAS Institute Inc, Cary, North Carolina) and WinNonlin (version 7.0; Certara LP, Princeton, New Jersey) for PKs only.
Results
A total of 594 patients were randomized between September 5, 2016, and November 24, 2017. Seven patients did not receive the study treatment (Fig. 1). The baseline characteristics of the full analysis set were similar across the placebo and fosnetupitant 81‐mg dose and 235‐mg dose groups (Table 1). In addition, 17 patients and 10 patients, respectively, in the fosnetupitant 81‐mg dose and 235‐mg dose groups were evaluated for PKs. The baseline characteristics of the PK population were similar to those in the full analysis set (data not shown).
Figure 1.
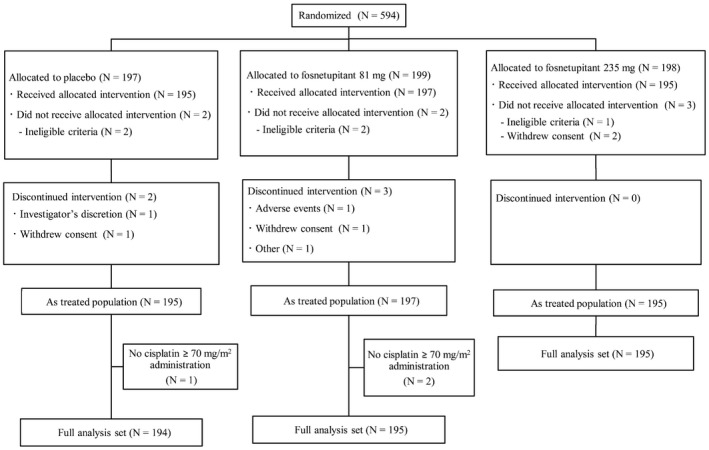
Consolidated Standards Of Reporting Trials (CONSORT) diagram.
Table 1.
Baseline Patient Characteristics: Full Analysis Set
| Placebo N = 194 | Fosnetupitant at a Dose of 81 mg N = 195 | Fosnetupitant at a Dose of 235 mg N = 195 | |
|---|---|---|---|
| Sex, no. (%) | |||
| Male | 147 (75.8) | 146 (74.9) | 148 (75.9) |
| Female | 47 (24.2) | 49 (25.1) | 47 (24.1) |
| Age, y | |||
| Median | 67.0 | 66.0 | 67.0 |
| Range | 36‐79 | 41‐76 | 37‐78 |
| Age category, no. (%) | |||
| <55 y | 24 (12.4) | 25 (12.8) | 22 (11.3) |
| ≥55 y | 170 (87.6) | 170 (87.2) | 173 (88.7) |
| Drinking history, no. (%) | |||
| No | 71 (36.6) | 70 (35.9) | 67 (34.4) |
| Rarely (once per mo) | 18 (9.3) | 22 (11.3) | 23 (11.8) |
| Occasionally | 18 (9.3) | 21 (10.8) | 24 (12.3) |
| Regularly | 87 (44.8) | 82 (42.1) | 81 (41.5) |
| Smoking history, no. (%) | |||
| Nonsmoker | 41 (21.1) | 33 (16.9) | 40 (20.5) |
| Stopped smoking prior to 180 d before registration | 82 (42.3) | 88 (45.1) | 87 (44.6) |
| Stopped smoking within 180 d before registration | 51 (26.3) | 63 (32.3) | 52 (26.7) |
| Smoker | 20 (10.3) | 11 (5.6) | 16 (8.2) |
| ECOG performance status, no. (%) | |||
| 0 | 124 (63.9) | 125 (64.1) | 113 (57.9) |
| 1 | 70 (36.1) | 70 (35.9) | 82 (42.1) |
| Cancer type, no. (%) | |||
| Lung | 184 (94.8) | 184 (94.4) | 187 (95.9) |
| Other | 10 (5.2) | 11 (5.6) | 8 (4.1) |
| Prior systemic drug therapies, no. (%) | |||
| No | 175 (90.2) | 175 (89.7) | 167 (85.6) |
| Yes | 19 (9.8) | 20 (10.3) | 28 (14.4) |
| Chemotherapy regimen, no. (%) | |||
| Cisplatin plus vinorelbine tartrate | 69 (35.6) | 66 (33.8) | 56 (28.7) |
| Cisplatin plus pemetrexed | 56 (28.9) | 57 (29.2) | 63 (32.3) |
| Cisplatin plus pemetrexed plus bevacizumab | 29 (14.9) | 32 (16.4) | 29 (14.9) |
| Cisplatin plus etoposide | 24 (12.4) | 21 (10.8) | 24 (12.3) |
| Cisplatin plus other cancer treatment | 16 (8.2) | 19 (9.7) | 23 (11.8) |
Abbreviation: ECOG, Eastern Cooperative Oncology Group.
