

Abstract
Introduction
BAY 94‐9027 is an extended‒half‐life, site‐specifically PEGylated, B‐domain‒deleted recombinant factor VIII (FVIII). The PROTECT VIII main study demonstrated efficacy of bleed control using extended‐interval prophylaxis with BAY 94‐9027 for 36 weeks.
Aim
To report long‐term efficacy and safety of prophylaxis with BAY 94‐9027 in a descriptive analysis of the ongoing PROTECT VIII extension with a total treatment time of up to >5 years.
Methods
Previously treated males aged 12‐65 years with severe haemophilia A who completed the PROTECT VIII main study were eligible for the open‐label extension. Patients received on‐demand treatment or prophylaxis (30‒40 IU/kg twice weekly, 45‒60 IU/kg every 5 days, or 60 IU/kg every 7 days) and could switch regimens as needed.
Results
Patients (N = 121; on demand, n = 14; prophylaxis, n = 107) accumulated a median (range) of 3.9 years (297‒1965 days) and 223 (23‒563) total exposure days by 31 January 2018. During the extension, median (quartile [Q]1; Q3) annualized bleeding rates (ABRs) for total bleeds were 1.6 (0.3; 4.6) for patients receiving prophylaxis and 34.1 (20.3; 36.6) for patients receiving on‐demand treatment. ABRs for twice‐weekly (n = 23), every‐5‐days (n = 33), every‐7‐days (n = 23) and variable frequency (n = 28) treatments were 1.7, 1.2, 0.7 and 3.1, respectively. Of prophylaxis patients, 20.6% were bleed‐free throughout the extension (median time, 3.2 years), and 51.0% were bleed‐free during the last 6 months. No patients developed FVIII inhibitors.
Conclusions
BAY 94‐9027 prophylaxis was efficacious and well tolerated with dosing intervals up to every 7 days for a median (range) of 3.9 years (0.8‐5.4 years).
Keywords: clinical trial, factor VIII, haemophilia A, intravenous infusions, recombinant proteins
1. INTRODUCTION
For patients with haemophilia A, replacement of factor VIII (FVIII) with routine prophylaxis can effectively reduce or prevent bleeds and ultimately preserve long‐term joint function.1, 2 Prophylaxis initiation is recommended as early as possible for patients with severe disease (FVIII:C < 1%),3, 4 but prophylaxis initiated at later ages and following onset of joint damage may also improve patient outcomes.5 Despite the established benefits of prophylaxis, several barriers persist, including lack of optimal adherence to a prescribed dosing regimen because of the time commitment and lifestyle adjustments needed to maintain frequent infusions.6 This barrier may limit treatment success.7
Individualized prophylaxis, a strategy of tailored dosing based on a patient's unique bleeding pattern and characteristics, pharmacokinetic (PK) profile, lifestyle and needs, may be a cost‐effective method of mitigating disease burden by decreasing the number of intravenous injections, reducing the frequency and number of bleeds, improving long‐term joint function, improving quality of life, and reducing overall FVIII consumption.8, 9, 10, 11, 12, 13, 14 Recently developed extended–half‐life products may further allow for individualization of treatment for patients with haemophilia A by maintaining appropriate FVIII levels with less frequent infusions.12, 15, 16
BAY 94‐9027 is a B‐domain–deleted recombinant FVIII (rFVIII) product that is site‐specifically conjugated with polyethylene glycol (PEG) to extend circulatory half‐life.17, 18 The efficacy and safety of BAY 94‐9027 for prophylaxis and treatment of bleeds were demonstrated in the partially randomized, open‐label, 36‐week pivotal phase 2/3 PROTECT VIII study (NCT01580293) in which patients were randomized to 45‐60 IU/kg every 5 days or 60 IU/kg every 7 days, or were assigned to prophylaxis dosing regimens of 30‐40 IU/kg twice weekly if randomization criterion of not more than 1 spontaneous bleed during a 10‐week run‐in period of twice‐weekly 25 IU/kg was not fulfilled or once the randomization arms were filled.19 Following the completion of PROTECT VIII, patients could continue to receive BAY 94‐9027 in an extension study that evaluated safety and efficacy for ≥ 100 exposure days (EDs) and examined long‐term experience with BAY 94‐9027 prophylaxis treatment. A January 2015 interim analysis was scheduled as this was when ≥ 100 ED data would be available from patients in PROTECT VIII. At that point in the extension, patients were receiving prophylaxis twice weekly (n = 24), every 5 days (n = 37), every 7 days (n = 29) or had switched regimens (n = 17). Median (quartile [Q]1; Q3) ABRs were 2.2 (0.4; 4.3), 1.2 (0.0; 5.6), 0.5 (0.0; 1.0) and 3.9 (1.2; 6.4) for patients receiving prophylaxis twice weekly, every 5 days, every 7 days or with variable frequency, respectively.20 Here, we present updated interim results from January 2018, after up to >5 years of treatment, to provide a descriptive analysis of extension study progress.
