

Abstract
The ACTN1 gene has been implicated in inherited macrothrombocytopenia. To decipher the spectrum of variants and phenotype of ACTN1‐related thrombocytopenia, we sequenced the ACTN1 gene in 272 cases of unexplained chronic or familial thrombocytopenia. We identified 15 rare, monoallelic, nonsynonymous and likely pathogenic ACTN1 variants in 20 index cases from 20 unrelated families. Thirty‐one family members exhibited thrombocytopenia. Targeted sequencing was carried out on 12 affected relatives, which confirmed presence of the variant. Twenty‐eight of 32 cases with monoallelic ACTN1 variants had mild to no bleeding complications. Eleven cases harbored 11 different unreported ACTN1 variants that were monoallelic and likely pathogenic. Nine variants were located in the α‐actinin‐1 (ACTN1) rod domain and were predicted to hinder dimer formation. These variants displayed a smaller increase in platelet size compared with variants located outside the rod domain. In vitro expression of the new ACTN1 variants induced actin network disorganization and led to increased thickness of actin fibers. These findings expand the repertoire of ACTN1 variants associated with thrombocytopenia and highlight the high frequency of ACTN1‐related thrombocytopenia cases. The rod domain, like other ACTN1 functional domains, may be mutated resulting in actin disorganization in vitro and thrombocytopenia with normal platelet size in most cases.
Keywords: actin, actinin, ACTN1, constitutional, platelet, rod domain, thrombocytopenia
1. INTRODUCTION
Inherited thrombocytopenia comprises a group of rare and genetically heterogeneous diseases that are sometimes difficult to distinguish from acquired thrombocytopenia (Nurden, Freson, & Seligsohn, 2012). As a result, diagnosis of the disease is often delayed, and patients may receive inappropriate treatment. Pathogenic variants of the ACTN1 gene, which encodes α‐actinin‐1 (ACTN1) protein, have recently been reported to cause ACTN1‐related thrombocytopenia (ACTN1‐RT) (MIM #102575) (Bottega et al., 2015; Boutroux et al., 2017; Guéguen et al., 2013; Kunishima et al., 2013; Westbury, Shoemark, & Mumford, 2017; Yasutomi et al., 2016). ACTN1‐RT is one of the most frequent forms of inherited thrombocytopenia (4.2–5.6%) and is characterized by irregular mild bleeding and slightly increased mean platelet volume (Bottega et al., 2015; Faleschini et al., 2018).
ACTN1 is an antiparallel, dimeric, actin cross‐linking protein composed of three domains: the N‐terminal actin‐binding (AB) domain composed of two calponin homology domains; the C‐terminal calmodulin (CaM) domain, which binds another actinin molecule, and the central rod domain composed of four spectrin‐like repeats (SRs) (Gimona & Mital, 1998; Murphy, Lindsay, McCaffrey, Djinović‐Carugo, & Young, 2016; Sjöblom, Salmazo, & Djinović‐Carugo, 2008). Twenty‐five ACTN1 variants have been described so far, of which 16 have been shown to alter the AB and CaM domains (Bottega et al., 2015; Faleschini et al., 2018; Guéguen et al., 2013; Kunishima et al., 2013) and induce varying degrees of actin network disorganization, while four variants were localized in the neck domains flanking the rod domain (Bottega et al., 2015; Kunishima et al., 2013). Variants in the AB domain have been demonstrated to induce increased ACTN1 binding to actin filaments (Murphy et al., 2016). However, functional data on variants affecting the rod domain are scarce. Only one variant p.(Leu395Gln) has been identified in the second SR of the rod domain. This variant was associated in vitro with disorganized, thicker, and shorter actin filaments (Yasutomi et al., 2016). Using cluster‐based analysis of high‐throughput sequencing data, the Bridge Consortium has described four novel ACTN1 rod domain variants: (p.(Arg320Gln), p.(Ala425Thr), p.(Leu547Pro), p.(Ala587Val) associated with macrothrombocytopenia and mild to no bleeding (Westbury et al., 2015). However, a functional study was not performed.
To better define the spectrum of ACTN1 gene variants and the ACTN1‐RT phenotype, we sequenced the ACTN1 gene in cases of unexplained constitutional thrombocytopenia. We identified 15 different ACTN1 variants associated with constitutional thrombocytopenia, including 11 novel genetic variants. This report describes the largest set of novel ACTN1 variants to date. In this study, we focused on nine of the new variants associated with alterations in the rod domain, thereby providing new insight into the role that the rod domain plays in ACTN1‐RT.
2. PATIENTS AND METHODS
2.1. Patients
From 2009 to 2017, we recruited 272 unrelated probands with inherited thrombocytopenia of unknown origin. Genetic analyses were performed at the French Reference Center for Inherited Platelet Disorders at Robert Debré University Hospital in Paris (n = 166) and at La Timone University Hospital in Marseille (n = 106). Relatives of ACTN1 variant carriers were also included in the study when possible.
Medical and family history data were obtained from medical reports and patient interviews. Bleeding tendency was evaluated according to the World Health Organization (WHO) bleeding score: grade 0, no bleeding; grade 1, petechiae; grade 2, mild blood loss; grade 3, gross blood loss; and grade 4, debilitating blood loss.
All cases were included after obtaining informed written consent in accordance with protocols approved by local institutional review boards and the Declaration of Helsinki principles.
2.2. Screening and prediction of variant pathogenicity
Genomic DNA was extracted from peripheral blood samples and used to screen the ACTN1 gene (NG_029480.1). Targeted ACTN1 DNA sequence analysis was performed on samples from 97 cases with MYH9‐negative thrombocytopenia, and targeted multigene sequencing was carried out on samples from 69 cases in Paris (50‐gene panel) and 106 cases in Marseille (308‐gene panel) (Saultier et al., 2017). The variants identified via gene‐panel sequencing were confirmed by targeted ACTN1 DNA sequence analysis. When an ACTN1 variant was identified via targeted ACTN1 DNA sequence analysis, the DNA sample was also analyzed using the 50‐gene panel to exclude the presence of other pathological variants. The multigene panels used are described in the Supporting Information data file. Once a variant was discovered, available samples from family members were tested for the variant using targeted ACTN1 sequencing. We denoted novel variants as those that were either absent or present at a very low level (<0.1%) in public variant repositories and the functional impact of each variant was predicted using various algorithms (Supplemental data file).
Nomenclature: All variants are described according to HGVS guidelines (http://varnomen.hgvs.org/bg-material/refseq/) numbering the A of the initiation methionine as +1 in reference sequence NM_001130004.1 and the corresponding amino acid as +1 in reference sequence NP_001123476.
Molecular modeling was carried out to predict modifications caused by the variants using PyMOL software (PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC) and the crystal structure of actinin isoform 2 (ACTN2, PDB code 4D1E). ACTN2 sequence alignment revealed a highly conserved sequence with 81.4% identity (without considering signal peptides and the large insertion from the ACTN1 residues 752–790). Therefore, ACTN2 is a good template to construct the ACTN1 structural model (Ribeiro et al., 2014).
2.3. Site‐directed mutagenesis
The wild‐type ACTN1 gene corresponded to a full‐length ACTN1 sequence from normal platelet complementary DNA (cDNA) subcloned in a pCDNA3.1‐N‐Myc plasmid as previously described (Kunishima et al., 2013). Site‐directed mutagenesis of the human expression plasmid pCDNA3.1‐ACTN1‐N‐Myc was performed using a GeneArt Site‐Directed Mutagenesis System (Thermo Fisher Scientific) according to the manufacturer's instructions. The primer sequences are available upon request. Mutagenesis efficiency was confirmed by sequencing (Beckman Coulter Genomics).
2.4. Immunofluorescence analysis
The pathogenic consequence of each new ACTN1 variant on actin structure was assessed using immunofluorescence in CHO cells transiently transfected with wild‐type or ACTN1 variant constructs (PolyJet In Vitro DNA Transfection Reagent, SignaGen Laboratories). Briefly, CHO cells were cultured in F12 Nutrient Mixture supplemented with 10% fetal bovine serum in glass coverslips coated with fibronectin (2 µg/ml). Forty‐eight hours after transfection, the cells were fixed with 1% paraformaldehyde for 10 min, permeabilized with 0.3% triton X‐100 in phosphate‐buffered saline (PBS) for 5 min, and blocked using 3% bovine serum albumin in PBS for 30 min. The cells were labeled using anti‐c‐Myc antibody (Santa Cruz), followed by incubation with Alexa 546‐labeled goat antirabbit IgG (Life Technologies A11010) and Alexa 488‐conjugated phalloidin (Life Technologies 12379). After washing the cells, the slides were mounted with DAPI Fluoromount (Southern Biotech), and images were obtained using an AXIO Imager M1 microscope (Carl Zeiss, Germany). Actin fibers were examined using the “analyze particle” plugin of the ImageJ program. Six 100‐pixel square areas with the greatest density of actin fibers were analyzed to determine the feret diameter of the actin fibers. Two to three cells were analyzed for each condition. Low levels of fluorescent signal attributed to low‐level autofluorescence and background noise were corrected using a 9.75% threshold value. The intergroup difference was evaluated using one‐way analysis of variance (ANOVA) with Dunnett's posthoc test.
2.5. Blood cell analysis
Platelet count was measured on venipuncture ethylenediaminetetraacetic acid (EDTA)‐anticoagulated blood samples using an automated cell counter. Blood smears were stained with May‐Grünwald‐Giemsa stain. Mean platelet diameter (MPD) was measured on peripheral blood films using optical microscopy and software‐assisted image analysis (Calopix, TRIBVN Healthcare, Châtillon, France) as previously described (Saposnik et al., 2014). The reference ranges for platelet count and diameter were 155–394 × 109 L−1 and 1.9–2.9 µm, respectively, as defined by the 2.5–97.5 percentile range of the control population (n = 64 for platelet count and n = 92 for platelet diameter).
2.6. Statistical analyses
Quantitative variables are expressed as the mean ± standard error of the mean. The intergroup difference was evaluated using an unpaired t test or one‐way ANOVA with Dunnett's posthoc test. The p < 0.05 was considered as significant.
3. RESULTS
3.1. Identification of novel ACTN1 variants
Targeted DNA sequence analysis and gene‐panel sequencing of the ACTN1 gene were carried out on samples derived from 272 cases with suspected inherited thrombocytopenia: 97 cases with MYH9‐negative thrombocytopenia and 175 cases with thrombocytopenia of unknown origin.
We identified 15 different monoallelic and nonsynonymous ACTN1 variants, which were likely pathogenic, in 20 index cases (Figure 1 and Table 1). Gene‐panel sequencing did not reveal potentially pathogenic variants in other genes among the carriers of ACTN1 variants nor other ACTN1 variants.
Figure 1.
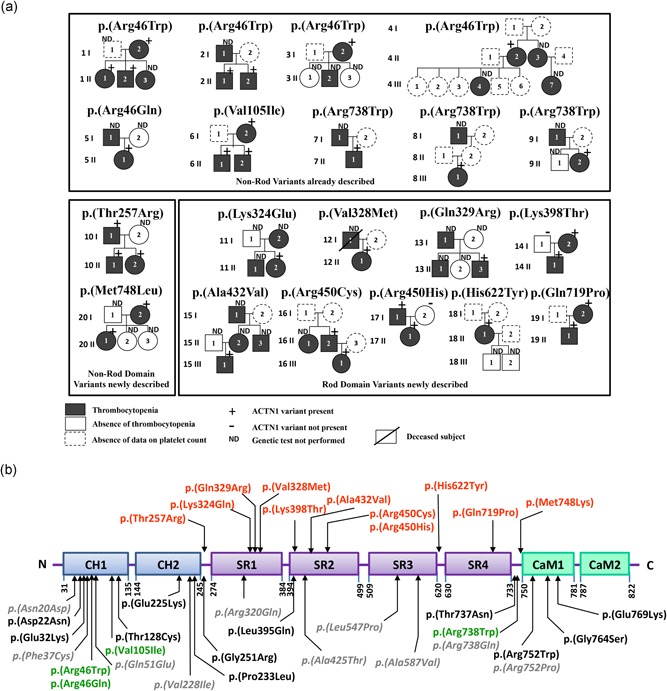
Identification and characterization of novel ACTN1 variants. (a) Pedigrees of the affected families Squares denote males, and circles denote females. Black filled, and empty symbols represent thrombocytopenic and nonthrombocytopenic family members, respectively. Dotted‐line symbols were used when platelet count was unavailable. ATCN1 gene constitutional variant status is indicated at the top of symbols representing family members. All identified variants were monoallelic. (b) Structural domains of ACTN1 and localization of ACTN1 variants: (i) previously described with proven functional impact (dark), (ii) previously described but untested for functional impact (gray), (iii) previously described with proven functional impact and reported here (green), and (iv) previously undescribed and reported here (red). The four SRs form the rod domain of the protein. CaM, calmodulin‐like domain; CH, calponin homology domain; SR, spectrin repeat
Table 1.
ACTN1 variants identified
| Mutation (cDNA) | VEP consequence | Exon | Protein effect | Bioinformatics analysis | Allele frequency (%) | ClinVar | In vitro actin fibers disorganization | Variants classification (ACMG) | References | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Poly Phen‐2 | SIFT | Provean | Mutation Taster | PhastCons | CADD score | 1000G | ExAc | dbSNP | ||||||||
| c.136C>T | Missense | 2 | p.(Arg46Trp) | D | D | D | D | 1 | 35 | 0 | 0.00081 | NA(rs747559032) | NA | Yes | Likely pathogenic | Bottega et al. (2015) |
| c.137G>A | Missense | 2 | p.(Arg46Gln) | D | D | D | D | 1 | 35 | 0 | 0 | NA (rs387907348) | Pathogenic | Yes | Likely pathogenic | Kunishima et al. (2013) |
| c.313G>A | Missense | 2 | p.(Val105Ile) | D | D | B | D | 1 | 34 | 0 | 0 | NA (rs387907345) | Pathogenic | Yes | Likely pathogenic | Kunishima et al. (2013) |
| c.2212C>T | Missense | 18 | p.(Arg738Trp) | B | D | D | D | 1 | 35 | 0 | 0 | NA (rs387907349) | Pathogenic | Yes | Likely pathogenic | Kunishima et al. (2013) |
| c.770C>G | Missense | 9 | p.(Thr257Arg) | D | D | D | D | 1 | 28.6 | 0 | 0 | 0 | NA | Yes | Likely pathogenic | This study |
| c.970A>G | Missense | 10 | p.(Lys324Glu) | D | D | D | D | 1 | 28.6 | 0 | 0 | 0 | NA | Yes | Likely pathogenic | This study |
| c.982G>A | Missense | 10 | p.(Val328Met) | D | D | B | D | 1 | 26.8 | 0 | 0 | 0 | NA | Yes | Likely pathogenic | This study |
| c.986A>G | Missense | 10 | p.(Gln329Arg) | B | D | B | D | 1 | 26.4 | 0 | 0 | 0 | NA | Yes | Likely pathogenic | This study |
| c.1193A>C | Missense | 11 | p.(Lys398Thr) | D | D | D | D | 1 | 29.8 | 0 | 0 | 0 | NA | Yes | Likely pathogenic | This study |
| c.1295C>T | Missense | 12 | p.(Ala432Val) | D | D | D | D | 1 | 34 | 0 | 0 | 0 | NA | Yes | Likely pathogenic | This study |
| c.1348C>T | Missense | 12 | p.(Arg450Cys) | D | D | D | D | 1 | 35 | 0 | 0 | 0 | NA | Yes | Likely pathogenic | This study |
| c.1349G>A | Missense | 12 | p.(Arg450His) | D | D | D | D | 1 | 35 | 0 | 0 | 0 | NA | Yes | Likely pathogenic | This study |
| c.1864C>T | Missense | 16 | p.(His622Tyr) | D | D | B | D | 1 | 20.9 | 0 | 0 | 0 | NA | Yes | Likely pathogenic | This study |
| c.[2156A>C; 2157G>C] | Missense | 18 | p.(Gln719Pro) | D | D | D | D | 1 | 25.7 | 0 | 0 | 0 | NA | Yes | Likely pathogenic | This study |
| c.2243T>A | Missense | 18 | p.(Met748Lys) | D | D | D | D | 1 | 24.8 | 0 | 0 | 0 | NA | Yes | Likely pathogenic | This study |
Note: The consequences of variations were analyzed using VEP (http://www.ensembl.org/Tools/VEP). The effect on the protein's function was analyzed using several bioinformatics tools (PolyPhen‐2, SIFT, Mutation Taster, PhastCons, and CADD scores). A PhastCons score of 1 reflects a total conservation of the residue among species, indicating then a probably pathogenic alteration. A scaled CADD score (http://cadd.gs.washington.edu/score) of 20 means that a variant is amongst the top 1% of deleterious variants in the human genome, while a score of 30 means that the variant is in the top 0.1%.The in‐population allele frequencies were derived from 1000 Genomes Project (1000G) and ExAc databases.
Abbreviations: B, benign, D, damaging, NA, not avalaible; VEP, variant effect predictor.
Four of these variants, which have been associated with ACTN1‐RT in previous studies (Bottega et al., 2015; Guéguen et al., 2013; Kunishima et al., 2013), were detected in nine families (Families 1–9: c.136C>T: p.(Arg46Trp); c.137G>A: p.(Arg46Gln); c.313G>A: p.(Val105Ile); and c.2212C>T: p.(Arg738Trp) (Figure 1).
The 11 remaining variants (Families 10–20) were novel and corresponded to single amino acid substitutions primarily located in the SR domains: c.770C>G: p.(Thr257Arg); c.970A>G: p.(Lys324Glu); c.982G>A: p.(Val328Met); c.986A>G: p.(Gln329Arg); c.1193A>C: p.(Lys398Thr); c.1295C>T: p.(Ala432Val); c.1348C>T: p.(Arg450Cys); c.1349G>A: p.(Arg450His); c.1864C>T: p.(His622Tyr); and c.2243T>A: p.(Met748Lys) (Figure 1). We found two variants located in the SR4 domain (c.[2156A>C; 2157G>C]) in Family 19. These two variants were also found in the mother of the propositus, thus indicating that the two variants are probably located on the same ACTN1 allele, resulting in a p.(Glu719Pro) amino acid substitution (Figure 1).
All new variants were absent from the dbSNP, ExAC and gnomAD databases (Table 1). Some of these variants (p.(Thr257Arg), p.(Lys398Thr), p.(Arg450Cys), p.(Arg450His), p.(Gln719Pro), and p.Met748Lys)) were found in seven relatives of six different families (Families 10, 14, 16, 17, 19, and 20) and were associated with thrombocytopenia (Figure 1a and Table 2). This finding reveals a strong probability of association with dominant inheritance.
Table 2.
Clinical and biological characteristics of families with ACTN1 variants
| Family | Patient | Age at diagnosis (year) | WHO BS | Platelet count (×109/l) | MPD (μm) | ACTN1 variant |
|---|---|---|---|---|---|---|
| 1 | I 2 | 20 | 0 | 39 | 3.1 | p.(Arg46Trp) |
| II 1 | 1 | 0 | 54 | 2.8 | p.(Arg46Trp) | |
| II 2 | 0 | 0 | NA | 2.7 | p.(Arg46Trp) | |
| 2 | II 1 | 29 | 0 | 54 | 3.0 | p.(Arg46Trp) |
| II 2 | 27 | 0 | 76 | 3.2 | p.(Arg46Trp) | |
| 3 | I 2 | 32 | 3 | 90 | 4.0 | p.(Arg46Trp) |
| 4 | II 2 | 18 | 0 | 95 | 3.4 | p.(Arg46Trp) |
| 5 | II 1 | 11 | 0 | 104 | 2.9 | p.(Arg46Gln) |
| 6 | I 2 | 16 | 0 | 86 | 2.7 | p.(Val105Ile) |
| II 1 | 19 | 1 | 87 | 3.0 | p.(Val105Ile) | |
| II 2 | 0 | 0 | 106 | 3.2 | p.(Val105Ile) | |
| 7 | II 1 | 15 | 1 | 80 | 3.5 | p.(Arg738Trp) |
| 8 | III 1 | 18 | 2 | 95 | 3.0 | p.(Arg738Trp) |
| 9 | II 2 | 13 | 0 | 45 | NA | p.(Arg738Trp) |
| 10 | I 1 | 66 | 0 | 72 | NA | p.(Thr257Arg) |
| II 1 | 43 | 0 | 120 | NA | p.(Thr257Arg) | |
| II 2 | 35 | 0 | 135 | NA | p.(Thr257Arg) | |
| 11 | II 2 | NA | 0 | 70 | NA | p.(Lys324Glu) |
| 12 | II 1 | 18 | 0 | 114 | 2.7 | p.(Val328Met) |
| 13 | II 3 | 31 | 0 | 124 | 2.9 | p.(Gln329Arg) |
| 14 | I2 | NA | 0 | 137 | NA | p.(Lys398Thr) |
| II 1 | 28 | 2 | 124 | 2.5 | p.(Lys398Thr) | |
| 15 | III 1 | 23 | 0 | 94 | 2.8 | p.(Ala432Val) |
| 16 | II 2 | 48 | 0 | 51 | 2.8 | p.(Arg450Cys) |
| III 1 | 20 | 0 | 99 | 3.2 | p.(Arg450Cys) | |
| 17 | I1 | NA | 0 | NA | NA | p.(Arg450His) |
| II 1 | NA | 0 | 140 | 2.5 | p.(Arg450His) | |
| 18 | II 1 | 48 | 3 | 78 | 3.2 | p.(His622Tyr) |
| 19 | I 2 | NA | 0 | 65 | NA | p.(Gln719Pro) |
| II 1 | 9 | 0 | 78 | 2.5 | p.(Gln719Pro) | |
| 20 | I 2 | 23 | 1 | 74 | 3.1 | p.(Met748Lys) |
| II 1 | 9 | 0 | 95 | 3.0 | p.(Met748Lys) |
Note: The cases are annotated with the identifiers described in Figure 1a. WHO BS: grade 0, no bleeding; grade 1, cutaneous bleeding only; grade 2, mild blood loss; grade 3, gross blood loss, requiring transfusion; and grade 4, debilitating blood loss, retinal or cerebral, associated with fatality. The platelet count value provided for each patient represents the mean of all platelet counts available for that patient. The reference ranges for platelet count and diameter were 155–394 × 109 L−1 and 1.9–2.9 µm, respectively, as defined by the 2.5–97.5 percentile range of the control population (n = 64 for platelet count and n = 92 for platelet diameter).
Abbreviations: MPD: mean platelet diameter; ND, not determined; WHO BS, World Health Organization Bleeding Score.
3.2. Bleeding diathesis
Among the 32 affected patients (propositi and relatives), 18 were female and 14 were male. Most index cases were diagnosed during adolescence or young adulthood (Table 2), at a mean age of 25.9 years (9–66 years). For each case, thrombocytopenia was identified incidentally during a blood cell count and outside any bleeding episode. None of the patients were diagnosed with immune thrombocytopenia. Patient examinations did not reveal any phenotypic abnormalities that may be associated with ACTN1 variants. Bleeding diathesis was absent in 22 patients and mild in six patients, consisting of either epistaxis, subcutaneous hemorrhage, or prolonged bleeding after surgery. Four of 13 patients who underwent at least one surgical procedure experienced bleeding after surgery, and severe bleeding was reported in two patients (WHO bleeding score = 3; Figure 2a and Table 2). Eight women gave birth to a total of 13 children without excessive bleeding reported. All patients displayed normal hemoglobin values and leukocyte counts (data not shown).
Figure 2.
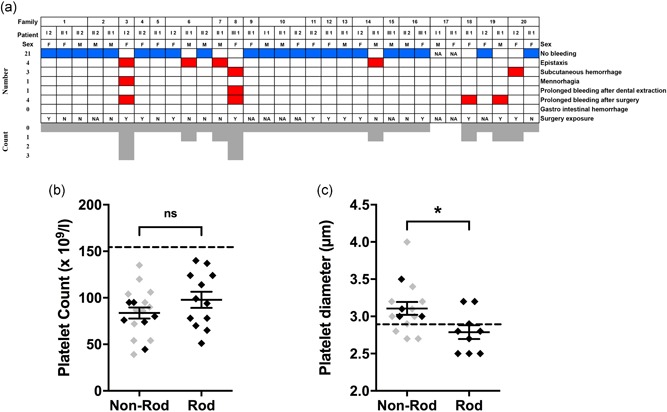
Bleeding, platelet counts and platelet diameters in ACTN1 variant carriers. (a) Characterization of bleeding phenotype in ACTN1 variant carriers. Bleeding episodes of the 30 affected patients were recorded. The cases are annotated with the identifiers described in Figure 1a. The number of patients suffering from a bleeding phenotype is reported on the left. The number of bleeding symptoms for each patient is indicated at the bottom. The type of bleeding disorder is indicated on the right. Exposure to surgery is indicated in the last line. (b,c) Platelet phenotype of ACTN1 variant carriers according to variant location (rod domain vs.nonrod domain variant): platelet count (×109 L−1) (b) and platelet diameter (µm) (c). Among nonrod domain ACTN1 variant carriers, those with variants in the AB domain or the AB/rod domain neck are depicted in gray and those with variants in the CaM domain or the rod domain/CaM domain neck are depicted in black. The mean ± standard error of the mean is shown for each group. MPD values represent the mean of 200 platelet diameter values. The intergroup difference was evaluated using an unpaired t test. *p < .05. The lower limit of the reference range for platelet counts (155 × 109 L−1) and the upper limit of the reference range for platelet diameters (2.9 µm) are represented with a dotted line. ACTN1, α‐actinin‐1; N, no; NA, not available; ns, nonsignificant; Y, yes
3.3. Platelet characteristics
Thrombocytopenia was moderate in the patient cohort (mean platelet count: 88 × 109 L−1 [range 39–135 × 109 L−1]). Twelve (37.5%) of the 32 patients had a mean platelet count below 80 × 109 L−1. Only two patients had a mean platelet count below 50 × 109 L−1 (Table 2). Patient blood smears did not reveal platelet poikilocytosis. Platelet counts did not differ between carriers of rod and nonrod domain variants (Figure 2b).
Overall, platelet diameter values were increased in carriers of ACTN1 variants. Fifteen (62.5%) of the 24 patients tested had a platelet diameter >2.9 µm (97.5th percentile of the control values). However, carriers of variants in the rod domain displayed platelets of smaller diameter compared with carriers of variants outside the rod domain (2.79 ± 0.28 µm vs. 3.11 ± 0.33 µm p = .026) (Figure 2c). This intergroup difference was even greater after merging MPD data from the present study with data from previously reported ACTN1 variant carriers (−0.5 µm; p < .01; n = 54; Figure S1), thereby indicating that some patients with ACTN1‐RT may not have large platelets, especially if the variant is localized in the rod domain.
3.4. In silico protein prediction analysis
Eight new variants were predicted as pathogenic using the six in silico prediction algorithms (Table 1).
All new variants involved residues that are highly conserved in ACTN1 orthologs in nine distantly‐related species (Table 3); all variants also had a PhastCons score of 1. The CADD scores were high, ranging from 20.9 to 35 (Table 1), within the top 1% of deleterious variants genomewide (Kircher et al., 2014), assuming the deleterious nature of the variants. We obtained conflicting prediction results for the variants c.986A>G: p.(Gln329Arg) (predicted as pathogenic using the Sift, PROVEAN and Mutation Taster algorithms and benign using the Polyphen‐2 algorithm) as well as c.982G>A: p.(Val328Met) and c.986A>G: p.(Gln329Arg) (both predicted as pathogenic using the Polyphen‐2, Sift and Mutation Taster algorithms and benign using the PROVEAN algorithm). We obtained such discrepancies for the previously reported deleterious ACTN1 variants c.2212C>T, p.(Arg738Trp) and c.313G>A:(Val105Ile).
Table 3.
Conservation of the human ACTN1 residues Thr257, Val328, Gln329, Lys324, Lys398, Ala432, Arg450, His622, Gln719, and Met748 in orthologs
| Species | UniProt ID | Residues | Identity rate (%) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ACTN1 | Rod domain | |||||||||||||
| Human | Homo sapiens | P12814 | T257 | V328 | K324 | Q329 | K398 | A432 | R450 | H622 | Q719 | M748 | Reference | Reference |
| Chimpanzee | Pan troglodytes | H2RDW7 | T244 | V315 | K311 | Q316 | K385 | A419 | R437 | H609 | Q706 | M735 | 99.8 | 100.0 |
| Macaque | Macaca mulatta | H9YUP4 | T257 | V328 | K324 | Q329 | K398 | A432 | R450 | H622 | Q719 | M748 | 99.9 | 100.0 |
| Dog | Canis lupus familiaris | E2QY08 | T257 | V328 | K324 | Q329 | K398 | A432 | R450 | H622 | Q719 | M748 | 99.3 | 99.3 |
| Cow | Bos taurus | Q3B7N2 | T257 | V328 | K324 | Q329 | K398 | A432 | R450 | H622 | Q719 | M748 | 99.4 | 99.3 |
| Mouse | Mus musculus | Q7TPR4 | T257 | V328 | K324 | Q329 | K398 | A432 | R450 | H622 | Q719 | M748 | 99.2 | 99.3 |
| Rat | Rattus norvegicus | Q6T487 | T257 | V328 | K324 | Q329 | K398 | A432 | R450 | H622 | Q719 | M748 | 99.3 | 99.3 |
| Zebrafish | Danio rerio | B8JHU4 | T267 | V338 | K334 | Q339 | K408 | A442 | R460 | H632 | Q729 | M758 | 93.4 | 91.9 |
| Xenopus | Xenopus tropicalis | F7B6G0 | T255 | V326 | K322 | Q327 | K396 | A430 | R448 | H620 | Q717 | M746 | 94.9 | 93.2 |
Note: The amino‐acid sequence of human ACTN1 protein was compared to orthologs using Clustal OMEGA (http://www.ebi.ac.uk/Tools/msa/clustalo/). The novel variations reported in this study alter the residues Thr257, Val328, Lys324, Gln329, Lys398, Ala432, Arg450, His622, Gln719, and Met748. The table gives the corresponding residue in eight other species. Identity rate is provided for the entire ACTN1 sequence (penultimate column) and for the rod domain sequence (last column), showing that ACTN1 sequence and its rod domain are highly conserved between these species.
The impact of the novel variants on ACTN1 structure and function was examined in silico using PyMOL software (Figure 3). Sequence alignment of ACTN1 and ACTN2 revealed a highly conserved sequence, enabling reliable localization of the ACTN1 variants against the ACTN2 crystal structure (PDB code 4D1E) (Ribeiro et al., 2014). The ACTN2 dimer displayed a cylindrical shape approximately 360 Å long and 60 Å wide. The antiparallel ACTN2 dimer assembled predominantly via the rod domain (SRs 1–4). Interestingly, seven variants (i.e., p.(Lys324Glu), p.(Val328Met), p.(Gln329Arg), p.(Lys398Thr), p.(Ala432Val), p.(Arg450Cys), and p.(Arg450His)) were located at the dimer interface and may thus impair dimerization (Foley & Young, 2013) and cause protein instability. Protein predictions revealed that the variants p.(Arg450Cys) and p.(Arg450His) have disruptive potential, as they result in loss of a salt bridge. The variant p.(Gln719Pro) was predicted to destabilize the structure of SR4 due to the introduction of a proline residue in a helical secondary structure. Such destabilization might alter the interaction of ACTN1 with numerous structural or signaling platelet proteins such as actin, integrins, talin, or vinculin (Sjöblom et al., 2008). Finally, p.(Met748Lys), which is located on the external face far from the dimer interface, may disrupt protein binding to partner molecules.
Figure 3.
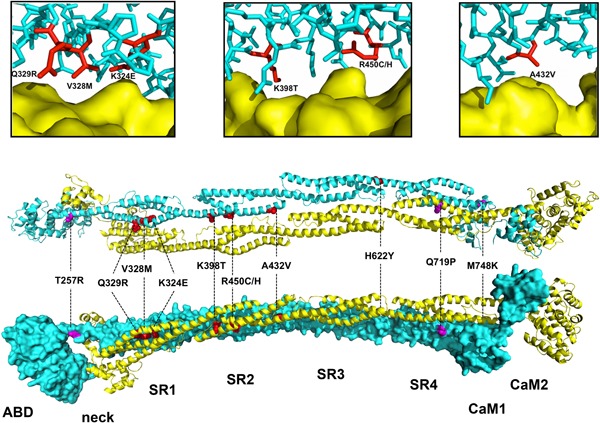
Visualization of the ACTN1 variant protein structures. The dimeric structure of ACTN2 was assembled from two halves of the ACTN2 protomer (AB domain‐neck‐SR1‐SR2‐SR3‐SR4‐CaM1‐CaM2) through a crystallographic twofold axis. One monomer is represented as a blue surface (bottom view) and the other is shown as a yellow ribbon (top view). The two views are rotated 90° around the horizontal axis. Residues corresponding to the variants in ACTN1 are indicated in red when located at the dimer interface and in purple when located elsewhere. The upper panels correspond to close‐up views of the p.(Lys324Glu), p.(Gln329Arg), p.(Val328Met), p.(Lys398Thr), p.(Arg450Cys), and p.(Ala432Val) variants. The monomer colored in blue is shown in stick representation with the residues corresponding to the variants colored in red. The other monomer is represented as a yellow surface. ACTN1, α‐actinin‐1; CaM, calmodulin
3.5. Functional evaluation of variant pathogenicity
Immunofluorescence analysis of ACTN1 was performed in CHO cells. The cells transfected with wild‐type ACTN1 displayed peripheral and stretched actin filaments, which were localized beneath the plasma membrane (Figure 4a), with colocalization of ACTN1 along actin filaments. By contrast, cells transfected with ACTN1 variants showed altered distribution of actin, resulting in thicker and disorganized branched fibers and cytoskeletal disorganization (Figure 4a). Actin network disorganization was quantified via image analysis using the ImageJ program after correction for autofluorescence and background noise. Mean fiber thickness was significantly increased in cells transfected with ACTN1 variants compared with wild‐type ACTN1‐transfected cells, with an average increase in actin fiber diameter of 1.8‐fold (range, 1.4‐ to 2.4‐fold) (Figure 4b). The same trend was observed for fiber perimeter (1.8‐fold increase; range, 1.4‐ to 2.4‐fold) and area (2.5‐fold increase; range, 1.8‐ to 4.0‐fold) (Figure 4b).
Figure 4.
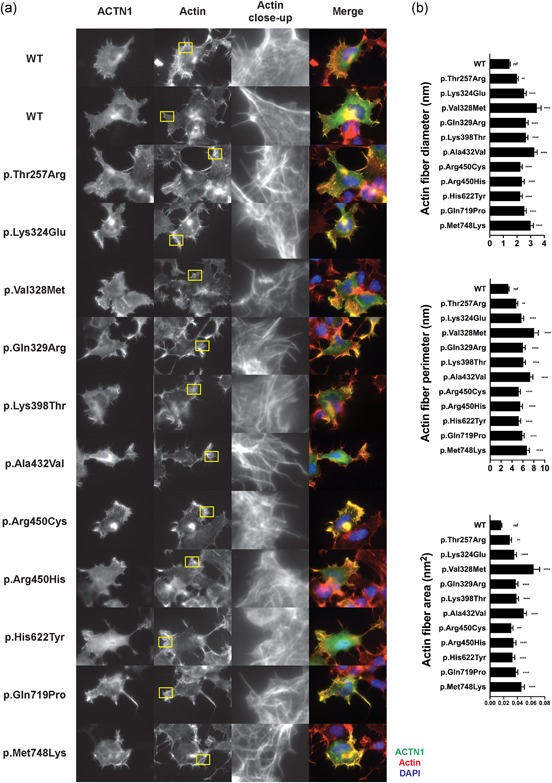
Novel ACTN1 variants alter ACTN1 localization and actin network organization (a) Representative immunofluorescence microscopy images of CHO cells transiently transfected with wild‐type or variant ACTN1 constructs using the standard procedure. The images were visualized after ACTN1 and actin staining, as previously described (Kunishima et al., 2013). Magnification: ×100. (b) Quantification of actin fiber diameter, perimeter and area. For each variant, six 100‐pixel square areas with the greatest density of actin fibers were analyzed to measure the feret diameter, perimeter and area of actin fibers within two or three cells using the “analyze particle” plugin of the ImageJ program. Low levels of fluorescent signal attributed to low‐level autofluorescence and background noise were corrected using a 9.75% threshold value. The intergroup difference was evaluated using one‐way ANOVA with Dunnett's posthoc test. The data are shown as the mean ± standard error of the mean. ref: reference (wild‐type). **p ≤ .01, ***p ≤ .001, ****p ≤ .0001. ACTN1, α‐actinin‐1
4. DISCUSSION
We identified 15 rare, monoallelic, non‐synonymous and likely pathogenic ACTN1 variants in 32 of 272 cases with thrombocytopenia. Eleven of the ACTN1 variants were novel. Our assessment of this comprehensive panel of ACTN1 variants suggests that ACTN1‐RT is an autosomal dominant and non‐syndromic inherited form of thrombocytopenia that is primarily associated with no bleeding.
The dominant pattern of disease inheritance and the association between variants and thrombocytopenia were confirmed in six families (for which segregation data was available), thus revealing an association between the indicator phenotype and presence of the variant. All novel variants were classified as pathogenic according to in silico analysis by at least four of six different prediction algorithms. The substituted residues were highly conserved in ACTN1 orthologs from nine distantly related species.
Transient overexpression of ACTN1 constructs (wild‐type and variants) in cell lines is a proven method to study the effect of ACTN1 variants on cytoskeleton organization (Bottega et al., 2015; Faleschini et al., 2018; Guéguen et al., 2013; Kunishima et al., 2013; Yasutomi et al., 2016). Some reported pathogenic variants display a deleterious effect on the actin fiber cytoskeleton, which appeared disorganized, while neutral ACTN1 variants have no effect (Bottega et al., 2015; Faleschini et al., 2018; Kunishima et al., 2013). Analysis of actin structure showed that each of the 11 new variants exerted a pathogenic effect on actin fiber organization and size. As a result, the variants were classified as “likely pathogenic” (Class 4) according to the criteria of the American College of Medical Genetics and Genomics/Association for Molecular Pathology (Richards et al., 2015). ACTN1 is a bundling protein that anchors actin to a variety of intracellular structures. Recently, Murphy et al. (2016) have reported that several disease‐associated variants located within the AB domain (p.(Arg46Gln), p.(Val105Ile), and p.(Glu225Lys)) cause increased binding of actinin‐1 to actin filaments. We hypothesize that ACTN1 variants induce more stable actin filaments, thereby impairing stretching and leading to shorter and thicker fibers in transfected CHO cells. Conversely, we speculate that some variants in the AB domain cause decreased binding to actin, thus leading to an unstable and disorganized actin network.
Several missense variants have previously been identified in the α‐helical rod domain of various genes, which were associated with heritable diseases (Chakravarty, Chakraborti, & Chakrabarti, 2015; Fatkin et al., 1999). The central rod domain has been proposed to act as a shock absorber, a force transducer or a spacer separating the N‐ and C‐terminal domains (Ribeiro et al., 2014) and to be the prominent protein interaction platform of ACTN1 (Djinovic‐Carugo, Gautel, Ylänne, & Young, 2002). Our in silico structural study shows that seven of the new variants in the rod domain may have the potential to hinder dimer formation (Foley & Young, 2013). The other variants in the rod domain may also exert protein destabilization through loss of a salt bridge or introduction of a proline residue in a helical secondary structure. These modifications may affect rigidity and flexibility of the ACTN1 protein and consequently of the actin fibers.
Most index cases had moderate thrombocytopenia and were incidentally diagnosed during adolescence or young adulthood. The reason why this mild phenotype is limited to platelets while ACTN1 has a ubiquitous expression is currently not well understood. Interestingly, a restricted tissue expression profile has been also reported for variants of ACTN4, another isoform of actinin, that cause an inherited dominant form of a kidney disease, the focal segmental glomerulosclerosis (Kaplan et al., 2000). In a model of mouse knock‐out for the ACTN4 gene (Actn4−/−), histological examination show abnormalities only in the kidneys (Kos et al., 2003), despite the widespread distribution of ACTN4.
We suppose that the presence of one wild‐type ACTN1 allele may preserve the majority of protein cell functions. Additionally, actinin isoforms have a high degree of sequence similarity (87% between ACTN1 and ACTN4) and it has been showed that ACTN1 and ACTN4 heterodimers are the most abundant form of actinin in many cell lines (Foley & Young, 2013). Platelets express ACTN2 and ACTN4 (Burkhart et al., 2012), and actinin heterodimers formation has been also demonstrated in platelets (Gache, Landon, & Olomucki, 1984). Actinin paralogues homodimers, as well as heterodimers, could then share overlapping functions in cells or on the contrary have isoform‐specific functions, via perhaps isoform‐specific interactions with other proteins. Further experimental approaches are needed for a better understand of these mechanisms and to verify these assumptions.
Bleeding diathesis was mild or absent in patients with ACTN1‐RT, as previously reported (Bottega et al., 2015; Faleschini et al., 2018). Nearly half of the patients (n = 10/22 available data) had never undergone surgery and may not have been sufficiently challenged to reveal bleeding complications. Two cases (Family 3‐I‐2 and Family 18‐II‐1) experienced severe bleeding episodes (WHO bleeding score = 3) during surgery despite moderate thrombocytopenia (90 and 78 × 109 L−1, respectively). However, it is difficult to establish with certainty the role that ACTN1 variants play in these severe episodes. These data highlight the need to carefully assess bleeding risk in ACTN1‐RT patients as a precautionary measure.
Our results are consistent with previous reports showing platelet macrocytosis in ACTN1‐RT patients (Bottega et al., 2015; Faleschini et al., 2018; Guéguen et al., 2013; Kunishima et al., 2013). Nevertheless, MPD values were at the upper normal limit in seven of nine cases with rod domain ACTN1 variants. Rod domain variant carriers showed identical platelet counts but reduced MPD values compared with non‐rod domain ACTN1 variant carriers. This observation suggests that ACTN1‐RT diagnosis should also be considered for inherited thrombocytopenia patients with normal platelet size. The functional consequences of rod domain variants may be less severe than those of the non‐rod domains. The different effects of rod and non‐rod variants in structurally homologous ACTN1 proteins, such as the lamin A/C, have been reported. Variants in the rod domain of lamin A/C cause dilated cardiomyopathy and conduction system disease without skeletal myopathy, whereas variants that do not affect the α‐helical rod domain cause more severe disease with muscular dystrophy (Fatkin et al., 1999). Perhaps variants in the AB and CaM domains directly alter AB and head‐to‐tail polymerization, whereas variants in the rod domain affect rigidity and flexibility, thus causing a milder defect (Golji, Collins, & Mofrad, 2009).
Assessing the blood smears of ACTN1‐RT patients, Faleschini et al. (2018) have recently described elongated and often curved platelets that sometimes had typical features of barbell‐shaped proplatelets, which were not observed in healthy subjects. Such atypical platelets were not observed in our patients. However, blood smears in our study were prepared with EDTA anticoagulated blood, while the blood smears assessed by Faleschini et al. were prepared with non‐anticoagulated blood.
This report of 11 new variants and 20 new pedigrees provides significant insight into the mechanisms of ACTN1‐RT and highlights the high frequency of congenital ACTN1 variants as a cause of inherited thrombocytopenia. The rod domain, like the other functional domains of ACTN1, may harbor variants resulting in actin disorganization in vitro and thrombocytopenia with normal platelet size in most cases. The ACTN1 gene should be analyzed in cases of constitutional thrombocytopenia with normal or increased platelet size.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
A. V. performed clinical and biological characterization of patients, analyzed the data, and wrote the paper. P. S. and M. P. designed and performed experiments (mutagenesis, immunofluorescence in CHO cells), analyzed the data, and wrote the paper. S. K. interpreted the data and revised the manuscript. M.F. H.‐R. performed clinical and biological characterization of patients and analyzed the data. A. R. performed the structure analysis. ACTN1 study coinvestigators recruited the patients and collected clinical data. N. S. and M. C. A. supervised the study and wrote the paper.
Supporting information
Supporting information
Supporting information
ACKNOWLEDGEMENTS
The authors thank Noémie Saut for performing genetic analyses, Sonia Poirault Chassac for performing immunofluorescence image analyses, Odile Fenneteau for helpful discussions, and Olivier René and Sylvie Binard for technical assistance. The authors also thank the patients and their families for their participation in the study. The study was funded in part by the “Département de la Recherche Clinique et du Développement (DRCD), Assistance Publique‐Hôpitaux de Paris” (ID No.: P070110), the “Fondation pour la Recherche Médicale” (Grant to P. S.: FDM20150633607) and the French Foundation for Rare Diseases (Grant WES 2012‐2001).
APPENDIX A.
ACTN1 STUDY CO‐INVESTIGATORS
Christine Biron‐Andreani,1 Jean‐François Claisse,2 Rémy Duléry,3,4 Eric Durot,5 Mathieu Fiore,6 Rémi Favier,7 Pierre Fenaux,8,9 Felipe Guerrero,10,11 Véronique Le Cam‐Duchez,12 Elisabeth Mazoyer,13 Stéphanie Muller,14 Bénédicte Neven,15 Catherine Pouymayou,16 Bruno Royer,17 Pierre Sie,10,18 Sandrine Thouvenin,19 Catherine Trichet,20 Nathalie Trillot21 and Karima Yakouben22
1Laboratory of Hematology, Regional Center for Hemophilia Treatment, University Hospital of Montpellier, Montpellier, France
2Laboratory of Hematology, Center of Human Biology, University Hospital of Amiens‐Picardie, Amiens, France
3CHU Saint Antoine, Hematology Department, APHP, Paris, France
4Pierre and Marie Curie University, Paris, France
5Clinical Hematology, University Hospital of Reims, Reims, France
6Laboratory of Hematology, University Hospital of Bordeaux, Bordeaux, France
7Biological Hematology and National Reference Center for Inherited Platelet Disorders, CHU Armand Trousseau, Paris, France
8 Hematology Seniors Service, CHU Saint Louis, Paris, France
9 Paris 7 University, Paris, France
10CHU Toulouse, Biological Hematology, Toulouse, France
11CHU Toulouse, National Reference Center for Inherited Platelet Disorders, Toulouse, France
12Rouen University Hospital, Department of Biological Hematology, F76000 Rouen, France
13Hôpital Avicenne, Biological Hematology, Bobigny, France
14CH Rambouillet, Pediatrics, Rambouillet, France
15CHU Necker‐Enfants Malades, Immunological and Pediatric Hematology, Paris, France
16CHU Timone, National Reference Center for Inherited Platelet Disorders, Marseille, France
17CHU Amiens, Clinical Hematology and Cellular Therapy, Amiens, France
18Université Paul Sabatier, Toulouse, France
19Pediatric Hematology, University Hospital of Saint‐Etienne, Saint‐Etienne, France
20Hematology Department, HUPNVS, APHP, Clichy, France
21CHU Lille, Hematology and Transfusion Institute, F‐59000 Lille, France
22CHU Robert Debré, Department of Pediatric Clinical Hematology and Immunology, APHP, Paris, France
Vincenot A, Saultier P, Kunishima S, et al. Novel ACTN1 variants in cases of thrombocytopenia. Human Mutation. 2019;40:2258–2269. 10.1002/humu.23840
References
REFERENCES
- Bottega, R. , Marconi, C. , Faleschini, M. , Baj, G. , Cagioni, C. , Pecci, A. , … Noris, P. (2015). ACTN1‐related thrombocytopenia: Identification of novel families for phenotypic characterization. Blood, 125(5), 869–872. 10.1182/blood-2014-08-594531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutroux, H. , David, B. , Guéguen, P. , Frange, P. , Vincenot, A. , Leverger, G. , & Favier, R. (2017). ACTN1‐related Macrothrombocytopenia: A novel entity in the progressing field of pediatric thrombocytopenia. Journal of Pediatric Hematology/Oncology, 39(8), e515–e518. 10.1097/MPH.0000000000000885 [DOI] [PubMed] [Google Scholar]
- Burkhart, J. M. , Vaudel, M. , Gambaryan, S. , Radau, S. , Walter, U. , Martens, L. , … Zahedi, R. P. (2012). The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood, 120(15), e73–e82. 10.1182/blood-2012-04-416594 [DOI] [PubMed] [Google Scholar]
- Chakravarty, D. , Chakraborti, S. , & Chakrabarti, P. (2015). Flexibility in the N‐terminal actin‐binding domain: Clues from in silico mutations and molecular dynamics. Proteins, 83(4), 696–710. 10.1002/prot.24767 [DOI] [PubMed] [Google Scholar]
- Djinovic‐Carugo, K. , Gautel, M. , Ylänne, J. , & Young, P. (2002). The spectrin repeat: A structural platform for cytoskeletal protein assemblies. FEBS Letters, 513(1), 119–123. [DOI] [PubMed] [Google Scholar]
- Faleschini, M. , Melazzini, F. , Marconi, C. , Giangregorio, T. , Pippucci, T. , Cigalini, E. , … Noris, P. (2018). ACTN1 mutations lead to a benign form of platelet macrocytosis not always associated with thrombocytopenia. British Journal of Haematology, 183(2), 276–288. 10.1111/bjh.15531 [DOI] [PubMed] [Google Scholar]
- Fatkin, D. , MacRae, C. , Sasaki, T. , Wolff, M. R. , Porcu, M. , Frenneaux, M. , … McDonough, B. (1999). Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction‐system disease. The New England Journal of Medicine, 341(23), 1715–1724. 10.1056/NEJM199912023412302 [DOI] [PubMed] [Google Scholar]
- Foley, K. S. , & Young, P. W. (2013). An analysis of splicing, actin‐binding properties, heterodimerization and molecular interactions of the non‐muscle α‐actinins. The Biochemical Journal, 452(3), 477–488. 10.1042/BJ20121824 [DOI] [PubMed] [Google Scholar]
- Gache, Y. , Landon, F. , & Olomucki, A. (1984). Polymorphism of alpha‐actinin from human blood platelets. Homodimeric and heterodimeric forms. European Journal of Biochemistry, 141(1), 57–61. [DOI] [PubMed] [Google Scholar]
- Gimona, M. , & Mital, R. (1998). The single CH domain of calponin is neither sufficient nor necessary for F‐actin binding. Journal of Cell Science, 111(Pt 13), 1813–1821. [DOI] [PubMed] [Google Scholar]
- Golji, J. , Collins, R. , & Mofrad, M. R. K. (2009). Molecular mechanics of the alpha‐actinin rod domain: Bending, torsional, and extensional behavior. PLoS Computational Biology, 5(5), e1000389 10.1371/journal.pcbi.1000389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guéguen, P. , Rouault, K. , Chen, J.‐M. , Raguénès, O. , Fichou, Y. , Hardy, E. , … Férec, C. (2013). A missense mutation in the alpha‐actinin 1 gene (ACTN1) is the cause of autosomal dominant macrothrombocytopenia in a large French family. PLoS One, 8(9), e74728 10.1371/journal.pone.0074728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan, J. M. , Kim, S. H. , North, K. N. , Rennke, H. , Correia, L. A. , Tong, H. Q. , … Pollak, M. R. (2000). Mutations in ACTN4, encoding alpha‐actinin‐4, cause familial focal segmental glomerulosclerosis. Nature Genetics, 24(3), 251–256. 10.1038/73456. [DOI] [PubMed] [Google Scholar]
- Kircher, M. , Witten, D. M. , Jain, P. , O'Roak, B. J. , Cooper, G. M. , & Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics, 46(3), 310–315. 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kos, C. H. , Le, T. C. , Sinha, S. , Henderson, J. M. , Kim, S. H. , Sugimoto, H. , … Pollak, M. R. (2003). Mice deficient in alpha‐actinin‐4 have severe glomerular disease. The Journal of Clinical Investigation, 111(11), 1683–1690. 10.1172/JCI17988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunishima, S. , Okuno, Y. , Yoshida, K. , Shiraishi, Y. , Sanada, M. , Muramatsu, H. , … Ogawa, S. (2013). ACTN1 mutations cause congenital macrothrombocytopenia. American Journal of Human Genetics, 92(3), 431–438. 10.1016/j.ajhg.2013.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy, A. C. H. , Lindsay, A. J. , McCaffrey, M. W. , Djinović‐Carugo, K. , & Young, P. W. (2016). Congenital macrothrombocytopenia‐linked mutations in the actin‐binding domain of α‐actinin‐1 enhance F‐actin association. FEBS Letters, 590(6), 685–695. 10.1002/1873-3468.12101 [DOI] [PubMed] [Google Scholar]
- Nurden, A. T. , Freson, K. , & Seligsohn, U. (2012). Inherited platelet disorders. Haemophilia, 18(Suppl 4), 154–160. 10.1111/j.1365-2516.2012.02856.x [DOI] [PubMed] [Google Scholar]
- Ribeiro, E. , Pinotsis, N. , Ghisleni, A. , Salmazo, A. , Konarev, P. , Kostan, J. , … Djinović‐Carugo, K. (2014). The structure and regulation of human muscle α‐actinin. Cell, 159(6), 1447–1460. 10.1016/j.cell.2014.10.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … ACMG Laboratory Quality Assurance Committee (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saposnik, B. , Binard, S. , Fenneteau, O. , Nurden, A. , Nurden, P. , Hurtaud‐Roux, M.‐F. , … French MYH9 networka (2014). Mutation spectrum and genotype‐phenotype correlations in a large French cohort of MYH9‐Related Disorders. Molecular Genetics and Genomic Medicine, 2(4), 297–312. 10.1002/mgg3.68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saultier, P. , Vidal, L. , Canault, M. , Bernot, D. , Falaise, C. , Pouymayou, C. , … Alessi, M.‐C. (2017). Macrothrombocytopenia and dense granule deficiency associated with FLI1 variants: Ultrastructural and pathogenic features. Haematologica, 102(6), 1006–1016. 10.3324/haematol.2016.153577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöblom, B. , Salmazo, A. , & Djinović‐Carugo, K. (2008). Alpha‐actinin structure and regulation. Cellular and Molecular Life Sciences, 65(17), 2688–2701. 10.1007/s00018-008-8080-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westbury, S. K. , Shoemark, D. K. , & Mumford, A. D. (2017). ACTN1 variants associated with thrombocytopenia. Platelets, 28(6), 625–627. 10.1080/09537104.2017.1356455 [DOI] [PubMed] [Google Scholar]
- Westbury, S. K. , Turro, E. , Greene, D. , Lentaigne, C. , Kelly, A. M. , Bariana, T. K. , … BRIDGE‐BPD Consortium (2015). Human phenotype ontology annotation and cluster analysis to unravel genetic defects in 707 cases with unexplained bleeding and platelet disorders. Genome Medicine, 7(1), 36 10.1186/s13073-015-0151-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasutomi, M. , Kunishima, S. , Okazaki, S. , Tanizawa, A. , Tsuchida, S. , & Ohshima, Y. (2016). ACTN1 rod domain mutation associated with congenital macrothrombocytopenia. Annals of Hematology, 95(1), 141–144. 10.1007/s00277-015-2517-6 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information