Efficacy
The overall CR rates were 54.7%, 63.8%, and 76.8%, respectively, in the placebo, fosnetupitant 81‐mg dose, and fosnetupitant 235‐mg dose groups. There was not a statistically significant difference noted between the fosnetupitant 81‐mg dose and placebo groups (adjusted difference, 9.1%; 95% CI, ‐0.4% to 18.6% [P = .061; 2‐sided significance level = .05]). The overall CR rate in the fosnetupitant 235‐mg dose group was statistically superior to that in the placebo group (adjusted difference, 22.0%; 97.5% CI, 11.7% to 32.3% [P < .001; 2‐sided significance level = .025]).
The CR rates for each observation period are shown in Figure 2. The CR rates for the acute and delayed phases and for the period of 24 to 168 hours were higher in the fosnetupitant 235‐mg dose group compared with the placebo group. The rates of all secondary efficacy endpoints were higher in the fosnetupitant 235‐mg dose group compared with the placebo group (see Supporting Table S3). The rate of “no nausea” in the delayed phase was approximately 15 percentage points higher in the fosnetupitant 235‐mg dose group (56.4%) compared with the placebo group (41.2%).
Figure 2.
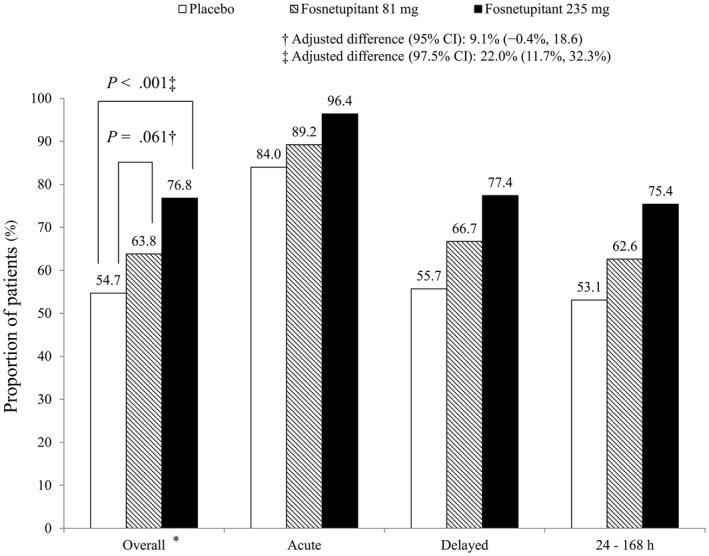
Percentage of patients achieving a complete response in the overall (0‐120 hours), acute (0‐24 hours), and delayed (24‐120 hours) phases and during 24 to 168 hours after cisplatin administration (full analysis set). * indicates the primary endpoint.
