

Abstract
Introduction
Physical inactivity significantly contributes to loss of muscle mass and performance in bed‐bound patients. Loss of skeletal muscle mitochondrial content has been well‐established in muscle unloading models, but the underlying molecular mechanism remains unclear. We hypothesized that apparent unloading‐induced loss of muscle mitochondrial content is preceded by increased mitophagy‐ and decreased mitochondrial biogenesis‐signaling during the early stages of unloading.
Methods
We analyzed a comprehensive set of molecular markers involved in mitochondrial‐autophagy, −biogenesis, −dynamics, and ‐content, in the gastrocnemius muscle of C57BL/6J mice subjected to 0‐ and 3‐days hind limb suspension, and in biopsies from human vastus lateralis muscle obtained before and after 7 days of one‐leg immobilization.
Results
In both mice and men, short‐term skeletal muscle unloading results in molecular marker patterns indicative of increased receptor‐mediated mitophagy and decreased mitochondrial biogenesis regulation, before apparent loss of mitochondrial content.
Discussion
These results emphasize the early‐onset of skeletal muscle disuse‐induced mitochondrial remodeling.
Keywords: inactivity, mitochondria, mitochondrial biogenesis, mitophagy, muscle unloading, skeletal muscle
Abbreviations
- 1‐RM
one‐repetition max
- ATP5A
ATP synthase, H+ transporting, mitochondrial F1 complex, alpha
- BL
baseline
- BNIP3
BCL2/adenovirus E1B 19 kDa protein interacting protein 3
- BNIP3L
BCL2/adenovirus E1B 19 kDa protein interacting protein 3 like
- CALCOCO2
calcium binding and coiled‐coil domain 2
- COX4I1
cytochrome c oxidase subunit 4I1
- CS
citrate synthase
- CSA
cross‐sectional area
- CT
computed tomography
- DNM1L
dynamin‐1‐like
- EDL
extensor digitorum longus
- FIS1
mitochondrial fission gene 1
- FUNDC1
FUN14 domain containing 1
- GABARAPL1
gamma‐aminobutyric acid (GABA) A receptor‐associated protein‐like 1
- HAD
β‐hydroxyacyl‐CoA dehydrogenase
- HLS
hind limb suspension
- MAP1LC3B
microtubule‐associated protein 1 light chain 3 beta
- MFN
mitofusin
- MT‐CO2
cytochrome c oxidase subunit 2
- MT‐COI
mitochondrially encoded cytochrome c oxidase I
- NDUFB8
NADH:ubiquinone oxidoreductase subunit B8
- NRF1
nuclear respiratory factor 1
- OPA1
mitochondrial dynamin like GTPase
- OPTN
optineurin
- OXPHOS
oxidative phosphorylation
- PARK2
Parkin
- PCR
polymerase chain reaction
- PF
pair‐fed
- PINK1
PTEN induced putative kinase 1
- PPAR
peroxisome proliferative activated receptor
- PPARGC1
PPAR gamma, coactivator 1
- SDHB
succinate dehydrogenase complex, subunit B
- SQSTM1
sequestosome‐1
- TFAM
transcription factor A, mitochondrial
- TOMM20
translocase of outer mitochondrial membrane 20
- UL
unloading
- UQCRC2
ubiquinol cytochrome c reductase core protein 2
1. INTRODUCTION
Physical inactivity significantly contributes to rapid loss of muscle mass and muscle performance in hospitalized and bed‐bound patients.1, 2 Moreover, the loss of skeletal muscle mitochondrial content and oxidative capacity, both determinants of muscle performance, has been reported in men exposed to 1 week of strict bed rest, and 2 weeks of one‐leg skeletal muscle immobilization,1, 2 and has been well‐established in different models of rat and murine skeletal muscle unloading (UL).3, 4, 5, 6, 7, 8, 9, 10 Unraveling the exact molecular regulation underlying this loss of mitochondrial content during UL will be instrumental for the generation of new therapies aimed to preserve muscle performance, and more specifically mitochondrial content.
The total mitochondrial content is determined by the balance between mitochondrial biogenesis and breakdown, of which mitochondrial breakdown primarily occurs through mitophagy.11 Mitochondrial biogenesis (ie, de novo synthesis of mitochondria) is mainly regulated by the peroxisome proliferator‐activated receptor (PPAR) co‐activator (PGC) signaling network,12 while mitophagy (ie, a mitochondria‐specific form of autophagy13) can be regulated by pathways like the receptor‐mediated and mitochondrial‐based PTEN‐induced putative kinase 1 (PINK1)/Parkin (PARK2)‐mediated mitophagy, although crosstalk between these pathways has been shown.14, 15 Furthermore, skeletal muscle mitochondria are highly dynamic organelles which constantly undergo fission (ie, separation of mitochondria) and fusion (ie, merging of mitochondria) events, of which mitochondrial fission is often considered a prerequisite for mitophagy as it isolates small mitochondria from the network.15, 16 Therefore, a net loss of mitochondrial content during muscle inactivity may be driven by modulations in mitochondrial biogenesis, mitophagy, dynamics, or a combination of these.
Mitochondrial biogenesis has been extensively studied in rodent models for skeletal muscle inactivity, and biogenesis markers are generally found to be decreased in these models3, 4, 5, 6, 7, 8, 17, 18 Moreover, modulations in protein or mRNA‐expression of mitophagy‐related markers have been found in several rodent models of skeletal muscle inactivity (ie, hind limb immobilization, suspension, and denervation), all performed with at least 7 days of inactivity when the loss in mitochondrial content has already occurred.4, 9, 10, 19 Due to the coinciding nature of changes in biogenesis, mitophagy, and mitochondrial content, it is difficult to establish if changes in biogenesis and mitophagy underlie the loss of mitochondrial content, or are simply the result of a renewed biogenesis/mitophagy balance set to maintain fewer mitochondria.
Of interest, mitophagy signaling has not been studied in the early phases of inactivity preceding apparent loss of mitochondrial content. Moreover, many studies have reported expression of mitochondrial dynamics markers in rodent models of muscle inactivity, but convincing evidence indicating increased fission is still lacking.3, 4, 6, 7, 8, 9, 18 In human studies, it has been shown that expression of key regulators of mitochondrial biogenesis are lower in the skeletal muscle of chronically inactive compared with active subjects,20, 21, 22 and that transcript abundance of some autophagy‐ and mitophagy‐related markers was lower in inactive subjects.22 However, there is only limited data available on the regulation of mitochondrial biogenesis and mitochondrial dynamics during acute muscle disuse,23, 24, 25 and none on regulation of mitophagy as far as we know.
Although previous studies have reported differences in the regulation of mitochondrial breakdown and biogenesis at time‐points when loss of mitochondrial content was already present, studies describing these mechanisms at a time‐point before the apparent loss of mitochondrial content remain limited to date in both in mice and humans. As total mitochondrial quantity itself is an independent determinant of both the basal mitochondrial breakdown and biogenesis levels, short‐term UL studies are, therefore, not only valuable to provide an indication of chronology, but also to omit decreases in mitochondrial content as a possible confounding factor.
Therefore, we hypothesized that an increase of mitophagy‐signaling and a decrease of mitochondrial biogenesis‐signaling is present during the early stages of muscle UL, preceding the apparent loss of mitochondrial content, in both the murine and human skeletal muscle. To study this, the current rodent study was performed with a 3‐day UL period, and the human study with a 7‐day UL period, both approximately half of the periods at which loss of mitochondrial content was previously reported. A comprehensive set of molecular markers involved in the execution and regulation of mitophagy, autophagy, mitochondrial biogenesis, and mitochondrial dynamics, as well as markers for mitochondrial content, was analyzed in both a murine and human model of leg muscle UL.
2. METHODS
2.1. Ethical approval
Animal procedures were performed in accordance with the European Directive 2010/63/EU guidelines and conform the journal regulations.26 The study protocol was approved by the institutional animal care and use committee of Maastricht University (DEC‐2014‐101) in accordance to the National Institutes of Health guide for the care and use of Laboratory animals. The human samples were used from a previously performed double‐blind, randomized, placebo‐controlled intervention trial, approved by the Medical Ethics Committee of the Maastricht University Medical Centre (registered at http://www.clinicaltrials.gov as NCT01894737) in accordance with the Declaration of Helsinki, with written informed consent obtained from all subjects.27
2.2. Animal study design
Male C57BL/6J wild‐type mice were generated, housed, and fed as described previously,28 with exception for the pair‐fed (PF) mice, who received a daily feed amount matched to the consumption of the animals subjected to hind limb suspension. Twelve‐week‐old, young adult mice were divided in three groups, and subjected to no experimental procedure (baseline [BL]; n = 8), 3 days of muscle UL by hind limb suspension (HLS; n = 9), or 3 days of paired feeding (PF; n = 9). The HLS protocol was performed as previously described28, 29 with the exception that isoflurane (Abbott, Abbott Park, IL) was used as an anesthetic. In short, the tail of the animals was wrapped in a tail suspension device and connected to a swivel hook to allow circular motility. This hook was attached to a polyvinyl chloride ring, which slid over an iron rod spanning the length of the cage to allow longitudinal motility. The mice were raised just until the hind limbs were unable to touch the ground or sides of the cage. Body weight and feed intake was monitored throughout the experiment. The PF group was included to control for the lower caloric intake in mice subjected to HLS compared with BL, and, therefore, received the matched amount of feed consumed by the HLS group. After euthanasia, intraperitoneal sodium pentobarbital (115 mg/kg) followed by exsanguination, lower leg muscles were excised, weighed, snap frozen in liquid nitrogen, and stored at −80°C. The murine gastrocnemius muscle was used in analyses, which is a mixed muscle with a fiber type composition that contains both fast‐twitch and slow‐twitch fibers.
2.3. Human study design
The study group consisted of 13 healthy young adult male subjects who participated in a previously published trial,27 which was primarily designed to test the potential protective effect of creatine supplementation on disuse‐induced loss of muscle mass and function, of which the outcome was negative.
Briefly, subjects were subjected to muscle UL through 7 days of one‐leg immobilization by means of a full leg cast. Vastus lateralis muscle biopsies of the casted leg were taken following an overnight fast at BL and after UL, and were frozen in liquid nitrogen and stored at −80°C. Muscle mass (cross‐sectional area; CSA) was assessed by computed tomography (CT) of the upper legs, muscle strength was estimated using a one‐repetition maximum (1‐RM) knee extension test, and body weight was measured with a digital balance. None of the subjects performed progressive resistance‐type exercise training, or took any medication interfering with muscle metabolism in the previous 6 months. Inclusion in the current study was based on the availability of muscle biopsy material before and after the immobilization period. Human vastus lateralis muscle has a mixed fiber type composition which is similar to the fiber type composition observed in the murine gastrocnemius muscle.
2.4. RNA extraction and polymerase chain reaction
Murine gastrocnemius and human vastus lateralis muscle were crushed while frozen. RNA was harvested from lysates of crushed murine and human tissue. cDNA was synthesized and polymerase chain reaction (PCR) was performed as described in the Supporting Information Methods, which are available online.
2.5. Western blotting
Protein was harvested from lysates of crushed murine gastrocnemius and human vastus lateralis muscle and Western blotting was performed as described in the Supporting Information Methods. All Western blotting image data are included in the Supporting Information Methods.
2.6. Enzyme activity assays
Enzymatic assays for citrate synthase (CS) (EC 2.3.3.1), β‐hydroxyacyl‐CoA dehydrogenase (HAD) (EC 1.1.1.35), and phosphofructokinase (PFK) (EC 2.7.1.11) were performed on lysates of crushed murine gastrocnemius muscle as described in the Supporting Information Methods.
2.7. Data presentation and statistics
Data from the murine study are depicted as mean + SEM in either absolute numbers or in case of semi‐quantitative measurements as fold change compared with BL. Group means were tested for significance using an analysis of variance with a Bonferroni correction. General characteristics and body composition from the human study are depicted as absolute numbers or as ΔUL‐BL as median and minimum maximum. For all molecular markers, Δ values of UL‐BL are depicted as a boxplot with median and interquartile range with whiskers indicating minimum and maximum. To test differences between UL and BL measurement, Wilcoxon signed‐rank tests were performed. All statistical analyses were performed using IBM SPSS 22 software and differences were considered to be significant when P < .05.
3. RESULTS
3.1. Whole body and muscle response to UL
Mouse body weights were lower in the HLS compared with both the PF and BL group after three days of UL (Figure 1A). Muscle weights of all assessed hind limb muscles were lower in the HLS group, while only the plantaris muscle weight was significantly lower in the PF group compared with BL (Figure 1B).
Figure 1.
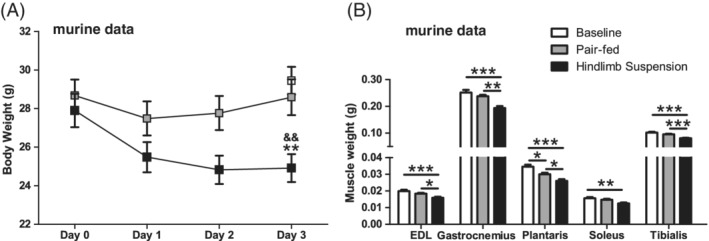
HLS‐induced body composition changes in mice. Body weight (A) and muscle weights of hind limb muscles (B) are depicted of the murine study. Statistical significance between indicated bars *P < .05, **P < .01, ***P < .001, or compared with PF && P < .01 are depicted
Human subjects did not lose body weight during 7 days of muscle UL. However, they lost muscle strength (as measured by 1‐RM), and muscle mass (as measured by CT‐derived thigh muscle CSA and quadriceps CSA) (Table 1).
Table 1.
Immobilization‐induced body composition changes in men
| Characteristic | Values (n = 13) |
|---|---|
| Age (years) | 22 [18‐28] |
| Height (m) | 1.78 [1.56‐1.89] |
| Body weight (kg) | 74 [55.8‐83.1] |
| BMI (kg m−2) | 23 [19.3‐25.2] |
| Δ Body weight (%) | −0.24 [−2.20‐2.07] |
| Δ 1‐RM (%) | −12.22 [−25.37‐8.11]** |
| Δ Thigh muscle CSA (%) | −3.43 [−6.54‐0.98]** |
| Δ Quadriceps CSA (%) | −4.86 [−8.48 to −1.04]** |
Abbreviations: BMI, body mass index.
Significant differences P < .01.
3.2. Mitochondrial content markers are unaltered during muscle UL
To investigate whether the loss of mitochondrial content is already initiated in an early stage of UL, we quantified several markers of mitochondrial content. None of the markers for mitochondrial content were significantly lower in HLS compared with PF or BL. To obtain a more functional overview of the skeletal muscle mitochondrial capacity, CS and HAD enzyme activity was determined in the murine study, as well as the more glycolytic PFK. In agreement with the mitochondrial protein expression data, no differences were found for any of these enzymes (Figure 2A,C). In line with the murine data, no significant differences were found for markers of mitochondrial content during UL of human skeletal muscles either (Figure 2B).
Figure 2.
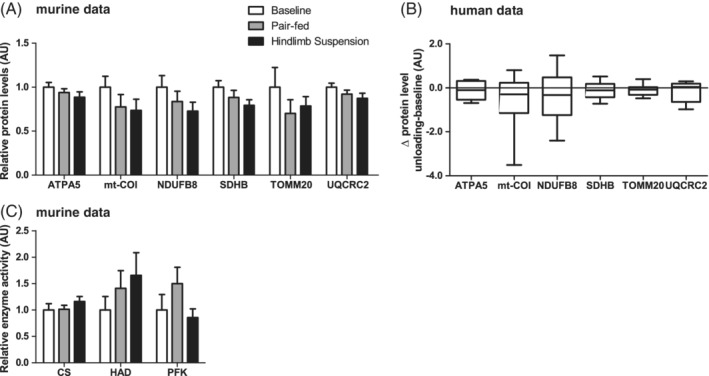
Mitochondrial quantity in response to muscle UL in murine gastrocnemius and human vastus lateralis muscle. Murine gastrocnemius muscle mitochondrial content‐associated protein levels (A) and enzyme activity (C) are depicted. Human vastus lateralis muscle ΔUL‐BL values of mitochondrial content‐associated protein levels are depicted (B). No statistical significance was found between indicated bars (A,C) or between ΔUL‐BL (B)
3.3. Modulations in receptor‐mediated mitophagy‐ and autophagy‐signaling during muscle UL
To investigate if mitophagy‐signaling is indicative of increased mitophagy in an early stage of UL, gene‐expression and protein levels of mitophagy‐receptors BCL2/Adenovirus E1B 19 kDa protein‐interacting protein 3 (BNIP3), BNIP3‐like (BNIP3L) and FUN14 domain‐containing protein 1 (FUNDC1), and of markers related to ubiquitin‐mediated mitophagy PARK2 and PINK1 were measured. In the murine study, HLS resulted in lower BNIP3L and higher BNIP3 and BNIP3L‐II protein levels compared with BL. Moreover, BNIP3 and BNIP3L mRNA‐expression were both higher in HLS compared with either BL or PF. FUNDC1 protein levels were lower in HLS compared with BL, while mRNA‐expression was higher in PF compared with both BL and HLS (Figure 3A,C). Furthermore, PARK2 protein levels were lower in HLS, while no differences were found in either PARK2 or PINK1 mRNA levels (Figure 3A,C).
Figure 3.
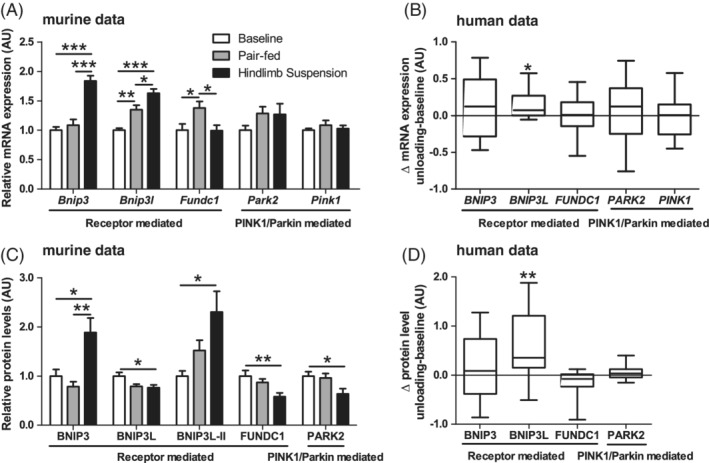
Mitophagy‐associated protein and mRNA‐expression in response to muscle UL in murine gastrocnemius and human vastus lateralis muscle. Murine gastrocnemius muscle mitophagy‐associated mRNA‐expression (A) and protein (C) levels are depicted. Human vastus lateralis muscle ΔUL‐BL values of mitophagy‐associated mRNA‐expression (B) and protein (D) levels are depicted. Statistical significance is indicated between indicated bars (A,C) or between ΔUL‐BL (B,D) *P < .05, **P < .01, ***P < .001
In the human subjects, BNIP3L protein levels and mRNA‐expression increased upon muscle UL, similar to the murine study. Moreover, no changes in BNIP3, FUNDC1, PARK2, or PINK1 protein or mRNA level were found in the human subjects upon muscle UL (Figure 3B,D).
To study if molecular signaling implicated in execution of autophagy was changed, gene‐expression and/or protein levels of autophagy‐modulators γ‐aminobutyric acid receptor‐associated protein‐like 1 (GABARAPL1), microtubule associated protein 1A/1B‐light chain 3 beta (LC3B), sequestosome 1 (SQSTM1), optineurin (OPTN), and calcium binding and coiled‐coil domain 2 (CALCOCO2) were measured. In the murine study, HLS resulted in higher GABARAPL1 protein levels compared with PF but not BL, while no differences were found in LC3B‐I, LC3B‐II, or LC3B‐II/LC3B‐I ratio. On mRNA‐expression level, LC3B was potently higher in HLS compared with both PF and BL, while GABARAPL1 was not. Moreover, SQSTM1 protein levels, and SQSTM1 and OPTN mRNA levels were higher in HLS (Figure 4A,C).
Figure 4.
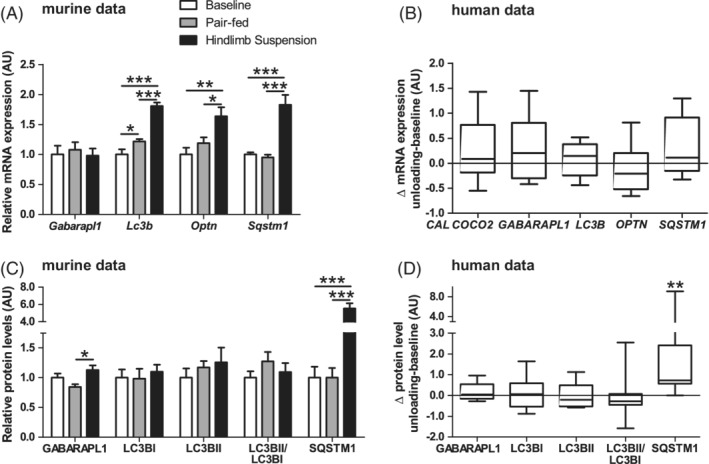
Autophagy‐associated protein and mRNA‐expression in response to muscle UL in murine gastrocnemius and human vastus lateralis muscle. Murine gastrocnemius muscle autophagy‐associated mRNA‐expression (A) and protein (C) levels are depicted. Human vastus lateralis muscle ΔUL‐BL values of autophagy‐associated mRNA‐expression (B) and protein (D) levels are depicted. Statistical significance is indicated between indicated bars (A,C) or between ΔUL‐BL (B,D) *P < .05, **P < .01, ***P < .001
In the human subjects, SQSTM1 protein levels were potently increased during muscle UL, which is in line with the data from the murine study. However, no significant changes were found in autophagy‐related mRNA expression in the human subjects (Figure 4B,D).
3.4. Transcriptional regulation of mitochondrial biogenesis is decreased during muscle UL
Because mitochondrial breakdown and synthesis together determine the mitochondrial content, we also investigated whether skeletal muscle UL resulted in alterations of mitochondrial biogenesis and the molecular regulation thereof. PPAR gamma, coactivator 1 alpha (PPARGC1A), transcription factor A, mitochondrial (TFAM), and nuclear respiratory factor 1 (NRF1) protein levels were not different in HLS compared with PF or BL, while PPARGC1A and PPAR gamma, coactivator 1 alpha (PPARGC1B) mRNA‐expression was found to be lower HLS, without corresponding differences in peroxisome proliferative activated receptor delta (PPARD), TFAM, and NRF1. TFAM and NRF1 mRNA‐expression was higher in PF compared with BL (Figure 5A,C). Moreover, lower mRNA‐expression of succinate dehydrogenase complex, subunit B (SDHB) (ie, nuclear‐transcribed subunit for oxidative complex II) was found, while the mRNA‐expression of cytochrome c oxidase subunit 2 (MT‐CO2) (ie, mitochondrial‐transcribed subunit for oxidative complex IV) was not different between groups (Figure 5E). In the human subjects, no changes in mitochondrial biogenesis related protein and mRNA levels were found during muscle UL in the human subjects (Figure 5B,D,F).
Figure 5.
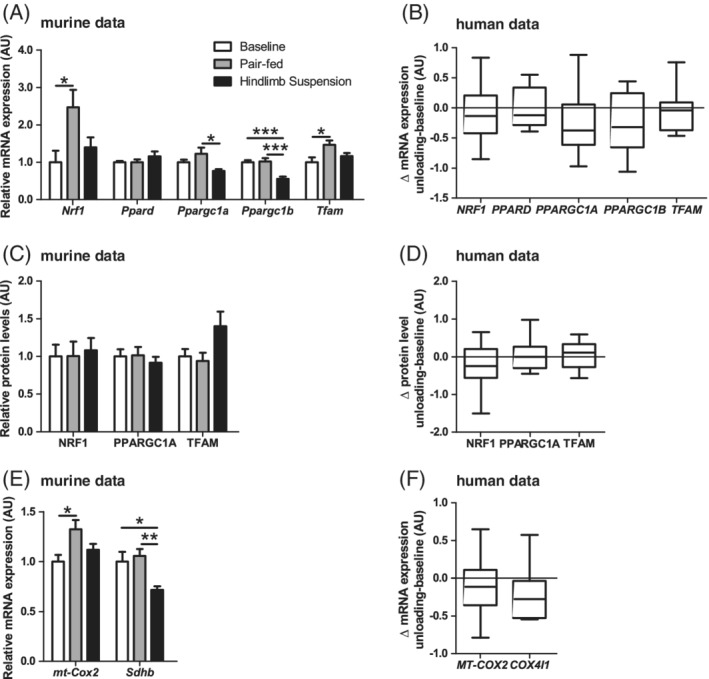
Mitochondrial biogenesis‐associated protein and mRNA‐expression in response to muscle UL in murine gastrocnemius and human vastus lateralis muscle. Murine gastrocnemius muscle mitochondrial biogenesis‐associated mRNA‐expression (A) and protein (C) levels are depicted. Human vastus lateralis muscle ΔUL‐BL values mitochondrial biogenesis‐associated mRNA‐expression (B) and protein (D) levels are depicted. Murine gastrocnemius (E) and human vastus lateralis muscle (F) ΔUL‐BL values of nuclear‐ and mitochondrial‐encoded mRNA‐expression of mitochondrial oxidative complexes are depicted. Statistical significance is indicated between indicated bars (A,C,E) or between ΔUL‐BL (B,D,F) *P < .05, **P < .01, ***P < .001
3.5. Mitochondrial dynamics related signaling is unaltered by muscle UL
Because mitochondrial fission is often described as a prerequisite for mitophagy, we investigated markers for both mitochondrial fission and fusion events. In the murine study, no differences were observed in mitochondrial dynamics‐associated protein levels or mRNA‐expression (Figure 6A,C). In concert with the murine data, no changes were found for any of the measured mitochondrial dynamics‐associated markers in the human subjects (Figure 6B,D).
Figure 6.
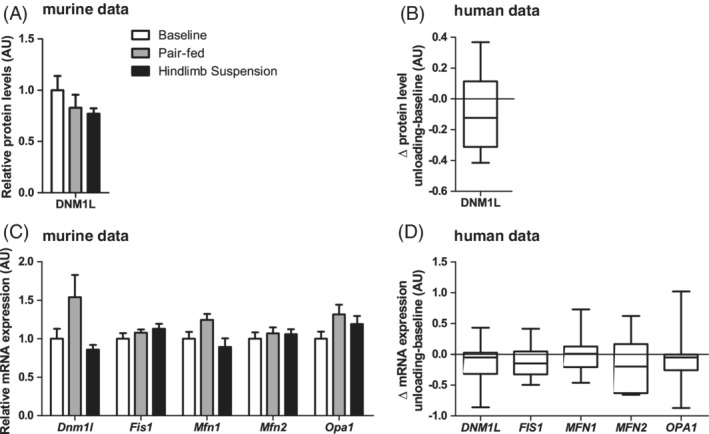
Mitochondrial dynamics‐associated protein and mRNA‐expression in response to muscle UL in murine gastrocnemius and human vastus lateralis muscle. Murine gastrocnemius muscle mitochondrial dynamics‐associated mRNA‐expression (A) and protein (C) levels are depicted. Human vastus lateralis muscle ΔUL‐BL values of mitochondrial dynamics‐associated mRNA‐expression (B) and protein levels (D) are depicted. No statistical significance was found between indicated bars (A,C) or between ΔUL‐BL (B,D)
4. DISCUSSION
The current study shows that skeletal muscle UL results in a pattern of early‐onset pro‐mitophagy signaling in murine skeletal muscle confirmed by a similar signaling pattern in unloaded human skeletal muscle, both still without an apparent decline of skeletal muscle mitochondrial content. These data suggest that the pro‐mitophagy signaling precedes the loss of mitochondrial content and, therefore, might play an important role in the onset of disuse‐induced loss of mitochondrial content. Moreover, these data show that the pathways of mitochondrial breakdown and biogenesis are regulated similarly in mice and humans during skeletal muscle UL.
The loss of mitochondrial content and oxidative capacity has been well‐established in different immobilization models in both rat and murine skeletal muscle.3, 4, 5, 6, 7, 8, 9, 10 More specifically, a decrease in muscle mitochondrial content was reported upon 14 days of UL in the exact same experimental setting as the current murine study, which subsequently recovered during muscle reloading.3 In line with this, 7 days of strict bedrest, and 14 but not 2 days of human limb immobilization resulted in decreased mitochondrial content markers.1, 2, 23 Currently, we report no changes in mitochondrial content markers in both the murine (3 days of HLS) and the human (7 days of UL) unloaded muscle.
Therefore, our results suggest that the timing of our study is before the well‐established loss of mitochondrial content during muscle UL. This made it possible to study the processes determining the turnover of mitochondrial content (eg, mitochondrial breakdown and biogenesis) without a decreased mitochondrial content as confounding factor.
Mitophagy can be initiated by several mitochondrial‐localized receptors (eg, BNIP3, BNIP3L, and FUNDC1), who stimulate their binding to proteins of the LC3/GABARAP family.30, 31, 32 Following this, mitochondria are engulfed in autophagosomal membranes and broken down by means of the lysosomal pathway. Previous studies provide mixed evidence, having found lower tibialis muscle protein levels of BNIP3,9 and lower gene‐expression but higher protein levels of BNIP3L10 after 7 days of hind limb immobilization by wrapping or denervation. Moreover, gastrocnemius muscle BNIP3 and BNIP3L gene‐expression was unchanged after prolonged UL.4 We report higher transcript and protein abundance of BNIP3 and BNIP3L during the first stage of UL in mice, indicating more pronounced mitophagy during the initial stages of UL. Of interest, the same pattern was found in human unloaded skeletal muscle, suggesting this molecular response is conserved between species.
FUNDC1‐mediated mitophagy, activated by posttranslational modifications (eg, by phosphorylation) of available FUNDC1 protein rather than increasing FUNDC1 protein abundance,33, 34, 35, 36 remains poorly described during muscle UL. In our study, we show that FUNDC1 protein levels are lower in skeletal muscle unloaded mice, while its mRNA transcript levels are unaltered, together suggesting posttranslational regulation of FUNDC protein abundance during the early phases of UL. Although it is likely that the observed FUNDC1 depletion contributes to subsequent mitochondrial quantity loss, the causality of this relationship remains to be established.
In a different pathway of mitophagy initiation, the PINK1/Parkin‐mediated mitophagy, a loss of mitochondrial membrane potential promotes the stabilization of PINK1, resulting in either the recruitment and activation of the E3 ubiquitin ligase PARK2 and downstream autophagy‐receptor SQSTM1, or in the PARK2 independent recruitment and activation of autophagy‐receptors like OPTN and CALCOCO2.37, 38, 39 Like mitophagy‐receptors, these autophagy receptors are able to bind LC3/GABARAP family proteins. In extension to our data which show no differences or changes for PINK1 protein or mRNA‐expression in both the murine and human model, PINK1 protein levels were found unaltered after 7 and 14 days of hind limb immobilization, but were found to be lower after 21 days of immobilization in murine tibialis anterior muscle.9 The same study reports higher PARK2 protein levels after 7 days of hind limb immobilization, while we found lower levels after 3 days.9 Because these studies show differences in timing, type of muscle analyzed, and protocol of muscle UL, the exact cause of this difference remains to be established. We did not find any differences in PINK1 or PARK2 protein or transcript expression in human skeletal muscle after UL, suggesting that this pathway does not play a major role in the loss of mitochondrial quantity during acute UL.
Autophagy‐related proteins are instrumental for the process of mitophagy, and their transcript levels were generally higher in the unloaded mice compared with the controls in our murine model. Although these differences were only marginally reflected by their representative protein levels, SQSTM1 protein levels were increased in both the unloaded murine and human skeletal muscle in our study, suggesting similar molecular autophagy‐signaling response between species. Combined with data from previous studies, generally showing increased presence of protein and/or transcript abundance of autophagy‐related proteins,4, 9, 10 autophagy‐related signaling is likely induced shortly after the start of muscle UL.
Many rodent studies have been performed to study mitochondrial biogenesis at various time‐points and in various different muscles during muscle UL.3, 4, 5, 6, 7, 8, 17, 18 In short, 1 day of hind limb UL resulted in decreased expression of many genes involved in mitochondrial biogenesis, except for increased PPARD transcript and protein levels in gastrocnemius muscle, and 3 days of hind limb UL resulted in lower PPARGC1A protein and transcript abundance in murine soleus but not in gastrocnemius muscle.8, 17, 18 These studies are in concert with our data, showing profoundly lower PPARGC1B mRNA‐expression in the unloaded muscle, while PPARGC1A mRNA‐expression was only marginally affected. In addition, it was previously reported that transcript levels of multiple mitochondrial biogenesis and metabolism markers were lower at 4‐11 days posthuman limb immobilization, while another study reported only marginal changes at 2 days.24, 40 Our results combined with literature suggest an early‐onset decreased transcriptional regulation of mitochondrial biogenesis regulation during muscle UL in rodents. More studies are needed to verify this observation in human muscle.
Skeletal muscle mitochondria are part of a highly dynamic network of organelles, constantly undergoing fission (ie, separation of mitochondria, regulated by, eg, Dynamin 1 Like [DNM1L] and Mitochondrial fission 1 protein [FIS1]) and fusion (ie, merging of mitochondria, regulated by, eg, Optic Atrophy 1 [OPA1] and Mitofusins [MFN1/2]) events.15 Because the autophagosomal size is limited, mitochondrial fission is often considered a prerequisite for mitophagy as it isolates small mitochondria from the network.15, 16 Although transcript and protein expression data related to mitochondrial fission and fusion is difficult to interpret, partly due to the posttranscriptional nature of their regulation, we included some measurements to obtain a general overview of major regulatory changes. In addition to our results, which show no differences in any of the fission and fusion markers for both the murine and human skeletal muscle UL models, many other rodent studies have been performed that generally report decreased or equal transcript and protein levels of the above mentioned markers.3, 4, 6, 7, 8, 9, 18 Together with the literature, our data make a profound shift from a fission/fusion homeostasis toward dominion of either fission or fusion unlikely in our models.
Although we report a comprehensive set of markers involved in the execution and regulation of mitophagy, autophagy, mitochondrial biogenesis, and mitochondrial dynamics measured in the early phase after muscle UL, there are some limitations in the current study. First, only muscle mass, and not actual muscle strength, was measured in the murine study, while both mass and strength were assessed in the human study. Moreover, no actual mitophagy or autophagy flux was measured in our samples. Therefore, the changed protein abundance of the mitophagy and autophagy‐related proteins might also be the result of impaired breakdown instead of increased synthesis. Moreover, we describe associations between changes in the processes of autophagy/mitophagy and mitochondrial biogenesis, but the causality of these associations remains unclear in our study. In addition, inclusion of electron microscopic analysis would be beneficial in future studies to obtain information on mitochondrial size and fractional area. Furthermore, the addition of more time‐points would highly improve the understanding of the temporal regulation of the different pathways discussed in this manuscript.
Despite these limitations, our data show that skeletal muscle UL results in a pattern of early‐onset pro‐mitophagy signaling in murine skeletal muscle confirmed by a similar signaling pattern in unloaded human skeletal muscle. These novel insights advance the current knowledge on disuse‐induced muscle pathology, and help to identify possible targets for future therapy to prevent muscle pathology in hospitalized and bed‐bound patients. Moreover, disuse‐induced muscle pathology is also present in inactive patients with chronic diseases41; therefore, these patients may benefit from development of a novel future therapy as well.
In conclusion, we now show that in both mice and humans skeletal muscle UL results in early‐onset molecular marker patterns indicative of increased receptor‐mediated mitophagy and decreased mitochondrial biogenesis regulation, before the loss of mitochondrial content. These results emphasize the early onset of skeletal muscle disuse‐induced mitochondrial remodeling, which is likely to underlie the loss of mitochondrial quantity in a later stage of muscle disuse. Moreover, we report similar expression patterns in the murine and human skeletal muscle, which suggests this molecular response is conserved between species and emphasize the early‐onset of human skeletal muscle disuse‐induced mitochondrial remodeling.
CONFLICT OF INTEREST
None of the authors has any conflict of interest to disclose.
ETHICAL PUBLICATION STATEMENT
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Appendix S1. Supporting Information.
Leermakers PA, Kneppers AEM, Schols AM, et al. Skeletal muscle unloading results in increased mitophagy and decreased mitochondrial biogenesis regulation. Muscle Nerve. 2019;60:769–778. 10.1002/mus.26702
Funding information The original work from which human muscle biopsies were obtained (Backx et al. Sports Med. 2017;47(8):1661‐1671) was funded by TI Food and Nutrition, a public‐private partnership on precompetitive research in food and nutrition. The mouse study was funded by a grant from the Lung Foundation/Netherlands Asthma Foundation (NAF 3.2.07.017) and the transnational University Limburg (tUL).
Funding information Lung Foundation/Netherlands Asthma Foundation, Grant/Award Number: NAF 3.2.07.017; TI Food and Nutrition
REFERENCES
- 1. Gram M, Vigelso A, Yokota T, et al. Two weeks of one‐leg immobilization decreases skeletal muscle respiratory capacity equally in young and elderly men. Exp Gerontol. 2014;58:269‐278. [DOI] [PubMed] [Google Scholar]
- 2. Dirks ML, Wall BT, van de Valk B, et al. One week of bed rest leads to substantial muscle atrophy and induces whole‐body insulin resistance in the absence of skeletal muscle lipid accumulation. Diabetes. 2016;65(10):2862‐2875. [DOI] [PubMed] [Google Scholar]
- 3. Remels AH, Pansters NA, Gosker HR, Schols AM, Langen RC. Activation of alternative NF‐kappaB signaling during recovery of disuse‐induced loss of muscle oxidative phenotype. Am J Physiol Endocrinol Metab. 2014;306(6):E615‐E626. [DOI] [PubMed] [Google Scholar]
- 4. Liu J, Peng Y, Cui Z, et al. Depressed mitochondrial biogenesis and dynamic remodeling in mouse tibialis anterior and gastrocnemius induced by 4‐week hindlimb unloading. IUBMB life. 2012;64(11):901‐910. [DOI] [PubMed] [Google Scholar]
- 5. Cassano P, Sciancalepore AG, Pesce V, et al. Acetyl‐L‐carnitine feeding to unloaded rats triggers in soleus muscle the coordinated expression of genes involved in mitochondrial biogenesis. Biochim Biophys Acta. 2006;1757(9‐10):1421‐1428. [DOI] [PubMed] [Google Scholar]
- 6. Qi Z, Zhang Y, Guo W, Ji L, Ding S. Increased insulin sensitivity and distorted mitochondrial adaptations during muscle unloading. Int J Mol Sci. 2012;13(12):16971‐16985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wagatsuma A, Kotake N, Kawachi T, Shiozuka M, Yamada S, Matsuda R. Mitochondrial adaptations in skeletal muscle to hindlimb unloading. Mol Cell Biochem. 2011;350(1‐2):1‐11. [DOI] [PubMed] [Google Scholar]
- 8. Cannavino J, Brocca L, Sandri M, Bottinelli R, Pellegrino MA. PGC1‐alpha over‐expression prevents metabolic alterations and soleus muscle atrophy in hindlimb unloaded mice. J Physiol. 2014;592(20):4575‐4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kang C, Yeo D, Ji LL. Muscle immobilization activates mitophagy and disrupts mitochondrial dynamics in mice. Acta Physiol. 2016;218(3):188‐197. [DOI] [PubMed] [Google Scholar]
- 10. Vainshtein A, Desjardins EM, Armani A, Sandri M, Hood DA. PGC‐1alpha modulates denervation‐induced mitophagy in skeletal muscle. Skelet Muscle. 2015;5:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Palikaras K, Tavernarakis N. Mitochondrial homeostasis: the interplay between mitophagy and mitochondrial biogenesis. Exp Gerontol. 2014;56:182‐188. [DOI] [PubMed] [Google Scholar]
- 12. Correia JC, Ferreira DM, Ruas JL. Intercellular: local and systemic actions of skeletal muscle PGC‐1s. Trends Endocrinol Metab. 2015;26(6):305‐314. [DOI] [PubMed] [Google Scholar]
- 13. Zhang T, Xue L, Li L, et al. BNIP3 protein suppresses PINK1 kinase proteolytic cleavage to promote mitophagy. J Biol Chem. 2016;291(41):21616‐21629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gao F, Chen D, Si J, et al. The mitochondrial protein BNIP3L is the substrate of PARK2 and mediates mitophagy in PINK1/PARK2 pathway. Hum Mol Genet. 2015;24(9):2528‐2538. [DOI] [PubMed] [Google Scholar]
- 15. Romanello V, Sandri M. Mitochondrial quality control and muscle mass maintenance. Front Physiol. 2015;6:422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gomes LC, Scorrano L. Mitochondrial morphology in mitophagy and macroautophagy. Biochim Biophys Acta. 2013;1833(1):205‐212. [DOI] [PubMed] [Google Scholar]
- 17. Mazzatti DJ, Smith MA, Oita RC, Lim FL, White AJ, Reid MB. Muscle unloading‐induced metabolic remodeling is associated with acute alterations in PPARdelta and UCP‐3 expression. Physiol Genomics. 2008;34(2):149‐161. [DOI] [PubMed] [Google Scholar]
- 18. Cannavino J, Brocca L, Sandri M, Grassi B, Bottinelli R, Pellegrino MA. The role of alterations in mitochondrial dynamics and PGC‐1alpha over‐expression in fast muscle atrophy following hindlimb unloading. J Physiol. 2015;593(8):1981‐1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Furuya N, Ikeda S, Sato S, et al. PARK2/Parkin‐mediated mitochondrial clearance contributes to proteasome activation during slow‐twitch muscle atrophy via NFE2L1 nuclear translocation. Autophagy. 2014;10(4):631‐641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cobley JN, Bartlett JD, Kayani A, et al. PGC‐1alpha transcriptional response and mitochondrial adaptation to acute exercise is maintained in skeletal muscle of sedentary elderly males. Biogerontology. 2012;13(6):621‐631. [DOI] [PubMed] [Google Scholar]
- 21. Safdar A, Hamadeh MJ, Kaczor JJ, Raha S, Debeer J, Tarnopolsky MA. Aberrant mitochondrial homeostasis in the skeletal muscle of sedentary older adults. PLoS One. 2010;5(5):e10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Drummond MJ, Addison O, Brunker L, et al. Downregulation of E3 ubiquitin ligases and mitophagy‐related genes in skeletal muscle of physically inactive, frail older women: a cross‐sectional comparison. J Gerontol A Biol Sci Med Sci. 2014;69(8):1040‐1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Abadi A, Glover EI, Isfort RJ, et al. Limb immobilization induces a coordinate down‐regulation of mitochondrial and other metabolic pathways in men and women. PLoS One. 2009;4(8):e6518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reich KA, Chen YW, Thompson PD, Hoffman EP, Clarkson PM. Forty‐eight hours of unloading and 24 h of reloading lead to changes in global gene expression patterns related to ubiquitination and oxidative stress in humans. J Appl Physiol (1985). 2010;109(5):1404‐1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Standley RA, Distefano G, Pereira SL, et al. Effects of beta‐hydroxy‐beta‐methylbutyrate on skeletal muscle mitochondrial content and dynamics, and lipids after 10 days of bed rest in older adults. J Appl Physiol (1985). 2017;123(5):1092‐1100. [DOI] [PubMed] [Google Scholar]
- 26. Grundy D. Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J Physiol. 2015;593(12):2547‐2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Backx EMP, Hangelbroek R, Snijders T, et al. Creatine loading does not preserve muscle mass or strength during leg immobilization in healthy, young males: a randomized controlled trial. Sports Med. 2017;47(8):1661‐1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pansters NA, Schols AM, Verhees KJ, et al. Muscle‐specific GSK‐3beta ablation accelerates regeneration of disuse‐atrophied skeletal muscle. Biochim Biophys Acta. 2015;1852(3):490‐506. [DOI] [PubMed] [Google Scholar]
- 29. Langen RC, Van Der Velden JL, Schols AM, Kelders MC, Wouters EF, Janssen‐Heininger YM. Tumor necrosis factor‐alpha inhibits myogenic differentiation through MyoD protein destabilization. FASEB J. 2004;18(2):227‐237. [DOI] [PubMed] [Google Scholar]
- 30. Yamaguchi O, Murakawa T, Nishida K, Otsu K. Receptor‐mediated mitophagy. J Mol Cell Cardiol. 2016;95:50‐56. [DOI] [PubMed] [Google Scholar]
- 31. Wei H, Liu L, Chen Q. Selective removal of mitochondria via mitophagy: distinct pathways for different mitochondrial stresses. Biochim Biophys Acta. 2015;1853(10 Pt B):2784‐2790. [DOI] [PubMed] [Google Scholar]
- 32. Stotland A, Gottlieb RA. Mitochondrial quality control: easy come, easy go. Biochim Biophys Acta. 2015;1853(10 Pt B):2802‐2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen M, Chen Z, Wang Y, et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy. 2016;12(4):689‐702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wu W, Tian W, Hu Z, et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014;15(5):566‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu L, Feng D, Chen G, et al. Mitochondrial outer‐membrane protein FUNDC1 mediates hypoxia‐induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14(2):177‐185. [DOI] [PubMed] [Google Scholar]
- 36. Lv M, Wang C, Li F, et al. Structural insights into the recognition of phosphorylated FUNDC1 by LC3B in mitophagy. Protein Cell. 2017;8(1):25‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Matsuda N. Phospho‐ubiquitin: upending the PINK‐Parkin‐ubiquitin cascade. Journal of biochemistry. 2016;159(4):379‐385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524(7565):309‐314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eiyama A, Okamoto K. PINK1/Parkin‐mediated mitophagy in mammalian cells. Curr Opin Cell Biol. 2015;33:95‐101. [DOI] [PubMed] [Google Scholar]
- 40. Chen YW, Gregory CM, Scarborough MT, Shi R, Walter GA, Vandenborne K. Transcriptional pathways associated with skeletal muscle disuse atrophy in humans. Physiol Genomics. 2007;31(3):510‐520. [DOI] [PubMed] [Google Scholar]
- 41. Leermakers PA, Gosker HR. Skeletal muscle mitophagy in chronic disease: implications for muscle oxidative capacity? Curr Opin Clin Nutr Metab Care. 2016;19(6):427‐433. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information.