

Abstract
We report herein a visible light‐mediated C−H hydroxyalkylation of quinolines and isoquinolines that proceeds via a radical path. The process exploits the excited‐state reactivity of 4‐acyl‐1,4‐dihydropyridines, which can readily generate acyl radicals upon blue light absorption. By avoiding the need for external oxidants, this radical‐generating strategy enables a departure from the classical, oxidative Minisci‐type pattern and unlocks a unique reactivity, leading to hydroxyalkylated heteroarenes. Mechanistic investigations provide evidence that a radical‐mediated spin‐center shift is the key step of the process. The method's mild reaction conditions and high functional group tolerance accounted for the late‐stage functionalization of active pharmaceutical ingredients and natural products.
Keywords: dihydropyridines, Minisci reaction, photochemistry, radical chemistry, synthetic methods
In a different light. A visible light‐mediated C−H hydroxyalkylation of quinolines and isoquinolines that proceeds via a radical path exploits the excited‐state reactivity of 4‐acyl‐1,4‐dihydropyridines 1, which can readily generate acyl radicals upon blue light absorption. By avoiding the need for external oxidants, this strategy enables a departure from the classical, oxidative Minisci‐type pattern and unlocks a unique reactivity, leading to hydroxyalkylated heteroarenes.
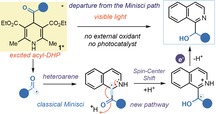
N‐Heteroarenes are ubiquitous motifs in natural products and active pharmaceutical ingredients.1 The venerable Minisci reaction2 (i.e. the addition of a carbon‐centered radical to a heteroarene followed by formal hydrogen atom loss) provides a rapid and direct method for heterocycle C−H alkylation.3 This direct functionalization of N‐heteroarenes avoids the need for de novo heterocycle synthesis, thus explaining the recent renewed interest in Minisci‐type chemistry.4
In their pioneering works, Minisci and co‐workers used silver oxidants at elevated temperatures to generate alkyl and acyl radicals from the corresponding carboxylic acids (Figure 1 a).2 Upon radical addition to the heteroarene, a stoichiometric amount of persulfate secured the aromaticity‐driven oxidation of the radical intermediate I, leading to the final product (the acylation5 adduct II is shown in Figure 1 a). Recent advances in radical generation strategies, which include photoredox catalysis6 and electrochemistry,7 have enabled the use of a wider variety of radical precursors that often operate under much milder conditions. These methods8 have facilitated the introduction of structurally different radicals into complex heteroarenes, and they have been quickly adopted by medicinal chemists for the late‐stage diversification of pharmaceutically relevant compounds.9
Figure 1.
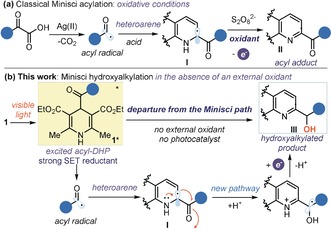
a) Classical Minisci reaction of acyl radicals with N‐heteroarenes where intermediate I is oxidized by an external oxidant to form the acylated product II. b) Diverting from the classical Minisci‐type chemistry: the photochemistry of 4‐acyl‐1,4‐dihydropyridines 1 secures the generation of acyl radicals in the absence of an external oxidant, thus triggering an unusual hydroxyalkylation of N‐heteroarenes; SET; single‐electron transfer.
Despite this progress, both classical and modern Minisci protocols have limitations. In particular, there is an intrinsic mechanistic tie in the need for an oxidant to promote the re‐aromatization of intermediates I and drive the overall reaction forward (Figure 1 a). The highly oxidative conditions of typical Minisci protocols may therefore limit the functional group tolerance and the application to more complex targets.
Herein, we report a rare example of radical C−H functionalization of quinolines and isoquinolines operating under reducing conditions, where the ability to generate acyl radicals without using external oxidants allowed a departure from the typical mechanistic pattern of the Minisci‐type chemistry (Figure 1 b). This study was motivated by our recent finding that 4‐acyl‐1,4‐dihydropyridines (acyl‐DHPs, 1) can unveil a rich photochemistry upon absorption of visible light.10 The excited acyl‐DHPs 1* can then act as strong single‐electron transfer (SET) reductants, thus securing reducing conditions, while generating nucleophilic acyl radicals.10, 11 Wondering if it was possible to harness the photochemistry of 1 in acyl radical additions to activated N‐heteroarenes, we found that the reaction yielded the hydroxyalkylated products III and not the typical Minisci‐type acylated adducts II. This is because the absence of an external oxidant disables the classical pattern, and the rearomatization of intermediate I takes place concomitantly to the formal reduction of the carbonyl moiety. In this communication, we detail the synthetic scope of this photochemical process, which allows the preparation of adducts that are difficult to access by traditional polar and Minisci‐type chemistry. We also discuss preliminary mechanistic studies that rationalize the observed reactivity.
