

Abstract
Background
Maturity‐onset diabetes of the young (MODY) is a form of monogenic diabetes with autosomal dominant inheritance. To date, mutations in 11 genes have been frequently associated with this phenotype. In Brazil, few cohorts have been screened for MODY, all using a candidate gene approach, with a high prevalence of undiagnosed cases (MODY‐X).
Methods
We conducted a next‐generation sequencing target panel (tNGS) study to investigate, for the first time, a Brazilian cohort of MODY patients with a negative prior genetic analysis. One hundred and two patients were selected, of which 26 had an initial clinical suspicion of MODY‐GCK and 76 were non‐GCK MODY.
Results
After excluding all benign and likely benign variants and variants of uncertain significance, we were able to assign a genetic cause for 12.7% (13/102) of the probands. Three rare MODY subtypes were identified (PDX1/NEUROD1/ABCC8), and eight variants had not been previously described/mapped in genomic databases. Important clinical findings were evidenced in some cases after genetic diagnosis, such as MODY‐PDX1/HNF1B.
Conclusion
A multiloci genetic approach allowed the identification of rare MODY subtypes, reducing the large percentage of MODY‐X in Brazilian cases and contributing to a better clinical, therapeutic, and prognostic characterization of these rare phenotypes.
Keywords: ACMG/AMP, MODY, MODY-X, targeted sequencing
Maturity‐onset diabetes of the young (MODY) is a form of monogenic diabetes with autosomal dominant inheritance and, to date, mutations in 11 genes have been frequently associated with this phenotype. We have conducted a NGS target panel (tNGS) study to investigate, for the first time, a Brazilian cohort of MODY patients with a negative prior genetic analysis. One hundred and two patients were selected and we were able to assign a genetic cause for 12.7% (13/102) of the probands, with three rare MODY subtypes were identified (PDX1/NEUROD1/ABCC8).

1. INTRODUCTION
Maturity‐onset diabetes of the young (MODY) (OMIM #606391) is characterized by defects in insulin secretion, autosomal dominant inheritance, early hyperglycemia onset, and negative anti‐beta cell antibodies (Fajans & Bell, 2011; Fajans, Bell, & Polonsky, 2001; McDonald et al., 2011; Vaxillaire & Froguel, 2008). To date, mutations in 11 genes have been frequently associated with MODY, some of which have recently been described (Bonnefond et al., 2012; Bowman et al., 2012; Prudente et al., 2015). Few cases, however, have been associated with the KLF11, PAX4, and BLK, and the validity of their association with the MODY phenotype is currently questioned (Flannick et al., 2013; Sanyoura, Philipson, & Naylor, 2018). Definitive diagnosis relies on genetic tests, traditionally by Sanger sequencing (Sanger & Coulson, 1975). However, given the genetic heterogeneity of this condition and the difficulty related to studying some genes due to their large size or lack of hotspots, next‐generation sequencing (NGS) seems promising for a cost‐effective genetic analysis upon clinical suspicion of MODY (American Diabetes Association, 2017; Colclough, Saint‐Martin, Timsit, Ellard, & Bellanné‐Chantelot, 2014). In Brazil, few cohorts have been screened for MODY, and all of them have been conducted using a candidate gene approach (mainly GCK/HNF1A) with a high prevalence of MODY‐X (unclear genetic diagnosis—46.2%–73.9%) (Furuzawa et al., 2008; Giuffrida et al., 2017; Maraschin et al., 2008; Moises et al., 2001; Santana et al., 2017), which could be explained by genes that are more rarely associated with MODY. The objective of this study was to use an NGS target panel (tNGS) to investigate a Brazilian cohort of MODY patients with negative prior genetic analysis by Sanger sequencing or Multiplex ligation ‐dependent probe amplification (MLPA).
2. MATERIALS AND METHODS
2.1. Subjects
Starting with a single‐gene approach previously published (Dotto et al., 2019; Santana et al., 2017) for three MODY subtypes: GCK (MODY‐GCK) (OMIM #125851) or HNF1A (OMIM #600496)/HNF1B (OMIM #137920) (MODY non‐GCK) (Figure 1 and supplemental material, cohort selection and data analysis), 102 patients with negative prior genetic analysis were selected for this targeted NGS study.
Figure 1.
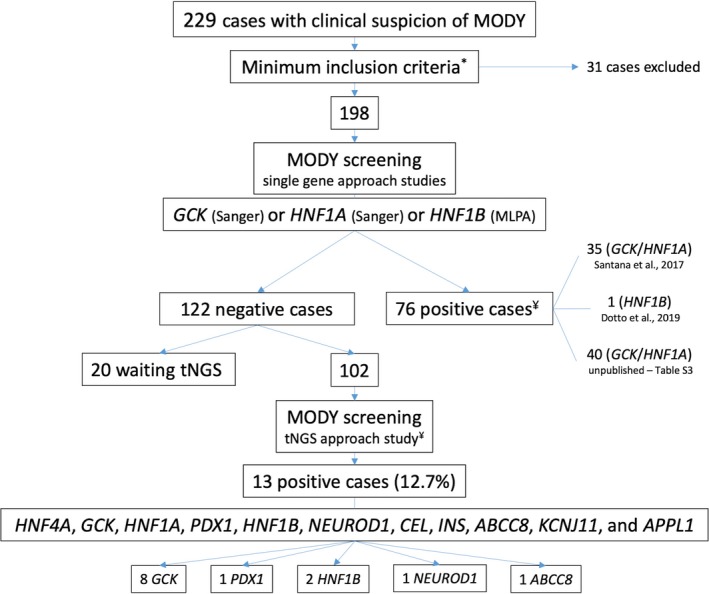
Flowchart of the study cohort selection. To select candidates for tNGS, we performed an initial genetic screening in 198 probands for 3 MODY subtypes, namely, GCK, HNF1A, and HNF1B. This candidate gene approach has been guided using clinical laboratory characteristics specific to each phenotype. Only 1 gene was investigated per patient, using Sanger sequencing (GCK OR HNF1A) OR MLPA (HNF1B). The cohort described in the current study is composed of patients who did not present any candidate variant in this initial approach (GCK, HNF1A, HNF1B) and were selected for tNGS. *Minimum inclusion criteria: Available in Supplemental Material ‐ Cohort Selection and Data Analysis; ¥ Number of patients presenting pathogenic / likely pathogenic variants
All the subjects were referred to the Monogenic Diabetes Group, School of Medicine, University of Sao Paulo (USP) using electronic forms (available at http://www.diabetesgeneticousp.com) or directly by the medical centers or universities from all regions of Brazil. The patients and/or legal guardians provided written informed consent.
2.2. Library preparation and sequencing
Samples from the 102 selected patients were subjected to genomic quality and integrity control (Bioanalyzer, Agilent Technologies) as well as DNA shearing (Covaris) prior to the enrichment protocol. The full region (upstream, exons, and introns) of 11 previously described genes frequently associated with MODY (HNF4A, GCK, HNF1A, PDX1, HNF1B, NEUROD1, CEL, INS, ABCC8, KCNJ11, and APPL1) was captured and enriched using a custom genetic panel (SureSelect XT, Agilent Technologies). Paired‐end sequencing was performed in a sequencing‐by‐synthesis assay (MiSeq System, Illumina). The manufacturer's instructions were followed in all steps.
2.3. Bioinformatics analysis
Each step performed and the programs used during preliminary processing, genomic mapping, variant calling, and annotation of the sequencing data are detailed in the supplemental material—cohort selection and data analysis.
2.4. Variant filtering workflow and pathogenicity interpretation
A prioritization process was applied to reduce the list of candidate variants for association with MODY. The allele frequency was evaluated using a public consortium and a local genomic database, namely, gnomAD (Lek et al., 2016) and ABraOM (Naslavsky et al., 2017), respectively. In addition to minor allele frequency < 1%, each final candidate variant was evaluated in relation to the MODY estimated frequency (0.01%) (Shields et al., 2010). This step was performed later to avoid exclusion of variants with border frequency in the initial filtering process. We interpreted the pathogenicity of each candidate variant according to the British Society for Genetic Medicine recommendations (Wallis et al., 2013) and the American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology (AMP) guidelines (Richards et al., 2015). To support familial cosegregation analysis, a mathematical model (Jarvik & Browning, 2016) was added to the ACMG/AMP ranking, allowing for the statistical quantification of familial aggregation data.
The location of each candidate variant was verified in the patients' binary alignment map file (BAM) using the Integrative Genomics Viewer (IGV) (Robinson et al., 2011). Sanger sequencing was performed as a confirmatory method. The details of the filtering workflow and pathogenicity interpretation are presented in the supplemental materials—cohort selection and data analysis.
3. RESULTS
3.1. Subjects
One hundred and two patients with a negative prior genetic analysis were selected for target NGS. Twenty‐six of these had an initial clinical suspicion of MODY‐GCK and 76 of non‐GCK MODY. The cohort was composed of 37.3% males. The clinical and laboratory data at the first evaluation are presented in Table 1.
Table 1.
Clinical and laboratory data for the cohort at the first evaluation
| Median (1st | 3rd quartile)f or Percentageg | |
|---|---|
| Age at diagnosis of diabetes or MFHa (years) | 20 (14 | 27.7) |
| Fasting plasma glucose (mg/dL) | 131 (104 | 213) |
| A1C (%) | 7.5 (6.0 | 9.2) |
| Glucose tolerance test (GTT) incrementb (mg/dL) | 109 (102 | 115) |
| Fasting C‐peptidec (ng/mL) | 1.7 (1.2 | 2.5) |
| Referred weight at diagnosisd | 14%‐low | 70%‐normal | 13%‐overweight | 3%‐obesity |
| Family history of diabetes/MFH | 94%‐positive | 5%‐negative | 1%‐unknown |
| Treatment at diagnosis | 24%‐diet | 38%‐OHAh | 38%‐insulin |
| Microvascular complicationse | 26%‐Retinopathy | 27%‐Microalbuminuria |
Normal range: Fasting plasma glucose (hexokinase) 70–99 mg/dL; A1C (HPLC) 4.1%–6.0%; Fasting C‐peptide (chemiluminescence) 1.1–4.4 ng/mL; Microalbuminuria (immunoturbidimetry) <14.0 mg/L; Glucose tolerance test (hexokinase) 0 min 70–99 mg/dL | 120 min < 140 mg/dL.
MFH: mild fasting hyperglycemia.
2 hr glucose‐fasting glucose (25 patients screened).
Fasting C‐peptide: 3 years after diagnosis.
Reported by the patient (“referred weight”).
Microvascular complications: Retinopathy (77 patients screened), Microalbuminuria (87 patients screened).
Continuous variables.
Discrete variables.
OHA: oral hypoglycemic agent.
3.2. Sequencing metrics
Table S1 summarizes the sequencing metrics, presenting data such as the number of reads obtained, the number of sequenced bases, and the target region coverage (%>10X, %>20X, and %>30X).
3.3. Genetic analysis
An average of 500 nonreference alleles per patient were identified during the variant calling protocol. After applying the prioritization criteria, 35/102 patients (34%) had at least one candidate variant, with one patient having two candidate variants in two different genes (Tables 2, 3, and Table S2).