Safety
The incidences of AEs and serious AEs were similar between the 3 groups (see Supporting Table S4). The incidences of AEs and treatment‐related AEs were similar between the 3 treatment groups, with no tendency noted for the rates of AEs to increase in a dose‐dependent manner (Table 2). Treatment‐related AEs with an incidence ≥5% were constipation (13.8%, 14.2%, and 16.4%, respectively, in the placebo and fosnetupitant 81‐mg dose and 235‐mg dose groups) and hiccups (4.6%, 7.1%, and 5.6%, respectively, in the placebo and fosnetupitant 81‐mg dose and 235‐mg dose groups). ISR‐related AEs were all grade 1 or 2, and there was no tendency for the rate to increase in a dose‐dependent manner (5.7%, 10.8%, and 7.9%, respectively, in the placebo and fosnetupitant 81‐mg dose and 235‐mg dose groups). The incidences of treatment‐related injection site thrombophlebitis and injection site discomfort were similar between the 3 treatment groups (Table 3).
Table 2.
AEs and Treatment‐Related AEs Occurring in at Least 5% of Patients in Each Treatment Group: As‐Treated Populationa
| AE | AE | Treatment‐Related AE | ||||
|---|---|---|---|---|---|---|
| Placebo N = 195 | Fosnetupitant at a Dose of 81 mg N = 197 | Fosnetupitant at a Dose of 235 mg N = 195 | Placebo N = 195 | Fosnetupitant at a Dose of 81 mg N = 197 | Fosnetupitant at a Dose of 235 mg N = 195 | |
| Anemia | 10 (5.1) | 17 (8.6) | 11 (5.6) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Febrile neutropenia | 3 (1.5) | 11 (5.6) | 6 (3.1) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Leukopenia | 8 (4.1) | 9 (4.6) | 14 (7.2) | 0 (0.0) | 0 (0.0) | 1 (0.5) |
| Neutropenia | 18 (9.2) | 28 (14.2) | 27 (13.8) | 0 (0.0) | 1 (0.5) | 1 (0.5) |
| Hyperglycemia | 5 (2.6) | 5 (2.5) | 10 (5.1) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Hyponatremia | 7 (3.6) | 20 (10.2) | 17 (8.7) | 0 (0.0) | 6 (3.0) | 3 (1.5) |
| Decreased appetite | 78 (40.0) | 78 (39.6) | 71 (36.4) | 2 (1.0) | 6 (3.0) | 8 (4.1) |
| Insomnia | 21 (10.8) | 20 (10.2) | 12 (6.2) | 1 (0.5) | 1 (0.5) | 1 (0.5) |
| Dysgeusia | 7 (3.6) | 11 (5.6) | 12 (6.2) | 0 (0.0) | 2 (1.0) | 0 (0.0) |
| Headache | 4 (2.1) | 9 (4.6) | 13 (6.7) | 0 (0.0) | 4 (2.0) | 4 (2.1) |
| Hiccups | 64 (32.8) | 65 (33.0) | 80 (41.0) | 9 (4.6) | 14 (7.1) | 11 (5.6) |
| Upper abdominal pain | 5 (2.6) | 13 (6.6) | 10 (5.1) | 0 (0.0) | 1 (0.5) | 0 (0.0) |
| Constipation | 120 (61.5) | 128 (65.0) | 129 (66.2) | 27 (13.8) | 28 (14.2) | 32 (16.4) |
| Diarrhea | 10 (5.1) | 19 (9.6) | 20 (10.3) | 1 (0.5) | 2 (1.0) | 1 (0.5) |
| Nausea | 25 (12.8) | 31 (15.7) | 23 (11.8) | 0 (0.0) | 2 (1.0) | 1 (0.5) |
| Stomatitis | 8 (4.1) | 11 (5.6) | 13 (6.7) | 0 (0.0) | 2 (1.0) | 0 (0.0) |
| Vomiting | 11 (5.6) | 3 (1.5) | 1 (0.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Injection site pain | 8 (4.1) | 14 (7.1) | 11 (5.6) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Malaise | 24 (12.3) | 25 (12.7) | 32 (16.4) | 2 (1.0) | 1 (0.5) | 3 (1.5) |
| Alanine aminotransferase increased | 8 (4.1) | 11 (5.6) | 11 (5.6) | 3 (1.5) | 5 (2.5) | 8 (4.1) |
| Blood creatinine increased | 10 (5.1) | 15 (7.6) | 6 (3.1) | 1 (0.5) | 1 (0.5) | 0 (0.0) |
| Neutrophil count decreased | 59 (30.3) | 56 (28.4) | 56 (28.7) | 0 (0.0) | 0 (0.0) | 1 (0.5) |
| Platelet count decreased | 17 (8.7) | 18 (9.1) | 16 (8.2) | 0 (0.0) | 1 (0.5) | 0 (0.0) |
| White blood cell count decreased | 42 (21.5) | 46 (23.4) | 43 (22.1) | 0 (0.0) | 0 (0.0) | 1 (0.5) |
Abbreviation: AE, adverse event.