For our initial explorations (Table 1), we selected the benzoyl derivative 1 a and isoquinoline 2 a as the model substrates. 1 a can absorb light in the visible region (see Figure S4 in the Supporting Information). The experiments were conducted in CH3CN using a blue (HP) LED (λ max=460 nm) with an irradiance of 30 mW cm−2, as controlled by an external power supply (details of the illumination set‐up are reported in Figure S1). When using TFA (2 equiv) to activate 2 a, the reaction exclusively afforded the hydroxyalkylated product 3 a in 72 % yield (entry 1). The reaction did not proceed without irradiation, confirming its photochemical character (entry 2). When the reaction was carried out without acid, a complex mixture of products was formed along with 3 a in 12 % yield (entry 3). As a testament to the method's robustness, the system tolerates traces of water (entry 4) and the reaction could be efficiently performed without degassing the solvent (entry 5). The process could also be scaled up without compromising the efficiency (entry 6).
Table 1.
Optimization studies and control experiments.[a]
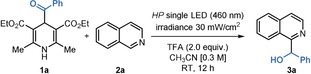
|
Entry |
Variation from standard conditions |
Yield (3 a) [%][b] |
|---|---|---|
|
1 |
none |
72 |
|
2 |
no light |
–[c] |
|
3 |
no acid |
12[d] |
|
4 |
2.0 equiv of water |
71 |
|
5 |
no solvent degassing |
68 |
|
6 |
1.5 mmol scale[e] |
70 |
[a] Reactions performed on a 0.2 mmol scale at 25 °C for 12 h using 0.6 mL of CH3CN under illumination by a single high‐power (HP) LED (λ max=465 nm, 30 mW cm−2) and using 1.2 equiv of 1 a and 2 equiv of TFA. [b] Yield of 3 a determined by 1H NMR analysis of the crude mixture using trichloroethylene as the internal standard. [c] Starting material only. [d] Complex mixture. [e] Reaction performed in EvoluChem™ 18W photoreactor (emission at 455 nm). The yield of the isolated 3 a is given. TFA: trifluoroacetic acid.
We then evaluated the synthetic potential of this photochemical strategy (Figure 2). The resulting hydroxyalkylated N‐heteroarene products 3 are important scaffolds,12 which are difficult to access by other synthetic methods. Polar pathways generally require multi‐step redox‐inefficient sequences and the use of pre‐functionalized heteroarenes,13 while only a few direct Minisci‐type strategies are available.14 Our method uses easily available substrates (acyl‐DHPs 1 are synthesized in a one‐step procedure, see section B in the Supporting Information) and works well with a variety of benzo‐fused azines, allowing the hydroxyalkylation of both isoquinoline and quinoline substrates (Figure 2 a). For the latter, the presence of a substituent directed the selective functionalization at either the C2 or C4 position, while unsubstituted quinoline substrates afforded a 1:1 regioisomeric mixture (details in Figure S2 of the Supporting Information).15 A remarkable functional group tolerance was observed, particularly for moieties prone to oxidation. For example, the reaction conditions tolerate both primary amines (‐NH2, product 3 c) and alcohols (‐OH, adduct 3 f), a phenol group (3 d), and a diarylamine moiety (3 k).
Figure 2.
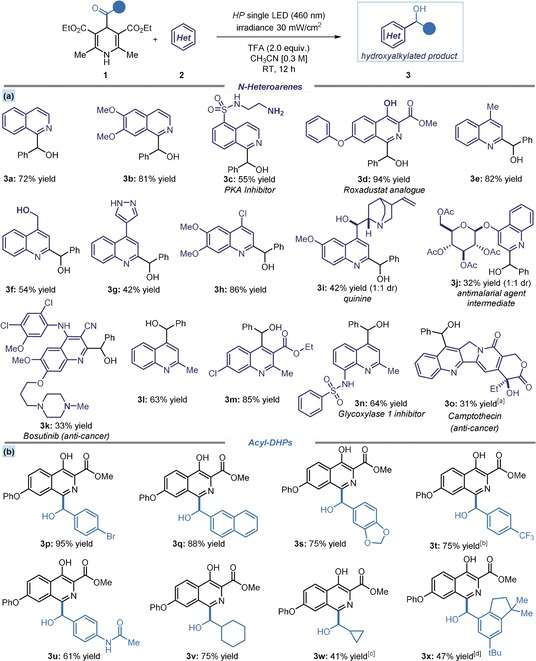
Substrate scope for the photochemical C−H hydroxyalkylation of N‐heteroarenes. Survey of the a) quinolines and isoquinolines, and b) radical precursors 1 that can participate in the reaction. Reactions performed on 0.2 mmol scale using 1.2 equiv of 1 and 0.6 mL of CH3CN. Yields refer to isolated products (average of two runs per substrate).[a] Performed in chloroform.[b] Reaction time: 48 h.[c] Performed at −10 °C for 64 h.[d] Performed in a CH3CN/DMSO mixture (1:1) for 48 h.
To demonstrate the potential utility for medicinal chemistry, we used our protocol on complex and bioactive molecules. We prepared advanced intermediates of a potent PKA inhibitor H‐89 and an antimalarial agent, which smoothly underwent hydroxyalkylation to form 3 c and the 4‐hydroxyquinoline‐β‐glucoside derivative 3 j, respectively. We also functionalized quinine (3 i), and the anticancer agents bosutinib (3 k) and camptothecin (3 o). The protocol also allowed the single‐step access to 4‐quinolinemethanol derivative (3 l), a purine receptor antagonist. One limitation is that pyridine substrates were poorly reactive (e.g. 2‐chloro‐4‐(trifluoromethyl)pyridine provided a complex mixture of products with 80 % of recovered starting material). A complete list of moderately successful and unsuccessful substrates for this strategy is reported in Figure S2 of the Supporting Information.
To evaluate the scope of the acyl‐DHP 1 radical precursors, we selected a functionalized intermediate used in the synthesis of HIF protyl‐hydroxylase inhibitor roxadustat (Figure 2 b). For aromatic moieties in 1, different substitution patterns were tolerated well, regardless of their electronic and steric properties, affording the corresponding hydroxyalkylated products 3 p–x in good yields. We also installed a naphthyl (3 q) and an amide functionality (3 u). Finally, alkyl substituents could be readily introduced (3 v), including a cyclopropyl moiety (3 w).
We then studied the reaction mechanism with experimental and theoretical tools. First, we checked if the Minisci‐type acylation product, the ketone 4 a, could be an intermediate of the process (Figure 3 a). When an independently prepared and authentic sample of 4 a was subjected to the reaction conditions, the reduced product 3 a was not formed at all in the absence of light, while traces of 3 a were observed (14 % yield) under irradiation. These experiments exclude the possibility that acyl‐DHP 1 a could serve as a reducing agent, and they suggest that Minisci‐type acylation product 4 a is not formed under our photochemical conditions.
Figure 3.
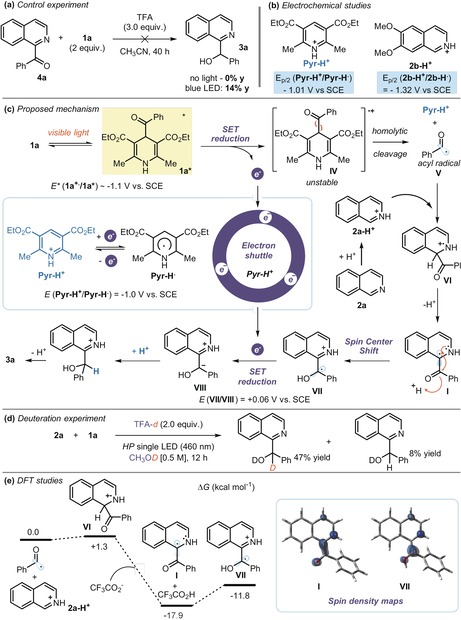
a) Control experiments. b) Redox properties of Pyr‐H+ (left) and 2 b‐H+ (right). c) Proposed mechanism. d) Deuteration experiment performed in CH3OD using TFA‐d. e) Computed free energy profile for the addition of the benzoyl radical V to isoquinolinium (2 a‐H+) and subsequent prototropy from VI to VII. Relative free energies are reported in kcal mol−1 at 298 K and 1 atm. Inset: spin density isosurfaces (value=0.009 e a0 −3) for I and VII, supporting the SCS process.
A plausible mechanism of the overall process is depicted in Figure 3 c. Visible‐light excitation turns 1 a into a strong reducing agent (E red (1 a+./1 a*)=−1.1 V vs. SCE in CH3CN, as estimated from electrochemical and spectroscopic measurements).10 The significantly lower redox potential of the protonated 6,7‐dimethoxyisoquinoline 2 b (E red(2 b‐H+/2 b‐H.)=−1.32 V vs. SCE in CH3CN, see Figure 3 b), which is a particularly effective substrate of this protocol, indicates that the SET from 1 a* to 2 b‐H+ is thermodynamically unfeasible.16 Electrochemical measurements also highlight that the pyridinium ion (Pyr‐H+), which arises from the photolysis of 1 a, has the right redox properties to accept an electron from 1 a* (E red (Pyr‐H+/PyrH.)=−1.01 V vs. SCE in CH3CN). This SET event triggers the formation of the unstable acyl‐DHP radical cation IV, which decomposes to deliver the acyl radical V. V then adds to the protonated heteroarene 2 a‐H+ to afford the radical intermediate VI. Deprotonation leads to the α‐amino radical of type I. Acid activation of the carbonyl moiety in I triggers the rearomatization of the azine ring, while the spin density is shifted by one atom and is localized at the benzylic position of the ensuing radical VII. This step formally corresponds to a spin‐center shift (SCS)‐type process (vide infra).17 SET reduction of VII (E red (VII/VIII)=+0.06 V vs. SCE, as estimated by DFT calculations (see section G for details) from PyrH. leads to the carbanion VIII, which is eventually protonated to form product 3 a. This step is supported by the deuterium incorporation experiment depicted in Figure 3 d (85 % D incorporation at α‐OH position of 3 a).
Two aspects of the proposed mechanism require further comment. Firstly, we propose that the Pyr‐H+/PyrH. couple acts as an electron mediator, effectively shuttling an electron from the highly reducing 1 a* to the fleeting intermediate VII.18 This scenario is consonant with the observation that the addition of Pyr‐H+ (50 mol %) to a CH3CN solution of 1 a increased the initial rate of radical generation via photolysis, as corroborated by EPR investigations (see section H in the Supporting Information). Secondly, the proposed SCS step, which converts I into VII (Figure 3 c), is supported by theoretical studies at the DFT level,19 which show that (i) prototropy converting VI to VII is thermodynamically favorable (ΔG=−13.1 kcal mol−1) and (ii) protonation of the carbonyl group in I induces a shift of the spin density to the adjacent benzylic carbon (Figure 3 e). Radical‐mediated SCS plays an important role in both a biochemical17b and synthetic context17c (Figure 4 a and b, respectively). Mechanistically, it is generally associated with the elimination of a leaving group (typically water) located at the atom adjacent to the radical center. Here, we propose extending the narrow definition of SCS17a to include a carbonyl group (and π‐electrons in general), which serves a similar purpose as the leaving group (Figure 4 c).
Figure 4.
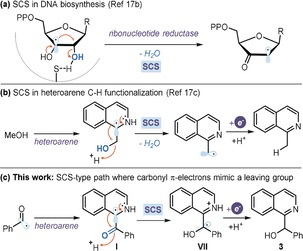
Spin‐center shift (SCS) pathways: water acting as a leaving group in the SCS‐based a) DNA biosynthesis and b) Minisci‐type alkylation of N‐heteroarenes. c) Proposed SCS where the carbonyl group facilitates the shift of spin density.
In summary, we have developed a photochemical method for the direct hydroxyalkylation of quinolines and isoquinolines. This transformation, which would be difficult to achieve by other direct methods, shows a mechanistic departure from the classical Minisci chemistry. This divergence is enabled by the acyl radical generation strategy, which avoids the need for external oxidants. The key step is a SCS process, where carbonyl π‐electrons act similarly to a leaving group.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support was provided by MICIU (CTQ2016‐75520‐P), the AGAUR (Grant 2017 SGR 981), and the European Research Council (ERC‐2015‐CoG 681840 – CATA‐LUX).
B. Bieszczad, L. A. Perego, P. Melchiorre, Angew. Chem. Int. Ed. 2019, 58, 16878.
Contributor Information
Dr. Bartosz Bieszczad, http://www.iciq.org/research/research_group/prof-paolo-melchiorre/.
Prof. Dr. Paolo Melchiorre, Email: pmelchiorre@iciq.es.
References
- 1.
- 1a. Pozharskii A. F., Soldatenkov A. T., Katritzky A. R., Heterocycles in Life and Society: An Introduction to Heterocyclic Chemistry, Biochemistry and Applications , 2nd ed., Wiley, Hoboken, 2011; [Google Scholar]
- 1b. Vitaku E., Smith D. T., Njardarson J. T., J. Med. Chem. 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Minisci F., Bernardi R., Bertini F., Galli R., Perchinummo M., Tetrahedron 1971, 27, 3575–3579; [Google Scholar]
- 2b. Caronna T., Gardini G. P., Minisci F., J. Chem. Soc. D 1969, 201; [Google Scholar]
- 2c. Caronna T., Fronza G., Minisci F., Porta O., Gardini G. P., J. Chem. Soc. Perkin Trans. 2 1972, 2, 1477–1481. [Google Scholar]
- 3.For reviews on classical Minisci reactions, see:
- 3a. Minisci F., Synthesis 1973, 1, 1–24; [Google Scholar]
- 3b. Minisci F., Vismara E., Fontana F., Heterocycles 1989, 28, 489–519; [Google Scholar]
- 3c. Minisci F., Fontana F., Vismara E., J. Heterocycl. Chem. 1990, 27, 79–96. [Google Scholar]
- 4.For recent developments, see: Proctor R. S. J., Phipps R. J., Angew. Chem. Int. Ed. 2019, 10.1002/anie.201900977; [DOI] [Google Scholar]; Angew. Chem. 2019, 10.1002/ange.201900977, and references therein. [DOI] [Google Scholar]
- 5.For classical acylation strategies:
- 5a. Caronna T., Fronza G., Minisci F., Porta O., J. Chem. Soc. Perkin Trans. 2 1972, 2035–2038; [Google Scholar]
- 5b. Fontana F., Minisci F., Nogueira Barbosa M. C., Vismara E., J. Org. Chem. 1991, 56, 2866–2869. For recent examples of Minisci-type acylation, see: [Google Scholar]
- 5c. Wang Q.-Q., Xu K., Jiang Y.-Y., Liu Y.-G., Sun B.-G., Zeng C.-C., Org. Lett. 2017, 19, 5517–5520; [DOI] [PubMed] [Google Scholar]
- 5d. Manna S., Prabhu K. R., J. Org. Chem. 2019, 84, 5067–5077; [DOI] [PubMed] [Google Scholar]
- 5e. Wang X.-Z., Zeng C.-C., Tetrahedron 2019, 75, 1425–1430. [Google Scholar]
- 6. Shaw M., Twilton J., MacMillan D. W. C., J. Org. Chem. 2016, 81, 6898–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yan M., Kuwamata Y., Baran P. S., Chem. Rev. 2017, 117, 13230–13319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For selected examples, see:
- 8a. Seiple I. B., Su S., Rodriguez R. A., Gianatassio R., Fujiwara Y., Sobel L. A., Baran P. S., J. Am. Chem. Soc. 2010, 132, 13194–13196; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Gutiérrez-Bonet A., Remeur C., Matsui J. K., Molander G. A., J. Am. Chem. Soc. 2017, 139, 12251–12258; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Matsui J. K., Primer D. N., Molander G. A., Chem. Sci. 2017, 8, 3512–3522; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8d. Jin J., MacMillan D. W. C., Angew. Chem. Int. Ed. 2015, 54, 1565–1569; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1585–1589; [Google Scholar]
- 8e. DiRocco D. A., Dykstra K., Krska S., Vachal P., Conway D. V., Tudge M., Angew. Chem. Int. Ed. 2014, 53, 4802–4806; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4902–4906; [Google Scholar]
- 8f. Klauck F. J. R., James M. J., Glorius F., Angew. Chem. Int. Ed. 2017, 56, 12336–12339; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12505–12509; [Google Scholar]
- 8g. Jatoi A. H., Pawar G. G., Robert F., Landais Y., Chem. Commun. 2019, 55, 466–469; [DOI] [PubMed] [Google Scholar]
- 8h. Proctor R. S. J., Davis H. J., Phipps R. J., Science 2018, 360, 419–422. [DOI] [PubMed] [Google Scholar]
- 9. Duncton M. A. J., MedChemComm 2011, 2, 1135–1161. [Google Scholar]
- 10. Goti G., Bieszczad B., Vega-Peñaloza A., Melchiorre P., Angew. Chem. Int. Ed. 2019, 58, 1213–1217; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1226–1230. [Google Scholar]
- 11.For the ability of 1,4-dihydropyridines to generate radicals upon visible-light absorption, see:
- 11a. Buzzetti L., Prieto A., Roy S. R., Melchiorre P., Angew. Chem. Int. Ed. 2017, 56, 15039–15043; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15235–15239; [Google Scholar]
- 11b. van Leeuwen T., Buzzetti L., Perego L. A., Melchiorre P., Angew. Chem. Int. Ed. 2019, 58, 4953–4957; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 5007–5011. [Google Scholar]
- 12.
- 12a. Bentley K. W., Isoquinoline alkaloids, Pergamon Press, Oxford, 1965; [Google Scholar]
- 12b. Luo G., Chen L., Pin S. S., Xu C., Kostich X., Kelley M., Conway C. M., Macor J. E., Dubuwchik G. M., Bioorg. Med. Chem. Lett. 2012, 22, 2917–2921; [DOI] [PubMed] [Google Scholar]
- 12c. Basak A., Abouelhassan Y., V. M. Norwood IV , Bai F., Nguyen M. T., Jin S., R. W. Huigens III , Chem. Eur. J. 2016, 22, 9181–9189. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Shankar R., More S. S., Madhubabu M. V., Vembu N., Syam Kumar U. K., Synlett 2012, 23, 1013–1014; [Google Scholar]
- 13b. Melzer B., Bracher F., Org. Biomol. Chem. 2015, 13, 7664–7672; [DOI] [PubMed] [Google Scholar]
- 13c. Liu Q., Wang C., Zhou H., Wang B., Lv J., Cao L., Fu Y., Org. Lett. 2018, 20, 971–974; [DOI] [PubMed] [Google Scholar]
- 13d. Gibson H. W., Dickson M. A. G., Berg J. C., Lecavalier P. R., Wang H., Merola J. S., J. Org. Chem. 2007, 72, 5759–5770. The reported transformation is an equivalent of the Emmert reaction: [DOI] [PubMed] [Google Scholar]
- 13e. Emmert B., Asendorf E., Ber. Dtsch. Chem. Ges. 1939, 72, 1188; [Google Scholar]
- 13f.“Emmert Reaction”: Wang Z. in Comprehensive Organic Name Reactions and Reagents, Wiley, Hoboken, 2010, pp. 993–996. [Google Scholar]
- 14.
- 14a. Citterio A., Gentile A., Minisci F., Serravalle M., Ventura S., Tetrahedron 1985, 41, 617–620; [Google Scholar]
- 14b. Correia C. A., Yang L., Li C.-J., Org. Lett. 2011, 13, 4581–4583; [DOI] [PubMed] [Google Scholar]
- 14c. Huff C. A., Cohen R. D., Dykstra K. D., Streckfuss E., DiRocco D. A., Krska S. W., J. Org. Chem. 2016, 81, 6980–6987; [DOI] [PubMed] [Google Scholar]
- 14d. Niu L., Liu J., Liang X.-A., Wang S., Lei A., Nat. Commun. 2019, 10, 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.For a discussion on the regioselectivity issues associated with Minisci-type reactions, see: O'Hara F., Blackmond D. G., Baran P. S., J. Am. Chem. Soc. 2013, 135, 12122–12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.SET reduction from 1 a* to other protonated azines, including isoquinoline 2 a (E (2 a-H+/2 a.)=−1.16 V vs. SCE) cannot be excluded as an alternate minor path.
- 17.For a review:
- 17a. Wessig P., Muehling O., Eur. J. Org. Chem. 2007, 2219–2232. For selected examples, see: [Google Scholar]
- 17b. Eklund H., Uhlin U., Färnegårdh M., Logan D. T., Nordlund P., Prog. Biophys. Mol. Biol. 2001, 77, 177–268; [DOI] [PubMed] [Google Scholar]
- 17c. Jin J., MacMillan D. W. C., Nature 2015, 525, 87–90; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17d. Nacsa E. D., MacMillan D. W. C., J. Am. Chem. Soc. 2018, 140, 3322–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.This hypothesis is reminiscent of our previous study on the direct excitation of 4-alkyl 1,4-dihydropyridines, in which a nickel complex facilitated the formation of the alkyl radical acting as electron mediator, see Ref. [11b].
- 19.Model chemistry: M06-2X/6–311+G(3df,2p)//M06-2X/6–31+G(d), IEF-PCM (CH3CN), see sections G and K in the Supporting Information for full computational details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary