

Abstract
Background
The current scenario of newborn screening is changing as DNA studies are being included in the programs of several countries. Severe combined immunodeficiency (SCID) disorders can be detected using quantitative PCR assays to measure T‐cell receptor excision circles (TRECs), a byproduct of correct T‐cell development. However, in addition to SCID, other T‐cell‐deficient phenotypes such as 22q11.2 deletion syndrome 22q11.2 duplication syndrome, CHARGE syndrome, and trisomy 21 are detected.
Methods
We present our experience with the detection of 22q11.2 deletion syndrome and 22q11.2 duplication syndrome in a series of 103,903 newborns included in the newborn screening program of Catalonia (Spain).
Results
Thirty newborns tested were positive (low TREC levels) and five were found to have copy number variations at the 22q11 region (4 deletions and 1 duplication) when investigated with array comparative genomic hybridization technology and MLPA.
Conclusion
Newborn screening for SCID enables detection of several conditions, such as 22q syndromes, which should be managed by prompt, proactive approaches with adequate counseling for families by a multidisciplinary team.
Keywords: 22q11.2 deletion, newborn screening, severe combined immunodeficiencye
Newborn screening (NBS) using T‐cell receptor excision circle (TREC) assays to detect severe combined immunodeficiency (SCID) has been established in several geographic areas. In addition to SCID, other T‐cell deficient conditions, such as 22q11.2 deletion syndrome (DS), CHARGE syndrome, and trisomy 21, can also be detected through NBS. An early diagnosis of 22q11.2 DS is associated with a better clinical outcome, mainly because of the possibility of early protocolled care, including rehabilitation and adapted school programs, an appropriate vaccination schedule, and adequate genetic counseling.

1. INTRODUCTION
Early detection and confirmation of several genetic disorders is of help to implement proactive measures, to improve the standard of care, to perform adequate genetic counseling and, in some diseases, to initiate therapy. Newborn screening (NBS) is the paradigm for presymptomatic detection of conditions that could have devastating consequences without an early intervention and treatment. NBS, initially implemented in the United States in the 1960s (Guthrie, 1996), has been carried out in most developed countries for decades, originally for the detection and early treatment of phenylketonuria and now for other endocrine and metabolic diseases, mainly using tandem mass spectrometry. DNA testing has been recently included in the NBS programs of some countries. Since its initial implementation in Wisconsin in 2008, NBS using T‐cell receptor excision circle (TREC) assays to detect severe combined immunodeficiency (SCID) has been established in several geographic areas (eg, most American states (Kwan et al., 2014), Taiwan (Chien et al., 2017), Israel (Rechavi, Lev, Simon, Stauber, & Daas, 2017), Qatar, several Canadian regions (King & Hammarström, 2018), and the Catalonian region in Spain), and has enabled early diagnosis and treatment, with better outcomes in SCID patients. Since 2017, SCID is one of the 24 diseases included in the universal NBS program of the Catalonian Department of Health.
In addition to SCID, other T‐cell deficient conditions, such as 22q11.2 deletion syndrome (DS) (Barry et al., 2017; Lingman Framme, Borte, Döbeln, Hammarström, & Oskarsdóttir, 2014), 22q11.2 duplication syndrome, CHARGE syndrome, and trisomy 21, can also be detected through NBS (Jyonouchi, Jongco, Puck, & Sullivan, 2017). 22q11.2 DS is the most common and well‐described microdeletion syndrome, with a prevalence of around 1 in 2000–4000 live births (Cancrini et al., 2014; Palmer et al., 2018; Swillen & McDonald‐McGinn, 2015). The prevalence of 22q11.2 microdeletion and microduplication in the low‐risk population is around 1/992 and 1/850, respectively (Grati, Molina Gomes, Ferreira, Dupont, & Alesi, 2015). 22q11.2 DS shows considerable inter‐ and intrafamilial clinical variability. Before the era of molecular diagnoses, this condition was named according to the main clinical manifestations as velocardiofacial syndrome (OMIM 192430), conotruncal anomaly face syndrome (OMIM 217095), or DiGeorge syndrome (OMIM 188400), honoring Angelo DiGeorge, who first described this syndrome in 1965 (DiGeorge, 1965). Because of this high clinical variability, delays in the diagnosis are common, particularly in patients who do not manifest the characteristic clinical symptoms (congenital heart disease, particularly conotruncal anomalies, and palatal abnormalities) and those with a wide spectrum of manifestations that involve different medical specialties (Bassett, McDonald‐McGinn, Devriendt, Digilio, & Goldenberg, 2011; Cancrini et al., 2014; Palmer et al., 2018; Swillen & McDonald‐McGinn, 2015). An early diagnosis is associated with a better clinical outcome, mainly because of the possibility of early protocolled care, including rehabilitation and adapted school programs, an appropriate vaccination schedule, and adequate genetic counseling.
We present our experience with NBS for SCID through TREC quantification, highlighting early presymptomatic detection of 22q11.2 deletion and duplication syndromes and the implications of proactive diagnostic and therapeutic measures, follow‐up, and counseling.
2. PATIENTS AND METHODS
In January 2017, NBS for SCID detection using the EnLite Neonatal TREC kit (Perkin Elmer, Turku, Finland) was universally implemented in Catalonia, Spain. A complex diagnostic decision algorithm was established, based on previous experience (Audrain, Léger, Hémont, Mirallié, & Cheillan, 2018) (Figure 1). In 2018, the retest cutoff was changed from 34 to 24 copies/µL, which decreased the retest rate from 3.34% to 1.4%. All cases considered positive were referred to the reference unit within 14 days and confirmation examinations were then carried out: flow cytometry of T, B and NK cells, including CD45+RO+/RA+, T CD4+CD8+, and HLA‐DR+, the T‐cell receptor repertoire, and lymphocyte mitogen proliferation. Depending on the results, a genetic panel for primary immunodeficiencies (PID) or consultation with a geneticist for clinical evaluation and array comparative genomic hybridization (CGH) studies were then carried out.
Figure 1.
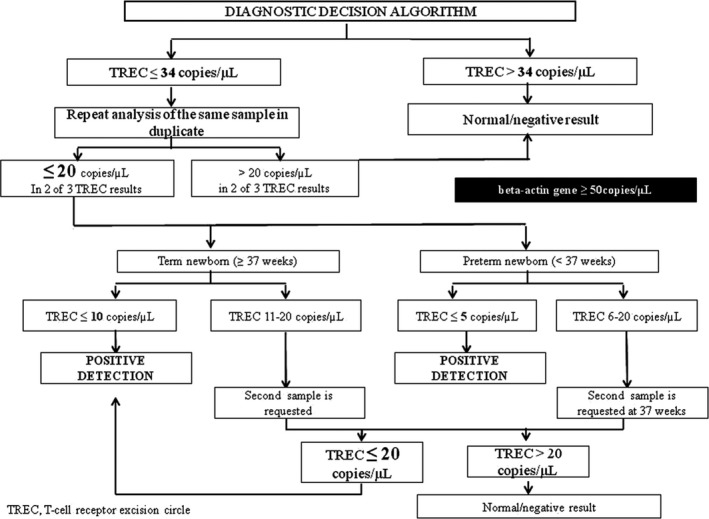
SCID NBS diagnostic decision algorithm
We are currently using a custom‐designed next‐generation sequencing (NGS)‐based panel that targets 323 genes, including most of the known PID‐causing genes according to the 2015 IUIS (International Union of Immunological Societies) classification and other more recently described genes causing PID (Picard, Al‐Herz, Bousfiha, Casanova, & Chatila, 2015). This panel has been successfully used in our laboratory for genetic diagnosis of PID (Dieli‐Crimi, Martínez‐Gallo, Franco‐Jarava, Antolin, & Blasco, 2018). DNA copy‐number variations were investigated with array CGH using the CytoSure Constitutional v3 array 8 × 60K (Oxford Gene Technology) according to the manufacturer's recommendations. Data analysis was performed with the Cytogenomics 2.1 software (Agilent company) with the ADM‐2 algorithm and a minimum of three consecutive probes to detect an anomaly. Array CGH was chosen despite its higher cost because of the relatively small number of expected patients to test and because FISH and QF‐PCR techniques fail to detect some patients with nested deletions or copy number anomalies outside the 22q11.2 region.
3. RESULTS
To date, 130,903 newborns have been screened, and 30 have tested positive. Nine of the 30 were considered false‐positive cases (initial normal lymphocyte count with normalization of TREC values between 3 and 6 months of life), four cases had transient lymphopenia at the beginning with recovery in the following months, and three patients are currently under study. The remaining 14 were true‐positive cases, classified as follows: one SCID (calculated incidence, 1:130,903), two preterm infant, two chylothorax, three idiopathic lymphopenia, one Down syndrome, four 22q11.2 DS, and one 22q11.2 duplication syndrome. These diagnoses are similar to those described in larger NBS SCID programs (Jyonouchi et al., 2017).
Among the 22q11.2 DS and 22q11.2 duplication syndrome patients (Table 1), two had been diagnosed prenatally, whereas the remaining three were diagnosed postnatally using array CGH studies. This technique was used in patients who did not fulfill the SCID criteria, but showed low TREC levels and mild lymphopenia, with no signs or symptoms (no congenital heart abnormalities, characteristic phenotype, or hypocalcemia). No clinical cases of complete classical DiGeorge syndrome were found during the first months of life in our cohort.
Table 1.
Clinical and laboratory data in patients with 22q11.2 deletion and duplication syndrome with abnormal TREC levels detected on NBS for SCID
| Patient | Weeks of gestation | Sex | Prenatal diagnosis | TREC levels, copies/μL | Lymphocyte count, x109/La |
Lymphocyte subsets: T cells/B cells/NK cells, x109/Lb |
Lymphoproliferation assay | Clinical manifestations at birth consistent with 22q11.2 |
Array CGH/MLPA |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 40 | Female | No | 17 | 4.2 | 2.1/19.9/1.1 | Normal | None | arr[GRCh37] 22q11.21(18894820_21457610)x1 22q11.2 (LCR22A‐LCR22D) deletion syndrome |
| 2 | 40 | Female | No | 5 | 5.4 | 1.6/1.1/2.3 | Normal | None | arr[GRCh37] 22q11.21(18894820_20311733)x1 22q11.2 (LCR22A‐LCR22B) deletion syndrome |
| 3 | 34 | Female | Yes | 5 | 2.6 | 1.1/0.6/0.7 | Normal | Congenital heart disease | arr[GRCh37] 22q11.21(18894820_21457610)x1 22q11.2 (LCR22A‐LCR22B) deletion syndrome |
| 4 | 39 | Female | Yes | 17 | 2 | 1.9/1.4/2 | Normal | Congenital heart disease and hypocalcemia due to hypoparathyroidism | rsa 22q11.2 (SALSA MLPA P250 DiGeorge)x1 22q11.2 (LCR22A‐LCR22D) deletion syndrome |
| 5 | 41 | Female | No | 17 | 1.7 | 1.2/0.04/0.35 | Normal | None | arr[GRCh37] 22q11.21(18894820_21457610)x3 22q11.2 (LCR22A‐LCR22D) duplication |
Abbreviation TREC, T‐cell receptor excision circle; NBS, newborn screening; SCID, severe combined immunodeficiency; CGH, comparative genomic hybridization.
Reference value: 3.4–7.6
Reference values: T cells, 1.8–5.9; B cells, 0.6–1.9; NK cells, 0.1–1.3
4. DISCUSSION
We report our experience with NBS for SCID through TREC quantification, which enables detection of presymptomatic or oligosymptomatic cases of 22q11.2 deletion and duplication syndromes in addition to SCID.
Several publications have mentioned the benefits of an early diagnosis in patients with 22q11.2 DS (Barry et al., 2017; Cancrini et al., 2014; Palmer et al., 2018; Swillen & McDonald‐McGinn, 2015), as a prompt intervention could anticipate complications. In addition to the patient's heart and kidney malformations, complications in the neonatal period can include severe hypocalcemia and immune dysfunction. Later, an apparently asymptomatic patient can reach school age with learning difficulties, speech problems, or velopharyngeal insufficiency, and during adolescence and adulthood manifest neurobehavioral or psychiatric disorders that could have been prevented or ameliorated with a prompt diagnosis (Fiksinski, Schneider, Murphy, Armando, & Vicari, 2018). Once 22q11.2 deletion or duplication syndrome has been established, multidisciplinary teams are required to manage the various clinical manifestations and avoid further complications.
22q11.2 duplication syndrome involving the same region and genes as the deletion counterpart shows considerable clinical variability (milder clinical course with a spectrum of symptoms, ranging from normality to hypotonia, learning difficulties, psychomotor retardation, or intellectual disability) and has a hereditary component. It is relatively common to secondarily discover a comparatively asymptomatic parent (Traynor, Butler, Cant, & Leahy, 2016). From the immunological viewpoint, the same evaluation is recommended in these patients as in those with the deletion syndrome (Traynor et al., 2016). Our patient with 22q11.2 duplication syndrome was clinically and immunologically indistinguishable from the 22q11.2 DS patients.
Two patients in our series had already received a prenatal diagnosis based on array CGH study, but the remaining three were only detected after TREC‐positive findings emerged on NBS. Despite the obvious benefit of the early diagnosis in these three patients, which enabled prompt initiation of protocolled care, avoidance of live vaccines when indicated (Lingman Framme et al., 2014), and opportune genetic counseling, the utility of NBS for 22q11.2 deletion and duplication syndromes remains somewhat controversial (Bales, Zaleski, & McPherson, 2010). It is known that several clinical manifestations of 22q11.2 DS such as hypocalcemia, immune dysfunction, learning difficulties, palate dysfunction, and feeding problems can benefit from early detection and a prompt intervention (Barry et al., 2017; Palmer et al., 2018) ultimately reducing morbidity. However, the 22q11.2 DS diagnosis through NBS places physicians in a clinical and ethical challenge, mainly regarding mild cases, as a curative treatment is not yet available. Thus, the parents’ anxiety and expectations must be considered when sharing the diagnostic information. Trained psychologists and clinical geneticists should always be included in the multidisciplinary team for this condition. This approach has led to good results in terms of the parents’ acceptance of the diagnosis, appropriate early proactive follow‐up and adequate genetic counseling. Long‐term experience with these patients should determine the benefits and limitations of this approach, as has been seen in cystic fibrosis (Mak, Sykes, Stephenson, & Lands, 2016), and will define whether this strategy should offered to other syndromic patients detected through NBS screening, such as those with CHARGE syndrome.
Gradual implementation of SCID NBS with detection of 22q11.2 deletion and duplication syndromes in other countries will provide additional observations on the baseline characteristics of these patients and improve our knowledge of the natural history of the disease, which is critical for designing and validating future therapeutic measures.
To conclude, NBS using TREC assays enables a prompt diagnosis of patients with 22q11.2 DS. Array CGH analysis should be considered in all infants with non‐SCID T‐cell lymphopenia even in the absence of phenotypic abnormalities and before next‐generation sequencing for PID. The percentage of 22q11.2 DS patients who show low TREC levels at birth is currently unknown, but it is likely far from 100%; hence, clinical suspicion should remain as the main practice to achieve an early diagnosis in these patients.
CONFLICT OF INTEREST
The authors have no conflict of interest to disclose.
ACKNOWLEDGMNTS
We are grateful to Dr. Marie Audrain (CHU de Nantes, Nantes, France) for contributing to the development of the algorithm. The authors thank Celine Cavallo for English language assistance.
Martin‐Nalda A, Cueto‐González AM, Argudo‐Ramírez A, et al. Identification of 22q11.2 deletion syndrome via newborn screening for severe combined immunodeficiency. Two years’ experience in Catalonia (Spain). Mol Genet Genomic Med. 2019;7:e1016 10.1002/mgg3.1016
EF Tizzano and P Soler‐Palacin are both senior contributing authors.
REFERENCES
- Audrain, M. A. P. , Léger, A. J. C. , Hémont, C. A. F. , Mirallié, S. M. , Cheillan, D. , & Rimbert, M. G. M. , … Thomas, C. J. C. , (2018). Newborn Screening for Severe Combined Immunodeficiency: Analytic and Clinical Performance of the T Cell Receptor Excision Circle Assay in France (DEPISTREC Study). Journal of Clinical Immunology, 38(7), 778–786. 10.1007/s10875-018-0550-7 [DOI] [PubMed] [Google Scholar]
- Bales, A. M. , Zaleski, C. A. , & McPherson, E. W. (2010). Newborn screening programs: Should 22q11 deletion syndrome be added? Genetics in Medicine, 12(3), 135–144. 10.1097/GIM.0b013e3181cdeb9a [DOI] [PubMed] [Google Scholar]
- Barry, J. C. , Crowley, T. B. , Jyonouchi, S. , Heimall, J. , Zackai, E. H. , Sullivan, K. E. , & McDonald‐McGinn, D. M. (2017). Identification of 22q11.2 Deletion Syndrome via Newborn Screening for Severe Combined Immunodeficiency. Journal of Clinical Immunology, 37(5), 476–485. 10.1007/s10875-017-0403-9 [DOI] [PubMed] [Google Scholar]
- Bassett, A. S. , McDonald‐McGinn, D. M. , Devriendt, K. , Digilio, M. C. , Goldenberg, P. , Habel, A. , … Vorstman, J. (2011). Practical guidelines for managing patients with 22q11.2 deletion syndrome. Journal of Pediatrics, 159(2), 332–339. 10.1016/j.jpeds.2011.02.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancrini, C. , Puliafito, P. , Digilio, M. C. , Soresina, A. , Martino, S. , Rondelli, R. , … Rossi, P. (2014). Clinical features and follow‐up in patients with 22q11.2 deletion syndrome. Journal of Pediatrics, 164(6), 1475–1480. 10.1016/j.jpeds.2014.01.056 [DOI] [PubMed] [Google Scholar]
- Chien, Y.‐H. , Yu, H.‐H. , Lee, N.‐C. , Ho, H.‐C. , Kao, S.‐M. , Lu, M.‐Y. , … Hwu, W.‐L. (2017). Newborn screening for severe combined immunodeficiency in Taiwan. International Journal of Neonatal Screening, 3(3), 16 10.3390/ijns3030016 [DOI] [Google Scholar]
- Dieli‐Crimi, R. , Martínez‐Gallo, M. , Franco‐Jarava, C. , Antolin, M. , Blasco, L. , & Paramonov, I. , … Colobran, R., (2018). Th1‐skewed profile and excessive production of proinflammatory cytokines in a NFKB1‐deficient patient with CVID and severe gastrointestinal manifestations. Clinical Immunology, 195, 49–58. 10.1016/j.clim.2018.07.015 [DOI] [PubMed] [Google Scholar]
- DiGeorge, A. M. (1965). Discussions on a new concept of the cellular basis of immunology. Journal of Pediatrics, 67, 907–908. https://link.springer.com/article/10.1007/BF00441974 [Google Scholar]
- Fiksinski, A. M. , Schneider, M. , Murphy, C. M. , Armando, M. , Vicari, S. , Canyelles, J. M. , … Vorstman, J. A. S. (2018). Understanding the pediatric psychiatric phenotype of 22q11.2 deletion syndrome. American Journal of Medical Genetics A, 176(10), 2182–2191. 10.1002/ajmg.a.40387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grati, F. R. , Molina Gomes, D. , Ferreira, J. C. , Dupont, C. , Alesi, V. , Gouas, L. , … Vialard, F. , (2015). Prevalence of recurrent pathogenic microdeletions and microduplications in over 9500 pregnancies. Prenatal Diagnosis, 35(8), 801–809. 10.1002/pd.4613 [DOI] [PubMed] [Google Scholar]
- Guthrie, R. (1996). The introduction of newborn screening for phenylketonuria. A personal history. European Journal of Pediatrics, 155(S1), S4–S5. 10.1007/pl00014247 [DOI] [PubMed] [Google Scholar]
- Jyonouchi, S. , Jongco, A. M. , Puck, J. , & Sullivan, K. E. (2017). Immunodeficiencies associated with abnormal newborn screening for T cell and B cell lymphopenia. Journal of Clinical Immunology, 37(4), 363–374. 10.1007/s10875-017-0388-4 [DOI] [PubMed] [Google Scholar]
- King, J. R. , & Hammarström, L. (2018). Newborn screening for primary immunodeficiency diseases: History, current and future practice. Journal of Clinical Immunology, 38(1), 56–66. 10.1007/s10875-017-0455-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan, A. , Abraham, R. S. , Currier, R. , Brower, A. , Andruszewski, K. , Abbott, J. K. , … Bonagura, V. R. (2014). Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA, 312(7), 729–738. 10.1001/jama.2014.9132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingman Framme, J. , Borte, S. , von Döbeln, U. , Hammarström, L. , & Oskarsdóttir, S. (2014). Retrospective analysis of TREC based newborn screening results and clinical phenotypes in infants with the 22q11 deletion syndrome. Journal of Clinical Immunology, 34(4), 514–519. 10.1007/s10875-014-0002-y [DOI] [PubMed] [Google Scholar]
- Mak, D. Y. , Sykes, J. , Stephenson, A. L. , & Lands, L. C. (2016). The benefits of newborn screening for cystic fibrosis: The Canadian experience. Journal of Cystic Fibrosis, 15(3), 302–308. 10.1016/j.jcf.2016.04.001 [DOI] [PubMed] [Google Scholar]
- Palmer, L. D. , Butcher, N. J. , Boot, E. , Hodgkinson, K. A. , Heung, T. , Chow, E. W. C. , … Bassett, A. S. (2018). Elucidating the diagnostic odyssey of 22q11.2 deletion syndrome. American Journal of Medical Genetics Part A, 176(4), 936–944. 10.1002/ajmg.a.38645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard, C. , Al‐Herz, W. , Bousfiha, A. , Casanova, J. L. , Chatila, T. , & Conley, M. E. , … Gaspar, H. B. , (2015). Primary immunodeficiency diseases: An update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Journal of Clinical Immunology, 35(8), 696–726. 10.1007/s10875-015-0201-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechavi, E. , Lev, A. , Simon, A. J. , Stauber, T. , Daas, S. , Saraf‐Levy, T. , … Somech, R. (2017). First Year of Israeli Newborn Screening for Severe Combined Immunodeficiency‐Clinical Achievements and Insights. Frontiers in Immunology, 6(8), 1448 10.3389/fimmu.2017.01448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swillen, A. , & McDonald‐McGinn, D. (2015). Developmental trajectories in 22q11.2 deletion. Am J Med Genet C Semin Med Genet, 169(2), 172–181. 10.1002/ajmg.c.31435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynor, R. , Butler, K. M. , Cant, A. J. , & Leahy, T. R. (2016). Immunodeficiency in a Child with 22q11.2 Microduplication Syndrome. Journal of Clinical Immunology, 36(5), 418–419. 10.1007/s10875-016-0286-1 [DOI] [PubMed] [Google Scholar]