Table 2.
Clinical, laboratory, and genetic characteristics of MODY probands with candidate variants
| Proband/Variant* | S.G approach | Age at diagnosis of diabetes or MFHa/age at genetic testing (years) | Referred weight at diagnosisb | Family history of diabetes/MFH (G/S/HT)c | Treatment at diagnosis | Current treatment | Range of FPGd (mg/dL) follow‐up | Range of A1Ce (%)follow‐up | C‐peptidef (ng/mL) follow‐up | Mic.C |
|---|---|---|---|---|---|---|---|---|---|---|
| HNF4A‐1/p.Ser3Glyk | HNF1A s | 8/14 | Normal | 1/NA/NA | Insulin | Insulin + Metformin | 82–140 | 6.8–9.0 | 1.2 | No |
| HNF4A‐2/p.His49Tyrk | HNF1A s | 17/37 | Normal | 2/4/1 | Insulin + Sulfonylurea | OHAs | 70–87 | 5.3–7.3 | 0.8 | No |
| HNF4A‐3/p.Ile441Vali | HNF1A s | 16/51 | Normal | 3/NA/NA | Sulfonylurea | Insulin + OHAs | 79–260 | 5.1–9.2 | 2.6 | R |
| GCK‐1/p.Val182Metg | HNF1A s | 8/19 | Low | 2/5/1 | Sulfonylurea | Diet | 89–176 | 6.0–8.2 | 2.1 | No |
| GCK‐2/p.Leu185Glnh | HNF1A s | 22/28 | Normal | 3/NA/NA | Metformin | Sulfonylurea | 102–141 | 6.4–8.7 | 1.7 | No |
| GCK‐3/p. Ala208Proh | HNF1A s | 12/32 | Normal | NA/NA/NA | Insulin | Diet | 128–138 | 7.1–7.4 | 3.0 | NA |
| GCK‐4/p.Met235Valg | HNF1A s | 10/16 | Normal | 3/5/2 | Insulin | Diet | 73–139 | 6.6–7.2 | 2.5 | No |
| GCK‐5/p.Gly318Argg | GCK s | 14/18 | Low | 3/5/1 | Insulin | Diet | 87–139 | 5.9–6.5 | 1.7 | No |
| GCK‐6/p.Gly318Argg | GCK s | 35/46 | Normal | 2/6/6 | Insulin | Diet | 110–146 | 5.4–6.0 | 1.3 | No |
| GCK‐7/p.Thr326Prog | GCK s | 28/54 | Normal | 3/4/3 | Diet | Diet | 89–112 | 5.9–6.6 | 3.0 | No |
| GCK‐8/p.ArgR447Lfs*2g | GCK s | 15/21 | Normal | 3/4/2 | Diet | Diet | 117–122 | 5.9–6.2 | 1.6 | No |
| HNF1A‐1/p.His505Asnk | HNF1A s | 17/43 | Normal | 3/3/1 | Diet | OHAs | 111–219 | 6.2–7.5 | 2.9 | No |
| PDX1‐1/p.Pro63Argfs*60g | HNF1A s | 14/53 | Normal | 3/6/1 | Insulin | Insulin + Sulfonylurea | 83–441 | 6.8–9.2 | 1.4 | R |
| PDX1‐2/p.Glu222Lysk | HNF1A s | 27/38 | Normal | 3/NA/NA | Insulin + Metformin | Insulin + Metformin | 58–193 | 8.5–9.7 | 1.1 | No |
| HNF1B‐1/p.Val61Glyi | HNF1A s | 25/39 | Normal | 2/NA/NA | Insulin | Insulin + Sulfonylurea | 500‐NA | 5.5–8.8 | 2.4 | No |
| HNF1B‐2/p.Gly76Cysk | HNF1A s | 34/44 | Normal | 2/NA/NA | Metformin | OHAs | 100–167 | 5.7–7.4 | 2.2 | No |
| HNF1B‐3/p.Ser148Leug | HNF1B m | 12/17 | NA | 2/NA/NA | Insulin | Insulin | 75–153 | 6.1–10.0 | NA | No |
| HNF1B‐4/p.Arg295Hish | HNF1A s | 12/20 | Normal | 1/1/1 | Diet | DPP4i | 88–120 | 5.7–6.8 | 2.4 | No |
| NEUROD1‐1/p.Tyr231Terg | HNF1A s | 20/47 | Normal | 2/3/0 | Sulfonylurea | OHAs | 137–NA | 6.1–10.0 | 1.6 | No |
| CEL‐1/p.Ala24Valk | HNF1A s | 12/17 | Overweight | 3/NA/NA | Diet | Sulfonylurea | 113–126 | 6.1–6.3 | 2.6 | No |
| CEL‐2/p.Asp102Alak | GCK s | 5/13 | Normal | 3/13/5 | Diet | Diet | 96–108 | 5.5–6.1 | 1.5 | M |
| CEL‐3/p.Val161Metk | HNF1A s | 27/32 | Normal | 3/NA/NA | OHAs | Sulfonylurea | 120–400 | 6.6–11.8 | 2.1 | No |
| CEL‐4/p.Pro718Thrj | HNF1A s | 30/34 | Obesity | 3/NA/NA | OHAs | Insulin + OHAs | 89–321 | 5.8–12.5 | NA | No |
| INS‐1/p.Arg6Hisk | HNF1A s | 19/61 | Low | 2/NA/NA | Sulfonylurea | Insulin + OHAs | 106–360 | 10.4–13.6 | 2.6 | R/M |
| INS‐2/p.Ala23Glnfs*3g | HNF1A s | 22/57 | NA | 2/NA/NA | OHAs | Insulin + OHAs | 89–255 | 6.3–9.7 | 1.9 | R/M |
| INS‐3/p.Ser76Asnj | HNF1A s | 30/39 | Overweight | 3/NA/NA | OHAs | Insulin + Metformin | 65–230 | 7.8–9.8 | 1.1 | No |
| ABCC8‐1/p.Ser53Cysk | HNF1A s | 30/39 | Low | 3/NA/NA | Metformin | Diet | 117–130 | 6.4–6.9 | 1.0 | No |
| ABCC8‐2/p.Ala235Thrj | HNF1A s | 34/53 | Normal | 2/3/1 | OHAs | Insulin | 68–202 | 6.2–8.1 | NA | R/M |
| ABCC8‐3/p.Val563Aspk | HNF1A s | 12/50 | NA | 3/NA/NA | Sulfonylurea | Insulin | 54–697 | 6.8–10.8 | 3.8 | R/M |
| ABCC8‐4/p.Gly658Valk | HNF1A s | 12/43 | NA | 3/NA/NA | Insulin | Insulin | 143–158 | 6.0–6.4 | 1.1 | No |
| ABCC8‐5/p.Asp673Asnk | HNF1A s | 14/27 | Normal | 3/1/0 | Insulin | Sulfonylurea | 67–270 | 6.0–13.7 | NA | No |
| ABCC8‐6/p.Arg825Trpg | HNF1A s | 21/52 | Low | 3/6/4 | Sulfonylurea | Sulfonylurea | 90–271 | 6.3–8.4 | 2.3 | R/M |
| ABCC8‐7/p.Leu1147Argk | HNF1B s,m | 17/51 | NA | 1/NA/NA | Insulin | Insulin | 45–234 | 5.1–7.6 | 0.4 | M |
| KCNJ11‐1/p.Ala96Thrk | HNF1A s | 20/49 | Normal | 4/6/4 | Insulin | Insulin + OHAs | 86–258 | 6.4–9.4 | 1.4 | No |
| D1/¢p.Lys266Gluk + ¥p.Ser1018Leuk | HNF1A s | 11/17 | Normal | 3/NA/NA | Metformin | Diet | 93–177 | 5.5–7.2 | 3.2 | NA |
Normal range: FPG (hexokinase) 70–99 mg/dL; A1C (HPLC) 4.1%–6.0%; Fasting C‐peptide (chemiluminescence) 1.1–4.4 ng/mL; Microalbuminuria (immunoturbidimetry) <14.0 mg/L.
RefSeq reference transcript: NM_175914.3 (HNF4A)/NM_000162.3 (GCK)/NM_000545.6 (HNF1A)/NM_000209.3 (PDX1)/NM_000458.2 (HNF1B)/NM_002500.3 (NEUROD1)/NM_001807.3 (CEL)/NM_000207.2 (INS)/NM_000352.3 (ABCC8)/NM_000525.3 (KCNJ11); S.G approach: Single‐gene approach (s Sanger sequencing/m MLPA); aMFH: mild fasting hyperglycemia; bReported by the patient (“referred weight”); cFamilial history (without proband)–G: generations, S: patients screened, HT: heterozygous patients for the family variant; dFPG: fasting plasma glucose; eA1C: Glycated hemoglobin; fFasting C‐peptide: 3 years after diagnosis; Mic.C: Microvascular complications (R‐Retinopathy, M‐Microalbuminuria); ACMG five‐tier system: gP (Pathogenic), hL.P (Likely pathogenic), iB (Benign), jL.B (Likely benign), kU.S (Uncertain significance); ¢ CEL; ¥ ABCC8; NA: not available; OHAs: more than one oral hypoglycemic agent (other than sulfonylurea).
Table 3.
