

Abstract
Studies on foraging partitioning in pollinators can provide critical information to the understanding of food‐web niche and pollination functions, thus aiding conservation. Metabarcoding based on PCR amplification and high‐throughput sequencing has seen increasing applications in characterizing pollen loads carried by pollinators. However, amplification bias across taxa could lead to unpredictable artefacts in estimation of pollen compositions. We examined the efficacy of a genome‐skimming method based on direct shotgun sequencing in quantifying mixed pollen, using mock samples (five and 14 mocks of flower and bee pollen, respectively). The results demonstrated a high level of repeatability and accuracy in identifying pollen from mixtures of varied species ratios. All pollen species were detected in all mocks, and pollen frequencies estimated from the number of sequence reads of each species were significantly correlated with pollen count proportions (linear model, R 2 = 86.7%, p = 2.2e−16). For >97% of the mixed taxa, pollen proportion could be quantified by sequencing to the correct order of magnitude, even for species which constituted only 0.2% of the total pollen. In addition, DNA extracted from pollen grains equivalent to those collected from a single honeybee corbicula was sufficient for genome‐skimming. We conclude that genome‐skimming is a feasible approach to identifying and quantifying mixed pollen samples. By providing reliable and sensitive taxon identification and relative abundance, this method is expected to improve our understanding in studies that involve plant–pollinator interactions, such as pollen preference in corbiculate bees, pollen diet analyses and identification of landscape pollen resource use from beehives.
Keywords: abundance, direct shotgun sequencing, metabarcoding, metagenomics, plastid genome, pollen identification
1. INTRODUCTION
Pollinator declines have been widely reported in the last decades, causing substantial losses in pollination services and subsequent reductions in crop yields (Potts et al., 2010). Over 80% of known flowering plants are pollinated by animals, which mainly comprise insects (Ollerton, Winfree, & Tarrant, 2011). Therefore, global conservation efforts have been carried out with a priority on biodiversity registration and monitoring of pollinators, such as bees, beetles, moths and flies (Burkle, Marlin, & Knight, 2013; Kremen, 2005; Winfree, Griswold, & Kremen, 2007). The potential drivers of pollinator decline include habitat loss and fragmentation, introduction of alien species, climate change and their synergistic effects (Potts et al., 2010; Tylianakis, Didham, Bascompte, & Wardle, 2008). Among these, nutritional stress caused by the lack of suitable foraging habitats and reduction in flower resources are important reasons for pollinator loss (Naug, 2009).
Pollen is a major part of the daily diet for bees, providing them with proteins, lipids, carbohydrates, vitamins and minerals (Brodschneider & Crailsheim, 2010). Noticeable variations in pollen nutrition have been reported for different plant species (Somerville & Nicol, 2006), whereas the diversity of pollen resources can affect foraging behaviour, disease resistance and longevity of the bees (Hanley, Franco, Pichon, Darvill, & Goulson, 2008; Standifer, 1964). Bees exposed to a range of flower resources in nature may have a better chance in maintaining a healthy population by developing stronger resistance to diseases and stresses. For instances, diversified pollen diets can enhance bee immunity and their resilience to pathogens (Di Pasquale et al., 2013) and insecticides (Schmehl, Teal, Frazier, & Grozinger, 2014). Thus, characterizing pollen use for the bees is a crucial path towards understanding their most suitable ecological niches, which serves as the foundation for long‐term monitoring and managing of the pollinating bees (Vaudo, Tooker, Grozinger, & Patch, 2015).
Furthermore, bee ecology and intra‐/interspecific interactions are largely correlated to pollen resource partitioning. For instance, bumblebees (Bombus spp.) with larger population sizes typically have a wider diet range than those of less abundant species (Goulson & Darvill, 2004). In addition, interspecific competition for limited resources can lead to reductions in survival, growth and/or reproduction (Paini, 2004). This is particularly relevant to the interactions between managed honeybees and wild bees, as well as those of the alien and native bees. The impact of domesticated bees on native pollinators can be trivial (Roubik & Wolda, 2001; Steffan‐Dewenter & Tscharntke, 2000), but others showed clear effects to native bees on their reproductive success and population size (Goulson & Sparrow, 2009). Similarly, Paini and Roberts (2005) found considerable overlap in plant resources between Australian native pollinators and alien invasive bees, which led to a decline of the local bees. Such impact seemed to be positively correlated to the level of food overlap (Stout & Morales, 2009). Therefore, understanding the diet range and food niche of bees is crucial to bee conservation.
Direct observation of flower visits has been used in estimating bees’ pollen resources, food range and niche overlap (Camillo & Garofalo, 1989; Connop, Hill, Steer, & Shaw, 2010). This approach is time‐consuming (Ranta & Lundberg, 1981), and the results can be coarse in resolution because it does not differentiate variations in pollen collection efficiencies among and within pollinator species (Barbir, Badenes‐Pérez, Fernández‐Quintanilla, & Dorado, 2015; Bosch, 1992; Woodcock et al., 2013). Alternatively, analysis of pollen loads from pollinators is more straightforward and may provide more accurate evaluation on pollen compositions (Connop et al., 2010). However, pollen identification based on classic palynology requires a specialized skill set. Pollen grains are usually stained and identified morphologically under a microscope, which is time‐ and labour‐consuming. Furthermore, rare species are prone to being overlooked in subsampling and microscopic examination. Alternatively, molecular identifications such as metabarcoding have seen increasing applications in bulk pollen characterizations (Bell et al., 2016, 2017; Cornman, Otto, Iwanowicz, & Pettis, 2015; Danner, Molitor, Schiele, Härtel, & Steffan‐Dewenter, 2016; Galimberti et al., 2014; Kamo et al., 2018; Keller et al., 2015; Pornon et al., 2016; Richardson et al., 2015). Metabarcoding employs high‐throughput sequencing (HTS) in analysing pooled amplicons obtained from mixed taxa (Cristescu, 2014; Ji et al., 2013). While PCR of target genes (e.g. DNA barcodes) helps to increase DNA quantity for HTS, this procedure tends to introducing taxonomic bias due to varied primer efficiencies across taxon lineages (Crampton‐Platt, Yu, Zhou, & Vogler, 2016; Lamb et al., 2018), although multiple optimization methods have been proposed (Nichols et al., 2018; Piñol, Senar, & Symondson, 2018). Recent studies have introduced a genome‐skimming approach, where the total DNA extracts from bulk samples are directly subject to shotgun sequencing, therefore providing better qualitative and quantitative results for pooled invertebrate samples (Arribas, Andújar, Hopkins, Shepherd, & Vogler, 2016; Bista et al., 2018; Choo, Crampton‐Platt, & Vogler, 2017; Tang et al., 2015; Zhou et al., 2013).
Quantitative composition of pollen mixtures, such as bee pollen grains and diets, is particularly important to the understanding of the flower diversity that bees forage. However, the feasibility of genome‐skimming in pollen samples has not been demonstrated. In particular, as pollen grains are typically small in size, they may not provide sufficient DNA for high‐throughput sequencing (HTS) library construction without PCR amplifications. In this study, we examined the potential of pollen genome‐skimming using mock samples consisting of known pollen at varied ratios. We show that our approach can provide accurate taxon identification for pollen mixtures and quantitative information for all member species, including those present at low abundances. We also demonstrate that our method is feasible with small amounts of pollen, where pollen pellets carried by individual bees can provide sufficient DNA for genome‐skimming. Finally, we discuss practical considerations in incorporating this new method into studies on pollen load and food niche, including analytical cost, operational complexity and perspectives on extended applications.
2. MATERIALS AND METHODS
2.1. Pollen samples
Flower pollen (FP) was collected from fresh flowers (Abutilon megapotamicum, Ab. pictum, Alstroemeria aurea, Antirrhinum majus, Lilium brownii, Nymphaea stellate and Schlumbergera truncates), which were purchased from a local flower market (Dasenlin Flower Market). Fresh flowers were identified morphologically by Dr. Lei Gu of Capital Normal University, China. Mature pollen grains were sampled with a sterile needle and preserved in a sterile vial from one or two individuals for each species. Before pollen maturation, stamens from each species were isolated in petri dishes separately to avoid cross‐contamination. Bee pollen (BP) of Brassica napus, Camellia japonica, Papaver rhoeas, Prunus armeniaca, Rhus chinensis and Vicia faba was purchased from Internet stores. These pollen pellets were collected from corbiculae (pollen baskets) of managed honeybees (Apis mellifera), then desiccated and bottled by the merchant. Pollen identity and composition were examined using DNA barcoding and shotgun sequencing (described in the following paragraphs). Two grams of each BP (c. 200–350 pollen pellets) was disaggregated with sterile water, then centrifuged at 14,000 g for 10 min and the supernatant was gently removed. FP and BP were suspended with 1 and 20 ml of 95% ethanol, respectively.
2.2. Pollen Counting
Subsamples of FP and BP pollen suspensions were added into the Fuchsin dilution (16% glycerol, 33% alcohol, 1% basic fuchsine dye and 50% deionized water, Alexander, 1969) for pollen counting, where the total volumes were adjusted so that individual pollen grains could be recognized under a microscope without overlapping (Appendix S1: Table S1). Pollen Fuchsin suspensions were homogenized by vortex shaking and a 5 μl or 10 μl subsample was then examined on a glass slide under a Nikon SMZ800N microscope or on a blood cell counting plate under a Nikon SMZ745T microscope, for FP and BP, respectively (almost all BP had smaller grains than FP in the studied species). To reduce stochastic errors during the process, the dilution procedure for each pollen species was repeated three times, whereas counting was repeated 3 times for each dilution. The average count from these nine replicates was considered as the final pollen count for that species. These counts were then used to calculate pollen numbers per unit volume for each species.
2.3. Pollen mixture mocks
Mock samples with species mixed at varied proportions each contained 200,000 to 5,000,000 pollen grains (Table 1), roughly reflecting those carried by an individual honeybee on one or two corbiculate legs (estimated by BP samples, Appendix S1: Table S1). Five fresh flower pollen mocks (M0001‐0005) were constructed. Among these, M0001 was made with an equal pollen ratio, which was used for calibrations of plastid genome copy numbers (See Section 2.7). Species ratios in FP mocks were set to test pollen‐number variation from a minimum of onefold (e.g. Al. aurea vs. L. brownii in M0003) to a maximum of 100‐fold (e.g. L. brownii vs. Ab. spp. in M0004). Fourteen bee pollen mocks were constructed, with M0014‐0018 and M0021 (equal species ratio at varied total counts) used for testing repeatability of the proposed protocol and for calibrations of plastid genome copy numbers of the relevant bee pollen species. M0006 and M0007 were mock sample replicates, while M0008 and M0009 were DNA replicates. Species ratios in the rest of BP mocks (M0010‐0013) were set to test pollen‐number variations from a minimum of onefold (e.g. B. napus vs. Pa. rhoeas in M0010) to a maximum of 300‐fold (e.g. C. japoica vs. Pr. armeniaca in M0010).
Table 1.
Total pollen counts and species ratios in mock samples
| Mock samples | Total pollen counts | Species ratios | |
|---|---|---|---|
| L. brownii: Ab. pictum: Al. aurea: S. truncates: Ab. megapotamicum: An. majus: N. stellata | |||
| FP | M0001 | 500,000 | 1: 1: 1: 1: 1: 1: 1 |
| M0002 | 500,000 | 91: 27: 9: 3: 1: 9: 1 | |
| M0003 | 500,000 | 1: 9: 1: 30: 9: 15: 30 | |
| M0004 | 500,000 | 10 :1,000: 100: 10: 1: 0: 0 | |
| M0005 | 500,000 | 4: 16: 32: 2: 1: 2: 8 | |
| R. chinensis: B. napus: Pa. rhoeas: V. faba: C. japoica: Pr. armeniaca | |||
| BP | M0006 | 2,500,000 | 5: 300: 125: 25: 5: 1 |
| M0007 | 2,500,000 | 5: 300: 125: 25: 5: 1 | |
| M0008 & M0009 | 5,000,000 | 5: 300: 125: 25: 5: 1 | |
| M0010 | 2,500,000 | 25: 5: 5: 125: 1: 300 | |
| M0011 | 2,500,000 | 180: 90: 30: 30: 1: 1 | |
| M0012 | 2,500,000 | 30: 90: 1: 1: 180: 30 | |
| M0013 | 2,500,000 | 4: 16: 2: 32: 1: 8 | |
| M0014 | 200,000 | 1: 1: 1: 1: 1: 1 | |
| M0015 | 500,000 | 1: 1: 1: 1: 1: 1 | |
| M0016 | 1,000,000 | 1: 1: 1: 1: 1: 1 | |
| M0017 | 2,500,000 | 1: 1: 1: 1: 1: 1 | |
| M0018 | 2,500,000 | 1: 1: 1: 1: 1: 1 | |
| M0021 | 5,000,000 | 1: 1: 1: 1: 1: 1 |
M0001–0005 are FP mixtures; M0006–0018 and M0021 are BP mixtures. M0006 and M0007 are sample replicates; and M0008 and M0009 are DNA replicates. M0014–0018 and M0021 contain pollen at equal ratios but varied total pollen counts.
2.4. Pollen DNA extractions
Pollen DNA was extracted using the modified Wizard method (Soares, Amaral, Oliveira, & Mafra, 2015). An equal volume of silica beads (consisting of both 0.5 mm and 0.1 mm beads in an equal proportion) and pollen mixture sample (0.01–0.1 g) were added to the tube containing 860 μl of TNE buffer, 100 μl of 5 M guanidine and 40 μl of proteinase K. The mixture was then smashed using a minibeadbeater for 2 min to break the cell wall. After incubation for 3 hr at 60°C, 1 ml of Wizard DNA purification resin was added and subsequently eluted through Genomic Spin Columns (TransGen Biotech). The retained resin was washed three times using 700 μl of isopropanol. The columns were dried and eluted for DNA using 100 μl of TE buffer, which was preheated at 70°C, and then centrifuged for 5 min at 14,000 g.
We also estimated the number of pollen grains needed to produce the regular DNA mass required for library construction on Illumina platforms (200 ng). The DNA yield per pollen grain is expected to vary among species due to differences in nuclear genome sizes and plastid genome copy numbers. We used 20,000, 40,000, 100,000 and 200,000 pollen grains for each of B. napus, C. japonica, Pa. rhoeas, Pr. Armeniaca, Rh. Chinensis and V. faba, respectively, and a mixture containing all six BP species at an equal ratio to extract total DNA. DNA extraction was repeated 3 times for each species at each pollen count (12 extracts for each species), each of which was quantified using an Invitrogen Qubit® 3.0 Fluorometer.
2.5. Bee pollen barcoding
The taxonomic identifications of BP were confirmed by Sanger sequencing of the rbcL barcode. Pollen DNA was extracted as described above. One microlitre of each primer (10 mmol/μl, 1F: 5′‐ATGTCACCACAAACAGAAAC‐3′ and 724R: 5′‐TCGCATGTACCTGCAGTAGC‐3′, Fay, Bayer, Alverson, De Bruijn, & Chase, 1998) was used in a PCR with a total volume of 20 μl, containing 2.5 μl of 10× TransStart Taq Buffer, 3.2 μl of dNTPs mix (Promega U1515, 2.5 mmol/μl), 0.2 μl of TransStart Taq DNA Polymerase and 1 μl of template DNA. The PCR programme was set as initial denaturation at 95°C for 2 min, 34 cycles of 94°C denaturation for 1 min, annealing at 55°C for 30 s and extension 72°C for 1 min, and a final extension step at 72°C for 7 min. Amplicons were sequenced using one‐direction Sanger sequencing at Ruibiotech, Beijing, China. Sanger sequences were blasted individually against the NCBI nucleotide database for taxonomic identifications.
2.6. Construction of a reference database for plastid genomes
About 100 mg of dried leaf tissues of each fresh flower species was ground with liquid nitrogen, treated with solution A following a modified CTAB method (Li, Wang, Jing, & Ling, 2013) and then extracted using a Plant Genomic DNA Kit (TIANGEN).
Dual‐indexed libraries with an insert size of 350 bp were prepared using leaf DNA extracts of Ab. pictum, Ab. megapotamicum, An. majus, N. stellate and S. truncates, following the manufacturer's instruction. DNA libraries were sequenced for 15 million reads per species with 150‐bp paired‐end (PE) reads using an Illumina HiSeq 4000 at BGI‐Shenzhen, China. Additionally, the pollen DNA of L. brownii was sequenced with the same sequencing strategy using a HiSeq X Ten at Novogene (Beijing, China).
Data were filtered by SOAPnuke 1.4.0 (‐l 20, ‐q 0.4, ‐n 0.1, ‐M 3, Chen et al., 2018), which removed reads containing adapter sequences, duplicates, poly‐Ns (>15 Ns) and those with >60 bases of quality score ≤32. Assemblies of chloroplast genomes were conducted using NOVOPlasty (Nicolas, Patrick, & Guillaume, 2017), except for L. brownii, which was assembled using SOAPdenovo‐Trans (K = 71, Xie et al., 2014). Protein‐coding genes (PCGs) were annotated by using perl scripts from Zhou et al. (2013), which blasted the assemblies against a database containing 1,552 angiosperm chloroplast genomes (Appendix S1: Table S2) downloaded from GenBank and predicted putative PCGs. All predicted plastid PCGs were aligned using MEGA 7.0 (Sudhir, Glen, & Koichiro, 2016), and then, PCGs shared by all FP and all BP taxa were concatenated, respectively, and used as respective reference sequences for pollen mixture analysis (Figure 1).
Figure 1.
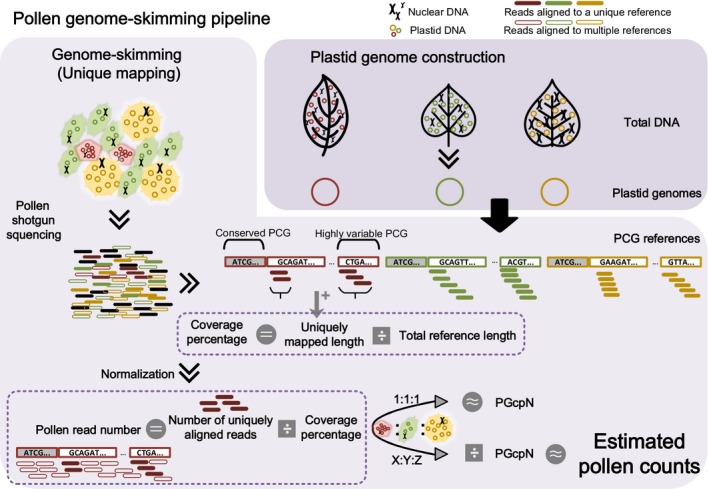
Pollen genome‐skimming pipeline. Plastid protein‐coding gene (PCG) references were obtained from ‘PLASTID GENOME CONSTRUCTION’ and used for unique mapping in ‘GENOME–SKIMMING’. Only reads mapped onto a unique reference (solid short bars) were retained for calculation of the coverage percentage, which was then used for normalization of pollen read number for each member species. Pollen read numbers of member species in mock samples mixed at equal pollen counts (1:1:1) were considered as the plastid genome copy number (PGcpN) for corresponding species. The PGcpN was then used in calculating pollen counts for all member species from sequence reads in regular mocks (X:Y:Z)
2.7. Genome‐skimming of mock pollen mixtures
For each mock, about 200 ng of DNA was used for library construction and high‐throughput sequencing. A 350‐bp insert‐size library was constructed for each mock pollen sample tagged with dual adapters and sequenced using 150 PE on an Illumina HiSeq 4000 platform, generating 30 million reads per sample. After data filtering as described above, clean reads for BP and FP samples were mapped onto reference PCGs using aln BWA 0.7.16 (Li & Durbin, 2010). Aligned reads were assigned to the mapped species only if they met all following criteria: 100% read coverage, ≤1 mismatch and mapped to no more than 1 reference (unique mapping). By the nature of the unique‐mapping algorithm, uniquely mapped reads would only represent highly variable regions among reference genomes. Additional reads would be expected to match multiple PCGs of low sequence divergence, which would not be assigned to any specific taxon (Tang et al., 2015). Therefore, the total pollen read number of a given species was defined as the number of uniquely mapped reads divided by the coverage percentage of its reference PCG sequence (Figure 1).
The copy number of plastid genomes in matured pollen shows notable variations between plant species but remains relatively conservative within species (personal communication with Dr. Sodmergen of Peking University, China). Mock samples M0001 (FP), M0014‐0018 (BP) and M0021 (BP) were constructed with all member species mixed at equal pollen ratios. Therefore, in the sequencing results, proportions of sequence read of the member species are expected to reflect natural copy number differences in plastid genomes among species. These seven mock samples were then used to estimate relative plastid genome copy number (PGcpN) ratios among BP and FP (Figure 1). The PGcpN of the species with the least number was set as 1, and the average values of BP replicates (M0014‐0018 and M0021) were adopted as PGcpN ratios for the member species. For other pollen mixture samples, the pollen read number for each species per sample was then weighted by its PGcpN ratio, resulting in its estimated pollen count in the mixture (Figure 1; Appendix S1: Table S3). Linear regression between pollen proportion from Table 1 and pollen frequency computed from sequencing reads was performed in r 3.4.4 base package (R Core Team, 2015), to estimate correlations between pollen counts and sequence reads for all species.
2.8. Examination for species mixture in BP
As honeybees are generalist pollinators, each pollen pellet collected from the corbicula is expected to contain pollen from multiple plants. To examine to what extent the purchased BP is ‘contaminated’ by nonlabelled pollen species, we sequenced each BP at 15 million reads with 150‐bp PE reads on Illumina platforms (B. napus, R. chinensis and Pa. rhoeas on a HiSeq X Ten at Novogene, Beijing, China; Pr. armeniaca, C. japoica and V. faba on a HiSeq 4000 at BGI‐Shenzhen, China). Cleaned data were uniquely mapped onto BP reference PCG sequences as described above and used to compute the read percentage for each of the mixed species.
3. RESULTS
3.1. DNA extraction from pollen samples
A total of 900–15,000 ng of DNA were obtained for the tested pollen mocks (Table S4). These results confirmed that one or two pollen pellets carried by a single honeybee can usually provide more than enough DNA for direct shotgun sequencing.
DNA yields were positively correlated to the number of grains used for extraction within each species (Figure 2; Appendix S1: Table S5). On the other hand, consistent differences in DNA yields were observed between species. For instances, c. 200,000 grains were needed to produce 200 ng DNA in Pr. armeniaca, whereas only c. 40,000 grains were needed to obtain the same DNA amount for C. japoica. These differences are caused by variations in genome sizes and plastid genome numbers in pollen across species. Approximately 60,000 pollen grains of the mixed sample (consisting of six species at equal ratios) were required for 200 ng DNA.
Figure 2.
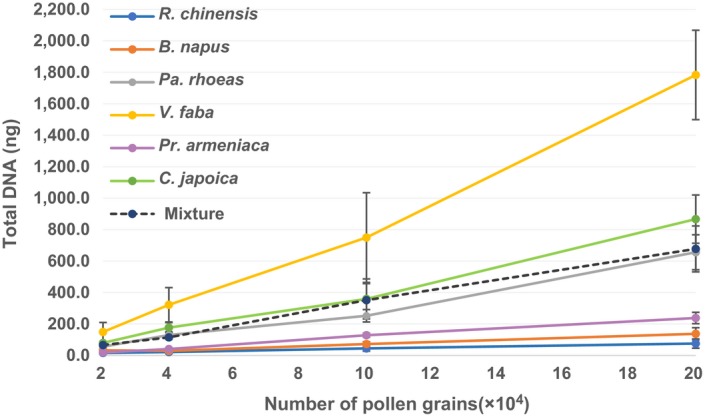
DNA yields from six bee pollen species. DNA was isolated, respectively, from 20,000, 40,000, 100,000 and 200,000 pollen grains of each BP species and the mixture contained six pollen species at equal ratios
3.2. Bee pollen identification by rbcL barcoding
Sanger sequences of the rbcL barcodes for the six BP species were obtained (Appendix S1: Table S6) with high quality. Barcodes confirmed the taxonomic identifications for the labelled species at ≥99% sequence identity.
3.3. Plastid reference genome
Plastid genomes were assembled into scaffolds of 120–171 kb for six plant species (Appendix S2: Figure S1). Reads assigned to plastid genomes account for 2.8%–19.6% of the total shotgun reads for varied species, with an exception in L. brownii that was extracted from pollen, which contained much fewer plastid genome copies than did leaf tissue (0.07%; Appendix S1: Table S7). In addition, plastid genomes of B. napus (NC_016734.1), C. japoica (NC_036830.1), R. chinensis (NC_033535.1), Pr. armeniaca (KY420025.1), Pa. rhoeas (MF943221.1), V. faba (KF042344.1) and Al. aurea (KC968976.1) were downloaded from GenBank and were included in the reference. A total of 65 PCGs shared by all FP species and 71 shared by all BP species were used as reference PCGs, respectively. However, Abutilon megapotamicum and Ab. pictum could not be differentiated from each other even using 65 PCGs, due to limited taxonomic resolution of the gene markers. These two species were pooled and treated as Abutilon spp. for downstream analysis.
3.4. Copy number variation of plastid genomes among pollen species
Mock samples were constructed with equal pollen counts for all member species, for both FP (M0001) and BP (M0014‐0018 and M0021). Shotgun reads were assigned to species using our unique‐mapping criteria, and pollen read number for each species per sample was calculated as described above. The species assigned with the least pollen read number was used as the standard, against which pollen read number of all other species was compared to produce relative copy numbers of plastid genomes (PGcpN). In our results, member species showed significant variations in plastid genome numbers (not the number of plastid organelles). For instance, Al. aurea had 27.8 times more plastid genomes per pollen than that of An. majus, and Pr. armeniaca had 57.7 times more plastid genomes per pollen than that of B. napus on average (Table 2). Nevertheless, plastid genome numbers were conserved within species as shown in BP samples M0014‐0018 and M0021 (Appendix S1: Table S3) and in proportion results in BP replicates M0006‐0009 (see Section 3.5).
Table 2.
Pollen proportions of mock samples calculated from pollen counts and sequences
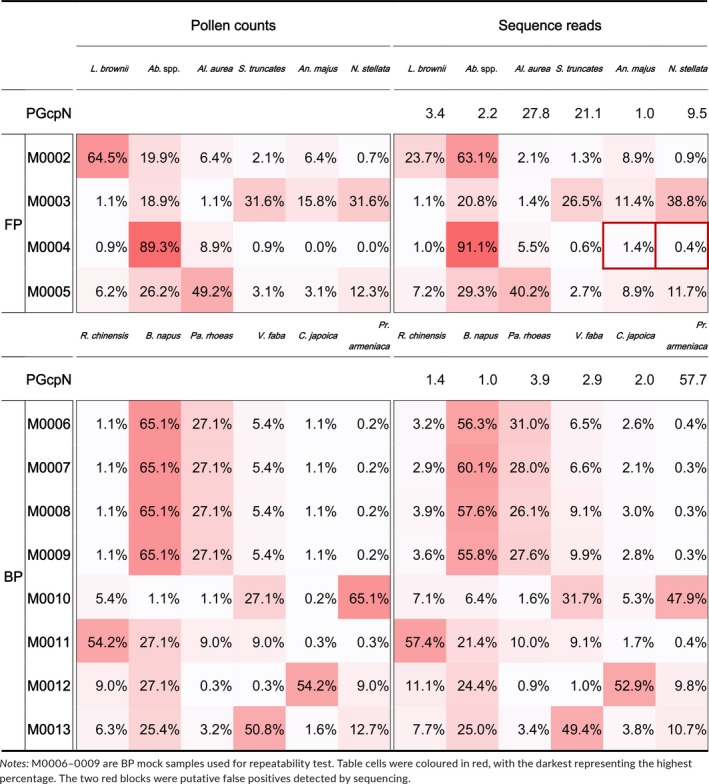
3.5. Repeatability
The sequencing results were highly repeatable in both sets of BP mock samples constructed at different species ratios (Figure 3, Table 2). It is worth noting that these results also reflect consistency in all relevant steps involved in the pipeline, including pollen counting, subsampling, pollen pooling, DNA extraction, library construction and sequencing. Although non‐negligible differences in pollen count were observed in our pollen mock replicates (Table S1), which might have contributed some variations in the final sequence numbers, overall consistency had been observed in most of our results.
Figure 3.
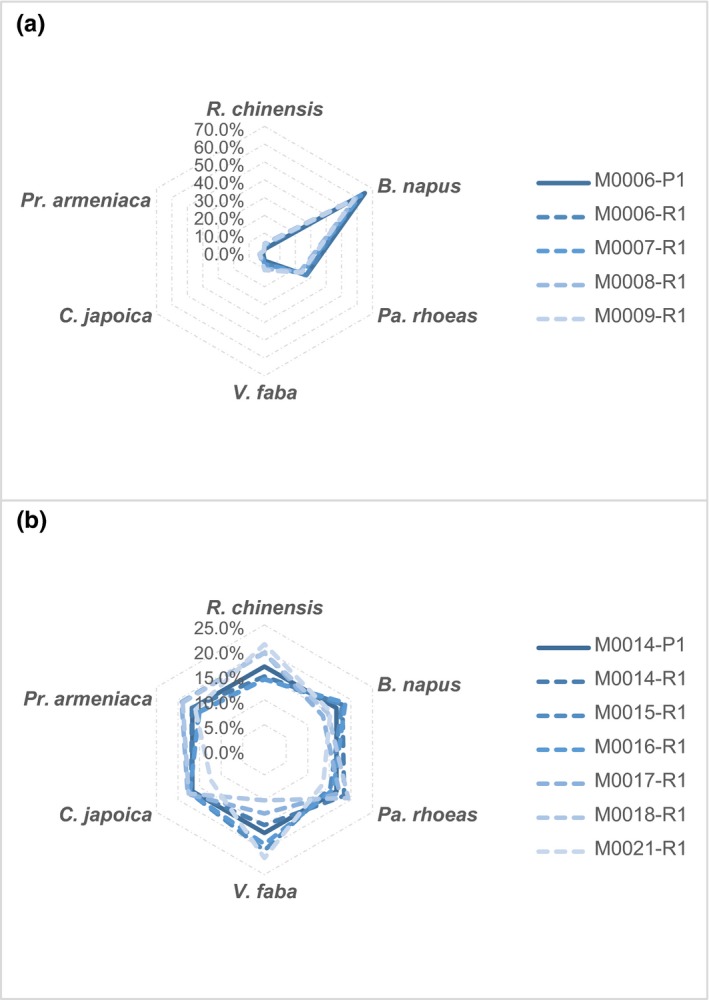
Repeatability of pollen genome‐skimming. Mock samples consisted of six species of bee pollen at given ratios: R. chinensis: B. napus: Pa. rhoeas: V. faba: C. japoica: Pr. armeniaca = 5:300:125:25:5:1 (panel A, four replicates) and 1:1:1:1:1:1 (panel B, six replicates). Solid lines and ‘–P1’ represent results from pollen counts; dashed lines and ‘–R1’ represent replicates of genome‐skimming sequencing
3.6. Species richness and abundance of pollen mocks
Genome‐skimming successfully detected all pollen species in all mock samples (Table 2; Appendix S1: Table S3), including species found at just 0.2% of the total abundance (5,000 pollen grains for C. japoica in M0010 and for Pr. armeniaca in M0006‐0009), 0.7% (3,500 pollen grains for N. stellata in M0002) and 0.9% (4,500 grains pollen for L. brownii and for S. truncates in M0004). In principle, our stringent unique‐mapping criteria would produce conservative results, where the reference species uniquely mapped by sequence reads would unlikely be an analytical artefact. This method was deliberately chosen to alleviate issues associated with the high sensitivity of high‐throughput sequencing technologies, where they tended to pick up minute traces of DNA from the environment, causing false positives. Following this method, low‐frequency species identified by sequencing are likely present in the real sample, providing confidence in detecting rare species in sample mixtures. In fact, all species with low abundances (e.g. with a relative abundance of 0.2% by pollen counting) were readily detected (Table 2; Appendix S1: Table S3). However, two species absent from pollen counting were also detected by sequencing (An. majus and N. stellata in M0004). These two species were not pooled in the mock sample but showed non‐negligible read depths and coverages (2.1X, 43.9% for An. majus and 5.1X, 23.2% for N. stellata, respectively), which were comparable to rare taxa truly present in M0002 (2.0X, 46.0% and 1.9X, 34.9% for the corresponding species, respectively; Appendix S1: Table S3). These results suggested that the two species detected by sequencing were likely a result of sample contamination rather than being analytical artefacts. However, the exact source of these unexpected taxa cannot be identified in our experimental setups. Therefore, the results from the genome‐skimming pipeline should be treated with caution, because pollen detected at low abundance (e.g. ~<1%) could also reflect contamination. Alternatively, these false positives may be caused by tag jumps during pooling of different indexed libraries before sequencing, which can lead to false assignment of sequences to samples and artificially inflate diversity (Schnell, Bohmann, & Gilbert, 2015).
Quantification results for nearly all FP and BP mock samples were highly congruent with those from pollen counting (Table 2, Figures 4, 5). The radar shapes representing species compositions inferred from sequencing (Figure 3, dashed lines) matched well with those from pollen counting (Figure 3, solid lines). The only exception was M0002, where the relative abundance (pollen ratio in mixture) of L. brownii was lower in the sequencing result than pollen counting, while that for Abutilon spp. was overestimated by the sequencing approach. This result was consistent in our repeat, which replicated the procedure since pollen pooling (Appendix S1: Table S3, Appendix S2: Fig. S2).
Figure 4.
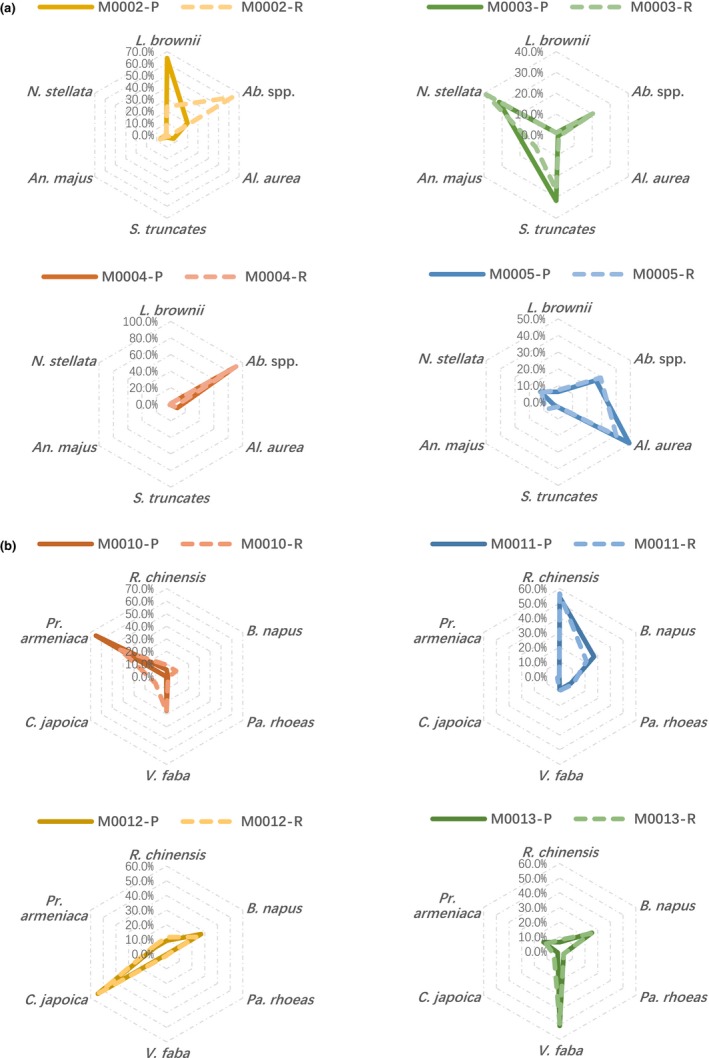
Coherence in pollen proportions between counting and sequencing. Solid lines represent results from pollen counting, and dashed lines represent replicates of genome‐skimming sequencing
Figure 5.
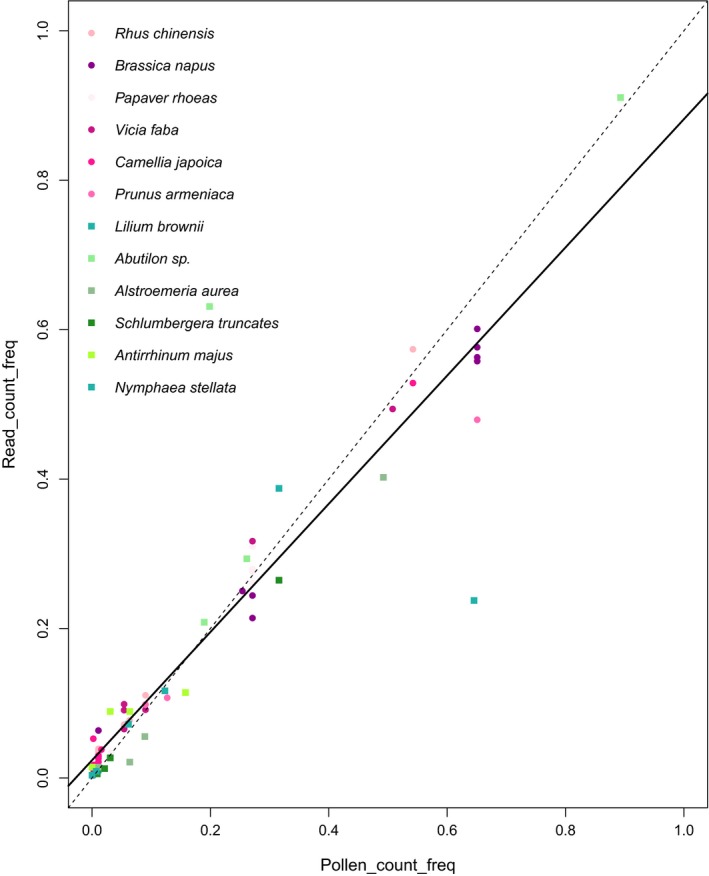
Scatter plots of pollen species frequencies from pollen counts versus genome‐skimming. The solid line is the linear regression (read_count_freq ~ 0.02367 + 0.85800*pollen_count_freq) and the dashed line is the 1:1 line, representing complete match between results from pollen counts and sequence reads
Overall, pollen frequencies computed from pollen reads of each species were significantly correlated with corresponding pollen count proportions (linear model, R 2 = 86.7%, p = 2.2e−16, Figure 5). The genome‐skimming approach demonstrated a high level of sensitivity and accuracy in discriminating pollen counts at a wide range of compositional differences. In 68 out of 70 cases (97.1%), the sequencing results were able to quantify pollen proportions to the correct order of magnitudes. Even when the maximum differences among pollen species had reached more than 100‐fold within mock samples (e.g. M0004, 0006–0012), genome‐skimming was still able to correctly quantify species at low abundances to the correct order of magnitudes with high consistency (e.g. R. chinensis vs. C. japoica in M0006‐0009, Pa. rhoeas vs. V. faba in M0012).
3.7. Pollen mixture in bee pollen
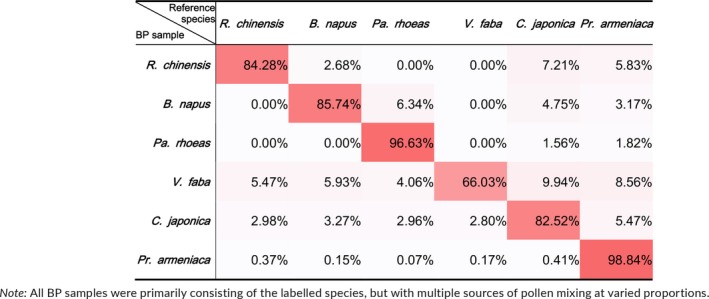
The majority of each BP data set was mapped back onto the corresponding reference genomes, indicating that the bee pollen pellets were mostly made up of the labelled pollen species. However, it was also clear that all BP samples were mixed with other pollen species at varied proportions (Table 3). BP V. faba contained the most nonlabelled pollen DNA (33.97%), while Pr. armeniaca showed the least mixture (1.16%). These results suggest that honeybees may regularly visit multiple flower species in a single trip, although we could not rule out the possibility of sample mixing during bee pollen preparation by the merchant.
Table 3.
Pollen mixing frequencies in bee pollen suggested by sequencing data
4. DISCUSSION
Diversified flower resources play a key role in maintaining bee health. For example, higher level of food varieties can mitigate local decline in bumblebees by providing elevated reproductive benefits when compared to mono‐floral diets (Tasei & Aupinel, 2008). Generalist bees also tend to access a variety of host plant species to avoid deleterious effects of the secondary metabolites from certain pollen (Eckhardt, Haider, Dorn, & Müller, 2014). Therefore, an effective approach to identifying pollen diversity carried or consumed by flower visitors would help to incorporate this important information into the understanding of biological and ecological adaptations in pollinators in terms of pollen resource partition and spatiotemporal dynamics.
Although some studies have suggested that metabarcoding is able to estimate valid abundances for pollen mixtures using amplicon frequencies (Pornon et al., 2016), others have shown less reliable correlations (Keller et al., 2015; Lamb et al., 2018; Richardson et al., 2015). These conflicting observations may imply that the success of amplicon‐based metabarcoding is dependent on species composition of the pollen sample, which is highly variable in natural systems. By bypassing target gene amplifications and by expanding sequence references, the genome‐skimming method can further provide quantitative compositions for pollen loads (corbicula pollen pellets) and diets for individual bees. In our results, a consistent positive correlation between pollen counts and pollen read numbers was established, in congruence with previous studies on macro‐invertebrates. In fact, the correlation of the linear model is much more significant in pollen than in invertebrate animals tested so far (Bista et al., 2018; Tang et al., 2015), which is likely due to a higher level of homogeneity in organelle genome copy numbers per sample unit (pollen grain vs. individual animal). As with previously studied animal samples, this individual‐read abundance correlation was significant independent of phylogenetic relationships among member pollen species (valid in both FP and BP samples) and levels of heterogeneity in pollen proportions (from onefold to 300‐fold). The only exception in our study was observed in M0002, where the proportions of L. brownii and Abutilon spp. were seemingly flipped in the sequencing results. This result was repeated in our second trial, for which we created a new mock community from scratch (Appendix S1: Table S3, Appendix S2: Fig. S2) excluding the likelihood of errors in sample contamination or mislabelling. We speculate that the structural nature of the Lilium pollen (more hydrophobic compared to other pollen) may have caused its reduced abundance in final pollen mixtures, in which case the sequencing results would be more reliable. In fact, Lilium pollen floated in the supernatants, which might have led to its low representation in the mock subsamples.
Our pipeline also expanded reference gene markers from standard DNA barcodes (e.g. matK, rbcL and trnL for plants) to dozens of PCGs selected from whole plastid genomes. This extended sequence reference can produce better taxonomic resolution by recruiting additional variable genes, although some closely related species may still remain indistinguishable, as demonstrated by Ab. megapotamicum and Ab. pictum in our study. As with classic DNA barcoding approaches, incomplete reference databases are often a key factor in causing false negatives. Fortunately, HTS‐based methods have promised feasible paths in producing both standard DNA barcodes (Hebert et al., 2018; Liu, Yang, Zhou, & Zhou, 2017; Srivathsan et al., 2018) and organelle genomes (Straub et al., 2011; Tang et al., 2014) at significantly reduced costs. Indeed, large sequencing efforts for plastid genomes have seen significant progress in China. For example, plastid genomes of 1,659 plants have been sequenced by the Kunming Institute of Botany, China (Li et al., 2019). And an ambitious plan is in place, with a goal to complete the sequencing of plastid genomes for 18,000 Chinese seed plant species by 2021, covering c. 2,750 genera (D. Dezhu Li, personal communication).
While gaining benefits in producing quantitative results, the genome‐skimming approach shows some compromise, where it requires higher DNA quantity for library construction and HTS sequencing (Zhou et al., 2013). Considering the low unit weight of pollen grains, DNA quantity may present a major challenge to a direct shotgun sequencing method. Current Illumina‐based sequencing protocols require 200 ng or less of genomic DNA for library construction, which is roughly the amount of DNA extracted from c. 60,000 mixed pollen grains of tested species (Figure 2). Regular pollen pellets collected from a single corbicula of the honeybees (Apis mellifera) were estimated to each contain more than 100,000 pollen grains (Appendix S1: Table S1), which therefore provide sufficient DNA for genome‐skimming. It is worth noting that although the proposed method has a minimum requirement for the total DNA quantity, pollen species represented by low DNA proportions can still be readily detected from the mixture. For instance, our pipeline was highly sensitive in that all pollen species in the mocks were detected, including those with very low abundances (e.g. 0.2% for C. japoica in M0010 or c. 100 pollen grains in a pollen mixture with c. 47,000 total grains; Appendix S1: Table S4). This observation would imply that very small amounts of pollen grains, such as those carried by smaller corbiculate bees and flies, can still be quantified using the genome‐skimming approach, if mixed with known pollen supplements.
Analytical cost is obviously variable depending on service carriers and technological advances. At the time we conducted our study, the sequencing of de novo plastid genomes and pollen mixtures costed c. 100 USD per sample (including DNA extraction, library construction and sequencing at c. 2–4 Gb data), with the major cost being DNA library construction. Costs on both sequencing and library construction have seen significant reduction in the past decade, although the latter is at a much slower pace (Feng, Costa, & Edwards, 2018). Given current chemistry cost and the fact that most of the laboratory pipelines have been standardized and can be accomplished at regular molecular laboratories, we expect future cost on pollen genome‐skimming can be further brought down to just a fraction of what we have now. Furthermore, global efforts on chloroplast genome sequencing coupled with more focused scrutiny of local flora will help to establish comprehend reference databases for molecular pollen identifications via metabarcoding or direct shotgun sequencing‐based genome‐skimming.
Finally, in addition to investigating food range and niche division in bees, the proposed pollen genome‐skimming method can be useful in diet analyses of other pollen consumers (hoverflies, bee flies, beetles, etc.), through sequencing of the gut contents. We also envision future applications in an extended range, such as in fraud control of honey products (Louveaux, Maurizio, & Vorwohl, 1978), forensic tracing for geographic origin (Bryant & Jones, 2006; Mathewes, 2006) and monitoring of airborne pollen allergens (Scheifinger et al., 2013). In these studies, DNA degradation may present a major challenge for a PCR‐based method, but less so for a direct shotgun sequencing approach, which is based on fragmented DNA libraries. Current constraints in the requirement on DNA quantity can already be solved by adding supplement pollen or DNA using the genome‐skimming approach described here. In the meantime, the ever‐developing technology in high‐throughput sequencing is expected to continuously reduce the minimum needs for input DNA. Admittedly, the current pipeline may potentially overestimate proportions for some rare taxa, especially when sequencing depth is low. We would call for cautions in interpreting results for rare taxa, especially if that is the main focus of the study, for example detections for invasive or toxic pollen species.
AUTHOR CONTRIBUTIONS
X.Z., D.D.L., M.T. and J.H.H. designed the study. D.D.L. and J.H.H. conducted the benchwork. D.D.L. and M.T. performed data analysis. All authors participated in writing and proofed the manuscript.
Supporting information
ACKNOWLEDGEMENTS
We thank Dr. Lei Gu of Capital Normal University and Dr. Sodmergen of Peking University for their advice in plant taxonomy and knowledge on the biology and molecular mechanism of plant organelles. XZ acknowledge Dr. Douglass W. Yu of University of East Anglia, UK, and Kunming Institute of Zoology, Chinese Academy of Sciences, for his helpful comments that improved the paper. XZ is supported by the National Natural Science Foundation of China (31772493), the Program of Ministry of Science and Technology of China (2018FY100403), and funding from the Beijing Advanced Innovation Center for Food Nutrition and Human Health through China Agricultural University.
Lang D, Tang M, Hu J, Zhou X. Genome‐skimming provides accurate quantification for pollen mixtures. Mol Ecol Resour. 2019;19:1433–1446. 10.1111/1755-0998.13061
Dandan Lang and Min Tang share the Co‐first authors.
DATA AVAILABILITY STATEMENT
The genomic data sets (6 FP, 6 BP and 20 pollen mixture samples) have been deposited in the NCBI Short Read Archive (PRJNA481636) and the CNSA (https://db.cngb.org/cnsa/) of CNGBdb with accession code CNP0000488.
REFERENCES
- Alexander, M. P. (1969). Differential staining of aborted and nonaborted pollen. Stain Technology, 44(3), 117–122. 10.3109/10520296909063335 [DOI] [PubMed] [Google Scholar]
- Arribas, P. , Andújar, C. , Hopkins, K. , Shepherd, M. , & Vogler, A. P. (2016). Metabarcoding and mitochondrial metagenomics of endogean arthropods to unveil the mesofauna of the soil. Methods in Ecology and Evolution, 7(9), 1071–1081. 10.1111/2041-210X.12557 [DOI] [Google Scholar]
- Barbir, J. , Badenes‐Pérez, F. R. , Fernández‐Quintanilla, C. , & Dorado, J. (2015). The attractiveness of flowering herbaceous plants to bees (Hymenoptera: Apoidea) and hoverflies (Diptera: Syrphidae) in agro‐ecosystems of Central Spain. Agricultural and Forest Entomology, 17(1), 20–28. 10.1111/afe.12076 [DOI] [Google Scholar]
- Bell, K. L. , De, V. N. , Keller, A. , Richardson, R. T. , Gous, A. , Burgess, K. S. , & Brosi, B. J. (2016). Pollen DNA barcoding: Current applications and future prospects. Genome, 59(9), 629–640. 10.1139/gen-2015-0200 [DOI] [PubMed] [Google Scholar]
- Bell, K. L. , Fowler, J. , Burgess, K. S. , Dobbs, E. K. , Gruenewald, D. , Lawley, B. , … Brosi, B. J. (2017). Applying pollen DNA metabarcoding to the study of plant–pollinator interactions1. Applications in Plant Sciences, 5(6), 1600124 10.3732/apps.1600124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bista, I. , Carvalho, G. R. , Tang, M. , Walsh, K. , Zhou, X. , Hajibabaei, M. , … Creer, S. (2018). Performance of amplicon and shotgun sequencing for accurate biomass estimation in invertebrate community samples. Molecular Ecology Resources, 10.1111/1755-0998.12888 [DOI] [PubMed] [Google Scholar]
- Bosch, J. (1992). Floral biology and pollinators of three co‐occurring Cistus species (Cistaceae). Botanical Journal of the Linnean Society, 109, 39–55. [Google Scholar]
- Brodschneider, R. , & Crailsheim, K. (2010). Nutrition and health in honey bees. Apidologie, 41(3), 278–294. 10.1051/apido/2010012 [DOI] [Google Scholar]
- Bryant, V. M. , & Jones, G. D. (2006). Forensic palynology: Current status of a rarely used technique in the United States of America. Forensic Science International, 163(3), 183–197. 10.1016/j.forsciint.2005.11.021 [DOI] [PubMed] [Google Scholar]
- Burkle, L. A. , Marlin, J. C. , & Knight, T. M. (2013). Plant‐pollinator interactions over 120 years: Loss of species, co‐occurrence, and function. Science, 339(6127), 1611–1615. [DOI] [PubMed] [Google Scholar]
- Camillo, E. , & Garofalo, C. A. (1989). Analysis of the niche of two sympatric species of bombus (Hymenoptera, Apidae) in southeastern Brazil. Journal of Tropical Ecology, 5(1), 81–92. [Google Scholar]
- Chen, Y. , Chen, Y. , Shi, C. , Huang, Z. , Zhang, Y. , Li, S. , … Chen, Q. (2018). SOAPnuke: A MapReduce acceleration supported software for integrated quality control and preprocessing of high‐throughput sequencing data. GigaScience, 7(1), 1–6. 10.1093/gigascience/gix120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo, L. Q. , Crampton‐Platt, A. , & Vogler, A. P. (2017). Shotgun mitogenomics across body size classes in a local assemblage of tropical Diptera: Phylogeny, species diversity and mitochondrial abundance spectrum. Molecular Ecology, 26(19), 5086–5098. 10.1111/mec.14258 [DOI] [PubMed] [Google Scholar]
- Connop, S. , Hill, T. , Steer, J. , & Shaw, P. (2010). The role of dietary breadth in national bumblebee (Bombus) declines: Simple correlation? Biological Conservation, 143(11), 2739–2746. 10.1016/j.biocon.2010.07.021 [DOI] [Google Scholar]
- Cornman, R. S. , Otto, C. R. , Iwanowicz, D. , & Pettis, J. S. (2015). Taxonomic characterization of honey bee (Apis mellifera) pollen foraging based on non‐overlapping paired‐end sequencing of nuclear ribosomal loci. PLoS ONE, 10(12), e145365 10.1371/journal.pone.0145365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crampton‐Platt, A. , Yu, D. W. , Zhou, X. , & Vogler, A. P. (2016). Mitochondrial metagenomics: Letting the genes out of the bottle. GigaScience, 5(1), 15 10.1186/s13742-016-0120-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristescu, M. E. (2014). From barcoding single individuals to metabarcoding biological communities: Towards an integrative approach to the study of global biodiversity. Trends in Ecology and Evolution, 29(10), 566–571. 10.1016/j.tree.2014.08.001 [DOI] [PubMed] [Google Scholar]
- Danner, N. , Molitor, A. M. , Schiele, S. , Härtel, S. , & Steffan‐Dewenter, I. (2016). Season and landscape composition affect pollen foraging distances and habitat use of honey bees. Ecological Applications, 26(6), 1920–1929. 10.1890/15-1840.1 [DOI] [PubMed] [Google Scholar]
- Di Pasquale, G. , Salignon, M. , Le Conte, Y. , Belzunces, L. P. , Decourtye, A. , Kretzschmar, A. , … Alaux, C. (2013). Influence of pollen nutrition on honey bee health: Do pollen quality and diversity matter? PLoS ONE, 8(8), 1–13. 10.1371/journal.pone.0072016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckhardt, M. , Haider, M. , Dorn, S. , & Müller, A. (2014). Pollen mixing in pollen generalist solitary bees: A possible strategy to complement or mitigate unfavourable pollen properties? Journal of Animal Ecology, 83(3), 588–597. 10.1111/1365-2656.12168 [DOI] [PubMed] [Google Scholar]
- Fay, M. F. , Bayer, C. , Alverson, W. S. , De Bruijn, A. Y. , & Chase, M. W. (1998). Plastid rbcL sequence data indicate a close affinity between Diegodendron and Bixa . Taxon, 47(1), 43–50. [Google Scholar]
- Feng, K. , Costa, J. , & Edwards, J. S. (2018). Next‐generation sequencing library construction on a surface. BMC Genomics, 19, 416 10.1186/s12864-018-4797-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galimberti, A. , De, F. M. , Bruni, I. , Scaccabarozzi, D. , Sandionigi, A. , Barbuto, M. , … Labra, M. (2014). A DNA barcoding approach to characterize pollen collected by honeybees. PLoS ONE, 9(10), e109363 10.1371/journal.pone.0109363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulson, D. , & Darvill, B. (2004). Niche overlap and diet breadth in bumblebees: Are rare species more specialized in their choice of flowers? Apidologie, 35(1), 55–63. [Google Scholar]
- Goulson, D. , & Sparrow, K. R. (2009). Evidence for competition between honeybees and bumblebees: Effects on bumblebee worker size. Journal of Insect Conservation, 13(2), 177–181. 10.1007/s10841-008-9140-y [DOI] [Google Scholar]
- Hanley, M. E. , Franco, M. , Pichon, S. , Darvill, B. , & Goulson, D. (2008). Breeding system, pollinator choice and variation in pollen quality in British herbaceous plants. Functional Ecology, 22(4), 592–598. 10.1111/j.1365-2435.2008.01415.x [DOI] [Google Scholar]
- Hebert, P. D. N. , Braukmann, T. W. A. , Prosser, S. W. J. , Ratnasingham, S. , deWaard, J. R. , Ivanova, N. V. , … Zakharov, E. V. (2018). A sequel to sanger: Amplicon sequencing that scales. BMC Genomics, 19(1), 219 10.1186/s12864-018-4611-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, Y. , Ashton, L. , Pedley, S. M. , Edwards, D. P. , Tang, Y. , Nakamura, A. , … Yu, D. W. (2013). Reliable, verifiable and efficient monitoring of biodiversity via metabarcoding. Ecology Letters, 16(10), 1245–1257. 10.1111/ele.12162 [DOI] [PubMed] [Google Scholar]
- Kamo, T. , Kusumoto, Y. , Tokuoka, Y. , Okubo, S. , Hayakawa, H. , Yoshiyama, M. , … Konuma, A. (2018). A DNA barcoding method for identifying and quantifying the composition of pollen species collected by European honeybees, Apis mellifera (Hymenoptera: Apidae). Applied Entomology and Zoology, 1–9. 10.1007/s13355-018-0565-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller, A. , Danner, N. , Grimmer, G. , Ankenbrand, M. , von der Ohe, K. , von der Ohe, W. , … Steffan‐Dewenter, I. (2015). Evaluating multiplexed next‐generation sequencing as a method in palynology for mixed pollen samples. Plant Biology, 17(2), 558–566. 10.1111/plb.12251 [DOI] [PubMed] [Google Scholar]
- Kremen, C. (2005). Managing ecosystem services: What do we need to know about their ecology? Ecology Letters, 8(5), 468–479. 10.1111/j.1461-0248.2005.00751.x [DOI] [PubMed] [Google Scholar]
- Lamb, P. D. , Hunter, E. , Pinnegar, J. K. , Creer, S. , Davies, R. G. , & Taylor, M. I. (2018). How quantitative is metabarcoding: A meta‐analytical approach. Molecular Ecology, 28, 420–430. 10.1111/mec.14920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2010). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 26, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Yi, T. , Gao, L. , Ma, P. , Zhang, T. , Yang, J. , … Li, D. (2019). Origin of angiosperms and the puzzle of the Jurassic gap. Nature Plants, 5(5), 461–470. 10.1038/s41477-019-0421-0 [DOI] [PubMed] [Google Scholar]
- Li, J. , Wang, S. , Jing, Y. , & Ling, W. (2013). A modified CTAB protocol for plant DNA extraction. Chinese Bulletin of Botany, 48(1), 72–78. [Google Scholar]
- Liu, S. , Yang, C. , Zhou, C. , & Zhou, X. (2017). Filling reference gaps via assembling DNA barcodes using high‐throughput sequencing—moving toward barcoding the world. GigaScience, 6(12), 1–8. 10.1093/gigascience/gix104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louveaux, J. , Maurizio, A. , & Vorwohl, G. (1978). Methods of melissopalynology. Bee World, 59(4), 139–157. 10.1080/0005772X.1978.11097714 [DOI] [Google Scholar]
- Mathewes, R. W. (2006). Forensic palynology in Canada: An overview with emphasis on archaeology and anthropology. Forensic Science International, 163(3), 198–203. 10.1016/j.forsciint.2006.06.069 [DOI] [PubMed] [Google Scholar]
- Naug, D. (2009). Nutritional stress due to habitat loss may explain recent honeybee colony collapses. Biological Conservation, 142(10), 2369–2372. 10.1016/j.biocon.2009.04.007 [DOI] [Google Scholar]
- Nichols, R. V. , Vollmers, C. , Newsom, L. A. , Wang, Y. , Heintzman, P. D. , Leighton, M. K. , … Shapiro, B. (2018). Minimizing polymerase biases in metabarcoding. Molecular Ecology Resources, 18(1), 927–939. 10.1111/1755-0998.12895 [DOI] [PubMed] [Google Scholar]
- Nicolas, D. , Patrick, M. , & Guillaume, S. (2017). NOVOPlasty: D e novo assembly of organelle genomes from whole genome data. Nucleic Acids Research, 45(4), e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollerton, J. , Winfree, R. , & Tarrant, S. (2011). How many flowering plants are pollinated by animals? Oikos, 120(3), 321–326. 10.1111/j.1600-0706.2010.18644.x [DOI] [Google Scholar]
- Paini, D. R. (2004). Impact of the introduced honey bee (Apis mellifera) (Hymenoptera: Apidae) on native bees: A review. Austral Ecology, 399–407. 10.1111/j.1442-9993.2004.01376.x [DOI] [Google Scholar]
- Paini, D. R. , & Roberts, J. D. (2005). Commercial honey bees (Apis mellifera) reduce the fecundity of an Australian native bee (Hylaeus alcyoneus). Biological Conservation, 123(1), 103–112. 10.1016/j.biocon.2004.11.001 [DOI] [Google Scholar]
- Piñol, J. , Senar, M. A. , & Symondson, W. O. (2018). The choice of universal primers and the characteristics of the species mixture determines when DNA metabarcoding can be quantitative. Molecular Ecology, 6, 1809–1813. [DOI] [PubMed] [Google Scholar]
- Pornon, A. , Escaravage, N. , Burrus, M. , Holota, H. , Khimoun, A. , Mariette, J. , … Andalo, C. (2016). Using metabarcoding to reveal and quantify plant‐pollinator interactions. Scientific Reports, 6, 1–12. 10.1038/srep27282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts, S. G. , Biesmeijer, J. C. , Kremen, C. , Neumann, P. , Schweiger, O. , & Kunin, W. E. (2010). Global pollinator declines: Trends, impacts and drivers. Trends in Ecology and Evolution, 25(6), 345–353. 10.1016/j.tree.2010.01.007 [DOI] [PubMed] [Google Scholar]
- R Core Team . (2015). R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; http://www.R-project.org/ [Google Scholar]
- Ranta, E. , & Lundberg, H. (1981). Food niche analyses of bumblebees: A comparison of three data collecting methods. Oikos, 36(1), 12–16. 10.2307/3544372 [DOI] [Google Scholar]
- Richardson, R. T. , Lin, C. H. , Sponsler, D. B. , Quijia, J. O. , Goodell, K. , & Johnson, R. M. (2015). Application of ITS2 metabarcoding to determine the provenance of pollen collected by honey bees in an agroecosystem. Applications in Plant Sciences, 3(1), 235–250. 10.3732/apps.1400066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roubik, D. W. , & Wolda, H. (2001). Do competing honey bees matter? Dynamics and abundance of native bees before and after honey bee invasion. Population Ecology, 43(1), 53–62. 10.1007/PL00012016 [DOI] [Google Scholar]
- Scheifinger, H. , Belmonte, J. , Buters, J. , Celenk, S. , Damialis, A. , Dechamp, C. , … Koch, E. (2013). Monitoring, modelling and forecasting of the pollen season. Allergenic Pollen (pp. 71–126). Dordrecht, The Netherlands: Springer. [Google Scholar]
- Schmehl, D. R. , Teal, P. E. A. , Frazier, J. L. , & Grozinger, C. M. (2014). Genomic analysis of the interaction between pesticide exposure and nutrition in honey bees (Apis mellifera). Journal of Insect Physiology, 71, 177–190. 10.1016/j.jinsphys.2014.10.002 [DOI] [PubMed] [Google Scholar]
- Schnell, I. B. , Bohmann, K. , & Gilbert, M. T. P. (2015). Tag jumps illuminated – reducing sequence‐to‐sample misidentifications in metabarcoding studies. Molecular Ecology Resources, 15(6), 1289–1303. 10.1111/1755-0998.12402 [DOI] [PubMed] [Google Scholar]
- Soares, S. , Amaral, J. S. , Oliveira, M. B. P. P. , & Mafra, I. (2015). Improving DNA isolation from honey for the botanical origin identification. Food Control, 48, 130–136. 10.1016/j.foodcont.2014.02.035 [DOI] [Google Scholar]
- Somerville, D. C. , & Nicol, H. I. (2006). Crude protein and amino acid composition of honey bee‐collected pollen pellets from south‐east Australia and a note on laboratory disparity. Australian Journal of Experimental Agriculture, 46(1), 141–149. 10.1071/EA03188 [DOI] [Google Scholar]
- Srivathsan, A. , Baloğlu, B. , Wang, W. , Tan, W. X. , Bertrand, D. , Ng, A. H. Q. , … Meier, R. (2018). A MinION‐based pipeline for fast and cost‐effective DNA barcoding. Molecular Ecology Resources, 1–36. 10.1111/1755-0998.12890 [DOI] [PubMed] [Google Scholar]
- Standifer, L. N. (1964). A comparison of the protein quality of pollens for growth‐stimulation of the hypopharyngeal glands and longevity of honey bees, Apis mellifera L. (Hymenopera: Apidae). Insectes Sociaux, 14(4), 415–426. [Google Scholar]
- Steffan‐Dewenter, I. , & Tscharntke, T. (2000). Resource overlap and possible competition between honey bees and wild bees in central Europe. Oecologia, 122(2), 288–296. 10.1007/s004420050034 [DOI] [PubMed] [Google Scholar]
- Stout, J. C. , & Morales, C. L. (2009). Ecological impacts of invasive alien species on bees. Apidologie, 40(3), 388–409. 10.1051/apido/2009023 [DOI] [Google Scholar]
- Straub, S. C. , Parks, M. , Weitemier, K. , Fishbein, M. , Cronn, R. C. , & Liston, A. (2011). Navigating the tip of the genomic iceberg: Next–generation sequencing for plant systematics. American Journal of Botany, 99, 349–364. 10.3732/ajb.1100335 [DOI] [PubMed] [Google Scholar]
- Sudhir, K. , Glen, S. , & Koichiro, T. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular Biology and Evolution, 33(7), 1870 10.1093/molbev/msw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, M. , Hardman, C. J. , Ji, Y. , Meng, G. , Liu, S. , Tan, M. , … Yu, D. W. (2015). High‐throughput monitoring of wild bee diversity and abundance via mitogenomics. Methods in Ecology and Evolution, 6(9), 1034–1043. 10.1111/2041-210X.12416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, M. , Tan, M. , Meng, G. , Yang, S. , Su, X. U. , Liu, S. , … Zhou, X. (2014). Multiplex sequencing of pooled mitochondrial genomes‐a crucial step toward biodiversity analysis using mito‐metagenomics. Nucleic Acids Research, 42(22), e166 10.1093/nar/gku917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasei, J. , & Aupinel, P. (2008). Nutritive value of 15 single pollens and pollen mixes tested on larvae produced by bumblebee workers (Bombus terrestris, Hymenoptera: Apidae). Apidologie, 39(4), 397–409. [Google Scholar]
- Tylianakis, J. M. , Didham, R. K. , Bascompte, J. , & Wardle, D. A. (2008). Global change and species interactions in terrestrial ecosystems. Ecology Letters, 11(12), 1351–1363. 10.1111/j.1461-0248.2008.01250.x [DOI] [PubMed] [Google Scholar]
- Vaudo, A. D. , Tooker, J. F. , Grozinger, C. M. , & Patch, H. M. (2015). Bee nutrition and floral resource restoration. Current Opinion in Insect Science, 10, 133–141. 10.1016/j.cois.2015.05.008 [DOI] [PubMed] [Google Scholar]
- Winfree, R. , Griswold, T. , & Kremen, C. (2007). Effect of human disturbance on bee communities in a forested ecosystem. Conservation Biology, 21(1), 213–223. 10.1111/j.1523-1739.2006.00574.x [DOI] [PubMed] [Google Scholar]
- Woodcock, B. A. , Edwards, M. , Redhead, J. , Meek, W. R. , Nuttall, P. , Falk, S. , … Pywell, R. F. (2013). Crop flower visitation by honeybees, bumblebees and solitary bees : Behavioural differences and diversity responses to landscape. Agriculture, Ecosystems and Environment, 171, 1–8. 10.1016/j.agee.2013.03.005 [DOI] [Google Scholar]
- Xie, Y. , Wu, G. , Tang, J. , Luo, R. , Patterson, J. , Liu, S. , … Wang, J. (2014). SOAPdenovo‐Trans: D e novo transcriptome assembly with short RNA‐Seq reads. Bioinformatics, 30(12), 1660–1666. 10.1093/bioinformatics/btu077 [DOI] [PubMed] [Google Scholar]
- Zhou, X. , Li, Y. , Liu, S. , Yang, Q. , Su, X. U. , Zhou, L. , … Huang, Q. (2013). Ultra‐deep sequencing enables high‐fidelity recovery of biodiversity for bulk arthropod samples without PCR amplification. GigaScience, 2(1), 4 10.1186/2047-217X-2-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The genomic data sets (6 FP, 6 BP and 20 pollen mixture samples) have been deposited in the NCBI Short Read Archive (PRJNA481636) and the CNSA (https://db.cngb.org/cnsa/) of CNGBdb with accession code CNP0000488.