Shown as the number of patients (%).
AEs reported by the investigator were coded using MedDRA/J (version 20.1). A treatment‐related AE was an AE judged by the investigator to be related, most likely related, or possibly related to the study drug.
Assessed using National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.03).
Table 3.
Summary of Infusion Site Reactions in Patients Without a Central Venous Access Device Among the As‐Treated Population
| Patients With AEs at the Infusion Site | AEsa | Treatment‐Related AEsa | ||||
|---|---|---|---|---|---|---|
| Placebo N = 193 | Fosnetupitant at a Dose of 81 mg N = 194 | Fosnetupitant at a Dose of 235 mg N = 191 | Placebo N = 193 | Fosnetupitant at a Dose of 81 mg N = 194 | Fosnetupitant at Dose of 235 mg N = 191 | |
| Total ISRs | 11 (5.7) | 21 (10.8) | 15 (7.9) | 0 (0.0) | 1 (0.5) | 2 (1.0) |
| Injection site pain | 8 (4.1) | 14 (7.2) | 11 (5.8) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Injection site erythema | 5 (2.6) | 3 (1.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Injection site induration | 3 (1.6) | 2 (1.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Injection site thrombophlebitis | 0 (0.0) | 3 (1.5) | 3 (1.6) | 0 (0.0) | 1 (0.5) | 1 (0.5) |
| Injection site reaction | 0 (0.0) | 2 (1.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Injection site discomfort | 0 (0.0) | 0 (0.0) | 1 (0.5) | 0 (0.0) | 0 (0.0) | 1 (0.5) |
Abbreviations: AE, adverse event; ISR, infusion site reaction.
Data are shown as the number of patients (%).
Assessed using National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.03).
Pharmacokinetics
The mean concentration‐time profiles of fosnetupitant and netupitant in plasma for the 2 fosnetupitant doses are illustrated in Supporting Figure S1. A summary of the PK parameters for each dose is presented in Supporting Table S2. Fosnetupitant was eliminated rapidly from plasma after administration. The plasma concentration of netupitant reached a maximum at the end of the infusion and then declined gradually, with a mean elimination half‐life of 69.5 to 82.7 hours. Exposure to netupitant increased in a dose‐dependent manner.
Discussion
In the current study, the addition of a single intravenous dose of 235 mg of fosnetupitant to palonosetron and dexamethasone demonstrated superiority to palonosetron and dexamethasone alone with respect to the overall CR rate for patients receiving cisplatin‐based HEC. Fosnetupitant at a dose of 235 mg consistently was found to be more effective than placebo and fosnetupitant at a dose of 81 mg across all secondary efficacy endpoints and demonstrated an acceptable safety profile. The adjusted difference in the overall CR rate between the fosnetupitant 81‐mg dose group and the placebo group was not statistically significant.
In a previous phase 2 study of oral netupitant that was conducted outside Japan, superiority versus placebo was demonstrated for all doses (100 mg, 200 mg, and 300 mg), but clear differences were not observed between the netupitant dose groups.11 There are several potential reasons for this difference between the 2 studies. First, the prescribed cisplatin dose was different. The previous phase 2 study of oral netupitant enrolled patients receiving cisplatin at a dose of ≥50 mg/m2,11 whereas the current study enrolled patients receiving cisplatin at a dose of ≥70 mg/m2. The difference in the treatment effect between (fos)netupitant doses might be more notable when administered with a cisplatin dose of ≥70 mg/m2 compared with a dose of ≥50 mg/m2.12 Second, the dose and formulation of palonosetron differed between the 2 studies (intravenous 0.75 mg in the current study vs an oral dose of 0.5 mg in the previous study11). Oral palonosetron at a dose of 0.5 mg is comparable to intravenous palonosetron at a dose of 0.25 mg.13 Palonosetron at a dose of 0.75 mg demonstrated better efficacy in the delayed and overall phases compared with the dose of 0.25 mg in the previous study conducted in Japan,14 and therefore a lower fosnetupitant dose of 81 mg might not provide sufficient additional efficacy to demonstrate superiority over the placebo group in this setting.
Although the CR rate in the acute phase in the group treated with fosnetupitant was comparable to that reported with other NK1 RA‐containing regimens, the additional effect of fosnetupitant on “no nausea” in the delayed phase was higher than that observed with other NK1 RAs. Although patients treated with aprepitant and fosaprepitant improved by 5.9 to 8.7 percentage points,15, 16 the patients treated with fosnetupitant at a dose of 235 mg improved by approximately 15 percentage points with regard to experiencing no nausea in the delayed phase versus patients receiving placebo. Given that >50% of patients enrolled in the current study were male, aged >55 years, and had an alcohol history, such a population might not be considered as group at an extremely high risk of developing CINV. However, the patient characteristics of the current study such as sex and age were not largely different from those of the previous studies of aprepitant and fosaprepitant.15, 16 A prospective registration study performed by the CINV Study Group of Japan reported that the incidence and severity of nausea peaked on days 4 to 5 among patients receiving cisplatin.17 Prophylaxis for delayed nausea remains an unmet medical need; the results of the current study suggest that regimens containing fosnetupitant at a dose of 235 mg could suppress such late‐onset nausea.
PK parameters demonstrated that fosnetupitant was eliminated rapidly from plasma and that netupitant reached a maximum plasma concentration at the end of infusion, suggesting that fosnetupitant is rapidly converted to netupitant, which is the active form. No remarkable differences in PK parameters were reported between patients in the United States and Japanese patients.18 Therefore, it appears that fosnetupitant can be administered just before the administration of cisplatin.
The incidence and severity of AEs and treatment‐related AEs were found to be similar between the treatment groups, and the incidence in the fosnetupitant groups did not increase in a dose‐dependent manner. The AEs reported generally were those known to occur in patients receiving chemotherapy and palonosetron. Treatment‐related ISRs occurred in ≤1.0% of patients in the current study and were not reported to occur in a phase 1 study of fosnetupitant in healthy adults7 conducted in Japan or in a phase 3 safety study of NEPA in initial and repeated cycles performed outside of Japan.6 Based on these data, we believe fosnetupitant is unlikely to cause ISRs and that intravenous fosnetupitant can be administered safely.
To our knowledge, the current study is the first to demonstrate superior efficacy with the addition of fosnetupitant to palonosetron at a dose of 0.75 mg and dexamethasone in patients receiving cisplatin. A limitation of the current study was that the relative clinical efficacy of a fosnetupitant‐based regimen was unclear because this study did not include an active comparator.
Fosnetupitant in combination with palonosetron at a dose of 0.75 mg and dexamethasone was superior to palonosetron at a dose of 0.75 mg and dexamethasone alone in patients receiving cisplatin‐based chemotherapy. The recommended dose of fosnetupitant in this regimen is 235 mg, which is the same dose that was approved for use with palonosetron at a dose of 0.25 mg as a fixed‐dose combination regimen in the United States. We are planning to conduct a confirmatory study to verify the efficacy and safety of fosnetupitant versus an existing active comparator regimen.
Funding Support
Funding for this trial was provided by Taiho Pharmaceutical Co Ltd.
Conflict of Interest Disclosures
Shunichi Sugawara received a lecture fee and writing support from Taiho Pharmaceutical Co Ltd and lecture fees from Chugai Pharmaceutical Co Ltd, AstraZeneca, Nippon Boehringer Ingelheim, MSD, Pfizer, Eli Lilly, Novartis, Kyowa Hakko Kirin, Bristol‐Myers Squibb, and Ono Pharmaceutical for work performed outside of the current study. Naoki Inui received a grant and lecture fee from and acted as a paid member of the advisory board for Taiho Pharmaceutical Co Ltd; received grants and lecture fees from Chugai Pharmaceutical Co Ltd and Boehringer Ingelheim; received a lecture fee from Ono Pharmaceutical; and received a grant from Novartis for work performed outside of the current study. Masashi Kanehara received a grant from Taiho Pharmaceutical Co Ltd for work performed as part of the current study. Masahiro Morise received a grant from Taiho Pharmaceutical Co Ltd for work performed as part of the current study and lecture fees from Taiho Pharmaceutical Co Ltd, Boehringer‐Ingelheim, Daiichi Sankyo, Eli Lilly, Chugai Pharmaceutical Co Ltd, AstraZeneca, Ono Pharmaceutical, and Pfizer for work performed outside of the current study. Kozo Yoshimori received lecture fees from Taiho Pharmaceutical Co Ltd, Chugai Pharmaceutical Co Ltd, Bristol‐Myers Squibb, MSD, AstraZeneca, Novartis, Pfizer, and Eli Lilly Japan for work performed outside of the current study. Toru Kumagai received a grant and lecture fee from Taiho Pharmaceutical Co Ltd and lecture fees from Teijin Pharma, AstraZeneca, Chugai Pharmaceutical Co Ltd, Boehringer Ingelheim, Eli Lilly Japan, Novartis, Ono Pharmaceutical, and MSD for work performed outside of the current study. Tomoya Fukui received lecture fees from Taiho Pharmaceutical Co Ltd, Boehringer‐Ingelheim Japan Inc, Bristol‐Myers Squibb, Ono Pharmaceutical, Novartis Pharma KK, Pfizer Japan Inc, and Chugai Pharmaceutical Co Ltd for work performed outside of the current study. Koichi Minato received grants from AstraZeneca and Bristol‐Myers Squibb for work performed outside of the current study. Akira Iwashima received grants and lecture fee from Taiho Pharmacetutical Co Ltd and lecture fee from Chugai Pharmaceutical Co Ltd, AstraZeneca, Bristol‐Myers Squibb, Eli Lilly Japan, Novartis, MSD, Pfizer Japan and Ono Pharemaceutical for work performed outside of the current study. Yuichiro Takeda received grants from Taiho Pharmacetutical Co Ltd, grants from Boehringer Ingelheim, and Chugai Pharmaceutical Co Ltd for work performed outside of the current study. Kaoru Kubota received grants, lecture fee, supervision fee and advisory boards fee from Taiho Pharmaceutical Co Ltd and grant and lecture fee from Ono Pharmaceutical Co Ltd and Boehringer Ingelheim, lecture fee form Chugai Pharmaceutical Co Ltd, MSD, AstraZeneca, Eli Lilly, Daiichi Sankyo, Bristol Myers Squibb, Novartis, Kyowa Hakko Kirin and Eisai for work performed outside of the current study. Toshiaki Saeki received grants and personal fee from Taiho Pharmaceutical Co Ltd, grant and honoraria from Ono, Chugai Pharmaceutical Co Ltd, AstraZeneca, Kyowa Hakko Kirin and Takeda, honoraria from Novartis, grant from EPS, Daiichi Sankyo, Eisai, Nihon Medi‐Physics, Nipponkayaku, MSD, Sawai, Fuji Pharma, Hamamatsu Photonics and Global Software for work performed outside of the current study. Tomohide Tamura received personal fee from Taiho Pharmaceutical Co Ltd, honoraria from Chugai Pharmaceutical Co Ltd, Eli Lilly, Boehringer Ingelheim, Bristol‐Myers Squibb, Ono Pharmaceutical, Kyowa Kirin, Shift Zero and MSD for work performed outside of the current study.
Author Contributions
Shunichi Sugawara: Investigation, writing–original draft, and writing–review and editing. Naoki Inui: Investigation, writing–original draft, and writing–review and editing. Masashi Kanehara: Investigation, writing–original draft, and writing–review and editing. Masahiro Morise: Investigation, writing–original draft, and writing–review and editing. Kozo Yoshimori: Investigation, writing–original draft, and writing–review and editing. Toru Kumagai: Investigation, writing–original draft, and writing–review and editing. Tomoya Fukui: Investigation, writing–original draft, and writing–review and editing. Koichi Minato: Investigation, writing–original draft, and writing–review and editing. Akira Iwashima: Investigation, writing–original draft, and writing–review and editing. Yuichiro Takeda: Investigation, writing–original draft, and writing–review and editing. Kaoru Kubota: Conceptualization, methodology, writing–original draft, writing–review and editing, and project administration. Toshiaki Saeki: Conceptualization, methodology, writing–original draft, writing–review and editing, and project administration. Tomohide Tamura: Conceptualization, methodology, writing–original draft, writing–review and editing, and project administration. All authors approved the final version of the article prior to submission.
Supporting information
We thank all the patients, their families, and the investigators who participated in the current study. We also thank Dr. Ali Nasermoaddeli (Taiho Pharmaceutical Co Ltd.) for his important contributions to the management of this study from a medical perspective.
Data Availability Statement: Data generated or analyzed during this study are on file with Taiho Pharmaceutical Co Ltd and are not publicly available. Inquiries for data access may be sent to the following email address: TOIAWASE@taiho.co.jp.
Data Availability Statement
Data generated or analyzed during this study are on file with Taiho Pharmaceutical Co Ltd and are not publicly available. Inquiries for data access may be sent to the following email address: TOIAWASE@taiho.co.jp.
References
- 1. Roila F, Molassiotis A, Herrstedt J, et al; participants of the MASCC/ESMO Consensus Conference Copenhagen 2015 . 2016 MASCC and ESMO guideline update for the prevention of chemotherapy‐ and radiotherapy‐induced nausea and vomiting and of nausea and vomiting in advanced cancer patients. Ann Oncol. 2016;27(suppl 5):v119‐v133. [DOI] [PubMed] [Google Scholar]
- 2. Hesketh PJ, Bohlke K, Lyman GH, et al; American Society of Clinical Oncology . Antiemetics: American Society of Clinical Oncology Focused Guideline Update. J Clin Oncol. 2016;34:381‐386. [DOI] [PubMed] [Google Scholar]
- 3. National Comprehensive Cancer Network . https://www.nccn.org/professionals/physician_gls/pdf/antiemesis.pdf. Accessed January 11, 2019.
- 4. Leal AD, Kadakia KC, Looker S, et al. Fosaprepitant‐induced phlebitis: a focus on patients receiving doxorubicin/cyclophosphamide therapy. Support Care Cancer. 2014;22:1313‐1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bernareggi A, Schultz A, Voisin D. Phase 1 study evaluating the pharmacokinetics (PK) and safety of intravenous (IV) fosnetupitant, and its bioequivalence to the netupitant component of oral Poster presented at: Annual Meeting of the American Society of Health‐System Pharmacists (ASHP); December 2‐6, 2018; Anaheim, CA. [Google Scholar]
- 6. Schwartzberg L, Roeland E, Andric Z, et al. Phase III safety study of intravenous NEPA: a novel fixed antiemetic combination of fosnetupitant and palonosetron in patients receiving highly emetogenic chemotherapy. Ann Oncol. 2018;29:1535‐1540. [DOI] [PubMed] [Google Scholar]
- 7. Taiho Pharamaceutical Co. Ltd . A Phase 1 Study of Fosnetupitant in Healthy Volunteers. Tokyo: Taiho Pharamaceutical Co. Ltd; 2015. [Google Scholar]
- 8. Osaki A, Inoue K, Sakai H, et al. A dose‐finding randomized phase II study of oral netupitant in combination with palonosetron.75 mg intravenous for the prevention of chemotherapy‐induced nausea and vomiting in Japanese patients receiving highly emetogenic chemotherapy. Jpn J Clin Oncol. 2019;49:121‐129. doi: 10.1093/jjco/hyy161 [DOI] [PubMed] [Google Scholar]
- 9. Saito M, Aogi K, Sekine I, et al. Palonosetron plus dexamethasone versus granisetron plus dexamethasone for prevention of nausea and vomiting during chemotherapy: a double‐blind, double‐dummy, randomised, comparative phase III trial. Lancet Oncol. 2009;10:115‐124. [DOI] [PubMed] [Google Scholar]
- 10. Muller CJ, MacLehose RF. Estimating predicted probabilities from logistic regression: different methods correspond to different target populations. Int J Epidemiol. 2014;43:962‐970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hesketh PJ, Rossi G, Rizzi G, et al. Efficacy and safety of NEPA, an oral combination of netupitant and palonosetron, for prevention of chemotherapy‐induced nausea and vomiting following highly emetogenic chemotherapy: a randomized dose‐ranging pivotal study. Ann Oncol. 2014;25:1340‐1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jordan K, Lanzarotti C, Olivari S, et al. Cisplatin dose‐dependent effect on nausea control: subset analysis from a phase 3 trial of NEPA versus aprepitant/granisetron Poster presented at: 2017 Multinational Association of Supportive Care in Cancer (MASCC)/International Society of Oral Oncology (ISOO) Annual Meeting on Supportive Care in Cancer; June 22‐24, 2017; Washington, DC. [Google Scholar]
- 13. Boccia R, Grunberg S, Franco‐Gonzales E, Rubenstein E, Voisin D. Efficacy of oral palonosetron compared to intravenous palonosetron for the prevention of chemotherapy‐induced nausea and vomiting associated with moderately emetogenic chemotherapy: a phase 3 trial. Support Care Cancer. 2013;21:1453‐1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Maemondo M, Masuda N, Sekine I, et al; PALO Japanese Cooperative Study Group . A phase II study of palonosetron combined with dexamethasone to prevent nausea and vomiting induced by highly emetogenic chemotherapy. Ann Oncol. 2009;20:1860‐1866. [DOI] [PubMed] [Google Scholar]
- 15. Takahashi T, Hoshi E, Takagi M, Katsumata N, Kawahara M, Eguchi K. Multicenter, phase II, placebo‐controlled, double‐blind, randomized study of aprepitant in Japanese patients receiving high‐dose cisplatin. Cancer Sci. 2010;101:2455‐2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Saito H, Yoshizawa H, Yoshimori K, et al. Efficacy and safety of single‐dose fosaprepitant in the prevention of chemotherapy‐induced nausea and vomiting in patients receiving high‐dose cisplatin: a multicentre, randomised, double‐blind, placebo‐controlled phase 3 trial. Ann Oncol. 2013;24:1067‐1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tamura K, Aiba K, Saeki T, et al; CINV Study Group of Japan . Testing the effectiveness of antiemetic guidelines: results of a prospective registry by the CINV Study Group of Japan. Int J Clin Oncol. 2015;20:855‐865. [DOI] [PubMed] [Google Scholar]
- 18. Helsinn Therapeutics Inc . AKYNZEO (fosnetupitant and palonosetron) for injection, for intravenous use [package insert]. Iselin, NJ: Helsinn Therapeutics Inc; 2018. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data generated or analyzed during this study are on file with Taiho Pharmaceutical Co Ltd and are not publicly available. Inquiries for data access may be sent to the following email address: TOIAWASE@taiho.co.jp.