Allelic candidate variants identified using a gene panel in MODY probands with a negative prior genetic analysis
| Gene | Nucleotide changea | Aminoacid changea | Region | Proband (s) | ACMGb | Reference (first report) |
|---|---|---|---|---|---|---|
| HNF4A | c.7A > G | p.Ser3Gly | Exon 1 | HNF4A‐1 | U.S | This studyc |
| c.145C > T | p.His49Tyr | Exon 2 | HNF4A‐2 | U.S | This study | |
| c.1321A > G | p.Ile441Val | Exon 10 | HNF4A‐3 | B | Malecki et al. (1999) | |
| GCK | c. 544G > A | p.Val182Met | Exon 5 | GCK‐1 | P | Froguel et al. (1993) |
| c.554T > A | p.Leu185Gln | Exon 5 | GCK‐2 | L.P | This study | |
| c.622G > C | p. Ala208Pro | Exon 6 | GCK‐3 | L.P | Garin et al. (2008) | |
| c.703A > G | p.Met235Val | Exon 7 | GCK‐4 | P | García‐Herrero et al. (2007) | |
| c.952G > A | p.Gly318Arg | Exon 8 | GCK‐5, GCK‐6 | P | Pruhova et al. (2003) | |
| c.976A > C | p.Thr326Pro | Exon 8 | GCK‐7 | P | Lorini et al. (2009) | |
| c.1340_1368del29 | p.ArgR447Lfs*2 | Exon 10 | GCK‐8 | P | Ziemssen, Bellanné‐Chantelot, Osterhoff, Schatz, and Pfeiffer, (2002) | |
| HNF1A | c.1513C > A | p.His505Asn | Exon 8 | HNF1A‐1 | U.S | Bellanne‐Chantelot et al. (2008) |
| PDX1 | c.188delC | p.Pro63Argfs*60 | Exon 1 | PDX1‐1 | P | Stoffers, Ferrer, Clarke, and Habener (1997) |
| c.664G > A | p.Glu222Lys | Exon 2 | PDX1‐2 | U.S | This studyc | |
| HNF1B | c.182T > G | p.Val61Gly | Exon 1 | HNF1B‐1 | B | Edghill, (2005) |
| c.226G > T | p.Gly76Cys | Exon 1 | HNF1B‐2 | U.S | Bellanne‐Chantelot et al. (2005) | |
| c.443C > T | p.Ser148Leu | Exon 2 | HNF1B‐3 | P | Edghill (2005) | |
| c.884G > A | p.Arg295His | Exon 4 | HNF1B‐4 | L.P | Bellanné‐Chantelot et al. (2004) | |
| NEUROD1 | c.693C > G | p.Tyr231Ter | Exon 2 | NEUROD1‐1 | P | This study |
| CEL | c.71C > T | p.Ala24Val | Exon 1 | CEL‐1 | U.S | This studyc |
| c.305A > C | p.Asp102Ala | Exon 3 | CEL‐2 | U.S | This study | |
| c.481G > A | p.Val161Met | Exon 4 | CEL‐3 | U.S | This studyc | |
| c.796A > G | p.Lys266Glu | Exon 7 | D1 | U.S | This study | |
| c.2152C > A | p.Pro718Thr | Exon 11 | CEL‐4 | L.B | Johansen et al. (2014) | |
| INS | c.17G > A | p.Arg6His | Exon 1 | INS‐1 | U.S | Meur et al. (2010) |
| c.65delC | p.Ala23Glnfs*3 | Exon 2 | INS‐2 | P | This study | |
| c.227G > A | p.Ser76Asn | Exon 3 | INS‐3 | L.B | This studyc | |
| ABCC8 | c.157A > T | p.Ser53Cys | Exon 2 | ABCC8‐1 | U.S | This study |
| c.703G > A | p.Ala235Thr | Exon 5 | ABCC8‐2 | L.B | This studyc | |
| c.1688T > A | p.Val563Asp | Exon 12 | ABCC8‐3 | U.S | This study | |
| c.1973G > T | p.Gly658Val | Exon 14 | ABCC8‐4 | U.S | This studyc | |
| c.2017G > A | p.Asp673Asn | Exon 14 | ABCC8‐5 | U.S | This studyc | |
| c.2473C > T | p.Arg825Trp | Exon 20 | ABCC8‐6 | P | Vaxillaire et al. (2007) | |
| c.3053C > T | p.Ser1018Leu | Exon 25 | D1 | U.S | This studyc | |
| c.3440T > G | p.Leu1147Arg | Exon 28 | ABCC8‐7 | U.S | Gussinyer et al. (2008) | |
| KCNJ11 | c.286G > A | p.Ala96Thr | Exon 1 | KCNJ11‐1 | U.S | Melikyan et al. (2012) |
RefSeq reference transcript: NM_175914.3 (HNF4A)/NM_000162.3 (GCK)/NM_000545.6 (HNF1A)/NM_000209.3 (PDX1)/NM_000458.2 (HNF1B)/NM_002500.3 (NEUROD1)/NM_001807.3 (CEL)/NM_000207.2 (INS)/NM_000352.3 (ABCC8)/NM_000525.3 (KCNJ11).
ACMG five‐tier system: B (Benign), L.B (Likely benign), P (Pathogenic), L.P (Likely pathogenic), U.S (Uncertain significance).
Reported in The Genome Aggregation Database (gnomAD).
3.3.1. Common MODY genes
Among a total of 34 patients with only one candidate variant, in 47% (16/34) of them, the change was identified in one of the genes most commonly associated with MODY (three in HNF4A, seven in GCK, one in HNF1A, and four in HNF1B). Only one variant was shared by two probands (GCK—c.952G > A/p.Gly318Arg).
Of these variants, three were novel (two in HNF4A/one in GCK) and 12 had been described previously. According to the ACMG/AMP guidelines, 60% (9/15) were considered pathogenic or likely pathogenic, 27% (4/15) of uncertain significance, and 13% (2/15) benign (already reported). A cosegregation analysis was possible in nine of the 16 families (Figure 2).
Figure 2.
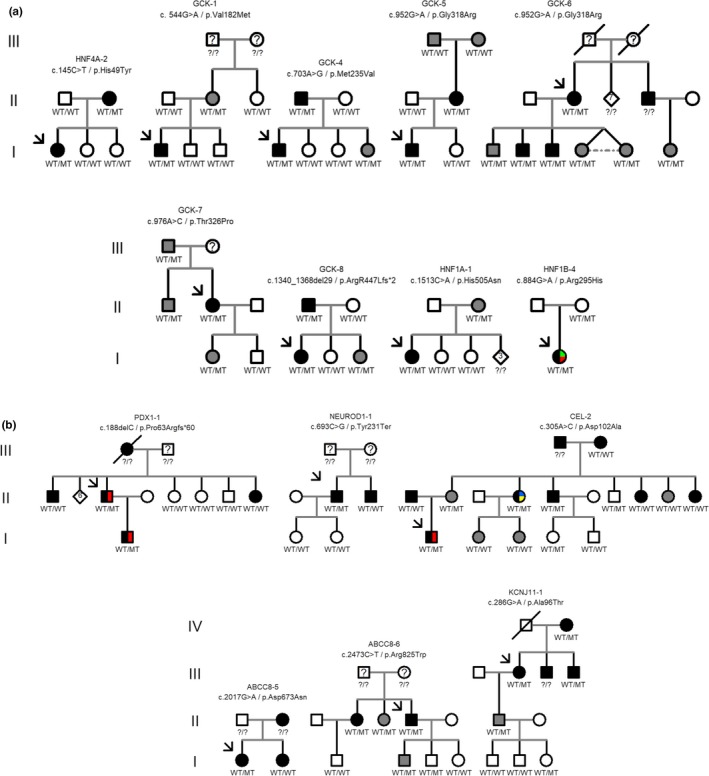
Pedigrees of screened MODY families with pathogenic/likely pathogenic/uncertain significance variants (A ‐ Common MODY genes, B ‐ Rare MODY genes). Square: male; Circle: female; Grey filled symbol: patient with prediabetes; Black filled symbol: patient with diabetes; Empty symbol: patient without diabetes nor prediabetes; Green filled symbol: renal cysts; Red filled symbol: pancreatic malformation; Blue filled symbol: low fecal elastase (LFE); Yellow filled symbol: LDL elevation; WT: wild‐type allele; MT: altered allele; ?/?: patient not genotyped
3.3.2. Rare MODY genes
Eighteen patients (53% (18/34)) had only one candidate variant in genes that are rarely associated with MODY (two PDX1; one NEUROD1; four CEL; three INS; seven ABCC8; one KCNJ11), most of which (94% (17/18)) had an initial clinical suspicion of non‐GCK MODY. No variant was shared by two or more probands.
Five variants (28% (5/18)) were described for the first time in this study. The remainder (72% (13/18)) had been reported in the literature and/or mapped in genomic databases (gnomAD/ABraOM). Only four (22% (4/18)) were classified as pathogenic by ACMG/AMP, three (17% (3/18)) were likely benign, and the majority (61% (11/18)) with uncertain significance.
Only one case (3%–1/35) presented candidate variants in two genes. In this patient, two novel missense changes of uncertain significance were present in CEL and ABCC8.
Table 4 presents clinical follow‐up data and additional information for patients with pathogenic/probably pathogenic/uncertain significance candidate variants in rare MODY genes. Six of the 19 families were available for cosegregation analysis (Figure 2).
Table 4.
Clinical and laboratory follow‐up data of probands with one pathogenic or uncertain significance candidate variant in a rare MODY gene
| MODY subtype | Number of probands | Age at diagnosis of diabetes or MFHa (years)e | FPGb (mg/dL)e follow‐up | A1Cc (%)e follow‐up | C‐peptided (ng/mL)e | Typical clinical findings/diabetes complications |
|---|---|---|---|---|---|---|
| Pathogenic | ||||||
| PDX1 | 1 | 14 | 83–441 | 6.8–9.2 | 1.4 | Pancreatic dysgenesis/NPDRf |
| NEUROD1 | 1 | 20 | 137–NA | 6.1–10.0 | 1.5 | — |
| INS | 1 | 22 | 89–255 | 6.3–9.7 | 1.9 | PDRg/microalbuminuria |
| ABCC8 | 1 | 21 | 90–271 | 6.3–8.4 | 2.3 | GSRh/PDRg/microalbuminuria |
| Uncertain significance | ||||||
| PDX1 | 1 | 27 | 58–193 | 8.5–9.7 | 1.1 | — |
| CEL | 4 | 5–30 | 89–400 | 5.5–12.5 | 1.5–2.6 | fecal elastase deficiency/dyslipidemia/microalbuminuria |
| INS | 1 | 19 | 106–360 | 10.4–13.6 | 2.6 | PDRg/microalbuminuria |
| KCNJ11 | 1 | 20 | 86–258 | 6.4–9.4 | 1.4 | — |
| ABCC8 | 5 | 12–30 | 45–697 | 5.1–13.7 | 0.4–3.8 | PDRg/microalbuminuria |
Normal range: FPG (hexokinase) 70–99 mg/dL Minor allele frequency ; A1C (HPLC) 4.1%–6.0%; Fasting C‐peptide (chemiluminescence) 1.1–4.4 ng/mL; Microalbuminuria (immunoturbidimetry) <14.0 mg/L; Fecal elastase (enzyme immunoassay ‐ ELISA) >200 µg/g.
MFH, mild fasting hyperglycemia.
FPG, fasting plasma glucose.
A1C, Glycated hemoglobin.
Fasting C‐peptide: 3 years after diagnosis.
Range if available.
NPDR, nonproliferative diabetic retinopathy.
PDR, proliferative diabetic retinopathy.
GSR, good sulfonylurea response.
4. DISCUSSION
The use of a genetic panel to investigate a multilocus disease with large associated genes (without hotspots), as in MODY, is extremely advantageous in relation to the gene‐specific approach by Sanger sequencing (Colclough et al., 2014; Ellard et al., 2013). In recent years, some cohorts of MODY patients have been investigated using gene panels (Alkorta‐Aranburu et al., 2014; Bonnefond et al., 2014; Ellard et al., 2013). However, these panels are not adequate for interrogating the genetic causes of MODY in many countries, such as Brazil, where the prevalence of MODY‐X (without genetic diagnosis) ranges from 46.2% to 73.9% (Furuzawa et al., 2008; Maraschin et al., 2008; Moises et al., 2001; Santana et al., 2017; Weinert et al., 2014). Thus, a thorough investigation of the rarer subtypes of MODY is needed to improve their genetic diagnosis.
It is important to note that the study of rare MODY genes in a cohort preselected for the most common subtypes would contribute to the clarification of a minority of cases. A whole‐exome sequencing (WES) approach would be more efficient for clarifying not only rare MODY subtypes but also possible new genotype‐phenotype associations. In centers located in emerging countries, however, the availability of financial resources is still a limiting factor and gene‐candidate approaches are routinely used in the selection of cases for broad genomic studies, such as WES.
With this work, and with reference to a previous publication (Santana et al., 2017), our group set out to investigate all the most frequent genes associated with MODY using a genetic panel in a cohort of Brazilian families. After excluding all benign, likely benign, and uncertain significance variants, besides those pathogenic/probably pathogenic ones that presented, despite their ACMG/AMP classification, some uncertain characteristic as to their definitive phenotypic association, we were able to assign a genetic cause for 12.7% (13/102) of the patients, consistent with the rate previously observed in similar studies (11%–33%) (Ellard et al., 2013; Pezzilli et al., 2018; Szopa et al., 2015).
Forty‐three percent of the candidate genetic findings (15/35), especially in rare MODY genes, were classified as variants of uncertain significance for having insufficient or conflicting evidence in the phenotypic association. Four of these variants of uncertain significance (4/15) have an allele frequency equal to or greater than 1/15,000 in gnomAD: p.His505Asn (HNF1A), p.Gly76Cys (HNF1B), p.Arg6His (INS), and p.Val161Met (CEL), a frequency similar to the risk of type 2 diabetes alleles mapped in common MODY genes, such as HNF1A (Najmi et al., 2017). This observation would support the notion of a complex/polygenic context of hyperglycemia, making them benign in the face of our monogenic hypothesis. Even for those variants with a populational frequency lower than expected for MODY, we cannot rule out the possibility of a spurious finding, as observed in rare nonsynonymous changes mapped in individuals without diabetes (Flannick et al., 2013).
If we consider the extended genetic approach of our entire cohort (combining the 2017 (Santana et al., 2017)/2019 (Dotto et al., 2019) studies, the present one, and added 40 MODY‐GCK/HNF1A unpublished cases (Table S3)), the overall positivity (pathogenic/likely pathogenic variants) would reach 50% (89/178), where 178 is the total number of cases already investigated by Sanger sequencing or MLPA and tNGS. As a consequence of this wider analytical approach, a more precise number of cases with MODY‐X can be obtained, reaching 50% (89/178), which is still higher than the average (currently 15%–20% (Chèvre et al., 1998; Fajans et al., 2001; Frayling et al., 2001; Frayling et al., 2003; Shepherd, Sparkes, & Hattersley, 2001; Shields et al., 2010)) reported in the literature worldwide. This number, however, may be lower since some of these unclarified cases may harbor genetic defects that were not investigated in this study (copy number variations, CNVs/large indels) or were located in regulatory and intronic regions, such as in genes that have not yet been related to the phenotype. In addition, we cannot rule out the possibility of patients who were investigated due to initial suspicion of MODY but presented another type of Diabetes Mellitus (DM) (overlapping clinical characteristics). Finally, the higher percentage of MODY‐X cases may be related to the genetically heterogeneous composition of our population, with centuries of colonization and immigration of people from dozens of countries, resulting in the observed mixing (Cardoso, de Oliveira, Paixão‐Côrtes, Castilla, & Schuler‐Faccini, 2019). This characteristic may have contributed, in some way, to the existence of a different set of MODY genes that are not found in European populations from which the current set of MODY genes have been identified. Thus, the use of multiloci genomic approaches (such as WES) could uncover new MODY gene–phenotype associations in this non‐European population.
Among patients with at least one candidate variant, 46% (16/35) presented with the nonreference allele in a common MODY gene. In five of them (31%–5/16), the same gene was previously analyzed by Sanger sequencing (Santana et al., 2017) (GCK or HNF1A), with a negative result (missed heterozygote) (Table 2). As these five heterozygotes that Sanger sequencing missed are from our previous study (Santana et al., 2017), with 35 cases (i.e., 40 true heterozygous genotypes), our missed heterozygote rate (MHR) reached 12.5% (5/40—uncalled heterozygotes/all heterozygous genotypes), consistent with the reported global MHR (9.1%–14.5%) (Quinlan & Marth, 2007).
In these patients, a false‐negative result occurred due to an allelic dropout or an analytic error. Allele dropout is a rare molecular event with an estimated occurrence rate of 0.3% (Blais et al., 2015). It has dependent and independent allele causal factors and has been reported during genetic research of numerous diseases (Fujimura, Northrup, Beaudet, & O’Brien, 1990; Lam & Mak, 2013; Schulze, Bettendorf, Maser‐gluth, Decker, & Schwabe, 1998; Somerville, Sprysak, Hicks, Elyas, & Vicen‐Wyhony, 1999; Wenzel et al., 2009), including MODY (Ellard et al., 1999; Raeder et al., 2006).
The three cases observed in our cohort (GCK‐5/p.Gly318Arg; GCK‐6/p.Gly318Arg; GCK‐7/p.Thr326Pro) occurred in exon 8 of the GCK. The allele was "lost" during PCR due to the variant rs76323047 present at the primer annealing site (NGRL Manchester, 2019), and it was recovered after oligo modification. This finding highlights an important Sanger sequencing limitation that is overcome by new sequencing technologies.
The remaining 11 common MODY cases represent the restrictions of a single‐gene approach, in addition to the application of only one molecular investigation method (Sequencing or MLPA).
Among these patients, a number of notable findings are further detailed below.
4.1. HNF4A | OMIM #125850
Of the three cases in which candidate variants were identified in HNF4A, only one comprised a previously described genetic finding (c.1321A > G/p.Ile441Val). The family reported by Malecki et al. (1999), however, had a genotype–phenotype discrepancy (unaffected bearer) in addition to cosegregation in half of the HNF4A variant bearers (as well as other affected family members), of a second variant (HNF1A—c.872dupC/p.Gly292Argfs*25—pathogenic). We must also point out that the HNF4A—c.1321A > G/p.Ile441Val was mapped at a frequency higher (0.08%) than that expected for MODY in a local cohort of 609 healthy Brazilian elderly individuals (ABraOM) (Naslavsky et al., 2017). All these findings resulted in a benign ACMG classification. Even with its isolated association with MODY is unproven, Malecki et al. (1999) noted that patients who carried both variants (HNF1A/HNF4A) presented an earlier age of diagnosis and the need for insulin therapy. Of the two remaining variants identified, neither had yet been described to be associated with the phenotype. However, one of them (c.A7G/p.Ser3Gly—uncertain significance) was mapped in a public genomic database (gnomAD) at an allele frequency lower (0.0004%) than expected for MODY. It is important to note that it was not possible to distinguish more typical MODY‐HNF4A characteristics, such as macrosomia, neonatal hypoglycemia (Pearson et al., 2007), or good sulfonylurea response (Pearson et al., 2003), in either of the two patients.
4.2. GCK
Three patients with initial diagnostic suspicion of MODY‐HNF1A presented previously described pathogenic/probably pathogenic variants in GCK. The diagnostic hypothesis error occurred due to the presence of an atypical glycemic pattern and/or a good therapeutic response to sulfonylurea (probands GCK‐1/GCK‐3/GCK‐4—Table 2). It is important to highlight that all of these patients currently control their glycemic levels by means of diet alone, demonstrating the importance of a genetic diagnosis with the correct MODY subtype identification.
4.3. HNF1B
Four probands were identified with candidate changes in HNF1B and to our surprise, only one had initial clinical manifestations specific to this MODY subtype (multiple renal cysts and pancreas body/tail agenesis). In one of the three remaining cases, the candidate variant c.182T > G/p.Val61Gly, which had been previously described, was disregarded at the end of its ACMG classification. Despite changes having been described in numerous cohorts (Bonnefond et al., 2014; Edghill, Bingham, Ellard, & Hattersley, 2006; Faguer et al., 2011; Granberg et al., 2012; Hoskins et al., 2007; Oram et al., 2010), in patients with DM and renal/pancreatic/genitourinary dysgenesis, a functional study (Granberg et al., 2012) showed no difference in comparison between the variant and its wild‐type allele. In addition, it was mapped at a high frequency for MODY in both the global (gnomAD) and local Brazilian cohorts (ABraOM), contributing to benign ACMG categorization. Although the last two cases initially did not show evident pancreatic/extra pancreatic manifestations, a phenotypic correlation with the HNF1B genetic finding was established. The proband bearer of the described likely pathogenic variant c.884G > A/p.Arg295His was investigated, after genetic testing, for the presence of typical dysgenesis. Magnetic resonance imaging (MRI) demonstrated pancreas body/tail agenesis in addition to the presence of renal cysts. Despite this finding, the patient had normal renal/liver function in addition to normomagnesemia. Finally, in the bearer of the described uncertain significance variant c.226G > T/p.Gly76Cys, a pancreas diffuse volume reduction was found after performing a CT scan. Additionally, an elevation of hepatic enzymes was observed without apparent functional impairment.
It is important to highlight how the use of a gene panel was a decisive factor for the correct identification of the MODY subtype in these cases. The genetic diagnoses of these patients could have been missed due to the absence of typical clinical findings during the first evaluation. In addition, because large deletions are a frequent finding in MODY‐HNF1B (in approximately 50% or more of the cases) (Edghill et al., 2013), the initial approach aimed to identify CNVs in the gene. Our three cases presented missense variants.
Over half (53% (18/34)) of the cases identified with only one candidate variant showed a change in a rare MODY gene, evidencing the importance of the investigating rare MODY subtypes.
4.4. PDX1 | OMIM #606392
In addition to the family with a new phenotype already described by our group (Caetano et al., 2018), a second proband was identified with the uncertain significance variant c.664G > A/p.Glu222Lys. Its population frequency, however, was high in the local control cohort (ABraOM), contributing to its ACMG classification. A familial cosegregation analysis, in addition to an investigation of possible pancreatic malformations (Caetano et al., 2018), will help to determine the pathogenicity of the variant.
4.5. NEUROD1 | OMIM #606394
The only nonsense modification in the cohort, the novel pathogenic c.693C > G/p.Tyr231Ter, was present in NEUROD1. Heterozygous variants in this gene are a rare cause of MODY, with a notable incomplete penetrance among the members of reported families (Demirbilek et al., 2018). Extrapancreatic manifestations have already been described, such as neurological abnormalities in homozygous individuals (permanent neonatal diabetes mellitus) (Demirbilek et al., 2018; Rubio‐Cabezas et al., 2010). Our patients did not present any other atypical clinical findings, besides the classic MODY remarks. Cosegregation analysis could help to reinforce the final interpretation of this variant, which compromises the NEUROD1 transactivation domain (Rubio‐Cabezas et al., 2010).
