

Abstract
Each year diagnostic laboratories in the Netherlands profile thousands of individuals for heritable disease using next‐generation sequencing (NGS). This requires pathogenicity classification of millions of DNA variants on the standard 5‐tier scale. To reduce time spent on data interpretation and increase data quality and reliability, the nine Dutch labs decided to publicly share their classifications. Variant classifications of nearly 100,000 unique variants were catalogued and compared in a centralized MOLGENIS database. Variants classified by more than one center were labeled as “consensus” when classifications agreed, and shared internationally with LOVD and ClinVar. When classifications opposed (LB/B vs. LP/P), they were labeled “conflicting”, while other nonconsensus observations were labeled “no consensus”. We assessed our classifications using the InterVar software to compare to ACMG 2015 guidelines, showing 99.7% overall consistency with only 0.3% discrepancies. Differences in classifications between Dutch labs or between Dutch labs and ACMG were mainly present in genes with low penetrance or for late onset disorders and highlight limitations of the current 5‐tier classification system. The data sharing boosted the quality of DNA diagnostics in Dutch labs, an initiative we hope will be followed internationally. Recently, a positive match with a case from outside our consortium resulted in a more definite disease diagnosis.
Keywords: data sharing, database, diagnostics, NGS, whole‐exome sequencing
1. INTRODUCTION
The introduction of next‐generation sequencing (NGS) technology in a clinical setting is a challenge for genome diagnostic laboratories. Two steps of the process are critical. First, the implementation and validation of the technology itself. Second, the interpretation of the increasing number of DNA variants detected, especially when going from small gene panel to whole exome/genome sequencing (WES/WGS). The latter creates a significantly larger workload for Clinical Laboratory Geneticists, spent mainly on variant interpretation.
The nine genome diagnostic labs in the Netherlands, organised in the VKGL (Vereniging Klinisch Genetische Laboratoriumdiagnostiek, http://www.vkgl.nl) test thousands of individuals every year using a standardized interpretation procedure, resulting in classifications on a 1–5 scale. So far, the classification data were stored locally in databases, each containing unique observations. These data facilitate interpretation of other patient's variants by providing classifications of variants that were previously assessed in this particular genome diagnostic lab. For other variants, after filtering for potential pathogenicity, various databases are consulted such as Human Gene Mutation Database (HGMD; Stenson et al., 2017), Leiden Open Variation Database (LOVD; Fokkema et al., 2011) and ClinVar (Landrum et al., 2016) as well as resources such as PubMed (Fiorini, Lipman, & Lu, 2017) and OMIM (Amberger, Bocchini, Schiettecatte, Scott, & Hamosh, 2015). In the absence of a positive hit or any clear in silico prediction of the variant effect, clinicians have to resort to contacting their peers in other diagnostic labs to determine whether they have seen these variants before. This approach does not scale with the current and ever‐increasing data volumes that have come with the introduction of NGS in the clinic. To overcome this limitation, the Dutch labs decided to share all interpreted variants and develop a platform to facilitate this process.
Implementation of a platform for sharing interpreted variants is far from trivial. Apart from foreseen technical challenges, such as the integration of novel software with existing diagnostic processes, there are also logistic demands; clinicians and laboratory personnel should not be burdened with the additional task of sharing variants and variant classification results. It also requires a significant amount of effort to agree on what data to share, under which conditions these may be shared, and for which purposes the data may be used, all in agreement with applicable laws and regulations on both national and international level. Finally, the resulting platform should not be limited by a specific technology that might exclude or otherwise obstruct participating laboratories and hinder further international collaboration.
The Dutch genome diagnostics labs from the Amsterdam UMC (Amsterdam University Medical Center, locations AMC [Academisch Medisch Centrum] and VUmc [Vrije Universiteit Medisch Centrum]), ErasmusMC (Erasmus Medical Center), UMCG (University Medical Center Groningen), LUMC (Leiden University Medical Center), Maastricht UMC+, NKI (Netherlands Cancer Institute), RadboudUMC and UMCU (University Medical Center Utrecht) have combined their efforts to create a national platform for sharing variants and their interpretations. Here, we present the platform we have developed using open source MOLGENIS software (MOLecular GENetics Information System; van der Velde et al., 2019), the problems we encountered and the solutions chosen. This platform minimizes the impact on the workload of the participating diagnostic labs by automating many labor‐intensive and repetitive tasks such as collecting variants and assigning consensus status, significantly improving data visibility, data quality and reliability of variant classifications on a national level. On an international level, the nonconflicting classifications have also been shared with the variant database LOVD (Leiden Open Variation Database), and consensus classifications have been shared with ClinVar, maximizing the utilisation of the collected data. Sharing on an international level resulted in a first example where the VKGL data sharing helped to resolve an initially unresolved case.
2. MATERIALS AND METHODS
2.1. Uploading variants from participating diagnostic centers
The variants provided by the participating labs were classified according to VKGL guidelines (Wallis et al., 2013). Software used to locally collect variant classifications include Agilent Alissa Interpret (Agilent Technologies, formerly Cartagenia) and LOVD+ whole‐exome analysis software. Tools and software supporting variant classification include Alamut (Interactive Biosoftware), in‐house databases, literature searches, functional effect prediction, population databases (1,000 G (1000 Genomes Project Consortium et al., 2015), ExAC (Lek et al., 2016), ESP6500 (Tennessen et al., 2012), GnomAD (Karczewski et al., 2019), GoNL (Francioli et al., 2014)), and Human Genome Mutation Database professional (HGMD; Stenson et al., 2017). We based the fields required to describe each variant on the VCF file format (Danecek et al., 2011). This format describes each sequence variant by their chromosome, genomic position, a reference string of the nucleotide(s) present in the reference sequence at the given position (REF), and the observed alternate nucleotide sequence (ALT). The given position refers to the first nucleotide of the reference string. In addition to these fields, each center provided the annotated HGNC gene symbol, the annotated transcript, the variant description based on the HGVS recommendations (den Dunnen et al., 2016) and the variant's classification using the common 5‐tier system (Plon et al., 2008; Richards et al., 2015; Wallis et al., 2013). All data were collected in an TSV or CSV file and uploaded to the central server.
The centers that could not provide the VCF fields because their analysis platform used the HGVS format, shared the HGVS‐based variant descriptions. These were then converted into VCF fields using the Mutalyzer service (https://mutalyzer.nl/).
2.2. Unique variant descriptions (VCF and HGVS)
While VCF files are adequate for exchanging variant data, various sequencing pipelines and analysis platforms may store and exchange the same variant in different formats. For example, a duplication of the G in AGCT could be described as position 1 A to AG, as position 2 G to GG, or as position 3 C to GC. HGVS nomenclature uses much stricter rules for variant descriptions, in this case allowing only the description g.2dup. To exclude the possibility that identical variants were described in more than one way, we decided to perform an additional quality check. All variants were sent to Mutalyzer's JSON API to generate unambiguous HGVS descriptions. As Mutalyzer expects HGVS descriptions as input, variants described using the VCF format were first converted into an HGVS description, after which they were submitted to Mutalyzer's API for normalization to nonambiguous HGVS variant descriptions.
2.3. Grouping the submissions and verifying consensus classification
After uploading the variants and checking the classifications, the data was processed by a program that compares the classifications and creates the consensus classification (https://github.com/molgenis/molgenis-projects/tree/master/VKGL/scripts/consensus). In this step, because subsequent clinical action does not differ between likely benign and benign or likely pathogenic and pathogenic, a 3‐tier classification system was applied rather than a 5‐tier system. Classifications in this 3‐tier system were: likely benign/benign, variant of uncertain significance (VUS) and likely pathogenic/pathogenic. When different centers provided opposite classifications (i.e., [likely] benign and [likely] pathogenic), the variant was marked as “opposite classification.” Variants classified as VUS by one center and either (likely) benign or (likely) pathogenic by another were marked as “no consensus.” Variants seen by more than one center and providing the same classification were marked as “consensus,” a status not given to variants submitted by only one participant. Variants only seen by one lab are set to ‘Classified by one lab.’
2.4. Setting up the national diagnostics variant database
The variant classifications from all VKGL centers were collected and shared via the national diagnostics variant database (http://molgenis.org/vkgl). The database was setup using the MOLGENIS software platform for scientific data (http://molgenis.org). After importing newly contributed data, the platform automatically generates statistics on the number of single‐lab submissions, consensus variants, and variants with no or conflicting classifications. Highlighting the conflicting classifications allows curators and labs to quickly pinpoint variants that require reinterpretation, where possible resolving the conflict status to a consensus status for the next release of the database. New releases are produced every 3 months. The database is updated by creating new tables for each lab with their latest data. From those tables a new consensus table is created. All tables are versioned with the date, and all previous versions of the tables are kept for future reference.
To aid variant classification, the Dutch diagnostic centers using Alissa Interpret received an extract of the central database to be imported in their analysis software. The diagnostic center in Leiden, using the LOVD + whole exome analysis software, received access to the consensus data and single lab submissions through the link with the LOVD variant sharing platform. Maastricht UMC+ and RadboudUMC received an export of the data and imported it in their shared classification database, which is connected to their interpretation software.
2.5. Sharing the variants with ClinVar and LOVD
Variants for which consensus was reached were shared with ClinVar (Landrum et al., 2016) and LOVD (Fokkema et al., 2011); additionally, single lab submissions were shared anonymously with LOVD. For ClinVar, each center created a submission account. The VKGL project data manager was added to the organization of all centers to be able to submit the variants for them. Submission sheets for ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/docs/submit/) were created from the MOLGENIS database using an export script (https://github.com/molgenis/molgenis-projects/tree/master/VKGL/scripts/clinvar_export). Only variants with one OMIM code attached to the related gene and for which consensus was reached were submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/?term=VKGL+Data-share+Consensus). Variants with multiple OMIM codes were not submitted because of their ambiguous association to disease. Variants from single lab submissions were not submitted to ClinVar to prevent identifiability; ClinVar does now allow anonymous submissions. To easily find the data in ClinVar and to emphasize that this classification was based on multiple evaluations, all submissions were labelled as “VKGL Data‐share Consensus.” The original 5‐tier classifications of the submitting centers were used for the ClinVar submission, rather than the 3‐tier classifications generated in the VKGL database consensus table. We intend to update the data in ClinVar when new data becomes available.
For sharing with LOVD, a submitter account was created for each participating lab on the “Global Variome shared LOVD” installation (http://LOVD.nl/shared). All nonconflicting variants, including single lab classifications, were shared and imported into LOVD using an import script (https://github.com/LOVDnl/VKGL_import), which also handles updates of the data in LOVD after every new data release. Consensus data had the data of each lab linked to their own account. To indicate their status, variants were labeled as “classification records”, not linked to an individual or a specific phenotype.
2.6. Classification assessment using ACMG2015 guidelines
We assessed the classifications made by the VKGL labs by comparing them with InterVar (Li & Wang, 2017) as a second‐opinion tool (version 2.0.2 20180118 downloaded from https://github.com/WGLab/InterVar). The InterVar tool is an automated implementation of the ACMG2015 guidelines. The InterVar classifications were compared to the VKGL classifications in two subsets: (a) only consensus variants, and (b) only variants submitted by one center. A discrepancy in classification is defined as when one variant is classified as likely benign/benign in one data set, and as likely pathogenic/pathogenic in the other. The output classification was compared with the VKGL classification with a program that checks discrepancies (available at https://github.com/joerivandervelde/vkgl).
3. RESULTS AND DISCUSSION
3.1. Setting up the national diagnostics variant database
The Dutch genome diagnostic laboratories decided to share their variant classifications to facilitate the clinical interpretation of variants encountered in daily genetic analyses. Each center's classified variants were uploaded to the national VKGL MOLGENIS database (http://www.molgenis.org/vkgl). Users can browse the database or query it using different options including gene name, variant description and genomic locations. For each variant the reporting labs are shown, and the classifications provided. This database is the bridgehead for dissemination of the variant classifications from the Dutch diagnostic labs, both nationally and internationally.
After uploading all variants, the database contained 97,801 unique variants (release 31‐05‐2018). Most of these, 82,898, were reported by one lab only. Fourteen thousand nine hundred and three variants were reported by more than one lab (from two to seven independent observations from eight potential sources). See Figure 1 for a graphical overview of the classifications in the VKGL database.
Figure 1.
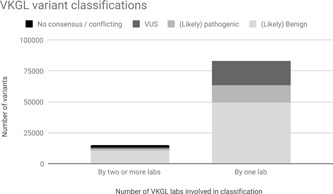
Overview of VKGL variant classifications. Data is split depending on whether a variant was classified and submitted to the database by two or more labs, or by only one lab. VKGL, Vereniging Klinisch Genetische Laboratoriumdiagnostiek
3.2. Unique variant descriptions (VCF and HGVS)
The variety in analysis tools in use (and those that may come in the future) requires a flexible approach to data sharing, as there is not one format supported by all tools, and not all formats require the same variant description fields. Here, we used the fields defined within the VCF file format to describe each variant, as VCF is the most commonly used file format for large‐scale genomic variant sharing. The variants were also converted into HGVS notation to allow consistency checking because VCF notation may be ambiguous.
A quality check was performed to verify the uniqueness of variant descriptions (e.g., g.2dup and g.2_3insG, see Section 2). We encountered 2,498 cases of alternative variant descriptions, leading to a drop of unique variants by 1,498 (1.5% of total) and an increase of 391 consensus variants (3.0% of consensus variants). As expected, most problematic were insertions that should be described as duplications and identical deletions or insertions where the nucleotide positions specified differed. One such example is NC_000019.9:g.13318710_13318712del, which was described by five centers in eight different ways. Other examples include substitutions described in the VCF file format as deletion‐insertion events. These figures show that when comparing multiple data sets, disambiguation is an important step that should not be overlooked. As such, a future release of our pipeline will automatically disambiguate all submitted variants.
It should be noted that when variant descriptions are not correct, queries to find them in public repositories will also fail, hampering correct and accurate variant classification. Although variant databases ClinVar and LOVD are aware of this problem, storing all alternative descriptions of a variant is not feasible as there are unlimited possibilities. Although variant databases could collect at least confirmed alternative notations for the same variant, as LOVD does in case there is a known alternative notation, this can never be exhaustive.
3.3. Consistency of classification in VKGL consortium
Comparing variant classifications (see Section 2) we observed that 12,965/14,903 (87%) variants with more than one submission had a consensus classification, 1,866 (12.5%) a nonconsensus classification and 72 (0.5%) a conflicting classification (see Figure 1).
After each release, all laboratories were notified of the classifications from their lab that conflicted with other classifications in the central database. Upon receiving this list, conflicts could be resolved by direct contact between the laboratories. Updated classifications were stored in each lab's own system, and the conflict was resolved in the next release. Since the implementation of the database, a total of 173 conflicting classifications have been resolved. In most cases, the date of classification turned out to be the culprit. Most laboratories have been performing diagnostic genetic tests for over 30 years. Classifications during the early days made use of far less information than we do now, for example by using a large body of reference literature and other resources such as ExAC/gnomAD, enabling a more specific classification.
Another category consists of variants that were classified as pathogenic by one lab but as VUS or even likely benign by another lab based on the fact that the variant has a low penetrance. In this case, reporting differed simply because clear guidelines for such cases are currently lacking. For example, the NM_025216.2:c.337C>T NP_079492.2:p.(Arg113Cys) variant in WNT10A is considered a pathogenic variant for the autosomal dominant and recessive inherited Tooth agenesis (MIM#150400). This is a low penetrant disorder; however, and the variant is present in 238 alleles (236 heterozygotes and one homozygote, out of 282,532 alleles observed) in the gnomAD database. Similar problems emerged when clearly deleterious variants are encountered in pharmacogenetic genes—should these be classified as benign or pathogenic, or should another class be added such as “risk factor, drug metabolism”?
We investigated the possibility of determining consensus classification by majority vote, where consensus could be reached when at least 75% (a ratio of 3:1) of the centers agreed. For this, at least four classifications were needed but very few variants in our data had been reported by four centers or more. We found that only an additional 0.15% of all variants could have been considered in consensus when applying majority vote, so this method was ultimately not used.
3.4. Assessing the quality of classifications provided
Since variant databases like ClinVar or LOVD are often consulted in Dutch genome diagnostic labs for existing classifications, they are not a good source for assessing the quality of the VKGL laboratory variant classifications. Moreover, many Dutch variants may not be present in these resources, or the variants are submitted by Dutch labs themselves. As such, to assess the classifications made by the VKGL laboratories, we compared them with those obtained through InterVar. We processed our data through InterVar, made sure that the reported variant was in agreement with the GRCh37/hg19 reference genome, matched VKGL gene names with InterVar gene names, and removed variants with nonconsensus VKGL classifications, resulting in 82,111 variants. Of these, 11,910 variants (a subset of the 12,965 variants mentioned above) were seen by multiple labs and 70,201 by single labs (see Figure 2).
Figure 2.
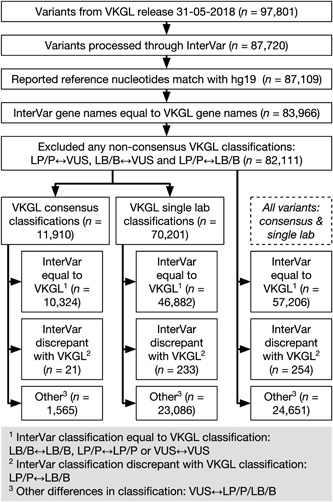
Flowchart describing the steps and results of the VKGL‐InterVar discrepancy analysis. VKGL, Vereniging Klinisch Genetische Laboratoriumdiagnostiek
The results show 99.7% overall consistency with InterVar, with only a few discrepancies. Significant discrepancies (LB/B‐LP/P swaps), were observed in only 0.15% of the consensus variants, 0.33% in case of single‐lab submissions and 0.3% across all variants combined. See Table 1. However, it is worth noting that differences between VUS and LB/B/LP/P were not counted as discrepancies, accounting for 30% of total variants included in the discrepancy analysis. As InterVar was used in a fully automated method and did not have access to any of the specifics such as the de novo status or segregation of a variant, it resorted to classifying a variant as VUS more often than the specialists manually curating the variant in the scope of a single patient.
Table 1.
Consensus and single lab classification discrepancies with InterVar
| VKGL variant interpretation | InterVar classification | VKGL B/LB | VKGL P/LP | VKGL VUS |
|---|---|---|---|---|
| Consensus only | InterVar B/LB | 8,892 | 18 | 121 |
| Consensus only | InterVar P/LP | 3 | 679 | 34 |
| Consensus only | InterVar VUS | 1,048 | 362 | 753 |
| Single‐lab only | InterVar B/LB | 26,195 | 164 | 1,814 |
| Single‐lab only | InterVar P/LP | 69 | 5,629 | 859 |
| Single‐lab only | InterVar VUS | 15,718 | 4,695 | 15,058 |
| Consensus + single‐lab | InterVar LB/B | 35,087 | 182 | 1,935 |
| Consensus + single‐lab | InterVar LP/P | 72 | 6,308 | 893 |
| Consensus + single‐lab | InterVar VUS | 16,766 | 5,057 | 15,811 |
Note: There are 11,910 variants submitted by multiple VKGL centers that reached full consensus classification (Consensus only), of which we found 0.15% LB/B‐LP/P discrepancies when comparing their classification with InterVar. Additionally, there are 70,201 variants classified by a single VKGL center (Single‐lab only), of which we found 0.33% LB/B‐LP/P discrepancies when comparing their classification with InterVar. In total, there are 82,111 variants classified by either single‐labs or in consensus by multiple VKGL centers (Consensus + single‐lab), of which we found 0.3% LB/B‐LP/P discrepancies when comparing their classification with InterVar.
Abbreviation: VKGL, Vereniging Klinisch Genetische Laboratoriumdiagnostiek.
The top VKGL‐InterVar discrepant genes are shown in Table 2. This table also indicates a number of opposing classifications for these genes within VKGL that were not used in the discrepancy analysis with InterVar. We investigated the overlap between genes that had none or at least one VKGL opposing classifications, with genes that had none or at least one VKGL‐InterVar discrepancy. We found a strong association (odds ratio of 21.94 with 95% confidence interval [CI] 11.34‐41.30 and p value 3.019e−16) for genes that are VKGL‐InterVar conflicting as these also have opposite classifications in the VKGL consensus list effort and vice versa in Table 3, showing that these 18 genes (SCN5A, SLCO1B1, VWF, ERCC2, SPTA1, ABCG8, WNT10A, MYBPC3, PKHD1, APOE, TTN, ATP7B, MITF, LPL, FIG. 4, MYO1A, LMF1, and SCN9A) are consistently problematic for variant interpretation.
Table 2.
Top VKGL‐InterVar conflicting genes
| Gene | VKGL total number of classifications | VKGL‐ Intervar conflicts | Consensus VKGL P → InterVar B | 1 VKGL lab B → InterVar P | 1 VKGL lab P → InterVar B | VKGL opposing classifications |
|---|---|---|---|---|---|---|
| APOB | 407 | 9 | 0 | 0 | 9 | 0 |
| PCSK9 | 163 | 9 | 0 | 0 | 9 | 0 |
| LDLR | 300 | 9 | 0 | 0 | 9 | 0 |
| NF1 | 908 | 5 | 0 | 2 | 3 | 0 |
| PAH | 115 | 5 | 0 | 0 | 5 | 0 |
| SCN5A | 352 | 5 | 0 | 1 | 4 | 1 |
| MEFV | 290 | 4 | 3 | 0 | 1 | 0 |
| SLCO1B1 | 76 | 4 | 0 | 1 | 3 | 1 |
| STS | 10 | 3 | 0 | 3 | 0 | 0 |
| APOA5 | 33 | 3 | 0 | 0 | 3 | 0 |
Note: The genes with the most VKGL‐InterVar classification conflicts are APOB, PCSK9, and LDLR, each with nine conflicts. Across 82,111 investigated variants we found 254 conflicts (shown here: 56), of which 21 originated from consensus variants (shown here: 3).
Abbreviation: VKGL, Vereniging Klinisch Genetische Laboratoriumdiagnostiek.
Table 3.
Contingency table
| One or more VKGL‐InterVar discrepant variants | No VKGL‐InterVar discrepant variants | |
|---|---|---|
| One or more VKGL opposing classifications | 18 | 32 |
| No VKGL opposing classifications | 155 | 6,059 |
Note: Fisher's exact test shows odds ratio of 21.94 (95% confidence interval: 11.34‐41.30, p value: 3.019e−16). This shows strong enrichment for genes that are VKGL‐InterVar conflicting to also contain opposite classifications in VKGL consensus data.
Abbreviation: VKGL, Vereniging Klinisch Genetische Laboratoriumdiagnostiek.
If the VKGL‐InterVar discrepancies were randomly distributed among all variant classifications, one would expect a linear relation between the number of variants and the number of discrepancies per gene. A regression analysis indeed shows a trend; however, the explained variation is less than 9% (see Figure 3). This means the discrepant classifications are not distributed fully randomly and may in part be explained by other causes. Closer examination attributes the discrepancies mainly to variants that had a clear functional consequence but were associated with low risk of disease, like hypercholesterolemia and familial Mediterranean fever or low penetrant variants in late onset disorders like cardiomyopathy. For instance, the pathogenic NM_000256.3:c.3628‐41_3628‐17del variant in MYBPC3 that leads to exon 33 skipping, has a MAF in the Indian population of 4% (Dhandapany et al., 2009), underlining the limitations of the 5‐tier classification system for low penetrant and late onset disorders. Nonetheless, the majority (99.7%) of interpretations did not conflict, thus we consider in silico interpretation tools such as InterVar a worthwhile second opinion for variant analysis pipelines. The 0.3% conflicting interpretations may be partially solved if more information such as familial segregation, family history, and de novo status would be included in a manually adjusted two‐Step InterVar analysis.
Figure 3.

Total number of VKGL classifications per gene plotted against the total number of discrepancies with InterVar classifications. A linear regression shows a significant but only slight trend, explaining <9% of the observed variation
3.5. Five‐tier classification limits
The classification conflicts discussed, both between Dutch labs and Dutch labs compared with ACMG, highlight the limits of the current 5‐tier classification system. Multiple studies have already published gene‐ or disease‐specific updates to the ACMG guidelines (Gelb et al., 2018; Kelly et al., 2018; Romanet et al., 2019). While these studies adapt the guidelines for low‐frequency disorders, also for more frequent or less‐penetrant disorders the classification system might be less unequivocal. ClinVar and LOVD already have additional terms such as “association,” “risk factor,” “protective,” “affects function,” “drug response,” “linked to nondisease phenotype.” One of the solutions could be to divide the classifications into two separate groups. A functional/molecular classification based on the consequence of the variant for the function of the gene, RNA or protein, and a clinical classification based on the consequences of the variant for the health of the individual. Revising or creating international guidelines on this topic would be welcome to prevent “conflicting” classifications and misdiagnoses.
3.6. Sharing with international databases
Key to increasing the value of shared data, is to increase the audience with which it is shared. In this light, the VKGL labs decided to share variant classifications internationally, with ClinVar and LOVD. These databases are well‐known international repositories used independently by clinics and researchers. Both have implemented a range of options to access the data, including API access and the GA4GH's beacon project (Global Alliance for Genomics & Health, 2016), and can be accessed using various genome browsers. Sharing variants via these platforms instantly connects our data with their users while efficiently using existing complex APIs and networks. Moreover, these international databases are used as sources for annotation in sequencing analysis pipelines and annotation services like Ensembl's Variant Effect Predictor (McLaren et al., 2016); by sharing variant classifications, the pool of classified variants that can be used as the training set for new or existing algorithms is also increased.
Upon depositing our data in LOVD, we quickly experienced a positive outcome of our sharing effort. A variant in the RPGRIP1 gene, linked to cone‐rod dystrophy and Leber congenital amaurosis, classified as VUS, was picked up by a clinical geneticist from Baltimore (Maryland), United States. Whole‐exome sequencing of a 10‐year old boy with severe retinopathy had revealed a homozygous NM_020366.3:c.1948C>T change in RPGRIP1. This single observation could not be used to classify this variant as pathogenic. However, since the variant was identified twice in the VKGL data set, and since the clinical features of all patients were similar, the classification could be changed from VUS to likely pathogenic. This first success of our data sharing efforts emphasize its importance, even on a global scale.
3.7. Evaluation of data sharing initiative by questionnaire
To obtain a human measure of success for our data sharing initiative, we sent out a questionnaire to VKGL members who use the shared data as part of their daily work. The questions were filled out anonymously except for affiliation. We received responses from 32 members representing all nine participating laboratories (see Supporting Information 1). We found that 87.5% of respondents use the VKGL shared variant classifications directly through the in‐house software used in their laboratory, such as Alissa, but that the central MOLGENIS database was also used by 40.7% of members. Respondents have a predominantly positive opinion about data sharing: 78.1% indicated it has helped their interpretation, 75.0% experienced the resolving of discrepancies and thereby improved variant classification, and 93.7% thinks that the initiative has some (score 2 out of 5) to exceptional (score 5 out of 5) added value for molecular diagnostics in the Netherlands. In addition, 81.2% of respondents indicated that a small (score 2 out of 5) to a huge (score 5 out of 5) amount of time was saved in classifying variants.
4. CONCLUSIONS
With the establishment of the sharing system presented, we have streamlined our diagnostic process, increased standardization, reduced time spent on data interpretation/variant classification and achieved an overall improved quality and reliability. We learnt how to ensure correct variant descriptions, reducing the chance of identical observations being missed, and experienced how the limitations of the current 5‐tier system led to seemingly conflicting classifications. Finally, our aim has been to improve overall diagnosis for patients and their families by internationally sharing our unpublished observations. Who will join our initiative?
Supporting information
Supporting information
ACKNOWLEDGMENTS
We thank ZonMW GGG project “WGS‐first: ‘One‐test‐fits‐all’ to diagnose rare genetic disorders” (nr. 846002003), NWO VIDI Grant number 917.164.455, and BBMRI‐NL for their voucher. BBMRI‐NL is a research infrastructure financed by the Netherlands Organization for Scientific Research (NWO), Grant number 184.033.111.
Fokkema IFAC, van der Velde KJ, Slofstra MK, et al. Dutch genome diagnostic laboratories accelerated and improved variant interpretation and increased accuracy by sharing data. Human Mutation. 2019;40:2230–2238. 10.1002/humu.23896
References
REFERENCES
- 1000 Genomes Project Consortium , Auton, A. , Brooks, L. D. , Durbin, R. M. , Garrison, E. P. , … Abecasis, G. R. (2015). A global reference for human genetic variation. Nature, 526(7571), 68–74. 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberger, J. S. , Bocchini, C. A. , Schiettecatte, F. , Scott, A. F. , & Hamosh, A. (2015). OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Research, 43(Database issue), D789–98. 10.1093/nar/gku1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C. A. , Banks, E. , DePristo, M. A. , … Durbin, R. (2011). The variant call format and VCF tools. Bioinformatics, 27(15), 2156–2158. 10.1093/bioinformatics/btr330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Dunnen, J. T. , Dalgleish, R. , Maglott, D. R. , Hart, R. K. , Greenblatt, M. S. , McGowan‐Jordan, J. , … Taschner, P. E. M. (2016). HGVS recommendations for the description of sequence variants: 2016 update. Human Mutation, 37(6), 564–569. 10.1002/humu.22981 [DOI] [PubMed] [Google Scholar]
- Dhandapany, P. S. , Sadayappan, S. , Xue, Y. , Powell, G. T. , Rani, D. S. , Nallari, P. , … Thangaraj, K. (2009). A common MYBPC3 (cardiac myosin binding protein C) variant associated with cardiomyopathies in South Asia. Nature Genetics, 41(2), 187–191. 10.1038/ng.309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorini, N. , Lipman, D. J. , & Lu, Z. (2017). Towards PubMed 2.0. eLife, 6, e28801 10.7554/eLife.28801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fokkema, I. F. A. C. , Taschner, P. E. M. , Schaafsma, G. C. P. , Celli, J. , Laros, J. F. J. , & den Dunnen, J. T. (2011). LOVD v.2.0: The next generation in gene variant databases. Human Mutation, 32(5), 557–563. 10.1002/humu.21438 [DOI] [PubMed] [Google Scholar]
- Francioli, L. C. , Menelaou, A. , Pulit, S. L. , Van Dijk, F. , Palamara, P. F. , Elbers, C. C. , … Wijmenga, C. (2014). Whole‐genome sequence variation, population structure and demographic history of the Dutch population. Nature Genetics, 46, 818–825. 10.1038/ng.3021 [DOI] [PubMed] [Google Scholar]
- Gelb, B. D. , Cavé, H. , Dillon, M. W. , Gripp, K. W. , Lee, J. A. , Mason‐Suares, H. , … Vincent, L. M. (2018). ClinGen's RASopathy Expert Panel consensus methods for variant interpretation. Genetics in Medicine, 20(11), 1334–1345. 10.1038/gim.2018.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Global Alliance for Genomics and Health (2016). A federated ecosystem for sharing genomic, clinical data. Science, 352(6291), 1278–1280. 10.1126/science.aaf6162 [DOI] [PubMed] [Google Scholar]
- Karczewski, K. J. , Francioli, L. C. , Tiao, G. , Cummings, B. B. , Alföldi, J. , Wang, Q. , … MacArthur, D. G. (2019). Variation across 141,456 human exomes and genomes reveals the spectrum of loss‐of‐function intolerance across human protein‐coding genes. BioRxiv, 10.1101/531210 [DOI] [Google Scholar]
- Kelly, M. A. , Caleshu, C. , Morales, A. , Buchan, J. , Wolf, Z. , Harrison, S. M. , … Funke, B. (2018). Adaptation and validation of the ACMG/AMP variant classification framework for MYH7‐associated inherited cardiomyopathies: Recommendations by ClinGen's Inherited Cardiomyopathy Expert Panel. Genetics in Medicine, 20(3), 351–359. 10.1038/gim.2017.218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum, M. J. , Lee, J. M. , Benson, M. , Brown, G. , Chao, C. , Chitipiralla, S. , … Maglott, D. R. (2016). ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Research, 44, D862–D868. 10.1093/nar/gkv1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek, M. , Karczewski, K. J. , Minikel, E. V. , Samocha, K. E. , Banks, E. , Fennell, T. , … MacArthur, D. G. (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Q. , & Wang, K. (2017). InterVar: Clinical interpretation of genetic variants by the 2015 ACMG‐AMP guidelines. The American Journal of Human Genetics, 100(2), 267–280. 10.1016/j.ajhg.2017.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren, W. , Gil, L. , Hunt, S. E. , Riat, H. S. , Ritchie, G. R. S. , Thormann, A. , … Cunningham, F. (2016). The ensembl variant effect predictor. Genome Biology, 17, 10.1186/s13059-016-0974-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plon, S. E. , Eccles, D. M. , Easton, D. , Foulkes, W. D. , Genuardi, M. , Greenblatt, M. S. , … Tavtigian, S. V. (2008). Sequence variant classification and reporting: Recommendations for improving the interpretation of cancer susceptibility genetic test results. Human Mutation, 29(11), 1282–1291. 10.1002/humu.20880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–423. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanet, P. , Odou, M.‐F. , North, M.‐O. , Saveanu, A. , Coppin, L. , Pasmant, E. , … Barlier, A. (2019). Proposition of adjustments to the ACMG‐AMP framework for the interpretation of MEN1 missense variants. Human Mutation, 40(6), 661–674. 10.1002/humu.23746 [DOI] [PubMed] [Google Scholar]
- Stenson, P. D. , Mort, M. , Ball, E. V. , Evans, K. , Hayden, M. , Heywood, S. , … Cooper, D. N. (2017). The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next‐generation sequencing studies. Human Genetics, 136, 665–677. 10.1007/s00439-017-1779-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen, J. A. , Bigham, A. W. , O'Connor, T. D. , Fu, W. , Kenny, E. E. , Gravel, S. , … Akey, J. M. (2012). Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science, 337(6090), 64–69. 10.1126/science.1219240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Velde, K. J. , Imhann, F. , Charbon, B. , Pang, C. , van Enckevort, D. , Slofstra, M. , … Swertz, M. A. (2019). MOLGENIS research: Advanced bioinformatics data software for non‐bioinformaticians. Bioinformatics, 35(6), 1076–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis, Y. , Payne, S. , McAnulty, C. , Bodmer, D. , Sistermans, E. , Robertson, K. , … Devereau, A. (2013). Practice guidelines for the evaluation of pathogenicity and the reporting of sequence variants in Clinical Molecular Genetics . Retrieved from http://www.acgs.uk.com/media/774853/evaluation_and_reporting_of_sequence_variants_bpgs_june_2013_-_finalpdf.pdf
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information