2. MATERIALS AND METHODS
2.1. Patients and study design
Patients aged 12‐65 years who completed the PROTECT VIII main study19 were eligible for inclusion in the multicentre, open‐label, uncontrolled, ongoing PROTECT VIII extension. The extension study took place across 52 treatment centres from 18 countries; it began in February 2013 on a rolling basis, (hereby referred to as extension enrolment) as each patient completed the main study. Patients treated on demand during PROTECT VIII had the option to either continue on‐demand treatment in the extension or switch to 1 of 3 prophylaxis groups (30‐40 IU/kg twice weekly, 45‐60 IU/kg every 5 days or 60 IU/kg every 7 days). Patients who received prophylaxis in the main study could either continue their regimen or switch regimens at the beginning or at any time during the extension; treatment adjustments were based on the clinical bleeding pattern, and as such, trough levels were not determined for the purposes of such treatment decisions. All patients who switched regimens at least once after the first week of the extension were analysed in a separate variable frequency group. The dose and number of infusions of BAY 94‐9027 needed to treat breakthrough bleeding were determined at physician discretion up to a maximum recommended dose of 60 IU/kg/infusion or 6000 IU/infusion. All patients provided written informed consent, and the protocol was approved by each site's independent ethics committee and/or institutional review board. Results are reported from an analysis of data collected by 31 January 2018 after up to >5 years in the study.
2.2. Efficacy and safety assessments
Efficacy of BAY 94‐9027 was assessed using electronic patient diaries (EPDs) to document number and dose of infusions, adherence with prophylaxis regimen, reasons for treatment, bleeding events, bleed characteristics and response to treatment of bleeds (subject or investigator assessment as excellent, good, moderate, poor). Annualized bleeding rates (ABRs) were calculated for each dosing group and for the variable frequency group. In a separate analysis, ABRs and other bleeding outcomes were calculated for the last 12 months of the extension in patients who had participated in the extension for ≥12 months by the January 2018 interim analysis period. Throughout the extension study, patients were closely monitored at visits every 6 months for the incidence of adverse events (AEs), including inhibitor development, which were documented in terms of type, severity and relationship to study drug. Inhibitor development was defined as a Nijmegen‐modified Bethesda assay measured titre of ≥0.6 BU that was confirmed in a second independent sample (ideally collected within 2 weeks of the first inhibitor detection).
2.3. Statistical analysis
ABRs were calculated for each dosing group (twice‐weekly, every‐5‐days, and, every‐7‐days‐patients who did not change regimen after the first week of the extension) as well as for the variable frequency group. The safety population comprised all participants in the extension study who received ≥1 dose of BAY 94‐9027. Efficacy variables were evaluated in the intent‐to‐treat (ITT) population for the extension period, which included patients in the safety population with available information regarding bleeds. Summary statistics and frequencies were calculated using SAS® version 9.2 (SAS Institute Inc) for continuous data and categorical data, respectively. Censoring was not used.
3. RESULTS
3.1. Patients
Of 134 patients who were treated in the PROTECT VIII main study, 121 patients (90.3%) chose to continue in the extension study (safety and ITT populations) receiving either on‐demand treatment (n = 14) or prophylaxis (n = 107). Baseline demographics for all patients (median [range] age at January 2018, 40 [15‐67] years) as well as regimens used during the main study are provided in Table 1. At baseline, 11/14 patients treated on demand (78.6%) and 77/107 patients receiving prophylaxis (72.0%) had target joints (defined as per the Scientific Standardized Committee of the International Society on Thrombosis and Hemostasis: 3 bleeds into the same joint within a 6‐month period21). Median (Q1; Q3) number of total bleeds and joint bleeds within 12 months before main study enrolment, respectively, was 25.5 (12.0; 47.0) and 19.5 (10.0; 47.0) for patients treated in the on‐demand arm in the extension; and 8.0 (2.0; 16.0) and 5.0 (1.0; 12.0) for patients in prophylaxis arms. Patients who received prophylaxis were continuously treated either twice weekly (n = 23), every 5 days (n = 33), every 7 days (n = 23) or switched regimens during the extension (variable frequency; n = 28). The variable frequency group included 20 patients who switched to a higher frequency regimen, 4 patients who switched to a lower frequency regimen, and 4 patients who switched more than once and were receiving their original regimen at the interim analysis (January 2018); further details of patient movement across treatment arms are shown in Figure 1. The most common reason for switching to a higher dosing frequency was increased bleeding (occurred in 12/28 patients in the variable frequency group). Most patients (79/107) remained with the regimen that was selected at the beginning of the extension period. The majority of patients were treated with extended intervals of every 5 days or every 7 days, and approximately 65% of patients who began the extension receiving every‐7‐days prophylaxis remained in this arm in the extension. At the time of the interim analysis, 81 patients (66.9%) had completed the extension, 33 patients (27.3%) were continuing in the study, and 7 patients (5.8%) had discontinued treatment (adverse event [n = 2], withdrawn consent [n = 3], lack of follow‐up [n = 1], or other [n = 1]).
Table 1.
Demographics of patients in the PROTECT VIII extension study
| Prophylaxis | On demand (n = 14) | Total (N = 121) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Twice weekly (n = 23) | Every 5 days (n = 33) | Every 7 days (n = 23) | Variable frequency* (n = 28) | Total prophylaxis (n = 107) | |||||
| Median (range) age at main study enrolment, y | 35.0 (12‒58) | 33.0 (14‒61) | 31.0 (13‒53) | 39.5 (13‒62) | 33.0 (12‒62) | 43.5 (22‒61) | 36.0 (12‒62) | ||
| Median (range) age at interim analysis, y | 36.0 (17‐61) | 38.0 (17‐66) | 36.0 (17‐59) | 43.0 (15‐67) | 38.0 (15‐67) | 47.5 (24‐66) | 40.0 (15‐67) | ||
| Age group at interim analysis, n (%) | |||||||||
| 12‐17 y | 2 (8.7) | 1 (3.0) | 1 (4.3) | 2 (7.1) | 6 (5.6) | 0 | 6 (5.0) | ||
| 18‐34 y | 7 (30.4) | 13 (39.4) | 10 (43.5) | 7 (25.0) | 37 (34.6) | 4 (28.6) | 41 (33.9) | ||
| 35‐59 y | 13 (56.5) | 16 (48.5) | 12 (52.2) | 16 (57.1) | 57 (53.3) | 6 (42.9) | 63 (52.1) | ||
| ≥60 y | 1 (4.3) | 3 (9.1) | 0 (0) | 3 (10.7) | 7 (6.5) | 4 (28.6) | 11 (9.1) | ||
| Race, n (%) | |||||||||
| White | 16 (69.6) | 22 (66.7) | 12 (52.2) | 21 (75.0) | 71 (66.4) | 7 (50.0) | 78 (64.5) | ||
| Black | 0 | 1 (3.0) | 2 (8.7) | 1 (3.6) | 4 (3.7) | 1 (7.1) | 5 (4.1) | ||
| Asian | 6 (26.1) | 9 (27.3) | 6 (26.1) | 4 (14.3) | 25 (23.4) | 5 (35.7) | 30 (24.8) | ||
| Not reported | 1 (4.3) | 1 (3.0) | 3 (13.0) | 2 (7.1) | 7 (6.5) | 1 (7.1) | 8 (6.6) | ||
| BMI, kg/m2 | |||||||||
| Median (range) | 22.6 (15‐30) | 24.6 (17‐34) | 23.4 (19‐34) | 24.6 (18‐42) | 23.9 (15‐42) | 26.0 (19‐31) | 24.0 (15‐42) | ||
| Treatment during main study, n (%) | |||||||||
| On demand | 2 (8.7) | 0 | 0 | 1 (3.6) | 3 (2.8) | 14 (100.0) | 17 (14.0) | ||
| Twice weekly | 19 (82.6) | 0 | 2 (8.7) | 0 | 21 (19.6) | 0 | 21 (17.4) | ||
| Every 5 days | 0 | 30 (90.9) | 3 (13.0) | 8 (28.6) | 41 (38.3) | 0 | 41 (33.9) | ||
| Every 7 days | 2 (8.7) | 3 (9.1) | 18 (78.3) | 19 (67.9) | 42 (39.3) | 0 | 42 (34.7) | ||
Abbreviation: BMI, body mass index.
Patients who switched regimens during the extension (switched to a higher frequency, n = 20; switched to a lower frequency, n = 4; switched twice and finished with their original frequency, n = 4).
Figure 1.
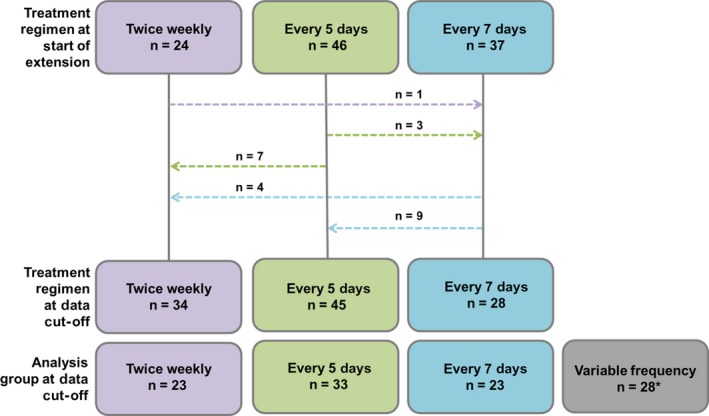
Patient movement across treatment regimens during the extension study. *The variable frequency group included all patients depicted who finished with a different regimen than their initial regimen (n = 24) as well as 1 patient in the every‐7‐days group and 3 patients in the every‐5‐days group who switched twice (ending up back on their original treatment regimen). The analysis group at data cut‐off refers to the group in which patients were included for the statistical analysis [Colour figure can be viewed at http://wileyonlinelibrary.com]
3.2. Treatment exposure
By 31 January 2018, patients had spent a median of 3.9 years (range, 0.8‐5.4) in the combined main study and extension (including patients ongoing in the extension), with an accumulated median of 223 (range, 23‐563) EDs; 110 patients (90.9%) had ≥100 EDs since enrolment in the main study, and 33 patients had been treated with BAY 94‐9027 for up to >5 years. For the extension only, median total time in extension study was 3.2 years (range, 0.1‐4.7 years) with a median of 211.0 EDs (range, 9‐476 EDs) for the prophylaxis group and 101.5 EDs (range, 13‒176 EDs) for the on‐demand group. Patients from Japan (n = 5) participated in the extension study for 16 weeks only, in order to accumulate data for a 1‐year treatment period for the main study, as per Japanese regulatory requirements. They did not continue further than this into the extension study. Median (Q1; Q3) total annual BAY 94‐9027 consumption was 3488 (3153; 4051) IU/kg for prophylaxis arms and 1394 (1059; 1715) IU/kg for the on‐demand arm; per‐dose and overall exposure varied by treatment arm and analysis group. Less frequent prophylactic treatment resulted in lower annual BAY 94‐9027 consumption, despite the higher individual dose per treatment (Table 2). Mean (median) infusion interval was 5.2 (5.0) days.
Table 2.
Treatment exposure during the PROTECT VIII extension study
| Prophylaxis | On demand (n = 14) | |||||
|---|---|---|---|---|---|---|
| Twice weekly (n = 23) | Every 5 days (n = 33) | Every 7 days (n = 23) | Variable frequency* (n = 28) | Total prophylaxis (n = 107) | ||
| Days in extension study | ||||||
| Median (range) | 480.0 (45‐1686) | 624.1 (112‐1686) | 1129.0 (110‐1695) | 1480.9 (182‐1707) | 1162.9 (45‐1707) | 1156.4 (203‐1484) |
| Exposure days in extension study | ||||||
| Median (range) | 168.0 (11‐476) | 129.0 (23‐352) | 163.0 (9‐247) | 292.5 (46‐408) | 211.0 (9‐476) | 101.5 (13‐176) |
| Dose per prophylaxis infusion, IU/kg | ||||||
| Median (Q1; Q3) | 37.5 (31.3; 40.0) | 46.2 (44.3; 49.2) | 58.9 (55.9; 62.5) | 51.6 (44.4; 57.6) | 47.8 (42.4; 57.1) | NA |
| Total consumption, IU/kg/y | ||||||
| Median (Q1; Q3) | 3917 (3241; 4289) | 3504 (3186; 4093) | 3120 (2901; 3256) | 3742 (3346; 4064) | 3488 (3153; 4051) | 1394 (1059; 1715) |
Abbreviations: NA, not applicable; Q, quartile.
Patients who switched regimens during the extension (switched to a higher frequency, n = 20; switched to a lower frequency, n = 4; switched twice and were receiving their original frequency at interim analysis, n = 4).
3.3. Efficacy
The median ABR (Q1; Q3) for total bleeds was 1.6 (0.3; 4.6) and 34.1 (20.3; 36.6) for patients who received prophylaxis and on‐demand treatment, respectively. In the total prophylaxis group, median (Q1; Q3) ABR for joint bleeds was 0.9 (0; 3.3). ABRs varied according to prophylaxis regimen (Figure 2), with patients who remained in the every‐7‐days group reporting median (Q1; Q3) ABRs for total bleeds of 0.7 (0; 1.6), for joint bleeds of 0.3 (0; 1.0), and for spontaneous bleeds of 0.2 (0; 0.8). For the variable frequency group, median (Q1; Q3) total ABR was 3.1 (1.2; 6.2); median ABR in this analysis group was 5.1 with their original regimen, and 2.0 with the regimen after adjustment. Bleeding outcomes in all patients, including those in the variable frequency group, were similar when patients who remained in the extension for ≥12 months were analysed only over the last 12 months in the extension (data not shown); most patients remained on a stable regimen over this period. However, total ABR was slightly lower for prophylaxis patients during the last 12 months compared with the entire duration of the extension; comparatively, this was particularly evident among patients who remained in the every‐7‐days arm, who had a median ABR of 0 during the last 12 months of the extension.
Figure 2.
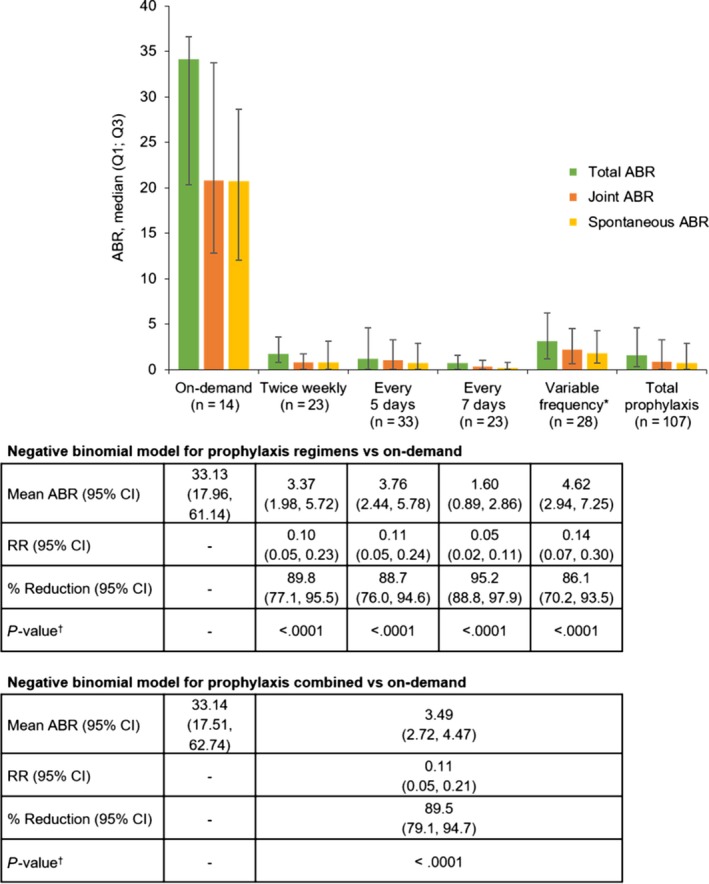
ABR by treatment regimen in the PROTECT VIII extension and negative binomial model. ABR = annualized bleeding rate; CI = confidence interval; Q, quartile; RR = rate ratio; *Patients who switched regimens during the extension (switched to a higher frequency, n = 20; switched to a lower frequency, n = 4; switched twice and were receiving their original frequency at interim analysis, n = 4). † P‐values were nominally derived from the negative binomial model, with no adjustments made for multiple comparisons [Colour figure can be viewed at http://wileyonlinelibrary.com]
Among patients receiving prophylaxis, 20.6% experienced 0 total bleeds, and 29.9% of patients experienced 0 joint bleeds during the total time in the extension study (Figure 3). Relative to the total time in the extension, the proportion of patients with 0 total bleeds and 0 joint bleeds was higher in the last 12 months of the extension for the total prophylaxis group (40.0% and 48.2%) and across all prophylaxis regimens (Figure 3), including every 7 days (64.7% and 70.6%); 60.4% of all prophylaxis patients had zero joint bleeds during the last 6 months of the extension.
Figure 3.
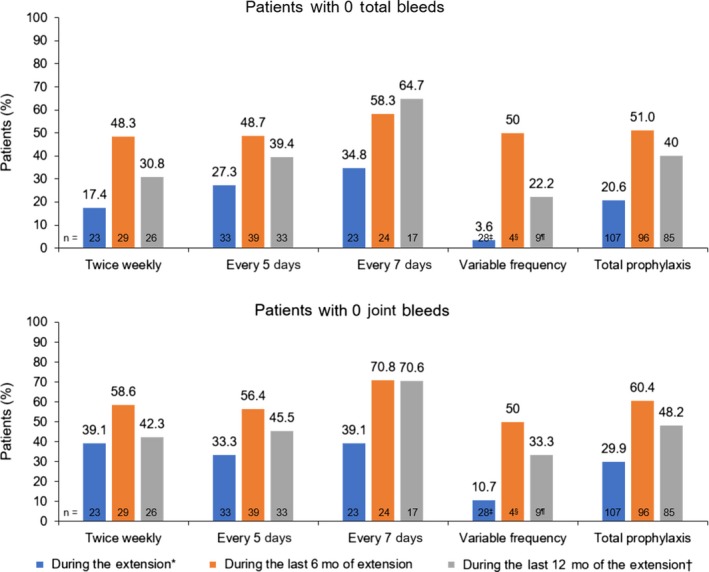
Patients with 0 total bleeds and 0 joint bleeds during prophylaxis. *Median (range) time spent in the extension, 3.2 (0.1‒4.7) years. †Calculated for the subset of patients who spent ≥12 months in the extension. ‡Patients who switched regimens during the extension (switched to a higher frequency, n = 20; switched to a lower frequency, n = 4; switched twice and were receiving their original frequency at interim analysis, n = 4). §Patients who switched regimens during the last 6 months of the extension. ¶Patients who switched regimens during the last 12 months of the extension [Colour figure can be viewed at http://wileyonlinelibrary.com]
Of 1850 total bleeds reported during the extension (on demand, n = 1086; prophylaxis, n = 764), 1739 bleeds (94.0%) were controlled with ≤ 2 infusions; most bleeds (84.8%) required only 1 infusion. The median (Q1; Q3) dose per infusion to treat bleeds was 37.9 (29.8; 47.2) IU/kg. Overall, the response to treatment of bleeds was similar regardless of treatment regimen, with 881 of 1086 bleeds (81.1%) assessed as having good or excellent haemostasis in patients receiving on‐demand treatment versus 646 of 764 bleeds (84.6%) in prophylaxis patients (Figure 4).
Figure 4.
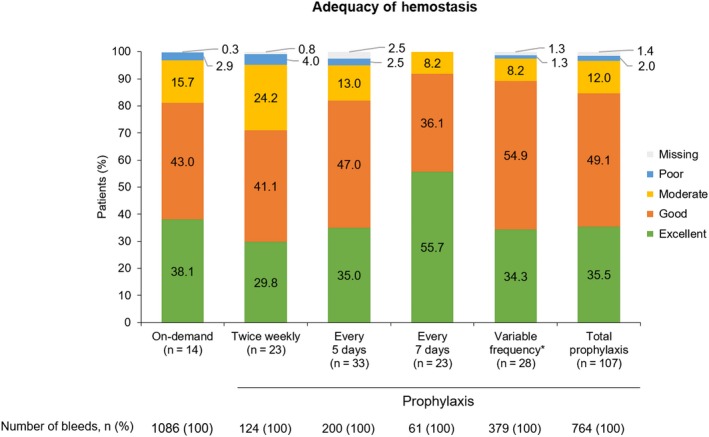
Assessment of response to treatment of bleeds and adequacy of haemostasis. *Patients who switched regimens during the extension (switched to a higher frequency, n = 20; switched to a lower frequency, n = 4; switched twice and were receiving their original frequency at interim analysis, n = 4) [Colour figure can be viewed at http://wileyonlinelibrary.com]
3.4. Safety
During the extension, 9 patients (7.4%; all receiving prophylaxis) experienced AEs that were considered by the investigator to be potentially related to BAY 94‐9027 treatment. AEs were mild in 4 patients (thrombocytopenia, injection site warmth, increased alanine aminotransferase in a patient with hepatitis C, increased β2 microglobulin in urine, arthralgia, pruritus), moderate in 4 patients (bone marrow oedema [originally recorded as a knee bleed], hepatic steatosis, elevated liver function test, meniscal degeneration, osteoarthritis, erythema multiforme), and severe in 1 patient (back pain requiring hospitalization). Of these AEs, 3 were classified as treatment‐related serious AEs in 2 patients (1.7%; both receiving twice‐weekly prophylaxis); elevated liver function test was reported in 1 patient with concomitant liver disease, and 2 episodes of back pain were reported in another patient. Both patients withdrew from the study. No patients developed inhibitors against FVIII.
4. DISCUSSION
Low bleeding rates were maintained for up to >5 years in the PROTECT VIII extension study with twice‐weekly, every‐5‐days and every‐7‐days prophylaxis. The majority of patients (72/107 patients) were treated with extended dosing intervals of every 5 days or every 7 days during the total time in extension. It should be noted that patients in the main study were originally randomized to the extended intervals of 5 or 7 days after a run‐in period of 10 weeks, only if they were controlled (defined as not more than 1 spontaneous bleed) with a twice‐weekly regimen. This pre‐selection therefore excluded high frequent bleeders from extended‐interval prophylaxis. Only a small group (13%) of severe patients did not qualify for randomization and continued on a twice‐weekly regimen. At start of the extension study, patients could choose to remain or switch to another regimen. However, most patients chose to continue with the same dosing regimen in the extension. The mean (median) treatment infusion interval for all prophylaxis patients was 5.2 (5.0) days. Median (Q1; Q3) ABR was 1.6 (0.3; 4.6) for all combined prophylaxis regimens, and 29.9% of patients remained joint bleed‐free up to January 2018. Both ABR for total bleeds and the percentage of patients who experienced ≥1 bleed were further reduced in the total prophylaxis group during the last 12 months of the extension, which may suggest that bleeding outcomes improved as regimens were adjusted to accommodate the individual needs of patients. This bleeding reduction may also reflect improvement in synovitis with fewer bleeds over time. Consistent with this, during the last 12 months of the extension, patients who remained in the every‐7‐days treatment arm had a median ABR of 0 with 64.7% of patients remaining bleed‐free. ABRs for all prophylaxis groups were also further reduced in the extension compared with results of the PROTECT VIII main study.19 No patients developed FVIII inhibitors, and the majority of AEs were mild or moderate in severity, indicating that BAY 94‐9027 was well tolerated. While the resolution of bleeds (Figure 4) showed that patients on prophylaxis who had bleeding episodes reported more “moderate” results which do not appear as favourable as the on‐demand treatment, which may be explained by “milder” bleeds on prophylaxis and less prominent relief after treatment with FVIII.
The unique design of the PROTECT VIII main study and extension, including 3 different prophylaxis regimens and allowing patients to switch infusion frequency, demonstrated that prophylaxis with BAY 94‐9027 can be tailored to the individual needs of each patient. This closely mimics a real‐world treatment setting, in which patients adjust regimens as needed to prevent bleeds. Patients in the variable frequency group had a median ABR of 3.1, but were able to switch regimens as necessary to manage bleeds (the reason for switching noted by 12 of 28 patients in this analysis group). Adjusting regimens to manage individual bleeding was an effective strategy, as evidenced by the majority of patients in this group experiencing fewer bleeds after switching their dosing frequency. Among patients who switched, the median ABR was reduced by >50% from 5.1 before adjustment to 2.0 after regimen adjustment. Because of the advantages of individualized prophylaxis and the potential for decreased dosing frequency to improve patient adherence and outcomes,12, 15 the data reported here suggest that BAY 94‐9027 may provide substantial clinical benefit for patients across a variety of bleeding phenotypes and treatment requirements.
Efficacy of BAY 94‐9027 was consistent with results from clinical trials investigating other extended‒half‐life products.22, 23 For 117 patients aged ≥12 years receiving individualized prophylaxis (beginning with 25 IU/kg twice weekly and adjusting dose and frequency as needed to maintain FVIII trough levels above 1%) with recombinant FVIII, Fc fusion protein (Eloctate®; Bioverativ, Cambridge, MA), median ABR for total bleeds was 1.6.22 In a clinical trial evaluating efficacy of antihaemophilic factor (recombinant), PEGylated (Adynovate®; Baxalta, Westlake Village, CA), median ABR for total bleeds was 1.9 among 120 patients aged ≥12 years receiving prophylaxis twice weekly.23 Results were similar in the current study, in which patients receiving up to every‐7‐days prophylaxis with BAY 94‐9027 had a median ABR for total bleeds of 1.6. Nevertheless, the mean interval between infusions was 5.2 days with BAY 94‐9027, as compared with 3.5 days for the other products.
Potential limitations of this study include the open‐label design and the subjective nature of the assessment of bleeds. However, the unique study design of the PROTECT VIII main study and extension allowed demonstration that BAY 94‐9027 can be successfully used for individualized prophylaxis in a setting that closely mimics real‐world treatment.
5. CONCLUSIONS
In this interim analysis of the ongoing PROTECT VIII extension study, BAY 94‐9027 was efficacious and well tolerated for up to >5 years of prophylactic treatment in adult and adolescent patients with severe haemophilia A. Most patients (67.3%) could be effectively treated with extended intervals of prophylaxis every 5 and 7 days, achieving low bleeding rates, and no major safety concerns were reported. BAY 94‐9027 was also efficacious in treatment of bleeds. These data support long‐term use of BAY 94‐9027 prophylaxis that can be tailored to individual patient needs.
6. ADDENDUM
S. Lalezari, M. T. Reding, I. Pabinger, P. A. Holme, C. Negrier and H.‐J. Shin are principal investigators, treated patients with study drug, and contributed to data acquisition. P. Chalasani is a principal investigator, treated patients with study drug, and contributed to data acquisition and interpretation. M. Wang was involved in design, analysis and interpretation of data. D. Tseneklidou‐Stoeter and M. Maas Enriquez were involved in analysis and interpretation of data. All authors contributed to the development of the manuscript, reviewed and commented on each draft, and approved the final draft.
DISCLOSURES
S. Lalezari: consultant for Bayer and Teva, has received honoraria/consultation fees from Teva, Pfizer and Bayer, and reimbursement for symposia/congresses from Bayer, Roche, Pfizer, Novo Nordisk, Alnylam/Sanofi, Grifols, Perrigo and Baxter.
M. T. Reding: has received honoraria for participation on advisory boards and/or speakers bureaus from Bayer, Bioverativ, Genentech, Novo Nordisk, Pfizer and Shire, and grant funding from Bayer and Biomarin.
I. Pabinger: has received honoraria for lectures and advisory board meetings from Bayer, CSL Behring, Pfizer, Novo Nordisk, Sobi and Shire, and has received unrestricted research grants from CSL Behring and Novo Nordisk.
P. A. Holme: has received grant/research support and speakers honorarium from Bayer.
C. Negrier: has received grant/research support, honoraria or consultation fees from Alnylam, Baxalta/Shire, Bayer, CSL Behring, LFB, Novo Nordisk, Octapharma, Roche, Pfizer and Sobi.
P. Chalasani: has received research support from Pfizer, not related to any haematologic products.
H. Shin: has no disclosures to report.
M. Wang, D. Tseneklidou‐Stoeter and M. Maas Enriquez are employees of Bayer.
ACKNOWLEDGEMENTS
This study and the PROTECT VIII main study were funded by Bayer. Medical writing assistance was provided by Anna Stern of Complete Healthcare Communications, LLC (North Wales, PA), and Rachel Price of Darwin Healthcare Communications (London, England) and was fully funded by Bayer.
Lalezari S, Reding MT, Pabinger I, et al. BAY 94‐9027 prophylaxis is efficacious and well tolerated for up to >5 years with extended dosing intervals: PROTECT VIII extension interim results. Haemophilia. 2019;25:1011–1019. 10.1111/hae.13853
REFERENCES
- 1. Manco‐Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357:535‐544. [DOI] [PubMed] [Google Scholar]
- 2. Gringeri A, Lundin B, Mackensen SV, et al. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study). J Thromb Haemost. 2011;9:700‐710. [DOI] [PubMed] [Google Scholar]
- 3. National Hemophilia Foundation . Medical and Scientific Advisory Council (MASAC) recommendations concerning prophylaxis (regular administration of clotting factor concentrate to prevent bleeding). Document #241. 2016; https://www.hemophilia.org/sites/default/files/document/files/241Prophylaxis.pdf. Accessed July 3, 2018.
- 4. Srivastava A, Brewer AK, Mauser‐Bunschoten EP, et al. Guidelines for the management of hemophilia. Haemophilia. 2013;19:e1‐e47. [DOI] [PubMed] [Google Scholar]
- 5. Manco‐Johnson MJ, Lundin B, Funk S, et al. Effect of late prophylaxis in hemophilia on joint status: a randomized trial. J Thromb Haemost 2017;15:2115‐2124. [DOI] [PubMed] [Google Scholar]
- 6. Geraghty S, Dunkley T, Harrington C, Lindvall K, Maahs J, Sek J. Practice patterns in haemophilia A therapy – global progress towards optimal care. Haemophilia. 2006;12:75‐81. [DOI] [PubMed] [Google Scholar]
- 7. Collins PW, Blanchette VS, Fischer K, et al. Break‐through bleeding in relation to predicted factor VIII levels in patients receiving prophylactic treatment for severe hemophilia A. J Thromb Haemost. 2009;7:413‐420. [DOI] [PubMed] [Google Scholar]
- 8. Feldman BM, Pai M, Rivard GE, et al. Tailored prophylaxis in severe hemophilia A: Interim results from the first 5 years of the Canadian Hemophilia Primary Prophylaxis Study. J Thromb Haemost. 2006;4:1228‐1236. [DOI] [PubMed] [Google Scholar]
- 9. Valentino LA, Mamonov V, Hellmann A, et al. A randomized comparison of two prophylaxis regimens and a paired comparison of on‐demand and prophylaxis treatments in hemophilia A management. J Thromb Haemost. 2012;10:359‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sun HL, McIntosh KA, Squire SJ, et al. Patient powered prophylaxis: a 12‐month study of individualized prophylaxis in adults with severe haemophilia A. Haemophilia. 2017;23:877‐883. [DOI] [PubMed] [Google Scholar]
- 11. Hilliard P, Zourikian N, Blanchette V, et al. Musculoskeletal health of subjects with hemophilia A treated with tailored prophylaxis: Canadian Hemophilia Primary Prophylaxis (CHPS) Study. J Thromb Haemost. 2013;11:460‐466. [DOI] [PubMed] [Google Scholar]
- 12. Petrini P, Valentino LA, Gringeri A, Re WM, Ewenstein B. Individualizing prophylaxis in hemophilia: a review. Expert Rev Hematol. 2015;8:237‐246. [DOI] [PubMed] [Google Scholar]
- 13. Fernandes S, Carvalho M, Lopes M, Araujo F. Impact of an individualized prophylaxis approach on young adults with severe hemophilia. Semin Thromb Hemost. 2014;40:785‐789. [DOI] [PubMed] [Google Scholar]
- 14. Stemberger M, Kallenbach F, Schmit E, et al. Impact of adopting population pharmacokinetics for tailoring prophylaxis in haemophilia a patients: A historically controlled observational study. Thromb Haemost. 2019;119:368‐376. [DOI] [PubMed] [Google Scholar]
- 15. Oldenburg J. Optimal treatment strategies for hemophilia: achievements and limitations of current prophylactic regimens. Blood. 2015;125:2038‐2044. [DOI] [PubMed] [Google Scholar]
- 16. Nagao A, Yeung C, Germini F, Suzuki T. Clinical outcomes in hemophilia A patients undergoing tailoring of prophylaxis based on population‐based pharmacokinetic dosing. Thromb Res. 2019;173:79‐84. [DOI] [PubMed] [Google Scholar]
- 17. Mei B, Pan C, Jiang H, et al. Rational design of a fully active, long‐acting PEGylated factor VIII for hemophilia A treatment. Blood. 2010;116:270‐279. [DOI] [PubMed] [Google Scholar]
- 18. Coyle TE, Reding MT, Lin JC, Michaels LA, Shah A, Powell J. Phase I study of BAY 94–9027, a PEGylated B‐domain‐deleted recombinant factor VIII with an extended half‐life, in subjects with hemophilia A. J Thromb Haemost. 2014;12:488‐496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Reding MT, Ng HJ, Poulsen LH, et al. Safety and efficacy of BAY 94–9027, a prolonged‐half‐life factor VIII. J Thromb Haemost. 2017;15:411‐419. [DOI] [PubMed] [Google Scholar]
- 20. Jivi . Jivi Public Assessment Report. European Medicines Agency. CHMP; 2018.
- 21. Blanchette VS, Key NS, Ljung LR, et al. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2014;12(11):1935‐1939. [DOI] [PubMed] [Google Scholar]
- 22. Mahlangu J, Powell JS, Ragni MV, et al. Phase 3 study of recombinant factor VIII Fc fusion protein in severe hemophilia A. Blood. 2014;123:317‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Konkle BA, Stasyshyn O, Chowdary P, et al. Pegylated, full‐length, recombinant factor VIII for prophylactic and on‐demand treatment of severe hemophilia A. Blood. 2015;126:1078‐1085. [DOI] [PMC free article] [PubMed] [Google Scholar]