4.6. CEL | OMIM #609812
CEL was the second, among rare MODY genes, with the largest number of candidate variants: five with uncertain significance missense changes. None of them had been previously described to be associated with the phenotype, and one was identified in one case with another candidate variant (ABCC8). Approximately, half of these CEL variants were mapped in the gnomAD consortium at an allele frequency lower than expected for MODY, which would not initially discard them from a probable association with the phenotype. The CEL analysis by first/second‐generation sequencing constituted a challenge due to its structure. The presence of its in‐tandem pseudogene (CELP) (Madeyski, Lidberg, Bjursell, & Nilsson, 1998) hindered the precise read alignment and variant calling process. In addition, it was extremely polymorphic and had a variable number of tandem repetitions (VNTR) in exon 11. Segments of approximately 33 bp may be repeated 7–23 times in the general population (Torsvik et al., 2010). Until now, only frameshift mutational events in this region were associated with a monogenic diabetes‐pancreatic exocrine dysfunction syndrome with typical fecal elastase deficiency (FED), mild abdominal pain, and loose stools (Ræder et al., 2006; Torsvik et al., 2010). All five candidate variants identified in our cohort were missense, and only one was located in exon 11. In addition, none of the patients showed any clinical signs of exocrine pancreatic deficiency. However, one of the CEL‐2 relatives had low fecal elastase. The proband of this family also showed a reduction in the volume of the pancreas on the MRI and microalbuminuria. Interestingly, three patients (variants in exons 1, 3, and 11) showed Low‐density lipoprotein elevations, one of which was diagnosed with aortic atheromatosis. A fraction of the CEL lipase has already been detected in the plasma, suggesting its interaction with cholesterol molecules and lipoproteins (Bengtsson‐Ellmark et al., 2004; Caillol et al., 1997). Although these variants may influence the lipid profile of these patients, their definitive association with MODY could not yet be established in our cohort. An extension of cosegregation analysis in these families, with the exception of that already performed with the CEL‐2 proband, would help to determine the degree of pathogenicity of these variants.
4.7. INS | OMIM #613370
With the exception of the probably benign and uncertain significance INS variants, only one (1/3) was classified as pathogenic according to ACMG/AMP guidelines. The novel heterozygous frameshift c.65delC/p.Ala23Glnfs*3 truncates the protein in preproinsulin signal peptide (Liu et al., 2015). Different molecular mechanisms (and inheritance models) have been previously described for the phenotypes caused by INS variants, that is, processing defects in preproinsulin, proinsulin misfolding, ER stress, and impaired insulin binding to its receptor (Liu et al., 2015). Since the INS does not suffer from haploinsufficiency, as previously demonstrated experimentally using knockout mice (Leroux et al., 2001), the molecular pathogenesis related to the dominant inheritance model is attributed to the mutated protein cytotoxicity (Oyadomari et al., 2002; Ron, 2002; Wang et al., 1999), rather than insulin deficiency (recessive inheritance) (Garin et al., 2010; Raile et al., 2011). Our variant c.65delC/p.Ala23Glnfs*3 is predicted to undergo nonsense‐mediated decay (NMD) due to the premature termination codon (PTC) at amino acid position 26. Thus, no protein would be generated and there is no toxic gain of function effect. Among the dominant‐inheritance INS variants already reported, there is only one null type: a heterozygous de novo stop‐gain p.Tyr108Ter. The variant was identified in a permanent neonatal diabetes patient (Colombo et al., 2008). Functional evidence suggests that the truncated protein is translated, and NMD is not involved, leading to a proinsulin misfolding with ER stress, in addition to improper disulfide bonds with little or no insulin secretion. In this case, an NMD escaping was possible due to PTC position (within the last exon of a gene) (Kurosaki & Maquat, 2016; Nagy & Maquat, 1998). This would apply to any PTC occurring at amino acid position 43 onwards in the INS. Despite the ACMG/AMP classification, so far, there is no functional evidence to suggest that c.65delC/p.Ala23Glnfs*3 will escape NMD. Hence, the role of this INS variant in heterozygous state in diabetes phenotype remains to be determined.
4.8. ABCC8 | OMIM *600509
ABCC8 had the largest number of candidate changes, with eight in total, among which one was in a patient with another candidate variant (CEL). Only one of the eight ABCC8 variants could be classified as pathogenic, the missense variant p.Arg825Trp/p.R825W, which was previously associated with MODY in a French subject (Riveline et al., 2012), in a study demonstrating the impairment of its function (de Wet et al., 2008), and which almost completely segregated with the phenotype in our family. The only observed genotype/phenotype discrepancy was most likely due to incomplete penetrance, as already described (Klupa et al., 2009). It is important to note that the proband was diabetic for more than 30 years and always controlled his hyperglycemia with low doses of sulfonylurea. This good therapeutic response is classically observed in neonatal diabetes cases due to sulfonylurea receptor 1 (SUR1) (encoded by ABCC8) pathogenic variants (Rafiq et al., 2008). Finally, an atypical finding caught our attention. The ABCC8‐7 proband, a typical initial suspicion of MODY‐HNF1B (multiple bilateral renal cysts associated with DM), presented an already described uncertain significance variant in ABCC8. Curiously, the change p.Leu1147Arg/p.L1147R was reported in compound heterozygosity associated with neonatal diabetes (OMIM #606176) (Alkorta‐Aranburu et al., 2014) and persistent hyperinsulinemic hypoglycemia of infancy (PHHI) (OMIM #256450) (evolving to glucose intolerance) (Gussinyer et al., 2008). Our patient did not present any typical neonatal characteristics of the previously mentioned phenotypes. We believe that cosegregation analysis could help to clarify the role of this heterozygosis variant in this family. It should be noted, however, that great clinical heterogeneity has already been described with ABCC8 variants, in the most diverse forms of inheritance (Klupa et al., 2009). In addition, the presence of renal cysts may only be an incidental finding unrelated to DM.
4.9. KCNJ11 | OMIM #616329
The only KCNJ11 candidate variant p.Ala96Thr was classified as uncertain significance and reported twice, namely, in homozygosis (Melikyan et al., 2012) and in an apparent focal case (somatic loss of heterozygosity (LOH)) (Mohnike et al., 2014) associated with congenital hyperinsulinism. A detailed clinical report about relative carriers is not available. A heterozygous father was cited in the focal case study; however, hyperglycemia was not mentioned (only congenital hyperinsulinism discarded). In our family, six members were screened. Genotype–phenotype discrepancy was observed in one individual. The unaffected proband's granddaughter carried the variant without any glycemic alteration. As with ABCC8, also a subunit encoder of the pancreatic beta‐cell ATP‐sensitive channel, diverse phenotypes with distinct inheritance patterns have been previously associated with KCNJ11 (neonatal diabetes | OMIM #606176 (Gloyn et al., 2004), MODY (Bonnefond et al., 2012), and hyperinsulinemic hypoglycemia | OMIM #601820 (Thomas, Ye, & Lightner, 1996)). This genetic‐molecular spectrum often hinders the interpretation of mono/biallelic carriers, as in the KCNJ11 p.Arg34Cys variant and transient neonatal diabetes/congenital hyperinsulinism, which is subsequently clarified using expression studies (Snider et al., 2013).
5. CONCLUSIONS
The gene panel used in this study allowed for the simultaneous analysis of 11 genes frequently associated with MODY for the first time in a Brazilian cohort of genetically unclarified cases. This approach allowed for the detection of variants in genes not usually studied for the genetic diagnosis of MODY at most Brazilian medical centers, contributing to the identification of rare subtypes. As a result, the large percentage of local "MODY‐X" cases commonly reported was reduced.
The identification of rare MODY subtypes is important to expand on existing reports. Given the small number of subjects and the often‐great phenotypic variability (even intrafamily), establishing a typical clinical laboratory pattern, in contrast to the established effects of the most prevalent ones, is challenging.
Therefore, the use of multiloci genomic approaches (such as targeted sequencing or WES) is of fundamental importance, allowing for the genetic diagnosis of typical unclarified cases. Moreover, it allows for a better clinical, therapeutic, and prognostic characterization of rare phenotypes, thus contributing to our understanding of the basic pathogenesis of common diseases.
CONFLICT OF INTEREST
Nothing to Disclose—authors LSS, LAC, ADCR, PCF, RPD, AFR, LSW, SPS, MFV, FAP, GCPA, AGFPA, MGRT, WRBG, ACSJ, BH, AALJ, MN, MGT.
ETHICAL STATEMENT
Study approved by the Ethics Committee for Analysis of Research Projects (CAPPesq) of the School of Medicine, University of Sao Paulo (USP) (#70637).
Supporting information
ACKNOWLEDGMENTS
We show our appreciation to those who gently referred their patients, making this study possible: Berenice Bilharinho de Mendonça, Denise Reis Franco, Márcia Silva Queiroz, Ana Cristina, Carlos Alberto Longui, Carlos, Guilherme Lyra, Caroline de Gouveia Buff Passone, Denise Ludovico Costra de Castro, Everlayny Fiorot Costalonga, Gustavo Arantes Rosa Maciel, Heidi Lui Reinhardt, Hermelinda Pedrosa, Katia Camarano Nogueira, Laura Fachin Greca, Lindiane Gomes Crisostomo, Lize Vargas Ferreria, Luciana Valadares Ferreira, Luciani Renata Silveira de Carvalho, Luis Eduardo Procopio Calliari, Maria Adelaide Albergaria Pereira, Maria Elizabeth Rossi da Silva, Maria Lucia Cardillo Corrêa Giannella, Marise Vilas Boas Pescador, Patricia Barretto Mory Del Vecchio, Paula Pires, Priscilla Cukier, Ricardo Ayello Guerra, Ricardo Vessoni Perez, Rodrigo Bastos Fóscolo, Rosana Martins, Sharon Nina Admoni, Sonir Roberto Rauber Antonini and Tania Maria Bulcão Lousada Ferraz. We also thank Flavio Galvão Ribeiro for the assistance during Sanger sequencing analysis.
de Santana LS, Caetano LA, Costa‐Riquetto AD, et al. Targeted sequencing identifies novel variants in common and rare MODY genes. Mol Genet Genomic Med. 2019;7:e962 10.1002/mgg3.962
Funding information
Sao Paulo Research Foundation (FAPESP) grants #2013/19920‐2 and #2017/15365‐5 awarded to MGT, #2017/14703‐4 awarded to LSS, #2015/05123‐9 awarded to AFR, and #2013/02162‐8 awarded to Multiusuário SELA (School of Medicine, University of Sao Paulo—USP). National Council for Scientific and Technological Development (CNPq) grant #160044/2013‐8 awarded to RPD.
REFERENCES
- Alkorta‐Aranburu, G. , Carmody, D. , Cheng, Y. W. , Nelakuditi, V. , Ma, L. , Dickens, J. T. , … del Gaudio, D. (2014). Phenotypic heterogeneity in monogenic diabetes: The clinical and diagnostic utility of a gene panel‐based next‐generation sequencing approach. Molecular Genetics and Metabolism, 113(4), 315–320. 10.1016/j.ymgme.2014.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Diabetes Association (2017). 2. Classification and diagnosis of diabetes. Diabetes Care, 40(Supplement 1), S11–S24. 10.2337/dc17-S005 [DOI] [PubMed] [Google Scholar]
- Bellanne‐Chantelot, C. , Carette, C. , Riveline, J.‐P. , Valero, R. , Gautier, J.‐F. , Larger, E. , … Timsit, J. (2008). The type and the position of HNF1A mutation modulate age at diagnosis of diabetes in patients with maturity‐onset diabetes of the young (MODY)‐3. Diabetes, 57(2), 503–508. 10.2337/db07-0859. [DOI] [PubMed] [Google Scholar]
- Bellanné‐Chantelot, C. , Chauveau, D. , Gautier, J.‐F. , Dubois‐Laforgue, D. , Clauin, S. , Beaufils, S. , … Timsit, J. (2004). Clinical spectrum associated with hepatocyte nuclear factor‐1β mutations. Annals of Internal Medicine, 140(7), 510 10.7326/0003-4819-140-7-200404060-00009. [DOI] [PubMed] [Google Scholar]
- Bellanne‐Chantelot, C. , Clauin, S. , Chauveau, D. , Collin, P. , Daumont, M. , Douillard, C. , … Timsit, J. (2005). Large genomic rearrangements in the hepatocyte nuclear factor‐1 (tcf2) gene are the most frequent cause of maturity‐onset diabetes of the young type 5. Diabetes, 54(11), 3126–3132. 10.2337/diabetes.54.11.3126. [DOI] [PubMed] [Google Scholar]
- Bengtsson‐Ellmark, S. H. , Nilsson, J. , Orho‐Melander, M. , Dahlenborg, K. , Groop, L. , & Bjursell, G. (2004). Association between a polymorphism in the carboxyl ester lipase gene and serum cholesterol profile. European Journal of Human Genetics, 12(8), 627–632. 10.1038/sj.ejhg.5201204 [DOI] [PubMed] [Google Scholar]
- Blais, J. , Lavoie, S. B. , Giroux, S. , Bussières, J. , Lindsay, C. , Dionne, J. , … Rousseau, F. (2015). Risk of misdiagnosis due to allele dropout and false‐positive PCR artifacts in molecular diagnostics. The Journal of Molecular Diagnostics, 17(5), 505–514. 10.1016/j.jmoldx.2015.04.004 [DOI] [PubMed] [Google Scholar]
- Bonnefond, A. , Philippe, J. , Durand, E. , Dechaume, A. , Huyvaert, M. , Montagne, L. , … Froguel, P. (2012). Whole‐exome sequencing and high throughput genotyping identified KCNJ11 as the thirteenth MODY gene. PLoS ONE, 7(6), e37423 10.1371/journal.pone.0037423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnefond, A. , Philippe, J. , Durand, E. , Muller, J. , Saeed, S. , Arslan, M. , … Froguel, P. (2014). Highly Sensitive diagnosis of 43 monogenic forms of diabetes or obesity through one‐step PCR‐based enrichment in combination with next‐generation sequencing. Diabetes Care, 37(2), 460–467. 10.2337/dc13-0698 [DOI] [PubMed] [Google Scholar]
- Bowman, P. , Flanagan, S. E. , Edghill, E. L. , Damhuis, A. , Shepherd, M. H. , Paisey, R. , … Ellard, S. (2012). Heterozygous ABCC8 mutations are a cause of MODY. Diabetologia, 55(1), 123–127. 10.1007/s00125-011-2319-x [DOI] [PubMed] [Google Scholar]
- Caetano, L. A. , Santana, L. S. , Costa‐Riquetto, A. D. , Lerario, A. M. , Nery, M. , Nogueira, G. F. , … Teles, M. G. (2018). PDX1 ‐MODY and dorsal pancreatic agenesis: New phenotype of a rare disease. Clinical Genetics, 93(2), 382–386. 10.1111/cge.13044 [DOI] [PubMed] [Google Scholar]
- Caillol, N. , Pasqualini, E. , Mas, E. , Valette, A. , Verine, A. , & Lombardo, D. (1997). Pancreatic bile salt‐dependent lipase activity in serum of normolipidemic patients. Lipids, 32(11), 1147–1153. 10.1007/s11745-997-0147-4 [DOI] [PubMed] [Google Scholar]
- Cardoso, G. C. , de Oliveira, M. Z. , Paixão‐Côrtes, V. R. , Castilla, E. E. , & Schuler‐Faccini, L. (2019). Clusters of genetic diseases in Brazil. Journal of Community Genetics, 10(1), 121–128. 10.1007/s12687-018-0369-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chèvre, J.‐C. , Hani, E. H. , Boutin, P. , Vaxillaire, M. , Blanché, H. , Vionnet, N. , … Froguel, P. (1998). Mutation screening in 18 Caucasian families suggest the existence of other MODY genes. Diabetologia, 41(9), 1017–1023. 10.1007/s001250051025 [DOI] [PubMed] [Google Scholar]
- Colclough, K. , Saint‐Martin, C. , Timsit, J. , Ellard, S. , & Bellanné‐Chantelot, C. (2014). Clinical utility gene card for: Maturity‐onset diabetes of the young. European Journal of Human Genetics, 22(9), 1153–1153. 10.1038/ejhg.2014.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo, C. , Porzio, O. , Liu, M. , Massa, O. , Vasta, M. , Salardi, S. , … Barbetti, F. ; Early Onset Diabetes Study Group of the Italian Society of Pediatric Endocrinology and Diabetes (SIEDP) (2008). Seven mutations in the human insulin gene linked to permanent neonatal/infancy‐onset diabetes mellitus. The Journal of Clinical Investigation, 118(6), 2148–2156. 10.1172/JCI33777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wet, H. , Proks, P. , Lafond, M. , Aittoniemi, J. , Sansom, M. S. P. , Flanagan, S. E. , … Ashcroft, F. M. (2008). A mutation (R826W) in nucleotide‐binding domain 1 of ABCC8 reduces ATPase activity and causes transient neonatal diabetes. EMBO Reports, 9(7), 648–654. 10.1038/embor.2008.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirbilek, H. , Hatipoglu, N. , Gul, U. , Tatli, Z. U. , Ellard, S. , Flanagan, S. E. , … Kurtoglu, S. (2018). Permanent neonatal diabetes mellitus and neurological abnormalities due to a novel homozygous missense mutation in NEUROD1. Pediatric Diabetes, 19(5), 898–904. 10.1111/pedi.12669 [DOI] [PubMed] [Google Scholar]
- Dotto, R. P. , Santana, L. S. D. , Lindsey, S. C. , Caetano, L. A. , Franco, L. F. , Moisés, R. C. M. S. , … Reis, A. F. (2019). Searching for mutations in the HNF1B gene in a Brazilian cohort with renal cysts and hyperglycemia. Archives of Endocrinology and Metabolism, 63(3), 250–257. 10.20945/2359-3997000000138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edghill, E. L. (2005). Mutations in hepatocyte nuclear factor‐1 and their related phenotypes. Journal of Medical Genetics, 43(1), 84–90. 10.1136/jmg.2005.032854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edghill, E. L. , Bingham, C. , Ellard, S. , & Hattersley, A. T. (2006). Mutations in hepatocyte nuclear factor‐1beta and their related phenotypes. Journal of Medical Genetics, 43(1), 84–90. 10.1136/jmg.2005.032854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edghill, E. L. , Stals, K. , Oram, R. A. , Shepherd, M. H. , Hattersley, A. T. , & Ellard, S. (2013). HNF1B deletions in patients with young‐onset diabetes but no known renal disease. Diabetic Medicine, 30(1), 114–117. 10.1111/j.1464-5491.2012.03709.x [DOI] [PubMed] [Google Scholar]
- Ellard, S. , Bulman, M. P. , Frayling, T. M. , Allen, L. I. , Dronsfield, M. J. , Tack, C. J. , & Hattersley, A. T. (1999). Allelic drop‐out in exon 2 of the hepatocyte nuclear factor‐1alpha gene hinders the identification of mutations in three families with maturity‐onset diabetes of the young. Diabetes, 48(4), 921–923. 10.2337/diabetes.48.4.921 [DOI] [PubMed] [Google Scholar]
- Ellard, S. , Lango Allen, H. , De Franco, E. , Flanagan, S. E. , Hysenaj, G. , Colclough, K. , … Caswell, R. (2013). Improved genetic testing for monogenic diabetes using targeted next‐generation sequencing. Diabetologia, 56(9), 1958–1963. 10.1007/s00125-013-2962-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faguer, S. , Decramer, S. , Chassaing, N. , Bellanné‐Chantelot, C. , Calvas, P. , Beaufils, S. , … Chauveau, D. (2011). Diagnosis, management, and prognosis of HNF1B nephropathy in adulthood. Kidney International, 80(7), 768–776. 10.1038/ki.2011.225 [DOI] [PubMed] [Google Scholar]
- Fajans, S. S. , & Bell, G. I. (2011). MODY: History, genetics, pathophysiology, and clinical decision making. Diabetes Care, 34(8), 1878–1884. 10.2337/dc11-0035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajans, S. S. , Bell, G. I. , & Polonsky, K. S. (2001). Molecular mechanisms and clinical pathophysiology of maturity‐onset diabetes of the young. New England Journal of Medicine, 345(13), 971–980. 10.1056/NEJMra002168 [DOI] [PubMed] [Google Scholar]
- Flannick, J. , Beer, N. L. , Bick, A. G. , Agarwala, V. , Molnes, J. , Gupta, N. , … Altshuler, D. (2013). Assessing the phenotypic effects in the general population of rare variants in genes for a dominant Mendelian form of diabetes. Nature Genetics, 45(11), 1380–1385. 10.1038/ng.2794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frayling, T. M. , Evans, J. C. , Bulman, M. P. , Pearson, E. , Allen, L. , Owen, K. , … Hattersley, A. T. (2001). Beta‐cell genes and diabetes: Molecular and clinical characterization of mutations in transcription factors. Diabetes, 50(Supplement 1), S94–S100. 10.2337/diabetes.50.2007.S94 [DOI] [PubMed] [Google Scholar]
- Frayling, T. M. , Lindgren, C. M. , Chevre, J. C. , Menzel, S. , Wishart, M. , Benmezroua, Y. , … Vaxillaire, M. (2003). A genome‐wide scan in families with maturity‐onset diabetes of the young: evidence for further genetic heterogeneity. Diabetes, 52(3), 872–881. 10.2337/diabetes.52.3.872 [DOI] [PubMed] [Google Scholar]
- Froguel, P. , Zouali, H. , Vionnet, N. , Velho, G. , Vaxillaire, M. , Sun, F. , … Cohen, D. (1993). Familial hyperglycemia due to mutations in glucokinase – definition of a subtype of diabetes mellitus. New England Journal of Medicine, 328(10), 697–702. 10.1056/NEJM199303113281005. [DOI] [PubMed] [Google Scholar]
- Fujimura, F. K. , Northrup, H. , Beaudet, A. L. , & O’Brien, W. E. (1990). Genotyping errors with the polymerase chain reaction. The New England Journal of Medicine, 322(1), 61 10.1056/NEJM199001043220117 [DOI] [PubMed] [Google Scholar]
- Furuzawa, G. K. , Giuffrida, F. M. A. , Oliveira, C. S. V. , Chacra, A. R. , Dib, S. , & Reis, A. F. (2008). Low prevalence of MODY2 and MODY3 mutations in Brazilian individuals with clinical MODY phenotype. Diabetes Research and Clinical Practice, 81(3), e12–e14. 10.1016/j.diabres.2008.06.011 [DOI] [PubMed] [Google Scholar]
- García‐Herrero, C. M. , Galán, M. , Vincent, O. , Flández, B. , Gargallo, M. , Delgado‐Alvarez, E. , … Navas, M. A. (2007). Functional analysis of human glucokinase gene mutations causing MODY2: exploring the regulatory mechanisms of glucokinase activity. Diabetologia, 50(2), 325–333. 10.1007/s00125-006-0542-7. [DOI] [PubMed] [Google Scholar]
- Garin, I. , Edghill, E. L. , Akerman, I. , Rubio‐Cabezas, O. , Rica, I. , Locke, J. M. , … Hattersley, A. T. (2010). Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis. Proceedings of the National Academy of Sciences, 107(7), 3105–3110. 10.1073/pnas.0910533107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garin, I. , Rica, I. , Estalella, I. , Oyarzabal, M. , Rodríguez‐Rigual, M. , Pedro, J. I. S. , … de Nanclares, G. P. (2008). Haploinsufficiency at GCK gene is not a frequent event in MODY2 patients. Clinical Endocrinology, 68(6), 873–878. 10.1111/j.1365-2265.2008.03214.x. [DOI] [PubMed] [Google Scholar]
- Giuffrida, F. M. A. , Moises, R. S. , Weinert, L. S. , Calliari, L. E. , Manna, T. D. , Dotto, R. P. , … Reis, A. F. ; Brazilian Monogenic Diabetes Study Group (BRASMOD) (2017). Maturity‐onset diabetes of the young (MODY) in Brazil: Establishment of a national registry and appraisal of available genetic and clinical data. Diabetes Research and Clinical Practice, 123, 134–142. 10.1016/j.diabres.2016.10.017 [DOI] [PubMed] [Google Scholar]
- Gloyn, A. L. , Pearson, E. R. , Antcliff, J. F. , Proks, P. , Bruining, G. J. , Slingerland, A. S. , … Hattersley, A. T. (2004). Activating mutations in the gene encoding the ATP‐sensitive potassium‐channel subunit Kir6.2 and permanent neonatal diabetes. New England Journal of Medicine, 350(18), 1838–1849. 10.1056/NEJMoa032922 [DOI] [PubMed] [Google Scholar]
- Granberg, C. F. , Harrison, S. M. , Dajusta, D. , Zhang, S. , Hajarnis, S. , Igarashi, P. , & Baker, L. A. (2012). Genetic basis of prune belly syndrome: Screening for HNF1β gene. Journal of Urology, 187(1), 272–278. 10.1016/j.juro.2011.09.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gussinyer, M. , Clemente, M. , Cebrian, R. , Yeste, D. , Albisu, M. , & Carrascosa, A. (2008). Glucose intolerance and diabetes are observed in the long‐term follow‐up of nonpancreatectomized patients with persistent hyperinsulinemic hypoglycemia of infancy due to mutations in the ABCC8 gene. Diabetes Care, 31(6), 1257–1259. 10.2337/dc07-2059 [DOI] [PubMed] [Google Scholar]
- Hoskins, B. E. , Cramer, C. H. , Tasic, V. , Kehinde, E. O. , Ashraf, S. , Bogdanovic, R. , … Hildebrandt, F. (2007). Missense mutations in EYA1 and TCF2 are a rare cause of urinary tract malformations. Nephrology Dialysis Transplantation, 23(2), 777–779. 10.1093/ndt/gfm685 [DOI] [PubMed] [Google Scholar]
- Jarvik, G. P. , & Browning, B. L. (2016). Consideration of cosegregation in the pathogenicity classification of genomic variants. American Journal of Human Genetics, 98(6), 1077–1081. 10.1016/j.ajhg.2016.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen, C. T. , Dubé, J. B. , Loyzer, M. N. , MacDonald, A. , Carter, D. E. , McIntyre, A. D. , … Hegele, R. A. (2014). LipidSeq: a next‐generation clinical resequencing panel for monogenic dyslipidemias. Journal of Lipid Research, 55(4), 765–772. 10.1194/jlr.D045963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klupa, T. , Kowalska, I. , Wyka, K. , Skupien, J. , Patch, A.‐M. , Flanagan, S. E. , … Malecki, M. T. (2009). Mutations in the ABCC8 (SUR1 subunit of the K ATP channel) gene are associated with a variable clinical phenotype. Clinical Endocrinology, 71(3), 358–362. 10.1111/j.1365-2265.2008.03478.x [DOI] [PubMed] [Google Scholar]
- Kurosaki, T. , & Maquat, L. E. (2016). Nonsense‐mediated mRNA decay in humans at a glance. Journal of Cell Science, 129(3), 461–467. 10.1242/jcs.181008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam, C. , & Mak, C. M. (2013). Allele dropout caused by a non‐primer‐site SNV affecting PCR amplification — A call for next‐generation primer design algorithm. Clinica Chimica Acta, 421, 208–212. 10.1016/j.cca.2013.03.014 [DOI] [PubMed] [Google Scholar]
- Lek, M. , Karczewski, K. J. , Minikel, E. V. , Samocha, K. E. , Banks, E. , Fennell, T. , … MacArthur, D. G. (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroux, L. , Desbois, P. , Lamotte, L. , Duvilli, B. , Cordonnier, N. , Jackerott, M. , … Joshi, R. J. (2001). Compensatory responses in mice carrying a null mutation for Ins1 or Ins2. Diabetes, 50(Supplement 1), S150–S153. 10.2337/diabetes.50.2007.S150 [DOI] [PubMed] [Google Scholar]
- Liu, M. , Sun, J. , Cui, J. , Chen, W. , Guo, H. , Barbetti, F. , & Arvan, P. (2015). INS‐gene mutations: From genetics and beta cell biology to clinical disease. Molecular Aspects of Medicine, 42(4), 3–18. 10.1016/j.mam.2014.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorini, R. , Klersy, C. , D’Annunzio, G. , Massa, O. , Minuto, N. , Iafusco, D. , … Barbetti, F. (2009). Maturity‐onset diabetes of the young in children with incidental hyperglycemia: A multicenter Italian study of 172 families. Diabetes Care, 32(10), 1864–1866. 10.2337/dc08-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeyski, K. , Lidberg, U. , Bjursell, G. , & Nilsson, J. (1998). Structure and organization of the human carboxyl ester lipase locus. Mammalian Genome, 9(4), 334–338. 10.1007/s003359900762 [DOI] [PubMed] [Google Scholar]
- Malecki, M. T. , Yang, Y. , Antonellis, A. , Curtis, S. , Warram, J. H. , & Krolewski, A. S. (1999). Identification of new mutations in the hepatocyte nuclear factor 4alpha gene among families with early onset Type 2 diabetes mellitus. Diabetic Medicine: A Journal of the British Diabetic Association, 16(3), 193–200. [DOI] [PubMed] [Google Scholar]
- Manchester, N. G. R. L. (2019). SNPCheck 3. Retrieved from https://genetools.org/SNPCheck/snpcheck.htm. [Google Scholar]
- Maraschin, J. D. F. , Kannengiesser, C. , Murussi, N. , Campagnolo, N. , Canani, L. H. , Gross, J. L. , … Silveiro, S. P. (2008). HNF1α mutations are present in half of clinically defined MODY patients in South‐Brazilian individuals. Arquivos Brasileiros De Endocrinologia & Metabologia, 52(8), 1326–1331. 10.1590/S0004-27302008000800020 [DOI] [PubMed] [Google Scholar]
- McDonald, T. J. , Colclough, K. , Brown, R. , Shields, B. , Shepherd, M. , Bingley, P. , … Ellard, S. (2011). Islet autoantibodies can discriminate maturity‐onset diabetes of the young (MODY) from Type 1 diabetes. Diabetic Medicine, 28(9), 1028–1033. 10.1111/j.1464-5491.2011.03287.x [DOI] [PubMed] [Google Scholar]
- Melikyan, M. , Kareva, M. , Petraykina, E. , Volkov, I. , Averyanova, J. , Gurevich, L. , … Christesen, H. (2012). Genotype‐phenotype associations in children with congenital hyperinsulinism. In Horm Res, 78(suppl 1), p. 216 10.1159/000343183 [DOI] [Google Scholar]
- Meur, G. , Simon, A. , Harun, N. , Virally, M. , Dechaume, A. , Bonnefond, A. , … Vaxillaire, M. (2010). Insulin gene mutations resulting in early‐onset diabetes: marked differences in clinical presentation, metabolic status, and pathogenic effect through endoplasmic reticulum retention. Diabetes, 59(3), 653–661. 10.2337/db09-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohnike, K. , Wieland, I. , Barthlen, W. , Vogelgesang, S. , Empting, S. , Mohnike, W. , … Zenker, M. (2014). Clinical and genetic evaluation of patients with K ATP channel mutations from the german registry for congenital hyperinsulinism. Hormone Research in Paediatrics, 81(3), 156–168. 10.1159/000356905 [DOI] [PubMed] [Google Scholar]
- Moises, R. S. , Reis, A. F. , Morel, V. , Chacra, A. R. , Dib, S. A. , Bellanne‐Chantelot, C. , & Velho, G. (2001). Prevalence of maturity‐onset diabetes of the young mutations in brazilian families with autosomal‐ dominant early‐onset type 2 diabetes. Diabetes Care, 24(4), 786–788. 10.2337/diacare.24.4.786 [DOI] [PubMed] [Google Scholar]
- Nagy, E. , & Maquat, L. E. (1998). A rule for termination‐codon position within intron‐containing genes: When nonsense affects RNA abundance. Trends in Biochemical Sciences, 23(6), 198–199. 10.1016/S0968-0004(98)01208-0 [DOI] [PubMed] [Google Scholar]
- Najmi, L. A. , Aukrust, I. , Flannick, J. , Molnes, J. , Burtt, N. , Molven, A. , … Njølstad, P. R. (2017). Functional investigations of HNF1A identify rare variants as risk factors for Type 2 diabetes in the general population. Diabetes, 66(2), 335–346. 10.2337/db16-0460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naslavsky, M. S. , Yamamoto, G. L. , de Almeida, T. F. , Ezquina, S. A. M. , Sunaga, D. Y. , Pho, N. , … Zatz, M. (2017). Exomic variants of an elderly cohort of Brazilians in the ABraOM database. Human Mutation, 38(7), 751–763. 10.1002/humu.23220 [DOI] [PubMed] [Google Scholar]
- Oram, R. A. , Edghill, E. L. , Blackman, J. , Taylor, M. J. O. , Kay, T. , Flanagan, S. E. , … Bingham, C. (2010). Mutations in the hepatocyte nuclear factor‐1β (HNF1B) gene are common with combined uterine and renal malformations but are not found with isolated uterine malformations. American Journal of Obstetrics and Gynecology, 203(4), 364.e1–364.e5. 10.1016/j.ajog.2010.05.022 [DOI] [PubMed] [Google Scholar]
- Oyadomari, S. , Koizumi, A. , Takeda, K. , Gotoh, T. , Akira, S. , Araki, E. , & Mori, M. (2002). Targeted disruption of the Chop gene delays endoplasmic reticulum stress–mediated diabetes. Journal of Clinical Investigation, 109(4), 525–532. 10.1172/JCI14550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson, E. R. , Boj, S. F. , Steele, A. M. , Barrett, T. , Stals, K. , Shield, J. P. , … Hattersley, A. T. (2007). Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLoS Medicine, 4(4), e118 10.1371/journal.pmed.0040118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson, E. R. , Starkey, B. J. , Powell, R. J. , Gribble, F. M. , Clark, P. M. , & Hattersley, A. T. (2003). Genetic cause of hyperglycaemia and response to treatment in diabetes. The Lancet, 362(9392), 1275–1281. 10.1016/S0140-6736(03)14571-0 [DOI] [PubMed] [Google Scholar]
- Pezzilli, S. , Ludovico, O. , Biagini, T. , Mercuri, L. , Alberico, F. , Lauricella, E. , … Prudente, S. (2018). Insights from molecular characterization of adult patients of families with multigenerational diabetes. Diabetes, 67(1), 137–145. 10.2337/db17-0867 [DOI] [PubMed] [Google Scholar]
- Prudente, S. , Jungtrakoon, P. , Marucci, A. , Ludovico, O. , Buranasupkajorn, P. , Mazza, T. , … Doria, A. (2015). Loss‐of‐function mutations in APPL1 in familial diabetes mellitus. The American Journal of Human Genetics, 97(1), 177–185. 10.1016/j.ajhg.2015.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruhova, S. , Ek, J. , Lebl, J. , Sumnik, Z. , Saudek, F. , Andel, M. , … Hansen, T. (2003). Genetic epidemiology of MODY in the Czech republic: new mutations in the MODY genes HNF‐4alpha, GCK and HNF‐1alpha. Diabetologia, 46(2), 291–295. 10.1007/s00125-002-1010-7. [DOI] [PubMed] [Google Scholar]
- Quinlan, A. R. , & Marth, G. T. (2007). Primer‐site SNPs mask mutations. Nature Methods, 4(3), 192–192. 10.1038/nmeth0307-192 [DOI] [PubMed] [Google Scholar]
- Raeder, H. , Bjorkhaug, L. , Johansson, S. , Mangseth, K. , Sagen, J. V. , Hunting, A. , … Njolstad, P. R. (2006). A Hepatocyte nuclear factor‐4 gene (HNF4A) P2 promoter haplotype linked with late‐onset diabetes: Studies of HNF4A variants in the Norwegian MODY Registry. Diabetes, 55(6), 1899–1903. 10.2337/db05-1677 [DOI] [PubMed] [Google Scholar]
- Ræder, H. , Johansson, S. , Holm, P. I. , Haldorsen, I. S. , Mas, E. , Sbarra, V. , … Njølstad, P. R. (2006). Mutations in the CEL VNTR cause a syndrome of diabetes and pancreatic exocrine dysfunction. Nature Genetics, 38(1), 54–62. 10.1038/ng1708 [DOI] [PubMed] [Google Scholar]
- Rafiq, M. , Flanagan, S. E. , Patch, A.‐M. , Shields, B. M. , Ellard, S. , & Hattersley, A. T. (2008). Effective treatment with oral sulfonylureas in patients with diabetes due to sulfonylurea receptor 1 (SUR1) mutations. Diabetes Care, 31(2), 204–209. 10.2337/dc07-1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raile, K. , O’Connell, M. , Galler, A. , Werther, G. , Kühnen, P. , Krude, H. , & Blankenstein, O. (2011). Diabetes caused by insulin gene (INS) deletion: Clinical characteristics of homozygous and heterozygous individuals. European Journal of Endocrinology, 165(2), 255–260. 10.1530/EJE-11-0208 [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–423. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riveline, J.‐P. , Rousseau, E. , Reznik, Y. , Fetita, S. , Philippe, J. , Dechaume, A. , … Vaxillaire, M. (2012). Clinical and metabolic features of adult‐onset diabetes caused by ABCC8 mutations. Diabetes Care, 35(2), 248–251. 10.2337/dc11-1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, J. T. , Thorvaldsdóttir, H. , Winckler, W. , Guttman, M. , Lander, E. S. , Getz, G. , & Mesirov, J. P. (2011). Integrative genomics viewer. Nature Biotechnology, 29(1), 24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron, D. (2002). Proteotoxicity in the endoplasmic reticulum: Lessons from the Akita diabetic mouse. Journal of Clinical Investigation, 109(4), 443–445. 10.1172/JCI15020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio‐Cabezas, O. , Minton, J. A. L. , Kantor, I. , Williams, D. , Ellard, S. , & Hattersley, A. T. (2010). Homozygous mutations in NEUROD1 are responsible for a novel syndrome of permanent neonatal diabetes and neurological abnormalities. Diabetes, 59(9), 2326–2331. 10.2337/db10-0011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger, F. , & Coulson, A. R. (1975). A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. Journal of Molecular Biology, 94(3), 441–448. 10.1016/0022-2836(75)90213-2 [DOI] [PubMed] [Google Scholar]
- Santana, L. S. , Caetano, L. A. , Costa‐Riquetto, A. D. , Quedas, E. P. S. , Nery, M. , Collett‐Solberg, P. , … Teles, M. G. (2017). Clinical application of ACMG‐AMP guidelines in HNF1A and GCK variants in a cohort of MODY families. Clinical Genetics, 92(4), 388–396. 10.1111/cge.12988 [DOI] [PubMed] [Google Scholar]
- Sanyoura, M. , Philipson, L. H. , & Naylor, R. (2018). Monogenic diabetes in children and adolescents: recognition and treatment options. Current Diabetes Reports, 18(8), 58 10.1007/s11892-018-1024-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze, E. , Bettendorf, M. , Maser‐gluth, C. , Decker, M. , & Schwabe, U. (1998). Allele‐dropout using pcr‐based diagnosis for the splicing mutation in intron‐2 of the CYP21B‐gene: Successful amplification with a TAQ/PWO‐polymerase mixture. Endocrine Research, 24(3–4), 637–641. 10.3109/07435809809032662 [DOI] [PubMed] [Google Scholar]
- Shepherd, M. , Sparkes, A. C. , & Hattersley, A. T. (2001). Genetic testing in maturity onset diabetes of the young (MODY): A new challenge for the diabetic clinic. Practical Diabetes International, 18(1), 16–21. 10.1002/pdi.108 [DOI] [PubMed] [Google Scholar]
- Shields, B. M. , Hicks, S. , Shepherd, M. H. , Colclough, K. , Hattersley, A. T. , & Ellard, S. (2010). Maturity‐onset diabetes of the young (MODY): How many cases are we missing? Diabetologia, 53(12), 2504–2508. 10.1007/s00125-010-1799-4 [DOI] [PubMed] [Google Scholar]
- Snider, K. E. , Becker, S. , Boyajian, L. , Shyng, S.‐L. , MacMullen, C. , Hughes, N. , … Ganguly, A. (2013). Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. The Journal of Clinical Endocrinology & Metabolism, 98(2), E355–E363. 10.1210/jc.2012-2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somerville, M. J. , Sprysak, K. A. , Hicks, M. , Elyas, B. G. , & Vicen‐Wyhony, L. (1999). An HFE intronic variant promotes misdiagnosis of hereditary hemochromatosis. The American Journal of Human Genetics, 65(3), 924–926. 10.1086/302550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoffers, D. A. , Ferrer, J. , Clarke, W. L. , & Habener, J. F. (1997). Early‐onset type‐II diabetes mellitus (MODY4) linked to IPF1. Nature Genetics, 17(2), 138–139. 10.1038/ng1097-138. [DOI] [PubMed] [Google Scholar]
- Szopa, M. , Ludwig‐Gałęzowska, A. , Radkowski, P. , Skupień, J. , Zapała, B. , Płatek, T. , … Małecki, M. T. (2015). Genetic testing for monogenic diabetes using targeted next‐generation sequencing in patients with maturity‐onset diabetes of the young. Polish Archives of Internal Medicine, 125(11), 845–851. 10.20452/pamw.3164. [DOI] [PubMed] [Google Scholar]
- Thomas, P. , Ye, Y. , & Lightner, E. (1996). Mutation of the pancreatic islet inward rectifier Kir6.2 also leads to familial persistent hyperinsulinemic hypoglycemia of infancy. Human Molecular Genetics, 5(11), 1809–1812. 10.1093/hmg/5.11.1809 [DOI] [PubMed] [Google Scholar]
- Torsvik, J. , Johansson, S. , Johansen, A. , Ek, J. , Minton, J. , Ræder, H. , … Njølstad, P. R. (2010). Mutations in the VNTR of the carboxyl‐ester lipase gene (CEL) are a rare cause of monogenic diabetes. Human Genetics, 127(1), 55–64. 10.1007/s00439-009-0740-8 [DOI] [PubMed] [Google Scholar]
- Vaxillaire, M. , Dechaume, A. , Busiah, K. , Cave, H. , Pereira, S. , Scharfmann, R. , … Polak, M. (2007). New ABCC8 mutations in relapsing neonatal diabetes and clinical features. Diabetes, 56(6), 1737–1741. 10.2337/db06-1540. [DOI] [PubMed] [Google Scholar]
- Vaxillaire, M. , & Froguel, P. (2008). Monogenic diabetes in the young, pharmacogenetics and relevance to multifactorial forms of type 2 diabetes. Endocrine Reviews, 29(3), 254–264. 10.1210/er.2007-0024 [DOI] [PubMed] [Google Scholar]
- Wallis, Y. , Payne, S. , Mcanulty, C. , Bodmer, D. , Sister‐, E. , Robertson, K. , … Deans, Z. (2013). Practice guidelines for the evaluation of pathogenicity and the reporting of sequence variants in clinical molecular genetics. Association for Clinical Genetic Science and the Dutch Society of Clinical Genetic Laboratory Specialists, (September). [Google Scholar]
- Wang, J. , Takeuchi, T. , Tanaka, S. , Kubo, S.‐K. , Kayo, T. , Lu, D. , … Izumi, T. (1999). A mutation in the insulin 2 gene induces diabetes with severe pancreatic β‐cell dysfunction in the Mody mouse. Journal of Clinical Investigation, 103(1), 27–37. 10.1172/JCI4431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert, L. S. , Silveiro, S. P. , Giuffrida, F. M. A. , Cunha, V. T. , Bulcão, C. , Calliari, L. E. , … Reis, A. F. (2014). Three unreported glucokinase (GCK) missense mutations detected in the screening of thirty‐two Brazilian kindreds for GCK and HNF1A‐MODY. Diabetes Research and Clinical Practice, 106(2), e44–e48. 10.1016/j.diabres.2014.08.006 [DOI] [PubMed] [Google Scholar]
- Wenzel, J. J. , Rossmann, H. , Fottner, C. , Neuwirth, S. , Neukirch, C. , Lohse, P. , … Lackner, K. J. (2009). Identification and prevention of genotyping errors caused by G‐quadruplex‐ and i‐motif‐like sequences. Clinical Chemistry, 55(7), 1361–1371. 10.1373/clinchem.2008.118661 [DOI] [PubMed] [Google Scholar]
- Ziemssen, F. , Bellanné‐Chantelot, C. , Osterhoff, M. , Schatz, H. , & Pfeiffer, A. F. H. (2002). ‐ to: Lindner T, Cockburn BN, Bell GI (1999) Molecular genetics of MODY in Germany. Diabetologia 42: 121–123. Diabetologia, 45(2), 286–287. 10.1007/s00125-001-0738-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials